
Retinal Ganglion Cell Loss and Microglial Activation in a SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis

 ,
,  , ,
, ,  ,
,  , ,
, ,  ,
,  and
and 
Abstract
1. Introduction
2. Results
2.1. Qualitative Study of Iba-1+ Retinal Microglial Cells
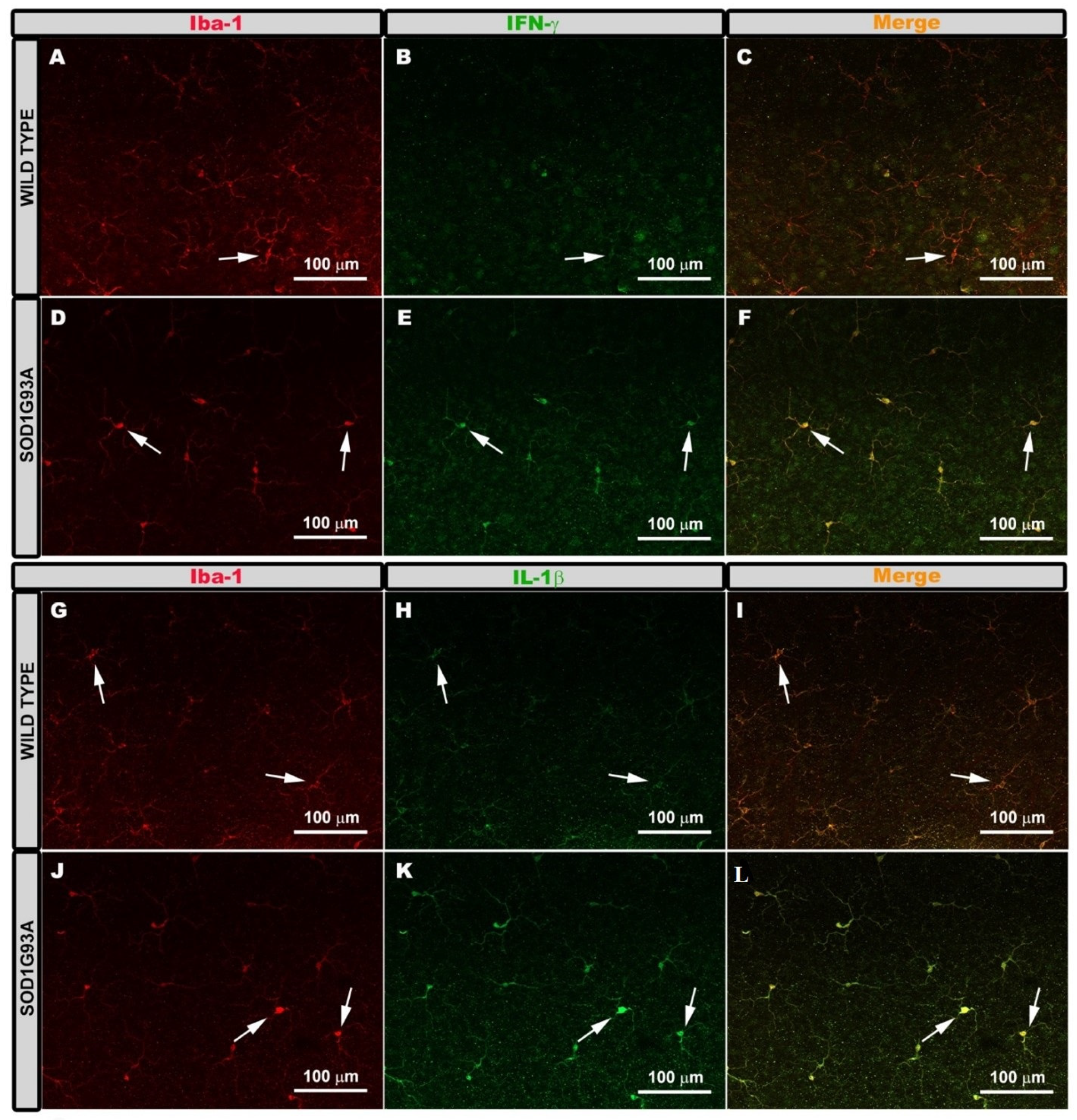
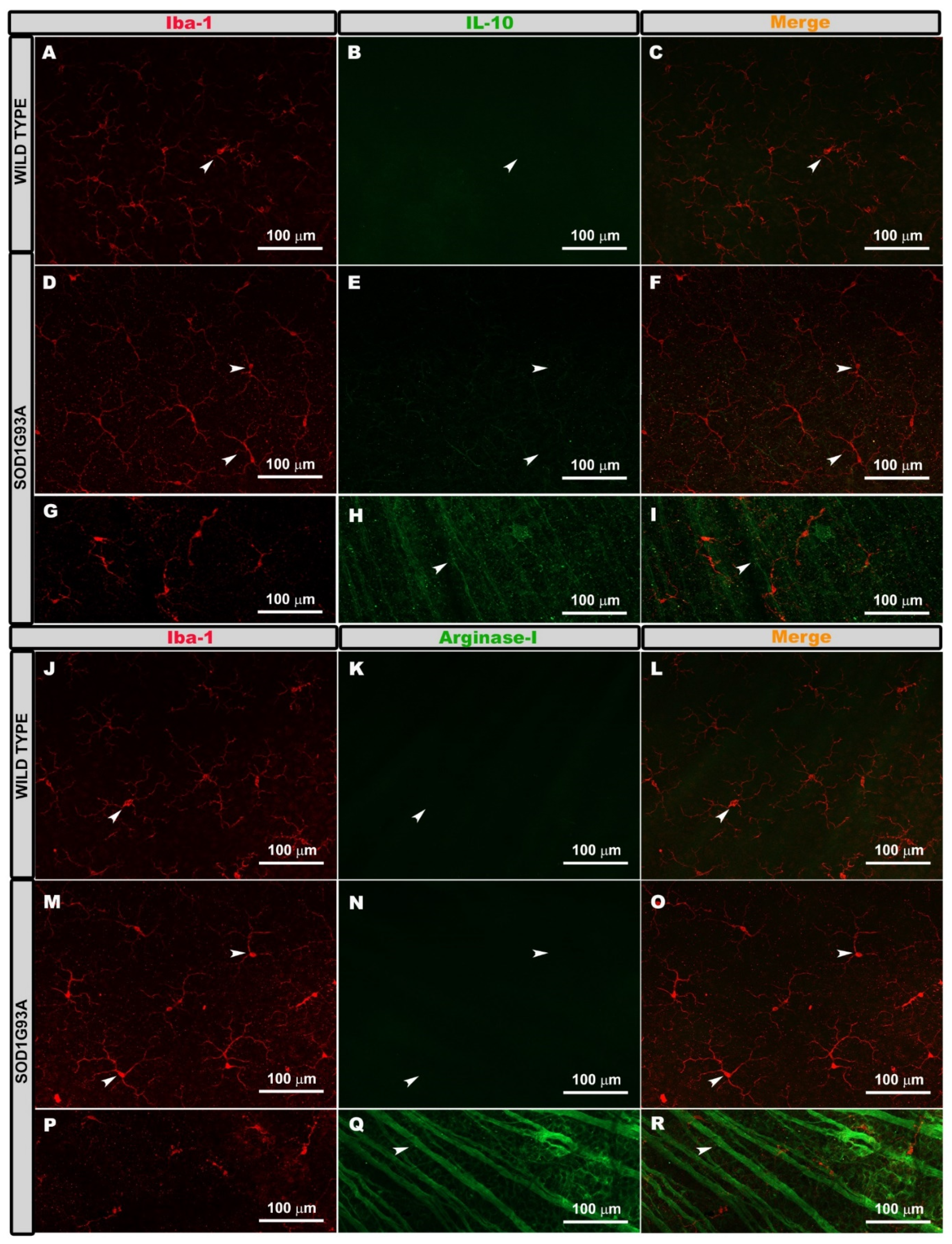
2.2. Expression of Microglial Phenotypes M1 or M2
2.3. Quantitative Study of Iba-1+ Microglial Cells
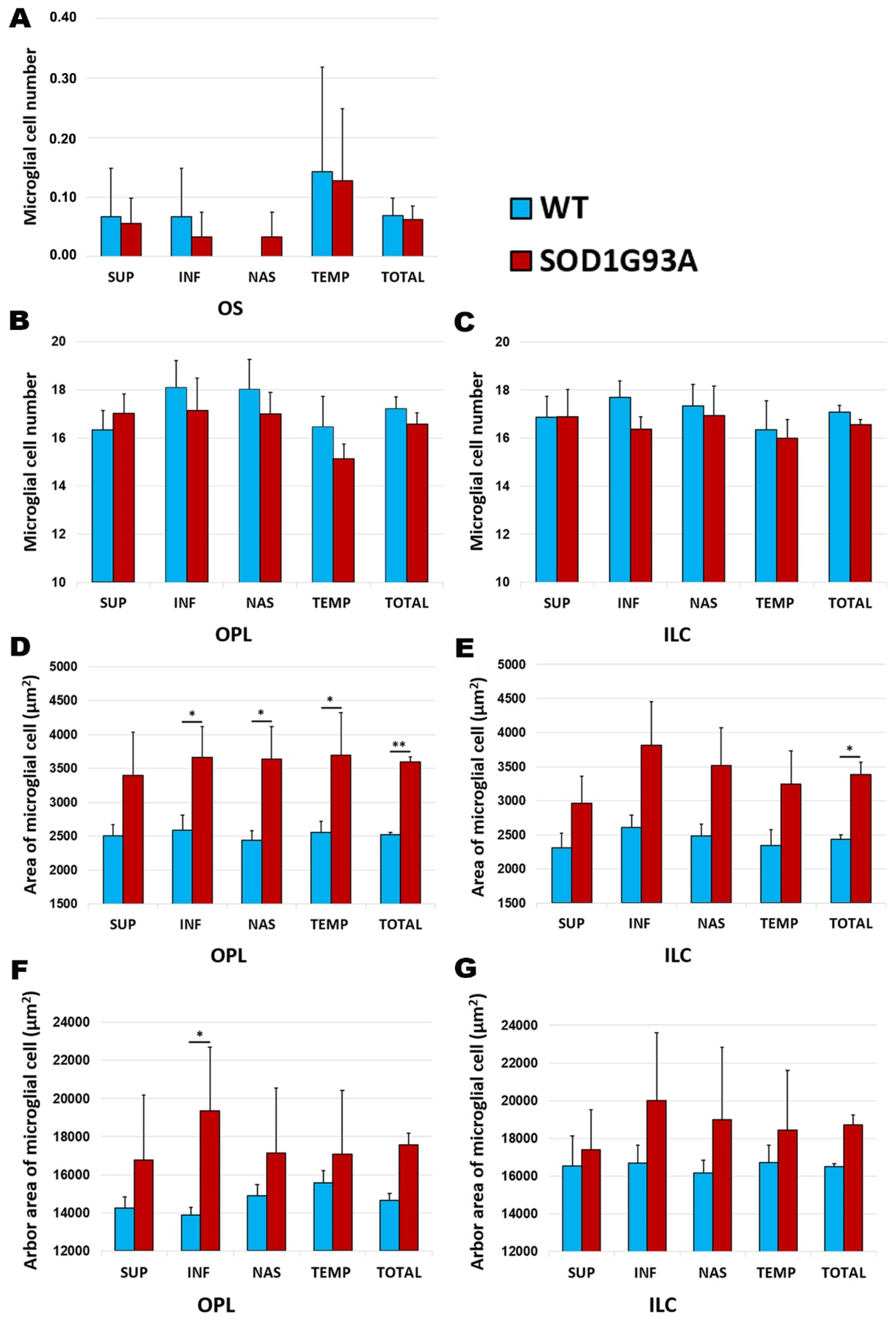
2.3.1. Number of Cells
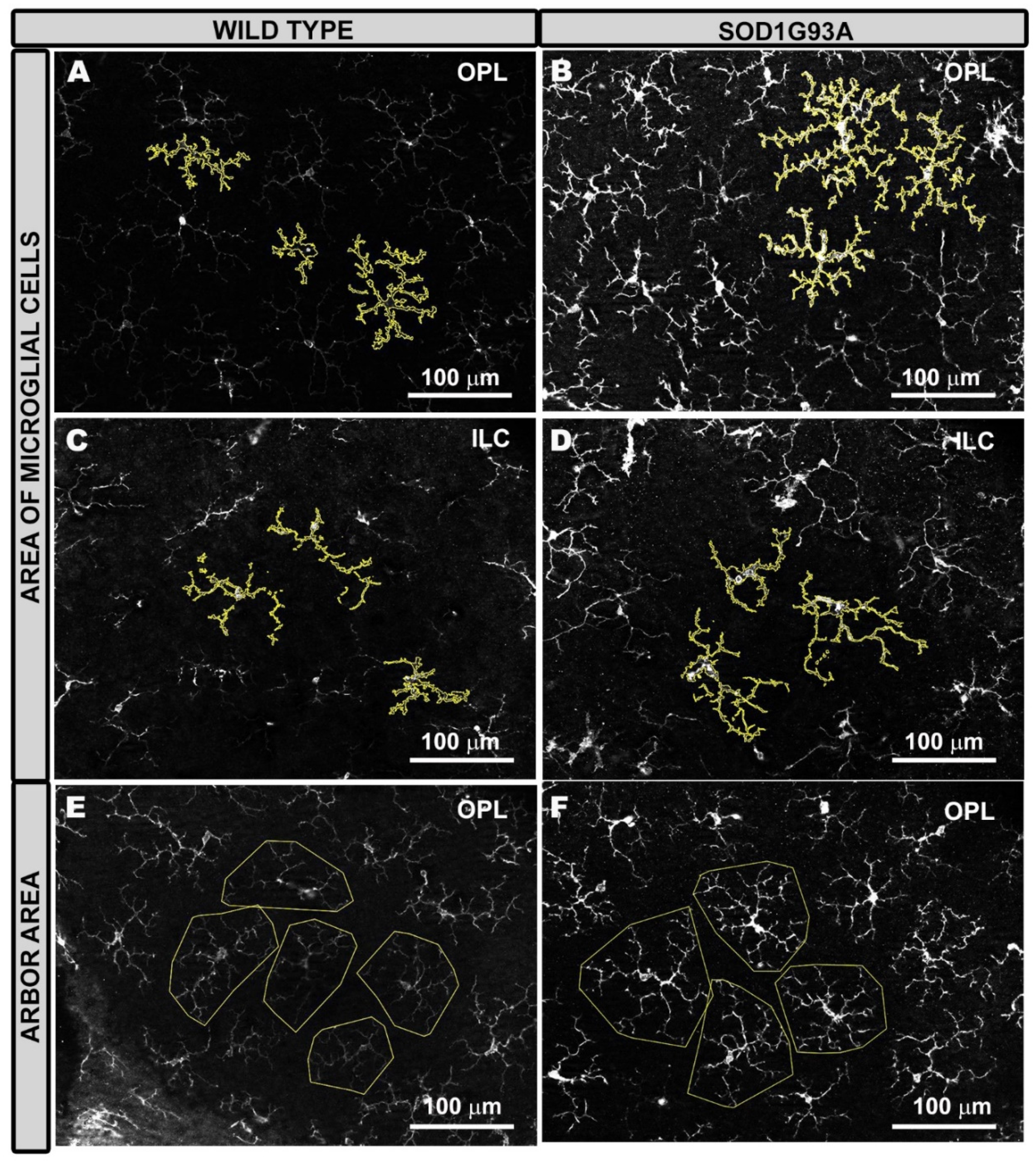
2.3.2. Area of Microglial Cells
2.3.3. Arbor Area of Microglial Cells
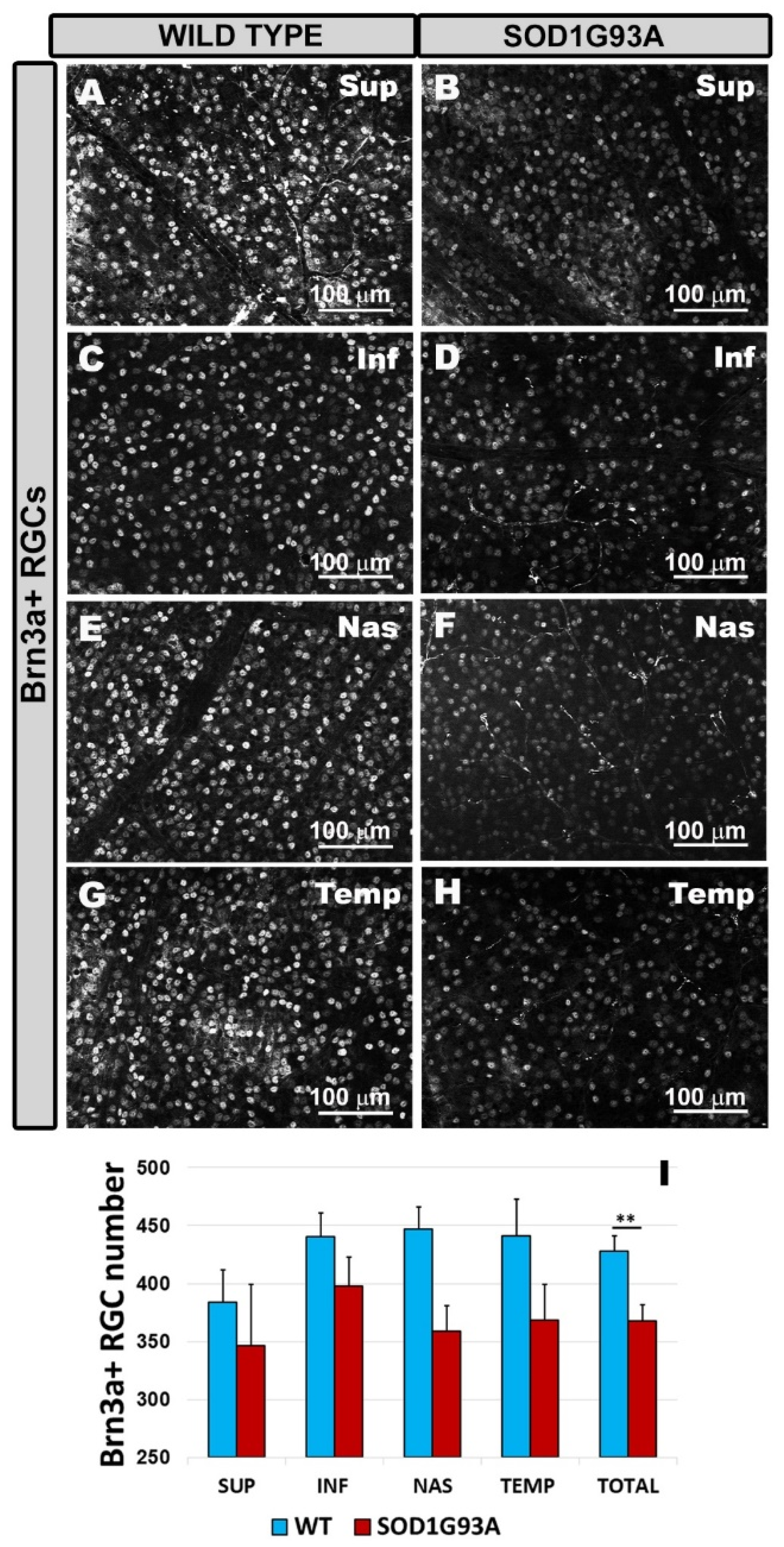
2.4. Quantitative Study of Brn3a+ RGCs
3. Discussion
4. Materials and Methods
4.1. Animals and Ethics
4.2. Experimental Groups
4.3. Immunohistochemistry
4.4. Quantitative Retina Analysis
4.4.1. Microglial Cell Quantification
- (1)
- Microglial Cell Number
- (2)
- Area Occupied by Each Microglial Cell
- (3)
- The Arbor Area of Microglial Cells
- (4)
- Mean IFN-γ and IL-1β Intensity Expression
4.4.2. Brn3a+ RGC Quantification
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic Lateral Sclerosis |
| BRB | Blood-Retinal Barrier |
| CNS | Central Nervous System |
| FALS | Familiar Amyotrophic Lateral Sclerosis |
| G93A | Glycine 93 into Alanine |
| GCL | Ganglion Cell Layer |
| GM-CSF | Macrophage Colony Stimulating Factor |
| Iba-1 | Ionized calcium binding adaptor molecule 1 |
| IFN-γ | Interferon γ |
| ILC | Inner Layer Complex |
| IPL | Inner Plexiform Layer |
| IL-1β | Interleukin-1β |
| M-CSF | Macrophage Colony Stimulating Factor |
| NFL | Nerve Fiber Layer |
| NOX2 | NADPH oxidase 2 |
| OCT | Optical Coherence Tomography |
| OPL | Outer Plexiform Layer |
| OS | Photoreceptor Outer Segment layer |
| PBS | Phosphate Buffer Saline |
| Ranbp2 | Ran-binding protein2 |
| RGCs | Retinal Ganglion Cells |
| SALS | Sporadic Amyotrophic Lateral Sclerosis |
| SOD1 | Superoxide Dismutase 1 |
| TNFα | Tumor Necrosis Factor α |
| UBQLN2 | Ubiquilin 2 |
| WT | Wild-Type |
References
