
Preventing Brain Injury in the Preterm Infant—Current Controversies and Potential Therapies

 , ,
, ,
Abstract
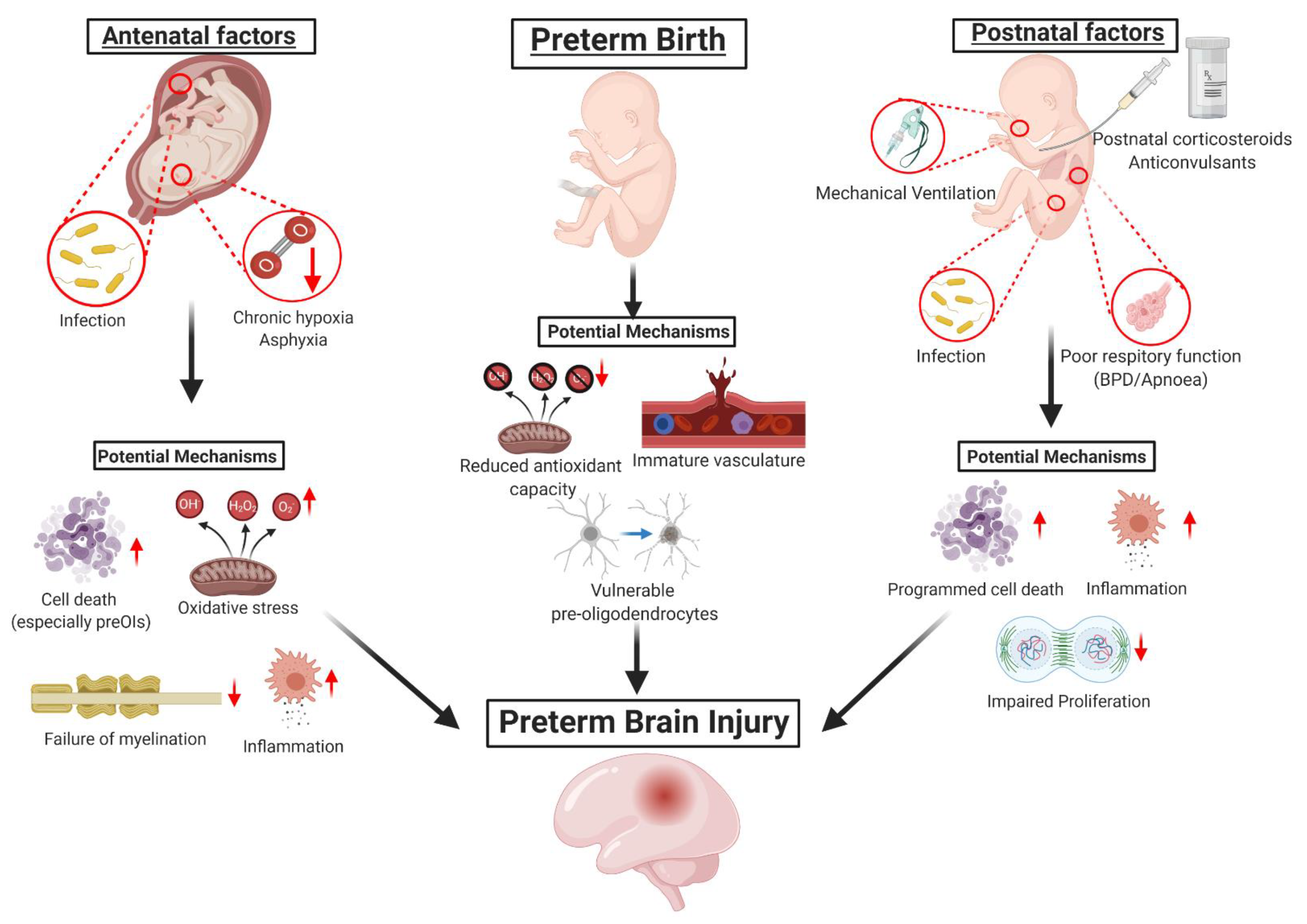
1. Introduction to Preterm Brain Injury
1.1. Brain Injury Associated with Hypoxia-Ischemia
1.2. Brain Injury Associated with Infection and/or Inflammation
1.3. Vulnerabilities of the Preterm Brain to Injury
2. Neurological Outcomes
The Quest to Develop Treatment Strategies to Reduce Preterm Brain Injury
3. Treatments Currently in Clinical Use for Preterm Infants
3.1. Antenatal Corticosteroids
3.2. Postnatal Glucocorticoids
3.3. Magnesium Sulfate (MgSO4)
3.4. Anticonvulsants
4. Potential Neuroprotective Treatments that Have Been Investigated in Clinical and Preclinical Trials for Use in Preterm Infants
4.1. Erythropoietin
4.2. Therapeutic Hypothermia
5. Potential Neuroprotective Treatments Showing Promise in Preclinical Studies of Preterm Brain Injury
5.1. Melatonin
5.2. Vitamin D Supplementation
5.3. Cell-Based Therapies
5.3.1. Human Amnion Epithelial Cells (hAECs)
5.3.2. Umbilical Cord Blood (UCB) Cells
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blencowe, H.; Cousens, S.; Oestergaard, M.Z.; Chou, D.; Moller, A.-B.; Narwal, R.; Adler, A.; Garcia, C.V.; Rohde, S.; Say, L.; et al. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: A systematic analysis and implications. Lancet 2012, 379, 2162–2172. [Google Scholar] [CrossRef]
- Laptook, A.R. Birth Asphyxia and Hypoxic-Ischemic Brain Injury in the Preterm Infant. Clin. Perinatol. 2016, 43, 529–545. [Google Scholar] [CrossRef]
- Gale, C.; Statnikov, Y.; Jawad, S.; Uthaya, S.N.; Modi, N. Neonatal brain injuries in England: Population-based incidence derived from routinely recorded clinical data held in the National Neonatal Research Database. Arch. Dis. Child. Fetal Neonatal Ed. 2017, 103, F301–F306. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, L.; Kang, B.; Zhu, T.; Li, Y.; Zhao, F.; Qu, Y.; Mu, D. Association between perinatal hypoxic-ischemia and periventricular leukomalacia in preterm infants: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0184993. [Google Scholar] [CrossRef]
- Low, J.A.; Killen, H.; Derrick, E.J. Antepartum fetal asphyxia in the preterm pregnancy. Am. J. Obstet. Gynecol. 2003, 188, 461–465. [Google Scholar] [CrossRef]
- Chalak, L.F.; Rollins, N.; Morriss, M.C.; Brion, L.P.; Heyne, R.; Sánchez, P.J. Perinatal Acidosis and Hypoxic-Ischemic Encephalopathy in Preterm Infants of 33 to 35 Weeks’ Gestation. J. Pediatr. 2012, 160, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.W.; Walsh, W.F. Hypoxic-ischemic encephalopathy in preterm infants. J. Neonatal-Perinat. Med. 2010, 3, 277–284. [Google Scholar] [CrossRef]
- Salhab, W.A.; Perlman, J.M. Severe fetal acidemia and subsequent neonatal encephalopathy in the larger premature infant. Pediatr. Neurol. 2005, 32, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Manuck, T.A.; Rice, M.M.; Bailit, J.L.; Grobman, W.A.; Reddy, U.M.; Wapner, R.J.; Thorp, J.M.; Caritis, S.N.; Prasad, M.; Tita, A.T.; et al. Preterm neonatal morbidity and mortality by gestational age: A contemporary cohort. Am. J. Obstet. Gynecol. 2016, 215, 103.e1–103.e14. [Google Scholar] [CrossRef]
- Ehrenkranz, R.A. Estimated fetal weights versus birth weights: Should the reference intrauterine growth curves based on birth weights be retired? Arch. Dis. Child. Fetal Neonatal Ed. 2007, 92, F161–F162. [Google Scholar] [CrossRef] [PubMed]
- Streimish, I.G.; Ehrenkranz, R.A.; Allred, E.N.; O’Shea, T.M.; Kuban, K.C.; Paneth, N.; Leviton, A. Birth weight- and fetal weight-growth restriction: Impact on neurodevelopment. Early Hum. Dev. 2012, 88, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L.; Huppi, P.S.; Mallard, C. The consequences of fetal growth restriction on brain structure and neurodevelopmental outcome. J. Physiol. 2016, 594, 807–823. [Google Scholar] [CrossRef]
- Hayes, B.C.; McGarvey, C.; Mulvany, S.; Kennedy, J.; Geary, M.P.; Matthews, T.G.; King, M.D. A case-control study of hypoxic-ischemic encephalopathy in newborn infants at >36 weeks gestation. Am. J. Obstet. Gynecol. 2013, 209, 29.e1–29.e19. [Google Scholar] [CrossRef]
- Takahashi, N.; Nishida, H.; Arai, T.; Kaneda, Y. Abnormal cardiac histology in severe intrauterine growth retardation infants. Pediatr. Int. 1995, 37, 341–346. [Google Scholar] [CrossRef]
- Schmidt, B.; Roberts, R.S.; Anderson, P.J.; Asztalos, E.V.; Costantini, L.; Davis, P.G.; Dewey, D.; D’Ilario, J.; Doyle, L.W.; Grunau, R.E.; et al. Academic Performance, Motor Function, and Behavior 11 Years After Neonatal Caffeine Citrate Therapy for Apnea of Prematurity: An 11-Year Follow-up of the CAP Randomized Clinical Trial. JAMA Pediatr. 2017, 171, 564–572. [Google Scholar] [CrossRef]
- Gallini, F.; Coppola, M.; De Rose, D.U.; Maggio, L.; Arena, R.; Romano, V.; Cota, F.; Ricci, D.; Romeo, D.M.; Mercuri, E.M.; et al. Neurodevelopmental outcomes in very preterm infants: The role of severity of Bronchopulmonary Dysplasia. Early Hum. Dev. 2021, 152, 105275. [Google Scholar] [CrossRef] [PubMed]
- Galinsky, R.; Polglase, G.R.; Hooper, S.B.; Black, M.J.; Moss, T.J.M. The Consequences of Chorioamnionitis: Preterm Birth and Effects on Development. J. Pregnancy 2013, 2013, 1–11. [Google Scholar] [CrossRef]
- Kim, C.J.; Romero, R.; Chaemsaithong, P.; Chaiyasit, N.; Yoon, B.H.; Kim, Y.M. Acute chorioamnionitis and funisitis: Definition, pathologic features, and clinical significance. Am. J. Obstet. Gynecol. 2015, 213, S29–S52. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Espinoza, J.; Gonçalves, L.F.; Kusanovic, J.P.; Friel, L.A.; Nien, J.K. Inflammation in preterm and term labour and delivery. Semin. Fetal Neonatal Med. 2006, 11, 317–326. [Google Scholar] [CrossRef]
- Arayici, S.; Simsek, G.K.; Öncel, M.Y.; Eras, Z.; Canpolat, F.E.; Oguz, S.S.; Uras, N.; Zergeroglu, S.; Dilmen, U. The effect of histological chorioamnionitis on the short-term outcome of preterm infants ≤32 weeks: A single-center study. J. Matern.-Fetal Neonatal Med. 2014, 27, 1129–1133. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, Q.; Wang, Q.-X.; Lu, J.-Y. Contribution of Histologic Chorioamnionitis and Fetal Inflammatory Response Syndrome to Increased Risk of Brain Injury in Infants with Preterm Premature Rupture of Membranes. Pediatr. Neurol. 2016, 61, 94–98.e1. [Google Scholar] [CrossRef]
- Korzeniewski, S.J.; Romero, R.; Cortez, J.; Pappas, A.; Schwartz, A.G.; Kim, C.J.; Kim, J.-S.; Kim, Y.M.; Yoon, B.H.; Chaiworapongsa, T.; et al. A “multi-hit” model of neonatal white matter injury: Cumulative contributions of chronic placental inflammation, acute fetal inflammation and postnatal inflammatory events. J. Périnat. Med. 2014, 42, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Soraisham, A.S.; Trevenen, C.; Wood, S.; Singhal, N.; Sauve, R. Histological chorioamnionitis and neurodevelopmental outcome in preterm infants. J. Perinatol. 2013, 33, 70–75. [Google Scholar] [CrossRef]
- Yap, V.; Perlman, J.M. Mechanisms of brain injury in newborn infants associated with the fetal inflammatory response syndrome. Semin. Fetal Neonatal Med. 2020, 25, 101110. [Google Scholar] [CrossRef]
- Malaeb, S.; Dammann, O. Fetal Inflammatory Response and Brain Injury in the Preterm Newborn. J. Child Neurol. 2009, 24, 1119–1126. [Google Scholar] [CrossRef]