- Carra, S.; Crippa, V.; Rusmini, P.; Boncoraglio, A.; Minoia, M.; Giorgetti, E.; Kampinga, H.H.; Poletti, A. Alteration of protein folding and degradation in motor neuron diseases: Implications and protective functions of small heat shock proteins. Prog. Neurobiol. 2012, 97, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Yedavalli, V.S.; Patil, A.; Shah, P. Amyotrophic Lateral Sclerosis and its mimics/variants: A comprehensive review. J. Clin. Imaging Sci. 2018, 8, 53. [Google Scholar] [CrossRef]
- Zarei, S.; Carr, K.; Reiley, L.; Diaz, K.; Guerra, O.; Altamirano, P.F.; Pagani, W.; Lodin, D.; Orozco, G.; Chinea, A. A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 2015, 6, 171. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Olsen, M.K.; Roberds, S.L.; Ellerbrock, B.R.; Fleck, T.J.; McKinley, D.K.; Gurney, M.E. Disease mechanisms revealed by transcription profiling in SOD1-G93A transgenic mouse spinal cord. Ann. Neurol. 2001, 50, 730–740. [Google Scholar] [CrossRef]
- Byrne, S.; Walsh, C.; Lynch, C.; Bede, P.; Elamin, M.; Kenna, K.; McLaughlin, R.; Hardiman, O. Rate of familial amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 623–627. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Ripps, M.E.; Huntleyt, G.W.; Hoft, P.R.; Morrisontt, J.H.; Gordont, J.W. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Genetics 1995, 92, 689–693. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef]
- Chiu, A.Y.; Zhai, P.; Dal Canto, M.C.; Peters, T.M.; Kwon, Y.W.; Prattis, S.M.; Gurney, M.E. Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol. Cell. Neurosci. 1995, 6, 349–362. [Google Scholar] [CrossRef]
- Mourelatos, Z.; Gonatas, N.K.; Stieber, A.; Gurney, M.E.; Dal Canto, M.C. The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu, Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc. Natl. Acad. Sci. USA 1996, 93, 5472–5477. [Google Scholar] [CrossRef]
- Ramirez, A.I.; de Hoz, R.; Salobrar-García, E.; Salazar, J.J.; Rojas, B.; Ajoy, D.; López-Cuenca, I.; Rojas, P.; Triviño, A.; Ramírez, J.M. The role of microglia in retinal neurodegeneration: Alzheimer’s disease, Parkinson, and glaucoma. Front. Aging Neurosci. 2017, 9, 214. [Google Scholar] [CrossRef]
- Ramírez, A.I.; Rojas, B.; de Hoz, R.; Salazar, J.J.; Gallego, B.; Triviño, A.; Ramírez, J.M. Microglia, inflammation, and glaucoma. In Glaucoma; SM Group Open Access eBooks: Dover, DE, USA, 2015; pp. 1–16. [Google Scholar]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Philips, T.; Robberecht, W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011, 10, 253–263. [Google Scholar] [CrossRef]