- Buser, J.R.; Segovia, K.N.; Dean, J.M.; Nelson, K.; Beardsley, D.; Gong, X.; Luo, N.L.; Ren, J.; Wan, Y.; Riddle, A.; et al. Timing of Appearance of Late Oligodendrocyte Progenitors Coincides with Enhanced Susceptibility of Preterm Rabbit Cerebral White Matter to Hypoxia-Ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 1053–1065. [Google Scholar] [CrossRef]
- Back, S.A.; Riddle, A.; McClure, M.M. Maturation-Dependent Vulnerability of Perinatal White Matter in Premature Birth. Stroke 2007, 38, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Levine, J.M.; Volpe, J.J.; Kinney, H.C. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J. Neurosci. 2001, 21, 1302–1312. [Google Scholar] [CrossRef]
- Back, S.A.; Han, B.H.; Luo, N.L.; Chricton, C.A.; Xanthoudakis, S.; Tam, J.; Arvin, K.L.; Holtzman, D.M. Selective Vulnerability of Late Oligodendrocyte Progenitors to Hypoxia–Ischemia. J. Neurosci. 2002, 22, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Volpe, J.J.; Kinney, H.C. Arrested Oligodendrocyte Lineage Progression during Human Cerebral White Matter Development: Dissociation between the Timing of Progenitor Differentiation and Myelinogenesis. J. Neuropathol. Exp. Neurol. 2002, 61, 197–211. [Google Scholar] [CrossRef]
- Riddle, A.; Luo, N.L.; Manese, M.; Beardsley, D.J.; Green, L.; Rorvik, D.A.; Kelly, K.A.; Barlow, C.H.; Kelly, J.J.; Hohimer, A.R.; et al. Spatial Heterogeneity in Oligodendrocyte Lineage Maturation and Not Cerebral Blood Flow Predicts Fetal Ovine Periventricular White Matter Injury. J. Neurosci. 2006, 26, 3045–3055. [Google Scholar] [CrossRef]
- Back, S.A.; Gan, X.; Li, Y.; Rosenberg, P.A.; Volpe, J.J. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J. Neurosci. 1998, 18, 6241–6253. [Google Scholar] [CrossRef] [PubMed]
- Gitto, E.; Pellegrino, S.; Gitto, P.; Barberi, I.; Reiter, R.J. Oxidative stress of the newborn in the pre- and postnatal period and the clinical utility of melatonin. J. Pineal Res. 2009, 46, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.L.; Wallace, E.M.; Walker, D.W. Antioxidant Therapies: A Potential Role in Perinatal Medicine. Neuroendocrinology 2012, 96, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Ballabh, P.; Braun, A.; Nedergaard, M. Anatomic Analysis of Blood Vessels in Germinal Matrix, Cerebral Cortex, and White Matter in Developing Infants. Pediatr. Res. 2004, 56, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Ballabh, P. Intraventricular Hemorrhage in Premature Infants: Mechanism of Disease. Pediatr. Res. 2010, 67, 1–8. [Google Scholar] [CrossRef]
- Özek, E.; Kersin, S.G. Intraventricular hemorrhage in preterm babies. Turk. Pediatri. Ars. 2020, 55, 215–221. [Google Scholar]
- Adams-Chapman, I.; Hansen, N.I.; Stoll, B.J.; Higgins, R. for the NICHD Research Network Neurodevelopmental Outcome of Extremely Low Birth Weight Infants with Posthemorrhagic Hydrocephalus Requiring Shunt Insertion. Pediatrics 2008, 121, e1167–e1177. [Google Scholar] [CrossRef]
- Bassan, H.; Limperopoulos, C.; Visconti, K.; Mayer, D.L.; Feldman, H.A.; Avery, L.; Benson, C.B.; Stewart, J.; Ringer, S.A.; Soul, J.S.; et al. Neurodevelopmental Outcome in Survivors of Periventricular Hemorrhagic Infarction. Pediatrics 2007, 120, 785–792. [Google Scholar] [CrossRef]
- Oskoui, M.; Coutinho, F.; Dykeman, J.; Jetté, N.; Pringsheim, T. An update on the prevalence of cerebral palsy: A systematic review and meta-analysis. Dev. Med. Child Neurol. 2013, 55, 509–519. [Google Scholar] [CrossRef]
- Arpino, C.; Compagnone, E.; Montanaro, M.L.; Cacciatore, D.; De Luca, A.; Cerulli, A.; Di Girolamo, S.; Curatolo, P. Preterm birth and neurodevelopmental outcome: A review. Childs Nerv. Syst. 2010, 26, 1139–1149. [Google Scholar] [CrossRef]
- Counsell, S.J.; Shen, Y.; Boardman, J.P.; Larkman, D.J.; Kapellou, O.; Ward, P.; Allsop, J.M.; Cowan, F.M.; Hajnal, J.V.; Edwards, A.D.; et al. Axial and Radial Diffusivity in Preterm Infants Who Have Diffuse White Matter Changes on Magnetic Resonance Imaging at Term-Equivalent Age. Pediatrics 2006, 117, 376–386. [Google Scholar] [CrossRef]
- Inder, T.E.; Warfield, S.K.; Wang, H.; Hüppi, P.S.; Volpe, J.J. Abnormal Cerebral Structure Is Present at Term in Premature Infants. Pediatrics 2005, 115, 286–294. [Google Scholar] [CrossRef]
- Iwata, S.; Nakamura, T.; Hizume, E.; Kihara, H.; Takashima, S.; Matsuishi, T.; Iwata, O. Qualitative Brain MRI at Term and Cognitive Outcomes at 9 Years After Very Preterm Birth. Pediatrics 2012, 129, e1138–e1147. [Google Scholar] [CrossRef]
- Dyet, L.E.; Kennea, N.; Counsell, S.J.; Maalouf, E.F.; Ajayi-Obe, M.; Duggan, P.J.; Harrison, M.; Allsop, J.M.; Hajnal, J.; Herlihy, A.H.; et al. Natural History of Brain Lesions in Extremely Preterm Infants Studied with Serial Magnetic Resonance Imaging from Birth and Neurodevelopmental Assessment. Pediatrics 2006, 118, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Galinsky, R.; Lear, C.A.; Dean, J.M.; Wassink, G.; Dhillon, S.K.; Fraser, M.; Davidson, J.O.; Bennet, L.; Gunn, A.J. Complex interactions between hypoxia-ischemia and inflammation in preterm brain injury. Dev. Med. Child Neurol. 2018, 60, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Chollat, C.; Enser, M.; Houivet, E.; Provost, D.; Bénichou, J.; Marpeau, L.; Marret, S. School-Age Outcomes following a Randomized Controlled Trial of Magnesium Sulfate for Neuroprotection of Preterm Infants. J. Pediatr. 2014, 165, 398–400.e3. [Google Scholar] [CrossRef]
- Doyle, L.W.; Anderson, P.J.; Haslam, R.; Lee, K.J.; Crowther, C. School-age Outcomes of Very Preterm Infants After Antenatal Treatment with Magnesium Sulfate vs Placebo. JAMA 2014, 312, 1105. [Google Scholar] [CrossRef]
- Khanna, A.; Walcott, B.; Kahle, K. Limitations of Current GABA Agonists in Neonatal Seizures: Toward GABA Modulation Via the Targeting of Neuronal Cl− Transport. Front. Neurol. 2013, 4, 78. [Google Scholar] [CrossRef]
- Foix-L’Helias, L.; Marret, S.; Ancel, P.; Marchand, L.; Arnaud, C.; Fresson, J.; Picaud, J.-C.; Rozé, J.-C.; Theret, B.; Burguet, A.; et al. Impact of the use of antenatal corticosteroids on mortality, cerebral lesions and 5-year neurodevelopmental outcomes of very preterm infants: The EPIPAGE cohort study. BJOG 2008, 115, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.; Brown, J.; Medley, N.; Dalziel, S.R. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst. Rev. 2017, 3, CD004454. [Google Scholar] [CrossRef]
- Salokorpi, T.; Sajaniemi, N.; Hällback, H.; Kari, A.; Rita, H.; Wendt, L. Randomized study of the effect of antenatal dexamethasone on growth and development of premature children at the corrected age of 2 years. Acta Paediatr. 1997, 86, 294–298. [Google Scholar] [CrossRef]
- Crowther, C.A.; McKinlay, C.J.D.; Middleton, P.; Harding, J.E. Repeat doses of prenatal corticosteroids for women at risk of preterm birth for improving neonatal health outcomes. Cochrane Database Syst. Rev. 2015, 2015, CD003935. [Google Scholar] [CrossRef]
- Crowther, C.A.; Ashwood, P.; Andersen, C.C.; Middleton, P.F.; Tran, T.; Doyle, L.W.; Robinson, J.S.; Harding, J.E.; Ball, V.; Holst, C.; et al. Maternal intramuscular dexamethasone versus betamethasone before preterm birth (ASTEROID): A multicentre, double-blind, randomised controlled trial. Lancet Child Adolesc. Health 2019, 3, 769–780. [Google Scholar] [CrossRef]