- Liao, B.; Zhao, W.; Beers, D.R.; Henkel, J.S.; Appel, S.H. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 2012, 237, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.R.; Johnson, J.A. Astrogliosis in Amyotrophic Lateral Sclerosis: Role and therapeutic potential of astrocytes. Neurotherapeutics 2010, 7, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Beers, D.R.; Henkel, J.S.; Zhang, W.; Urushitani, M.; Julien, J.P.; Appel, S.H. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 2010, 58, 231–243. [Google Scholar] [CrossRef]
- Roth, N.M.; Saidha, S.; Zimmermann, H.; Brandt, A.U.; Oberwahrenbrock, T.; Maragakis, N.J.; Tumani, H.; Ludolph, A.C.; Meyer, T.; Calabresi, P.A.; et al. Optical coherence tomography does not support optic nerve involvement in amyotrophic lateral sclerosis. Eur. J. Neurol. 2013, 20, 1170–1176. [Google Scholar] [CrossRef]
- Ringelstein, M.; Albrecht, P.; Südmeyer, M.; Harmel, J.; Müller, A.K.; Keser, N.; Finis, D.; Ferrea, S.; Guthoff, R.; Schnitzler, A.; et al. Subtle retinal pathology in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2014, 1, 290–297. [Google Scholar] [CrossRef]
- Volpe, N.J.; Simonett, J.; Fawzi, A.A.; Siddique, T. Opthalmic manifestations of amyotrophic lateral sclerosis (an American Ophthalmological Society thesis). Trans. Am. Ophthalmol Soc. 2015, 113, 1–15. [Google Scholar]
- Fawzi, A.A.; Simonett, J.M.; Purta, P.; Moss, H.E.; Lowry, J.L.; Deng, H.-X.; Siddique, N.; Sufit, R.; Bigio, E.H.; Volpe, N.J.; et al. Clinicopathologic report of ocular involvement in ALS patients with C9orf72 mutation. Amyotroph. Lateral Scler. Frontotemporal Degener. 2014, 15, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Hübers, A.; Müller, H.P.; Dreyhaupt, J.; Böhm, K.; Lauda, F.; Tumani, H.; Kassubek, J.; Ludolph, A.C.; Pinkhardt, E.H. Retinal involvement in amyotrophic lateral sclerosis: A study with optical coherence tomography and diffusion tensor imaging. J. Neural Transm. 2016, 123, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Simonett, J.M.; Huang, R.; Siddique, N.; Farsiu, S.; Siddique, T.; Volpe, N.J.; Fawzi, A.A. Macular sub-layer thinning and association with pulmonary function tests in Amyotrophic Lateral Sclerosis. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Rojas, P.; de Hoz, R.; Ramírez, A.I.; Ferreras, A.; Salobrar-Garcia, E.; Muñoz-Blanco, J.L.; Urcelay-Segura, J.L.; Ramirez, J.M. Changes in retinal OCT and their correlations with neurological disability in early ALS patients, a follow-up study. Brain Sci. 2019, 9, 337. [Google Scholar] [CrossRef]
- Ringer, C.; Weihe, E.; Schütz, B. SOD1 G93A mutant mice develop a neuroinflammation-independent dendropathy in excitatory neuronal subsets of the olfactory bulb and retina. J. Neuropathol. Exp. Neurol. 2017, 76, 769–778. [Google Scholar] [CrossRef]
- Cho, K.; Yoon, D.; Yu, M.; Peachey, N.S.; Ferreira, P.A. Microglial activation in an amyotrophic lateral sclerosis-like model caused by Ranbp2 loss and nucleocytoplasmic transport impairment in retinal ganglion neurons. Cell. Mol. Life Sci. 2019, 76, 3407–3432. [Google Scholar] [CrossRef]
- Ward, M.E.; Chen, R.; Huang, H.Y.; Ludwig, C.; Telpoukhovskaia, M.; Taubes, A.; Boudin, H.; Minami, S.S.; Reichert, M.; Albrecht, P.; et al. Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Vinsant, S.; Mansfield, C.; Jimenez-Moreno, R.; Moore, V.D.G.; Yoshikawa, M.; Hampton, T.G.; Prevette, D.; Caress, J.; Oppenheim, R.W.; Milligan, C. Characterization of early pathogenesis in the SOD1G93A mouse model of ALS: Part I, background and methods. Brain Behav. 2013, 3, 335–350. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Rodent models of amyotrophic lateral sclerosis. Curr. Protoc. Pharmacol. 2015, 69, 5.67.1–5.67.21. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Boillee, S.; Roberts, E.A.; Garcia, M.L.; McAlonis-Downes, M.; Mikse, O.R.; Cleveland, D.W.; Goldstein, L.S.B. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl. Acad. Sci. USA 2008, 105, 7594–7599. [Google Scholar] [CrossRef]
- Appel, S.H.; Zhao, W.; Beers, D.R.; Henkel, J.S. The microglial-motoneuron dialogue in ALS. Acta Myol. 2011, 30, 4–8. [Google Scholar] [PubMed]
- Lampson, L.A.; Kushner, P.D.; Sobel, R.A. Strong expression of class II major histocompatibility complex (MHC) antigens in the absence of detectable T cell infiltration in amyotrophic lateral sclerosis (ALS) spinal cord. J. Neuropathol. Exp. Neurol. 1988, 47, 353. [Google Scholar]