- Ciapponi, A.; Klein, K.; Colaci, D.; Althabe, F.; Belizán, J.M.; Deegan, A.; Veroniki, A.A.; Florez, I.D. Dexamethasone vs. betamethasone for preterm birth: A systematic review and network meta-analysis. Am. J. Obstet. Gynecol. MFM 2021, 100312. [Google Scholar] [CrossRef]
- Antonow-Schlorke, I.; Helgert, A.; Gey, C.; Coksaygan, T.; Schubert, H.; Nathanielsz, P.W.; Witte, O.W.; Schwab, M. Adverse Effects of Antenatal Glucocorticoids on Cerebral Myelination in Sheep. Obstet. Gynecol. 2009, 113, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Quaedackers, J.S.L.T.; George, S.A.; Gunn, A.J.; Bennet, L. Maternal dexamethasone and EEG hyperactivity in preterm fetal sheep. J. Physiol. 2011, 589, 3823–3835. [Google Scholar] [CrossRef]
- Parikh, N.A.; Lasky, R.E.; Kennedy, K.A.; McDavid, G.; Tyson, J.E. Perinatal Factors and Regional Brain Volume Abnormalities at Term in a Cohort of Extremely Low Birth Weight Infants. PLoS ONE 2013, 8, e62804. [Google Scholar] [CrossRef]
- Parikh, N.A.; Kennedy, K.A.; Lasky, R.E.; McDavid, G.E.; Tyson, J.E. Pilot Randomized Trial of Hydrocortisone in Ventilator-Dependent Extremely Preterm Infants: Effects on Regional Brain Volumes. J. Pediatr. 2013, 162, 685–690.e1. [Google Scholar] [CrossRef] [PubMed]
- Koome, M.E.; Davidson, J.O.; Drury, P.P.; Mathai, S.; Booth, L.C.; Gunn, A.J.; Bennet, L. Antenatal Dexamethasone after Asphyxia Increases Neural Injury in Preterm Fetal Sheep. PLoS ONE 2013, 8, e77480. [Google Scholar] [CrossRef]
- Lear, C.A.; Koome, M.E.; Davidson, J.O.; Drury, P.P.; Quaedackers, J.S.; Galinsky, R.; Gunn, A.J.; Bennet, L. The effects of dexamethasone on post-asphyxial cerebral oxygenation in the preterm fetal sheep. J. Physiol. 2014, 592, 5493–5505. [Google Scholar] [CrossRef]
- Lear, C.; Davidson, J.O.; Mackay, G.R.; Drury, P.P.; Galinsky, R.; Quaedackers, J.S.; Gunn, A.J.; Bennet, L. Antenatal dexamethasone before asphyxia promotes cystic neural injury in preterm fetal sheep by inducing hyperglycemia. J. Cereb. Blood Flow Metab. 2018, 38, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.P.; Inder, T.E.; Hüppi, P.S.; Warfield, S.K.; Zientara, G.P.; Kikinis, R.; Jolesz, F.A.; Volpe, J.J. Impaired cerebral cortical gray matter growth after treatment with dexamethasone for neonatal chronic lung disease. Pediatrics 2001, 107, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Parikh, N.; Lasky, R.E.; Kennedy, K.A.; Moya, F.R.; Hochhauser, L.; Romo, S.; Tyson, J.E. Postnatal Dexamethasone Therapy and Cerebral Tissue Volumes in Extremely Low Birth Weight Infants. Pediatrics 2007, 119, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.W.; Cheong, J.L.; Ehrenkranz, R.A.; Halliday, H.L. Early (<8 days) systemic postnatal corticosteroids for prevention of bronchopulmonary dysplasia in preterm infants. Cochrane Database Syst. Rev. 2017, 10, CD001146. [Google Scholar] [PubMed]
- Doyle, L.W.; Cheong, J.L.; Ehrenkranz, R.A.; Halliday, H.L. Late (>7 days) systemic postnatal corticosteroids for prevention of bronchopulmonary dysplasia in preterm infants. Cochrane Database Syst. Rev. 2017, 10, CD001145. [Google Scholar] [CrossRef]
- Onland, W.; Offringa, M.; De Jaegere, A.P.; Van Kaam, A.H. Finding the Optimal Postnatal Dexamethasone Regimen for Preterm Infants at Risk of Bronchopulmonary Dysplasia: A Systematic Review of Placebo-Controlled Trials. Pediatrics 2009, 123, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.; Crowther, C.; Middleton, P.; Marret, S. Antenatal Magnesium Sulfate and Neurologic Outcome in Preterm Infants: A Systematic Review. Obstet. Gynecol. 2009, 113, 1327–1333. [Google Scholar] [CrossRef]
- Conde-Agudelo, A.; Romero, R. Antenatal magnesium sulfate for the prevention of cerebral palsy in preterm infants less than 34 weeks’ gestation: A systematic review and metaanalysis. Am. J. Obstet. Gynecol. 2009, 200, 595–609. [Google Scholar] [CrossRef]
- Song, J.; Sun, H.; Xu, F.; Kang, W.; Gao, L.; Guo, J.; Zhang, Y.; Xia, L.; Wang, X.; Zhu, C. Recombinant human erythropoietin improves neurological outcomes in very preterm infants. Ann. Neurol. 2016, 80, 24–34. [Google Scholar] [CrossRef]
- Juul, S.E.; Comstock, B.A.; Wadhawan, R.; Mayock, D.E.; Courtney, S.E.; Robinson, T.; Ahmad, K.A.; Bendel-Stenzel, E.; Baserga, M.; LaGamma, E.F.; et al. A Randomized Trial of Erythropoietin for Neuroprotection in Preterm Infants. N. Engl. J. Med. 2020, 382, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Natalucci, G.; Latal, B.; Koller, B.; Rüegger, C.; Sick, B.; Held, L.; Bucher, H.U.; Fauchère, J.C.; The Swiss EPO Neuroprotection Trial Group. Effect of early prophylactic high-dose recombinant human erythropoietin in very preterm infants on neurodevelopmental outcome at 2 Years: A randomized clinical irial. JAMA 2016, 315, 2079–2085. [Google Scholar] [CrossRef] [PubMed]
- Natalucci, G.; Latal, B.; Koller, B.; Rüegger, C.; Sick, B.; Held, L.; Fauchère, J.-C. Swiss EPO Neuroprotection Trial Group Neurodevelopmental Outcomes at Age 5 Years After Prophylactic Early High-Dose Recombinant Human Erythropoietin for Neuroprotection in Very Preterm Infants. JAMA 2020, 324, 2324–2327. [Google Scholar] [CrossRef]
- Rao, R.; Trivedi, S.; Vesoulis, Z.A.; Liao, S.M.; Smyser, C.D.; Mathur, A.M. Safety and Short-Term Outcomes of Therapeutic Hypothermia in Preterm Neonates 34-35 Weeks Gestational Age with Hypoxic-Ischemic Encephalopathy. J. Pediatr. 2017, 183, 37–42. [Google Scholar] [CrossRef]
- Herrera, T.I.; Edwards, L.; Malcolm, W.F.; Smith, P.B.; Fisher, K.A.; Pizoli, C.; Gustafson, K.E.; Goldstein, R.F.; Cotten, C.M.; Goldberg, R.N.; et al. Outcomes of preterm infants treated with hypothermia for hypoxic-ischemic encephalopathy. Early Hum. Dev. 2018, 125, 1–7. [Google Scholar] [CrossRef]
- Costeloe, K.L.; Hennessy, E.M.; Haider, S.; Stacey, F.; Marlow, N.; Draper, E.S. Short term outcomes after extreme preterm birth in England: Comparison of two birth cohorts in 1995 and 2006 (the EPICure studies). BMJ 2012, 345, e7976. [Google Scholar] [CrossRef]
- Doyle, L.W.; Ranganathan, S.; Cheong, J.L. Ventilation in Preterm Infants and Lung Function at 8 Years. N. Engl. J. Med. 2017, 377, 1601–1602. [Google Scholar] [CrossRef] [PubMed]
- Yoder, B.A.; Harrison, M.; Clark, R.H. Time-Related Changes in Steroid Use and Bronchopulmonary Dysplasia in Preterm Infants. Pediatrics 2009, 124, 673–679. [Google Scholar] [CrossRef]
- Flagel, S.B.; Vázquez, D.M.; Watson, S.J.; Neal, C.R. Effects of tapering neonatal dexamethasone on rat growth, neurodevelopment, and stress response. Am. J. Physiol. Integr. Comp. Physiol. 2002, 282, R55–R63. [Google Scholar] [CrossRef]
- Gramsbergen, A.; Mulder, E.J.H. The Influence of Betamethasone and Dexamethasone on Motor Development in Young Rats. Pediatr. Res. 1998, 44, 105–110. [Google Scholar] [CrossRef]
- Ferguson, S.; Holson, R.R. Neonatal Dexamethasone on Day 7 Causes Mild Hyperactivity and Cerebellar Stunting. Neurotoxicology Teratol. 1999, 21, 71–76. [Google Scholar] [CrossRef]
- Brummelte, S.; Pawluski, J.L.; Galea, L.A. High post-partum levels of corticosterone given to dams influence postnatal hippocampal cell proliferation and behavior of offspring: A model of post-partum stress and possible depression. Horm. Behav. 2006, 50, 370–382. [Google Scholar] [CrossRef]