- Kawamata, T.; Akiyama, H.; Yamada, T.; Mcgeer, P.L. Immunologic reactions in Amyotrophic Lateral Sclerosis brain and spinal cord tissue. Am. J. Pathol. 1992, 140, 691–707. [Google Scholar] [PubMed]
- Sargsyan, S.A.; Monk, P.N.; Shaw, P.J. Microglia as potential contributors to motor neuron injury in amyotrophic lateral sclerosis. Glia 2005, 51, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Boillé, S.; Vande Velde, C.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef] [PubMed]
- King, A.E.; Dickson, T.C.; Blizzard, C.A.; Woodhouse, A.; Foster, S.S.; Chung, R.S.; Vickers, J.C. Neuron-glia interactions underlie ALS-like axonal cytoskeletal pathology. Neurobiol. Aging 2011, 32, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Ince, P.G.; Shaw, P.J.; Slade, J.Y.; Jones, C.; Hudgson, P. Familial amyotrophic lateral sclerosis with a mutation in exon 4 of the Cu/Zn superoxide dismutase gene: Pathological and immunocytochemical changes. Acta Neuropathol. 1996, 92, 395–403. [Google Scholar] [CrossRef]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillée, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Solomon, J.N.; Lewis, C.A.B.; Ajami, B.; Corbel, S.Y.; Rossi, F.M.V.; Krieger, C. Origin and distribution of bone marrow-derived cells in the central nervous system in a mouse model of amyotrophic lateral sclerosis. Glia 2006, 53, 744–753. [Google Scholar] [CrossRef]
- Ringer, C.; Luisa-Sybille, B.; Schäfer, M.K.; Eiden, L.E.; Weihe, E.; Schütz, B. PACAP signaling exerts opposing effects on neuroprotection and neuroinflammation during disease progression in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 54, 32–42. [Google Scholar] [CrossRef]
- Ringer, C.; Tune, S.; Bertoune, M.A.; Schwarzbach, H.; Tsujikawa, K.; Weihe, E.; Schütz, B. Disruption of calcitonin gene-related peptide signaling accelerates muscle denervation and dampens cytotoxic neuroinflammation in SOD1 mutant mice. Cell. Mol. Life Sci. 2017, 74, 339–358. [Google Scholar] [CrossRef] [PubMed]
- Ringer, C.; Weihe, E.; Schütz, B. Pre-symptomatic alterations in subcellular βCGRP distribution in motor neurons precede astrogliosis in ALS mice. Neurobiol. Dis. 2009, 35, 286–295. [Google Scholar] [CrossRef] [PubMed]