- Haynes, L.; Griffiths, M.; Hyde, R.; Barber, D.; Mitchell, I. Dexamethasone induces limited apoptosis and extensive sublethal damage to specific subregions of the striatum and hippocampus: Implications for mood disorders. Neuroscience 2001, 104, 57–69. [Google Scholar] [CrossRef]
- Baud, O.; Foix-L’Helias, L.; Kaminski, M.; Audibert, F.; Jarreau, P.-H.; Papiernik, E.; Huon, C.; Lepercq, J.; Dehan, M.; Lacaze-Masmonteil, T. Antenatal Glucocorticoid Treatment and Cystic Periventricular Leukomalacia in Very Premature Infants. N. Engl. J. Med. 1999, 341, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Shinwell, E.S.; Karplus, M.; Reich, D.; Weintraub, Z.; Blazer, S.; Bader, D.; Yurman, S.; Dolfin, T.; Kogan, A.; Dollberg, S.; et al. Early postnatal dexamethasone treatment and increased incidence of cerebral palsy. Arch. Dis. Child. Fetal Neonatal Ed. 2000, 83, F177–F181. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.W.; Davis, P.G.; Morley, C.J.; McPhee, A.; Carlin, J.B.; the DART Study Investigators. Outcome at 2 Years of Age of Infants from the DART Study: A Multicenter, International, Randomized, Controlled Trial of Low-Dose Dexamethasone. Pediatrics 2007, 119, 716–721. [Google Scholar] [CrossRef]
- Doyle, L.W.; Davis, P.G.; Morley, C.J.; McPhee, A.; Carlin, J.B. Low-Dose Dexamethasone Facilitates Extubation among Chronically Ventilator-Dependent Infants: A Multicenter, International, Randomized, Controlled Trial. Pediatrics 2006, 117, 75–83. [Google Scholar] [CrossRef]
- Yates, N.J.; Feindel, K.W.; Mehnert, A.; Beare, R.; Quick, S.; Blache, D.; Pillow, J.J.; Hunt, R.W. Ex Vivo MRI Analytical Methods and Brain Pathology in Preterm Lambs Treated with Postnatal Dexamethasone. Brain Sci. 2020, 10, 211. [Google Scholar] [CrossRef]
- Walsh, M.C.; Morris, B.H.; Wrage, L.A.; Vohr, B.R.; Poole, W.K.; Tyson, J.E.; Wright, L.L.; Ehrenkranz, R.A.; Stoll, B.J.; Fanaroff, A.A. Extremely Low Birthweight Neonates with Protracted Ventilation: Mortality and 18-Month Neurodevelopmental Outcomes. J. Pediatr. 2005, 146, 798–804. [Google Scholar] [CrossRef]
- Doyle, L.W.; Halliday, H.L.; Ehrenkranz, R.A.; Davis, P.G.; Sinclair, J.C. Impact of Postnatal Systemic Corticosteroids on Mortality and Cerebral Palsy in Preterm Infants: Effect Modification by Risk for Chronic Lung Disease. Pediatrics 2005, 115, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.W.; Halliday, H.L.; Ehrenkranz, R.A.; Davis, P.G.; Sinclair, J.C. An Update on the Impact of Postnatal Systemic Corticosteroids on Mortality and Cerebral Palsy in Preterm Infants: Effect Modification by Risk of Bronchopulmonary Dysplasia. J. Pediatr. 2014, 165, 1258–1260. [Google Scholar] [CrossRef] [PubMed]
- Galinsky, R.; Bennet, L.; Groenendaal, F.; Lear, C.A.; Tan, S.; Van Bel, F.; Juul, S.E.; Robertson, N.J.; Mallard, C.; Gunn, A.J. Magnesium Is Not Consistently Neuroprotective for Perinatal Hypoxia-Ischemia in Term-Equivalent Models in Preclinical Studies: A Systematic Review. Dev. Neurosci. 2014, 36, 73–82. [Google Scholar] [CrossRef]
- Galinsky, R.; Dean, J.M.; Lingam, I.; Robertson, N.J.; Mallard, C.; Bennet, L.; Gunn, A.J. A Systematic Review of Magnesium Sulfate for Perinatal Neuroprotection: What Have We Learnt from the Past Decade? Front. Neurol. 2020, 11, 449. [Google Scholar] [CrossRef]
- Cahill, A.G.; Caughey, A.B. Magnesium for neuroprophylaxis: Fact or fiction? Am. J. Obstet. Gynecol. 2009, 200, 590–594. [Google Scholar] [CrossRef]
- Doyle, L.W. Antenatal magnesium sulfate and neuroprotection. Curr. Opin. Pediatr. 2012, 24, 154–159. [Google Scholar] [CrossRef]
- Annegers, J.F.; Hauser, W.A.; Lee, J.R.-J.; Rocca, W.A. Incidence of Acute Symptomatic Seizures in Rochester, Minnesota, 1935-1984. Epilepsia 1995, 36, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Cowan, L.D. The epidemiology of the epilepsies in children. Ment. Retard. Dev. Disabil. Res. Rev. 2002, 8, 171–181. [Google Scholar] [CrossRef]
- Ronen, G.M.; Penney, S.; Andrews, W. The epidemiology of clinical neonatal seizures in Newfoundland: A population-based study. J. Pediatr. 1999, 134, 71–75. [Google Scholar] [CrossRef]
- Silverstein, F.S.; Jensen, F.E. Neonatal seizures. Ann. Neurol. 2007, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Ronen, G.M.; Buckley, D.; Penney, S.; Streiner, D.L. Long-term prognosis in children with neonatal seizures: A population-based study. Neurology 2007, 69, 1816–1822. [Google Scholar] [CrossRef] [PubMed]
- Glass, H.C.; Kan, J.; Bonifacio, S.L.; Ferriero, D.M. Neonatal Seizures: Treatment Practices among Term and Preterm Infants. Pediatr. Neurol. 2012, 46, 111–115. [Google Scholar] [CrossRef]
- Booth, D.; Evans, D.J. Anticonvulsants for neonates with seizures. Cochrane Database Syst. Rev. 2004, 2004, CD004218. [Google Scholar] [CrossRef]
- Evans, D.J.; Levene, M.I.; Tsakmakis, M. Anticonvulsants for preventing mortality and morbidity in full term newborns with perinatal asphyxia. Cochrane Database Syst. Rev. 2007, CD001240. [Google Scholar] [CrossRef]
- Bittigau, P.; Sifringer, M.; Genz, K.; Reith, E.; Pospischil, D.; Govindarajalu, S.; Dzietko, M.; Pesditschek, S.; Mai, I.; Dikranian, K.; et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc. Natl. Acad. Sci. USA 2002, 99, 15089–15094. [Google Scholar] [CrossRef]
- Endesfelder, S.; Weichelt, U.; Schiller, C.; Winter, K.; Von Haefen, C.; Bührer, C. Caffeine Protects Against Anticonvulsant-Induced Impaired Neurogenesis in the Developing Rat Brain. Neurotox. Res. 2018, 34, 173–187. [Google Scholar] [CrossRef]
- Forcelli, P.A.; Janssen, M.J.; Vicini, S.; Gale, K. Neonatal exposure to antiepileptic drugs disrupts striatal synaptic development. Ann. Neurol. 2012, 72, 363–372. [Google Scholar] [CrossRef]
- Kim, J.; Kondratyev, A.; Gale, K. Antiepileptic Drug-Induced Neuronal Cell Death in the Immature Brain: Effects of Carbamazepine, Topiramate, and Levetiracetam as Monotherapy versus Polytherapy. J. Pharmacol. Exp. Ther. 2007, 323, 165–173. [Google Scholar] [CrossRef]
- Khazipov, R.; Khalilov, I.; Tyzio, R.; Morozova, E.; Ben-Ari, Y.; Holmes, G.L. Developmental changes in GABAergic actions and seizure susceptibility in the rat hippocampus. Eur. J. Neurosci. 2004, 19, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Payne, J.A.; Rivera, C.; Voipio, J.; Kaila, K. Cation–chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003, 26, 199–206. [Google Scholar] [CrossRef]
- Pond, B.B.; Berglund, K.; Kuner, T.; Feng, G.; Augustine, G.J.; Schwartz-Bloom, R.D. The Chloride Transporter Na+-K+-Cl− Cotransporter Isoform-1 Contributes to Intracellular Chloride Increases after In Vitro Ischemia. J. Neurosci. 2006, 26, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Jantzie, L.L.; Getsy, P.M.; Denson, J.L.; Firl, D.J.; Maxwell, J.R.; Rogers, D.A.; Wilson, C.G.; Robinson, S. Prenatal Hypoxia–Ischemia Induces Abnormalities in CA3 Microstructure, Potassium Chloride Co-Transporter 2 Expression and Inhibitory Tone. Front. Cell. Neurosci. 2015, 9, 347. [Google Scholar] [CrossRef]
- Kahle, K.T.; Staley, K.J. The bumetanide-sensitive Na-K-2Cl cotransporter NKCC1 as a potential target of a novel mechanism-based treatment strategy for neonatal seizures. Neurosurg. Focus 2008, 25, E22. [Google Scholar] [CrossRef] [PubMed]