- De Hoz, R.; Ramírez, A.I.; González-Martín, R.; Ajoy, D.; Rojas, B.; Salobrar-García, E.; Valiente-Soriano, F.J.; Avilés-Trigueros, M.; Villegas-Pérez, M.P.; Vidal-Sanz, M.; et al. Bilateral early activation of retinal microglial cells in a mouse model of unilateral laser-induced experimental ocular hypertension. Exp. Eye Res. 2018, 171, 12–29. [Google Scholar] [CrossRef]
- Rojas, B.; Gallego, B.I.; Ramírez, A.I.; Salazar, J.J.; de Hoz, R.; Valiente-Soriano, F.J.; Avilés-Trigueros, M.; Villegas-Perez, M.P.; Vidal-Sanz, M.; Triviño, A.; et al. Microglia in mouse retina contralateral to experimental glaucoma exhibit multiple signs of activation in all retinal layers. J. Neuroinflamm. 2014, 11, 133. [Google Scholar] [CrossRef]
- Fernández-Albarral, J.A.; Ramírez, A.I.; de Hoz, R.; López-Villarín, N.; Salobrar-García, E.; López-Cuenca, I.; Licastro, E.; Inarejos-García, A.M.; Almodóvar, P.; Pinazo-Durán, M.D.; et al. Neuroprotective and anti-inflammatory effects of a hydrophilic saffron extract in a model of glaucoma. Int. J. Mol. Sci. 2019, 20, 4110. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, A.I.; de Hoz, R.; Fernández-Albarral, J.A.; Salobrar-García, E.; Rojas, B.; Valiente-Soriano, F.J.; Avilés-Trigueros, M.; Villegas-Pérez, M.P.; Vidal-Sanz, M.; Triviño, A.; et al. Time course of bilateral microglial activation in a mouse model of laser-induced glaucoma. Sci. Rep. 2020, 10, 4890. [Google Scholar] [CrossRef] [PubMed]
- Gallego, B.I.; Salazar, J.J.; de Hoz, R.; Rojas, B.; Ramírez, A.I.; Salinas-Navarro, M.; Ortín-Martínez, A.; Valiente-Soriano, F.J.; Avilés-Trigueros, M.; Villegas-Perez, M.P.; et al. IOP induces upregulation of GFAP and MHC-II and microglia reactivity in mice retina contralateral to experimental glaucoma. J. Neuroinflamm. 2012, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Salobrar-García, E.; Rodrigues-Neves, A.C.; Ramírez, A.I.; de Hoz, R.; Fernández-Albarral, J.A.; López-Cuenca, I.; Ramírez, J.M.; Ambrósio, A.F.; Salazar, J.J. Microglial Activation in the Retina of a Triple-Transgenic Alzheimer’s Disease Mouse Model (3xTg-AD). Int. J. Mol. Sci. 2020, 21, 816. [Google Scholar] [CrossRef]
- Perry, V.H. The influence of systemic inflammation on inflammation in the brain: Implications for chronic neurodegenerative disease. Brain. Behav. Immun. 2004, 18, 407–413. [Google Scholar] [CrossRef]
- Moisse, K.; Strong, M.J. Innate immunity in amyotrophic lateral sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Mutant superoxide dismutase 1-induced IL-1β accelerates ALS pathogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 13046–13050. [Google Scholar] [CrossRef] [PubMed]
- Munder, M. Arginase: An emerging key player in the mammalian immune system: Review. Br. J. Pharmacol. 2009, 158, 638–651. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11. [Google Scholar] [CrossRef]
- Laffer, B.; Bauer, D.; Wasmuth, S.; Busch, M.; Jalilvand, T.V.; Thanos, S.; Meyer zu Hörste, G.; Loser, K.; Langmann, T.; Heiligenhaus, A.; et al. Loss of IL-10 Promotes Differentiation of Microglia to a M1 Phenotype. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Langmann, T. Microglia activation in retinal degeneration. J. Leukoc. Biol. 2007, 81, 1345–1351. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Chiba, K. Role of microglial M1/M2 polarization in relapse and remission of psychiatric disorders and diseases. Pharmaceuticals 2014, 7, 1028–1048. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef]
- Shapiro, L.A.; Perez, Z.D.; Foresti, M.L.; Arisi, G.M.; Ribak, C.E. Morphological and ultrastructural features of Iba1-immunolabeled microglial cells in the hippocampal dentate gyrus. Brain Res. 2009, 1266, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Salinas-Navarro, M.; Alarcón-Martínez, L.; Valiente-Soriano, F.J.; Ortín-Martínez, A.; Jiménez-López, M.; Avilés-Trigueros, M.; Villegas-Pérez, M.P.; de la Villa, P.; Vidal-Sanz, M. Functional and morphological effects of laser-induced ocular hypertension in retinas of adult albino Swiss mice. Mol. Vis. 2009, 15, 2578–2598. [Google Scholar] [PubMed]
- De Gracia, P.; Gallego, B.I.; Rojas, B.; Ramírez, A.I.; de Hoz, R.; Salazar, J.J.; Triviño, A.; Ramírez, J.M. Automatic counting of microglial cells in healthy and glaucomatous mouse retinas. PLoS ONE 2015, 10, e0143278. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Color | Primary Antibody | Conc. | Secondary Antibody | Conc. |
|---|---|---|---|---|
| GREEN | Rabbit polyclonal anti IL1β (ref. ab9722, Abcam plc, Cambridge, UK) | 1:125 | Goat anti rabbit Alexa Fluor 488 (ref. ab150077, Abcam plc, Cambridge, UK) | 1:150 |
| Rabbit polyclonal anti IFN-γ (ref. ab9657, Abcam plc, Cambridge, UK) | 1:150 | |||