- Pressler, R.M.; Boylan, G.B.; Marlow, N.; Blennow, M.; Chiron, C.; Cross, J.H.; De Vries, L.S.; Hallberg, B.; Hellström-Westas, L.; Jullien, V.; et al. Bumetanide for the treatment of seizures in newborn babies with hypoxic ischaemic encephalopathy (NEMO): An open-label, dose finding, and feasibility phase 1/2 trial. Lancet Neurol. 2015, 14, 469–477. [Google Scholar] [CrossRef]
- Jantzie, L.; Getsy, P.M.; Firl, D.J.; Wilson, C.; Miller, R.; Robinson, S. Erythropoietin attenuates loss of potassium chloride co-transporters following prenatal brain injury. Mol. Cell. Neurosci. 2014, 61, 152–162. [Google Scholar] [CrossRef]
- Ohlsson, A.; Aher, S.M. Early erythropoietin for preventing red blood cell transfusion in preterm and/or low birth weight infants. Cochrane Database Syst. Rev. 2006, CD004863. [Google Scholar] [CrossRef]
- Juul, S.E.; Comstock, B.A.; Heagerty, P.J.; Mayock, D.E.; Goodman, A.M.; Hauge, S.; González, F.; Wu, Y.W. High-Dose Erythropoietin for Asphyxia and Encephalopathy (HEAL): A Randomized Controlled Trial—Background, Aims, and Study Protocol. Neonatology 2018, 113, 331–338. [Google Scholar] [CrossRef]
- Kellert, B.A.; McPherson, R.J.; Juul, S.E. A Comparison of High-Dose Recombinant Erythropoietin Treatment Regimens in Brain-Injured Neonatal Rats. Pediatr. Res. 2007, 61, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Wassink, G.; Davidson, J.O.; Dhillon, S.K.; Fraser, M.; Galinsky, R.; Bennet, L.; Gunn, A.J. Partial white and grey matter protection with prolonged infusion of recombinant human erythropoietin after asphyxia in preterm fetal sheep. J. Cereb. Blood Flow Metab. 2017, 37, 1080–1094. [Google Scholar] [CrossRef]
- Iwai, M.; Stetler, R.A.; Xing, J.; Hu, X.; Gao, Y.; Zhang, W.; Chen, J.; Cao, G. Enhanced Oligodendrogenesis and Recovery of Neurological Function by Erythropoietin After Neonatal Hypoxic/Ischemic Brain Injury. Stroke 2010, 41, 1032–1037. [Google Scholar] [CrossRef]
- Robinson, S.; Corbett, C.J.; Winer, J.L.; Chan, L.A.; Maxwell, J.R.; Anstine, C.V.; Yellowhair, T.R.; Andrews, N.A.; Yang, Y.; Sillerud, L.O.; et al. Neonatal erythropoietin mitigates impaired gait, social interaction and diffusion tensor imaging abnormalities in a rat model of prenatal brain injury. Exp. Neurol. 2018, 302, 1–13. [Google Scholar] [CrossRef]
- Kumral, A.; Baskin, H.; Yesilirmak, D.C.; Ergur, B.U.; Aykan, S.; Genc, S.; Genc, K.; Yilmaz, O.; Tugyan, K.; Giray, O.; et al. Erythropoietin Attenuates Lipopolysaccharide-Induced White Matter Injury in the Neonatal Rat Brain. Neonatology 2007, 92, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Rees, S.; Hale, N.; De Matteo, R.; Cardamone, L.; Tolcos, M.; Loeliger, M.; Mackintosh, A.; Shields, A.; Probyn, M.; Greenwood, D.; et al. Erythropoietin Is Neuroprotective in a Preterm Ovine Model of Endotoxin-Induced Brain Injury. J. Neuropathol. Exp. Neurol. 2010, 69, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.S.; Reibel, N.J.; Bührer, C.; Dame, C. Prophylactic Early Erythropoietin for Neuroprotection in Preterm Infants: A Meta-analysis. Pediatrics 2017, 139, e20164317. [Google Scholar] [CrossRef]
- Wu, Y.W.; Bauer, L.A.; Ballard, R.A.; Ferriero, D.M.; Glidden, D.V.; Mayock, D.E.; Chang, T.; Durand, D.J.; Song, D.; Bonifacio, S.L.; et al. Erythropoietin for Neuroprotection in Neonatal Encephalopathy: Safety and Pharmacokinetics. Pediatrics 2012, 130, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Baserga, M.; Beachy, J.C.; Roberts, J.K.; Ward, R.M.; DiGeronimo, R.J.; Walsh, W.F.; Ohls, R.K.; Anderson, J.; Mayock, D.E.; Juul, S.E.; et al. Darbepoetin administration to neonates undergoing cooling for encephalopathy: A safety and pharmacokinetic trial. Pediatr. Res. 2015, 78, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Ohls, R.K.; Kamath-Rayne, B.D.; Christensen, R.D.; Wiedmeier, S.E.; Rosenberg, A.; Fuller, J.; Lacy, C.B.; Roohi, M.; Lambert, D.K.; Burnett, J.J.; et al. Cognitive Outcomes of Preterm Infants Randomized to Darbepoetin, Erythropoietin, or Placebo. Pediatrics 2014, 133, 1023–1030. [Google Scholar] [CrossRef]
- Wassink, G.; Gunn, E.R.; Drury, P.P.; Bennet, L.; Gunn, A.J. The mechanisms and treatment of asphyxial encephalopathy. Front. Neurosci. 2014, 8, 40. [Google Scholar] [CrossRef]
- Jacobs, S.E.; Berg, M.; Hunt, R.; Tarnow-Mordi, W.O.; Inder, T.E.; Davis, P.G. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst. Rev. 2013, 1, CD003311. [Google Scholar] [CrossRef]
- Drury, P.P.; Gunn, E.R.; Bennet, L.; Gunn, A.J. Mechanisms of Hypothermic Neuroprotection. Clin. Perinatol. 2014, 41, 161–175. [Google Scholar] [CrossRef]
- Guillet, R.; Edwards, A.D.; on behalf of the CoolCap Trial Group; Thoresen, M.; Ferriero, D.M.; Gluckman, P.D.; Whitelaw, A.; Gunn, A.J. Seven- to eight-year follow-up of the CoolCap trial of head cooling for neonatal encephalopathy. Pediatr. Res. 2011, 71, 205–209. [Google Scholar] [CrossRef]
- Azzopardi, D.; Strohm, B.; Marlow, N.; Brocklehurst, P.; Deierl, A.; Eddama, O.; Goodwin, J.; Halliday, H.L.; Juszczak, E.; Kapellou, O.; et al. Effects of Hypothermia for Perinatal Asphyxia on Childhood Outcomes. N. Engl. J. Med. 2014, 371, 140–149. [Google Scholar] [CrossRef]
- Shankaran, S.; Pappas, A.; McDonald, S.A.; Vohr, B.R.; Hintz, S.R.; Yolton, K.; Gustafson, K.E.; Leach, T.M.; Green, C.; Bara, R.; et al. Childhood Outcomes after Hypothermia for Neonatal Encephalopathy. N. Engl. J. Med. 2012, 366, 2085–2092. [Google Scholar] [CrossRef]
- Bennet, L.; Roelfsema, V.; George, S.; Dean, J.M.; Emerald, B.S.; Gunn, A.J. The effect of cerebral hypothermia on white and grey matter injury induced by severe hypoxia in preterm fetal sheep. J. Physiol. 2007, 578, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Wassink, G.; Barrett, R.D.; Davidson, J.O.; Bennet, L.; Galinsky, R.; Dragunow, M.; Gunn, A.J. Hypothermic Neuroprotection Is Associated with Recovery of Spectral Edge Frequency After Asphyxia in Preterm Fetal Sheep. Stroke 2015, 46, 585–587. [Google Scholar] [CrossRef] [PubMed]
- Gunn, A.J.; Gunn, T.R.; Gunning, M.I.; Williams, C.E.; Gluckman, P.D. Neuroprotection with Prolonged Head Cooling Started Before Postischemic Seizures in Fetal Sheep. Pediatrics 1998, 102, 1098–1106. [Google Scholar] [CrossRef]
- Gunn, A.J.; Bennet, L. Brain Cooling for Preterm Infants. Clin. Perinatol. 2008, 35, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.J.; Tan, S.; Groenendaal, F.; Van Bel, F.; Juul, S.E.; Bennet, L.; Derrick, M.; Back, S.A.; Valdez, R.C.; Northington, F.; et al. Which Neuroprotective Agents are Ready for Bench to Bedside Translation in the Newborn Infant? J. Pediatr. 2012, 160, 544–552.e4. [Google Scholar] [CrossRef]
- Drury, P.P.; Davidson, J.O.; Bennet, L.; Booth, L.C.; Tan, S.; Fraser, M.; Heuij, L.G.V.D.; Gunn, A.J. Partial Neural Protection with Prophylactic Low-Dose Melatonin after Asphyxia in Preterm Fetal Sheep. J. Cereb. Blood Flow Metab. 2014, 34, 126–135. [Google Scholar] [CrossRef]
- Yawno, T.; Mahen, M.; Miller, S.L.; Fahey, M.C.; Jenkin, G.; Miller, S.L. The Beneficial Effects of Melatonin Administration Following Hypoxia-Ischemia in Preterm Fetal Sheep. Front. Cell. Neurosci. 2017, 11, 296. [Google Scholar] [CrossRef]