| Mouse monoclonal anti Arginase-I ref. 610708; BD Biosciences, San Jose, CA, USA) | 1:25 | Goat anti mouse Alexa Fluor 488 (ref. A11001 Invitrogen, Paisley, UK). | 1:150 | |
| Rat monoclonal anti IL10 (ref. ab189392, Abcam plc, Cambridge, UK) | 1:100 | Goat anti rat Alexa Fluor 488 (ref. ab150165, Abcam plc, Cambridge, UK) | 1:150 | |
| Mouse monoclonal anti Brn-3a (ref. MAB1585, Sigma-Aldrich, Darmstadt, Germany) | 1:300 | Goat anti mouse IgG1 Alexa Fluor 594 (ref. A21125, Invitrogen, Carlsbad, CA, USA) | 1:150 | |
| RED | Rabbit polyclonal anti Iba1 Red Fluorochrome 635 conjugated (ref. 5100756, Wako Chemicals GmbH, Neuss, Germany) | 1:100 | - | - |
| Rabbit polyclonal anti-Iba-1 (ref. 01919741, Fujifilm Wako pure chemical corporation, Osaka, Japan) | 1:600 | Donkey anti rabbit IgG Alexa Fluor 594 (ref. A21207; Invitrogen, Paisley, UK). | 1:800 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas, P.; Ramírez, A.I.; Cadena, M.; Fernández-Albarral, J.A.; Salobrar-García, E.; López-Cuenca, I.; Santos-García, I.; de Lago, E.; Urcelay-Segura, J.L.; Ramírez, J.M.; et al. Retinal Ganglion Cell Loss and Microglial Activation in a SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 1663. https://doi.org/10.3390/ijms22041663
Rojas P, Ramírez AI, Cadena M, Fernández-Albarral JA, Salobrar-García E, López-Cuenca I, Santos-García I, de Lago E, Urcelay-Segura JL, Ramírez JM, et al. Retinal Ganglion Cell Loss and Microglial Activation in a SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2021; 22(4):1663. https://doi.org/10.3390/ijms22041663
Chicago/Turabian StyleRojas, Pilar, Ana I. Ramírez, Manuel Cadena, José A. Fernández-Albarral, Elena Salobrar-García, Inés López-Cuenca, Irene Santos-García, Eva de Lago, José L. Urcelay-Segura, José M. Ramírez, and et al. 2021. "Retinal Ganglion Cell Loss and Microglial Activation in a SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 22, no. 4: 1663. https://doi.org/10.3390/ijms22041663
APA StyleRojas, P., Ramírez, A. I., Cadena, M., Fernández-Albarral, J. A., Salobrar-García, E., López-Cuenca, I., Santos-García, I., de Lago, E., Urcelay-Segura, J. L., Ramírez, J. M., de Hoz, R., & Salazar, J. J. (2021). Retinal Ganglion Cell Loss and Microglial Activation in a SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 22(4), 1663. https://doi.org/10.3390/ijms22041663