- Robertson, N.J.; Lingam, I.; Meehan, C.; Martinello, K.A.; Avdic-Belltheus, A.; Stein, L.; Tachrount, M.; Price, D.; Sokolska, M.; Bainbridge, A.; et al. High-Dose Melatonin and Ethanol Excipient Combined with Therapeutic Hypothermia in a Newborn Piglet Asphyxia Model. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Robertson, N.J.; Faulkner, S.; Fleiss, B.; Bainbridge, A.; Andorka, C.; Price, D.; Powell, E.; Lecky-Thompson, L.; Thei, L.; Chandrasekaran, M.; et al. Melatonin augments hypothermic neuroprotection in a perinatal asphyxia model. Brain 2013, 136, 90–105. [Google Scholar] [CrossRef]
- Robertson, N.J.; Martinello, K.; Lingam, I.; Avdic-Belltheus, A.; Meehan, C.; Alonso-Alconada, D.; Ragab, S.; Bainbridge, A.; Sokolska, M.; Tachrount, M.; et al. Melatonin as an adjunct to therapeutic hypothermia in a piglet model of neonatal encephalopathy: A translational study. Neurobiol. Dis. 2019, 121, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Song, H.; Dash, O.; Park, M.; Shin, N.E.; McLane, M.W.; Lei, J.; Hwang, J.Y.; Burd, I. Administration of melatonin for prevention of preterm birth and fetal brain injury associated with premature birth in a mouse model. Am. J. Reprod. Immunol. 2019, 82, e13151. [Google Scholar] [CrossRef]
- Shimizu, S.; Simon, R.P.; Graham, S.H. Dimethylsulfoxide (DMSO) treatment reduces infarction volume after permanent focal cerebral ischemia in rats. Neurosci. Lett. 1997, 239, 125–127. [Google Scholar] [CrossRef]
- Di Giorgio, A.M.; Hou, Y.; Zhao, X.; Zhang, B.; Lyeth, B.G.; Russell, M.J. Dimethyl sulfoxide provides neuroprotection in a traumatic brain injury model. Restor. Neurol. Neurosci. 2008, 26, 501–507. [Google Scholar] [PubMed]
- Gitto, E.; Reiter, R.J.; Sabatino, G.; Buonocore, G.; Romeo, C.; Gitto, P.; Bugge, C.; Trimarchi, G.; Barberi, I. Correlation among cytokines, bronchopulmonary dysplasia and modality of ventilation in preterm newborns: Improvement with melatonin treatment. J. Pineal Res. 2005, 39, 287–293. [Google Scholar] [CrossRef] [PubMed]
- El-Gendy, F.M.; El-Hawy, M.A.; Hassan, M.G. Beneficial effect of melatonin in the treatment of neonatal sepsis. J. Matern. Fetal Neonatal Med. 2018, 31, 2299–2303. [Google Scholar] [CrossRef]
- Aly, H.; Elmahdy, H.; Eldib, M.; Rowisha, M.; Awny, M.; Elgohary, T.; Elbatch, M.; Hamisa, M.; El-Mashad, A.-R. Melatonin use for neuroprotection in perinatal asphyxia: A randomized controlled pilot study. J. Perinatol. 2015, 35, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.L.; Baggerly, C.; McDonnell, S.; Hamilton, S.; Winkler, J.; Warner, G.; Rodriguez, C.; Shary, J.; Smith, P.; Hollis, B. Post-hoc comparison of vitamin D status at three timepoints during pregnancy demonstrates lower risk of preterm birth with higher vitamin D closer to delivery. J. Steroid Biochem. Mol. Biol. 2015, 148, 256–260. [Google Scholar] [CrossRef]
- Qin, L.-L.; Lu, F.-G.; Yang, S.-H.; Xu, H.-L.; Luo, B.-A. Does Maternal Vitamin D Deficiency Increase the Risk of Preterm Birth: A Meta-Analysis of Observational Studies. Nutrients 2016, 8, 301. [Google Scholar] [CrossRef]
- Merewood, A.; Mehta, S.D.; Chen, T.C.; Bauchner, H.; Holick, M.F. Association between Vitamin D Deficiency and Primary Cesarean Section. J. Clin. Endocrinol. Metab. 2009, 94, 940–945. [Google Scholar] [CrossRef]
- Bodnar, L.M.; Platt, R.W.; Simhan, H.N. Early-Pregnancy Vitamin D Deficiency and Risk of Preterm Birth Subtypes. Obstet. Gynecol. 2015, 125, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.-Q.; Qi, H.-P.; Luo, Z.-C.; Fraser, W.D. Maternal vitamin D status and adverse pregnancy outcomes: A systematic review and meta-analysis. J. Matern.-Fetal Neonatal Med. 2013, 26, 889–899. [Google Scholar] [CrossRef]
- Wagner, C.L.; Baggerly, C.; McDonnell, S.L.; Baggerly, K.; French, C.; Hamilton, S.A.; Hollis, B.W. Post-hoc analysis of vitamin D status and reduced risk of preterm birth in two vitamin D pregnancy cohorts compared with South Carolina March of Dimes 2009–2011 rates. J. Steroid Biochem. Mol. Biol. 2016, 155, 245–251. [Google Scholar] [CrossRef]
- Burris, H.H.; Van Marter, L.J.; McElrath, T.F.; Tabatabai, P.; Litonjua, A.A.; Weiss, S.T.; Christou, H. Vitamin D status among preterm and full-term infants at birth. Pediatr. Res. 2014, 75, 75–80. [Google Scholar] [CrossRef]
- Monangi, N.; Slaughter, J.L.; Dawodu, A.; Smith, C.; Akinbi, H.T. Vitamin D status of early preterm infants and the effects of vitamin D intake during hospital stay. Arch. Dis. Child. Fetal Neonatal Ed. 2014, 99, F166–F168. [Google Scholar] [CrossRef]
- Treiber, M.; Mujezinović, F.; Balon, B.P.; Gorenjak, M.; Maver, U.; Dovnik, A. Association between umbilical cord vitamin D levels and adverse neonatal outcomes. J. Int. Med. Res. 2020, 48, 300060520955001. [Google Scholar] [CrossRef]
- Manzon, L.; Altarescu, G.; Tevet, A.; Schimmel, M.S.; Elstein, D.; Samueloff, A.; Grisaru-Granovsky, S. Vitamin D receptor polymorphism FokI is associated with spontaneous idiopathic preterm birth in an Israeli population. Eur. J. Obstet. Gynecol. Reprod. Biol. 2014, 177, 84–88. [Google Scholar] [CrossRef]
- Eyles, D.W.; Burne, T.; McGrath, J. Vitamin D in fetal brain development. Semin. Cell Dev. Biol. 2011, 22, 629–636. [Google Scholar] [CrossRef]
- Yates, N.J.; Crew, R.C.; Wyrwoll, C.S. Vitamin D deficiency and impaired placental function: Potential regulation by glucocorticoids? Reproduction 2017, 153, R163–R171. [Google Scholar] [CrossRef]
- Tesic, D.; Hawes, J.E.; Zosky, G.R.; Wyrwoll, C.S. Vitamin D Deficiency in BALB/c Mouse Pregnancy Increases Placental Transfer of Glucocorticoids. Endocrinology 2015, 156, 3673–3679. [Google Scholar] [CrossRef] [PubMed]
- Yates, N.J.; Tesic, D.; Feindel, K.W.; Smith, J.T.; Clarke, M.W.; Wale, C.; Crew, R.C.; Wharfe, M.D.; Whitehouse, A.J.O.; Wyrwoll, C.S. Vitamin D is crucial for maternal care and offspring social behaviour in rats. J. Endocrinol. 2018, 237, 73–85. [Google Scholar] [CrossRef]
- McGrath, J.J.; Burne, T.H.; Féron, F.; Mackay-Sim, A.; Eyles, D.W. Developmental Vitamin D Deficiency and Risk of Schizophrenia: A 10-Year Update. Schizophr. Bull. 2010, 36, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, A.J.; Holt, B.J.; Serralha, M.; Holt, P.G.; Hart, P.H.; Kusel, M.M. Maternal vitamin D levels and the autism phenotype among offspring. J. Autism. Dev. Disord. 2013, 43, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, A.J.O.; Holt, B.J.; Serralha, M.; Holt, P.G.; Kusel, M.M.H.; Hart, P.H. Maternal Serum Vitamin D Levels During Pregnancy and Offspring Neurocognitive Development. Pediatrics 2012, 129, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Vuillermot, S.; Luan, W.; Meyer, U.; Eyles, D.W. Vitamin D treatment during pregnancy prevents autism-related phenotypes in a mouse model of maternal immune activation. Mol. Autism 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Mazahery, H.; Camargo, C.A.; Conlon, C.; Beck, K.L.; Kruger, M.C.; Von Hurst, P.R. Vitamin D and Autism Spectrum Disorder: A Literature Review. Nutrients 2016, 8, 236. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zheng, Y.; Wu, Y.; Ni, B.; Shi, S. Imbalance between IL-17A-Producing Cells and Regulatory T Cells during Ischemic Stroke. Mediat. Inflamm. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Y.; Yu, F.; Wang, Y.M.; Zhang, C.; Hu, C.; Wu, Z.; Xu, X.; Hu, S. Peripheral Th17/Treg imbalance in patients with atherosclerotic cerebral infarction. Int. J. Clin. Exp. Pathol. 2013, 6, 1015–1027. [Google Scholar]
- Evans, M.A.; Kim, H.A.; Ling, Y.H.; Uong, S.; Vinh, A.; De Silva, T.M.; Arumugam, T.V.; Clarkson, A.N.; Zosky, G.R.; Drummond, G.R.; et al. Vitamin D3 Supplementation Reduces Subsequent Brain Injury and Inflammation Associated with Ischemic Stroke. NeuroMolecular Med. 2018, 20, 147–159. [Google Scholar] [CrossRef]
- Won, S.; Sayeed, I.; Peterson, B.L.; Wali, B.; Kahn, J.S.; Stein, D.G. Vitamin D Prevents Hypoxia/Reoxygenation-Induced Blood-Brain Barrier Disruption via Vitamin D Receptor-Mediated NF-kB Signaling Pathways. PLoS ONE 2015, 10, e0122821. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Song, S.; Cui, J.; Feng, Y.; Gao, J.; Jiang, P. Vitamin D Receptor Activation Influences NADPH Oxidase (NOX2) Activity and Protects against Neurological Deficits and Apoptosis in a Rat Model of Traumatic Brain Injury. Oxidative Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.A.; The Committee on Nutrition; Bhatia, J.J.S.; Corkins, M.R.; De Ferranti, S.D.; Golden, N.H.; Silverstein, J. Calcium and Vitamin D Requirements of Enterally Fed Preterm Infants. Pediatrics 2013, 131, e1676–e1683. [Google Scholar] [CrossRef]
- Bennet, L.; Tan, S.; Msc, L.V.D.H.; Derrick, M.; Groenendaal, F.; Van Bel, F.; Juul, S.; Back, S.A.; Northington, F.; Robertson, N.J.; et al. Cell therapy for neonatal hypoxia-ischemia and cerebral palsy. Ann. Neurol. 2011, 71, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Melendez, M.; Yawno, T.; Jenkin, G.; Miller, S.L. Stem cell therapy to protect and repair the developing brain: A review of mechanisms of action of cord blood and amnion epithelial derived cells. Front. Neurosci. 2013, 7, 194. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Kim, Y.; Chang, J.W.; Yang, Y.S.; Oh, W.; Lee, J.M.; Park, H.R.; Kim, D.G.; Paek, S.H. Effect of Single and Double Administration of Human Umbilical Cord Blood-Derived Mesenchymal Stem Cells Following Focal Cerebral Ischemia in Rats. Exp. Neurobiol. 2017, 26, 55–65. [Google Scholar] [CrossRef]
- Zhu, D.; Wallace, E.M.; Lim, R. Cell-based therapies for the preterm infant. Cytotherapy 2014, 16, 1614–1628. [Google Scholar] [CrossRef]
- Yoshimoto, M.; Koenig, J.M. Stem Cells: Potential Therapy for Neonatal Injury? Clin. Perinatol. 2015, 42, 597–612. [Google Scholar] [CrossRef]
- Antoniadou, E.; David, A.L. Placental stem cells. Best Pract. Res. Clin. Obstet. Gynaecol. 2016, 13–29, 13–29. [Google Scholar] [CrossRef]
- Evans, M.A.; Lim, R.; Kim, H.A.; Chu, H.X.; Gardiner-Mann, C.V.; Taylor, K.W.; Chan, C.T.; Brait, V.H.; Lee, S.; Dinh, Q.N.; et al. Acute or Delayed Systemic Administration of Human Amnion Epithelial Cells Improves Outcomes in Experimental Stroke. Stroke 2018, 49, 700–709. [Google Scholar] [CrossRef]
- McDonald, C.A.; Payne, N.L.; Sun, G.; Moussa, L.; Siatskas, C.; Lim, R.; Wallace, E.M.; Jenkin, G.; Bernard, C.C. Immunosuppressive potential of human amnion epithelial cells in the treatment of experimental autoimmune encephalomyelitis. J. Neuroinflammation 2015, 12, 1–14. [Google Scholar] [CrossRef]
- Yawno, T.; Schuilwerve, J.; Moss, T.J.; Vosdoganes, P.; Westover, A.J.; Afandi, E.; Jenkin, G.; Wallace, E.M.; Miller, S.L. Human Amnion Epithelial Cells Reduce Fetal Brain Injury in Response to Intrauterine Inflammation. Dev. Neurosci. 2013, 35, 272–282. [Google Scholar] [CrossRef]
- Barton, S.K.; Melville, J.M.; Tolcos, M.; Polglase, G.R.; McDougall, A.R.; Azhan, A.; Crossley, K.; Jenkin, G.; Moss, T.J.M. Human Amnion Epithelial Cells Modulate Ventilation-Induced White Matter Pathology in Preterm Lambs. Dev. Neurosci. 2015, 37, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Hodges, R.J.; Lim, R.; Jenkin, G.; Wallace, E.M. Amnion Epithelial Cells as a Candidate Therapy for Acute and Chronic Lung Injury. Stem Cells Int. 2012, 2012, 1–7. [Google Scholar] [CrossRef]
- Vosdoganes, P.; Lim, R.; Koulaeva, E.; Chan, S.T.; Acharya, R.; Moss, T.J.; Wallace, E.M. Human amnion epithelial cells modulate hyperoxia-induced neonatal lung injury in mice. Cytotherapy 2013, 15, 1021–1029. [Google Scholar] [CrossRef]
- van den Heuij, L.G.; Fraser, M.; Miller, S.L.; Jenkin, G.; Wallace, E.M.; Davidson, J.O.; Lear, C.A.; Lim, R.; Wassink, G.; Gunn, A.J.; et al. Delayed intranasal infusion of human amnion epithelial cells improves white matter maturation after asphyxia in preterm fetal sheep. J. Cereb. Blood Flow Metab. 2017, 39, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Heuij, L.G.V.D.; Fraser, M.; Wassink, G.; Miller, S.L.; Lim, R.; Wallace, E.M.; Jenkin, G.; Gunn, A.J.; Bennet, L. Window of opportunity for human amnion epithelial stem cells to attenuate astrogliosis after umbilical cord occlusion in preterm fetal sheep. STEM CELLS Transl. Med. 2020. [Google Scholar] [CrossRef]
- Nott, F.; Pillow, J.J.; Dahl, M.; Kelly, S.B.; Melville, J.; McDonald, C.; Nitsos, I.; Lim, R.; Wallace, E.M.; Jenkin, G.; et al. Brain inflammation and injury at 48 h is not altered by human amnion epithelial cells in ventilated preterm lambs. Pediatr. Res. 2020, 88, 27–37. [Google Scholar] [CrossRef]
- Ali, H.; Bahbahani, H. Umbilical cord blood stem cells—Potential therapeutic tool for neural injuries and disorders. Acta Neurobiol. Exp. 2010, 70, 316–324. [Google Scholar]
- Tiwari, A.; Tursky, M.L.; Mushahary, D.; Wasnik, S.; Collier, F.M.; Suma, K.; Kirkland, M.A.; Pande, G. Ex vivo expansion of haematopoietic stem/progenitor cells from human umbilical cord blood on acellular scaffolds prepared from MS-5 stromal cell line. J. Tissue Eng. Regen. Med. 2013, 7, 871–883. [Google Scholar] [CrossRef]
- Pimentel-Coelho, P.M.; Rosado-De-Castro, P.H.; Da Fonseca, L.M.B.; Mendez-Otero, R. Umbilical cord blood mononuclear cell transplantation for neonatal hypoxic–ischemic encephalopathy. Pediatr. Res. 2012, 71, 464–473. [Google Scholar] [CrossRef]
- Wasielewski, B.; Jensen, A.; Roth-Härer, A.; Dermietzel, R.; Meier, C. Neuroglial activation and Cx43 expression are reduced upon transplantation of human umbilical cord blood cells after perinatal hypoxic-ischemic injury. Brain Res. 2012, 1487, 39–53. [Google Scholar] [CrossRef]
- Krakowiak, P.; Walker, C.K.; Bremer, A.A.; Baker, A.S.; Ozonoff, S.; Hansen, R.L.; Hertz-Picciotto, I. Maternal Metabolic Conditions and Risk for Autism and Other Neurodevelopmental Disorders. Pediatrics 2012, 129, e1121–e1128. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-L.; Zhao, Y.-S.; Hu, M.-Y.; Sun, Y.-Q.; Chen, Y.-X.; Bi, X.-H. Umbilical cord blood cells regulate endogenous neural stem cell proliferation via hedgehog signaling in hypoxic ischemic neonatal rats. Brain Res. 2013, 1518, 26–35. [Google Scholar] [CrossRef]
- Li, J.; Yawno, T.; Sutherland, A.; Loose, J.; Nitsos, I.; Bischof, R.; Castillo-Melendez, M.; McDonald, C.A.; Wong, F.; Jenkin, G.; et al. Preterm white matter brain injury is prevented by early administration of umbilical cord blood cells. Exp. Neurol. 2016, 283, 179–187. [Google Scholar] [CrossRef]
- Li, J.; Yawno, T.; Sutherland, A.E.; Gurung, S.; Paton, M.; McDonald, C.; Tiwari, A.; Pham, Y.; Castillo-Melendez, M.; Jenkin, G.; et al. Preterm umbilical cord blood derived mesenchymal stem/stromal cells protect preterm white matter brain development against hypoxia-ischemia. Exp. Neurol. 2018, 308, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yawno, T.; Sutherland, A.; Loose, J.; Nitsos, I.; Allison, B.J.; Bischof, R.; McDonald, C.A.; Jenkin, G.; Miller, S.L. Term vs. preterm cord blood cells for the prevention of preterm brain injury. Pediatr. Res. 2017, 82, 1030–1038. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Treatment | Study Details | Clinical Setting | Main Findings | Reference |
|---|---|---|---|---|
| Antenatal corticosteroids | Observational study 2323 infants (22–32 weeks) (EPIPAGE study) | Extreme or very preterm birth | Antenatal corticosteroids greatly increased survival with little evidence for effects on neurodevelopmental and behavioural outcomes at 5 years of age | [50] |
| Meta-analysis of 30 trials 7774 women and 8158 infants | Preterm birth | Antenatal corticosteroids were associated with a reduction in perinatal death, neonatal death, respiratory distress syndrome, intraventricular haemorrhage, need for mechanical ventilation, and infection therapy | [51] | |
| Double-blind randomized controlled trial 1509 fetuses who received dexamethasone or betamethasone | Preterm birth | No significant difference in the incidence of survival without neurosensory disability at 2 years of age between dexamethasone and betamethasone | [54] | |
| Meta-analysis of forty-five trials (11,227 women, 11,878 infants) dexamethasone vs. betamethasone | Preterm birth | No difference between dexamethasone and betamethasone on neonatal death, neurodevelopmental disability, IVH and birthweight | [55] | |
| Postnatal corticosteroids | 18 premature infants (23–31 weeks) 7 treated with postnatal dexamethasone 11 not treated | Preterm infants with chronic lung disease | Postnatal dexamethasone was associated with impaired brain growth, particularly in cerebral cortical grey matter | [63] |
| 53 extremely low birthweight infants with high-quality MRI 11 infants received postnatal dexamethasone 30 infants received no treatment | Extremely low birthweight infants | Postnatal dexamethasone use was associated with smaller total and regional cerebral tissue volumes | [64] | |
| Cochrane Database systematic review of 32 randomised controlled trials including 4395 preterm infants with BPD who received early systemic corticosteroid treatment (< 8 days) | High-risk preterm infants | Early postnatal corticosteroids were associated with increased risk of abnormal findings on neurological examination and increased risk of cerebral palsy on long-term follow up | [65] | |
| Cochrane Database systematic review of 21 randomised controlled trials including 1424 preterm infants with BPD who received late systemic postnatal corticosteroids treatment (> 7 days) | Preterm infants with BPD | Postnatal steroids were associated with increased retinopathy of prematurity but not blindness, a trend towards a reduction in severe IVH and death but a trend towards increased cerebral palsy or abnormal neurological examination | [66] | |
| Meta-analysis of 16 randomised controlled trials comparing 1136 ventilated preterm infants > 7 days who received dexamethasone or placebo | Ventilated preterm infants | Higher cumulative doses of dexamethasone after the first week of life may decrease the risk of BPD without increasing the risk for neurodevelopmental impairment | [67] | |
| Magnesium sulfate | Cochrane Database systematic review of 5 randomised controlled trials of antenatal magnesium sulfate therapy in women threatening preterm birth at less than 37 weeks gestational age including 6145 babies | Preterm birth | Antenatal magnesium sulfate therapy was associated with a small reduction in the incidence of cerebral palsy, with a number needed to treat of 63 | [68] |
| Systematic review of 6 trials involving Antenatal magnesium sulphate administration to women threatening preterm delivery before 34 weeks gestation including 5357 infants | Preterm birth | Antenatal magnesium sulphate therapy was associated with a significant reduction in the risk of cerebral palsy and substantial gross motor dysfunction | [69] | |
| Long-term follow up of a randomised controlled trial in women threatening preterm birth before 30 weeks gestation 535 received magnesium sulphate 527 received placebo | Preterm birth | Magnesium sulfate was not associated with improvements in neurological, cognitive, behavioral, growth, or functional outcomes in their children at school age (6–11 years) | [48] | |
| Long-term follow up of a randomised controlled trial of magnesium sulfate in women threatening preterm birth before 33 weeks gestation, including 503 children (7–14 years) | Preterm birth | Magnesium sulfate was not associated with any detrimental effects nor any significant effects on neurological outcome | [47] | |
| Human recombinant Epo | Randomized control trial of rEpo versus placebo 800 infants (≤32 weeks) | Extreme or very preterm birth rhEPO at 500 U/kg i.v. every other day for 2 weeks starting within 72 h of birth | rhEpo decreased risk of death and moderate/severe disability at 18 month (13%) compared to control (26.9%) | [70] |
| Randomized, double-blind trial 741 infants (24 weeks 0 days to 27 weeks 6 days) | Extremely preterm birth rEpo started <24 h of birth 1000 U/Kg every 48 h for 6 doses followed by 400 U/Kg 3 times/week | rhEpo did not reduce risk of severe developmental impairment at 2 years of age | [71] | |
| 448 infants (26 weeks 0 days’ and 31 weeks 6 days) | Very preterm birth 3000 IU/kg iv within 3, at 12–18, and at 36–42 postnatal hours | rhEpo had no effect on neurodevelopmental outcomes at 2 years or 5 years | [72,73] | |
| Therapeutic hypothermia | Retrospective study 31 preterm infants (34–35 weeks) 32 term infants | HIE | HT-associated complications in 90% of preterm infants and 81.3% term infants (p = 0.3) Preterms showed increased risk of hyperglycemia, early rewarming, death and white matter injury compared to term infants after cooling | [74] |
| Retrospective uncontrolled cohort analysis 30 infants (33–35 weeks) | HIE | High incidence of complications, including coagulopathy (50%), thrombocytopenia (20%) and death (18.2%). Death or moderate to severe neurodevelopmental impairment occurred in 50% of infants with known outcomes | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yates, N.; Gunn, A.J.; Bennet, L.; Dhillon, S.K.; Davidson, J.O. Preventing Brain Injury in the Preterm Infant—Current Controversies and Potential Therapies. Int. J. Mol. Sci. 2021, 22, 1671. https://doi.org/10.3390/ijms22041671
Yates N, Gunn AJ, Bennet L, Dhillon SK, Davidson JO. Preventing Brain Injury in the Preterm Infant—Current Controversies and Potential Therapies. International Journal of Molecular Sciences. 2021; 22(4):1671. https://doi.org/10.3390/ijms22041671
Chicago/Turabian StyleYates, Nathanael, Alistair J. Gunn, Laura Bennet, Simerdeep K. Dhillon, and Joanne O. Davidson. 2021. "Preventing Brain Injury in the Preterm Infant—Current Controversies and Potential Therapies" International Journal of Molecular Sciences 22, no. 4: 1671. https://doi.org/10.3390/ijms22041671
APA StyleYates, N., Gunn, A. J., Bennet, L., Dhillon, S. K., & Davidson, J. O. (2021). Preventing Brain Injury in the Preterm Infant—Current Controversies and Potential Therapies. International Journal of Molecular Sciences, 22(4), 1671. https://doi.org/10.3390/ijms22041671