
Origin and Expansion of the Serine Protease Repertoire in the Myelomonocyte Lineage

 , and
, and 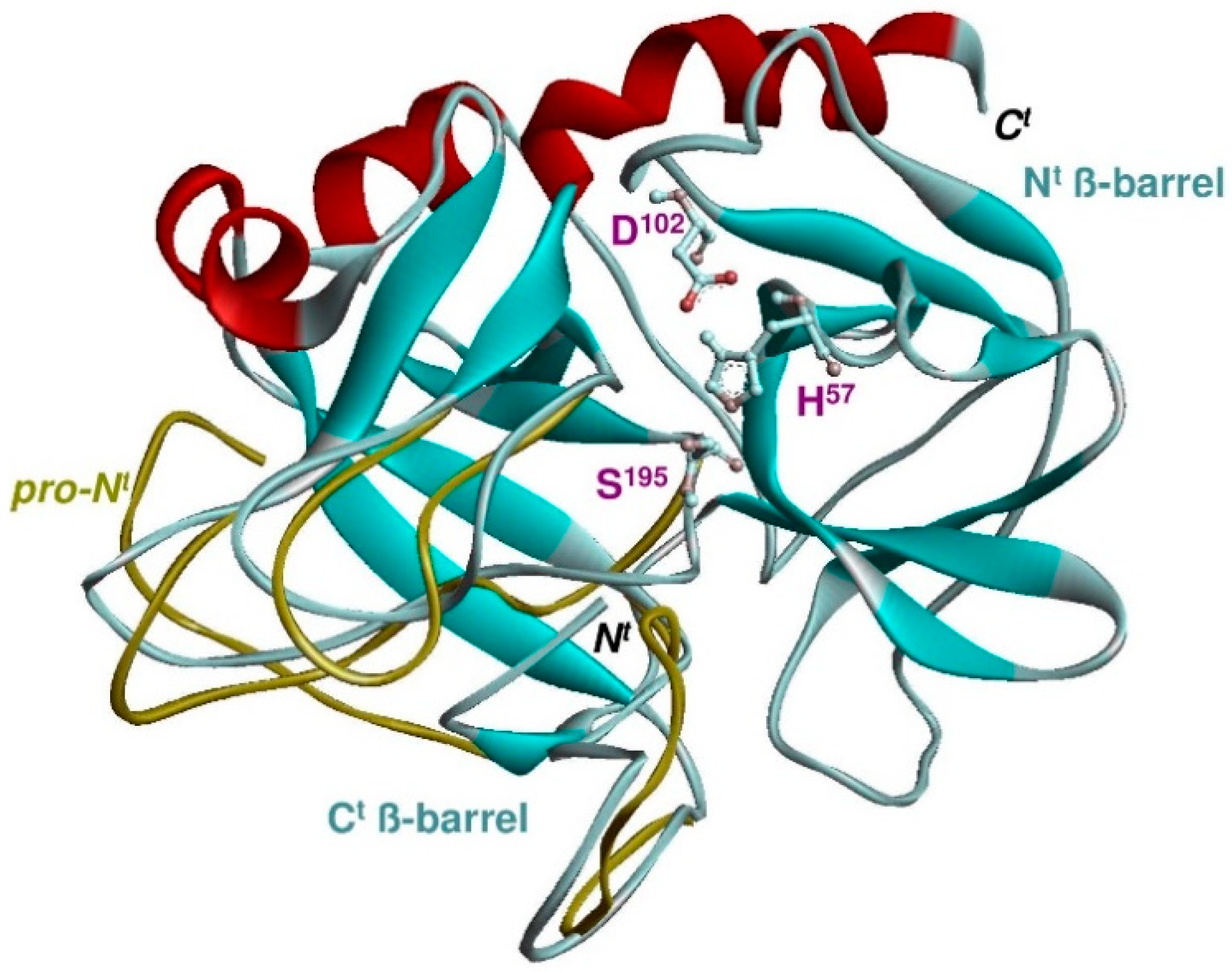{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction: Distinction between an Ancient Trypsin and Chymotrypsin Cluster
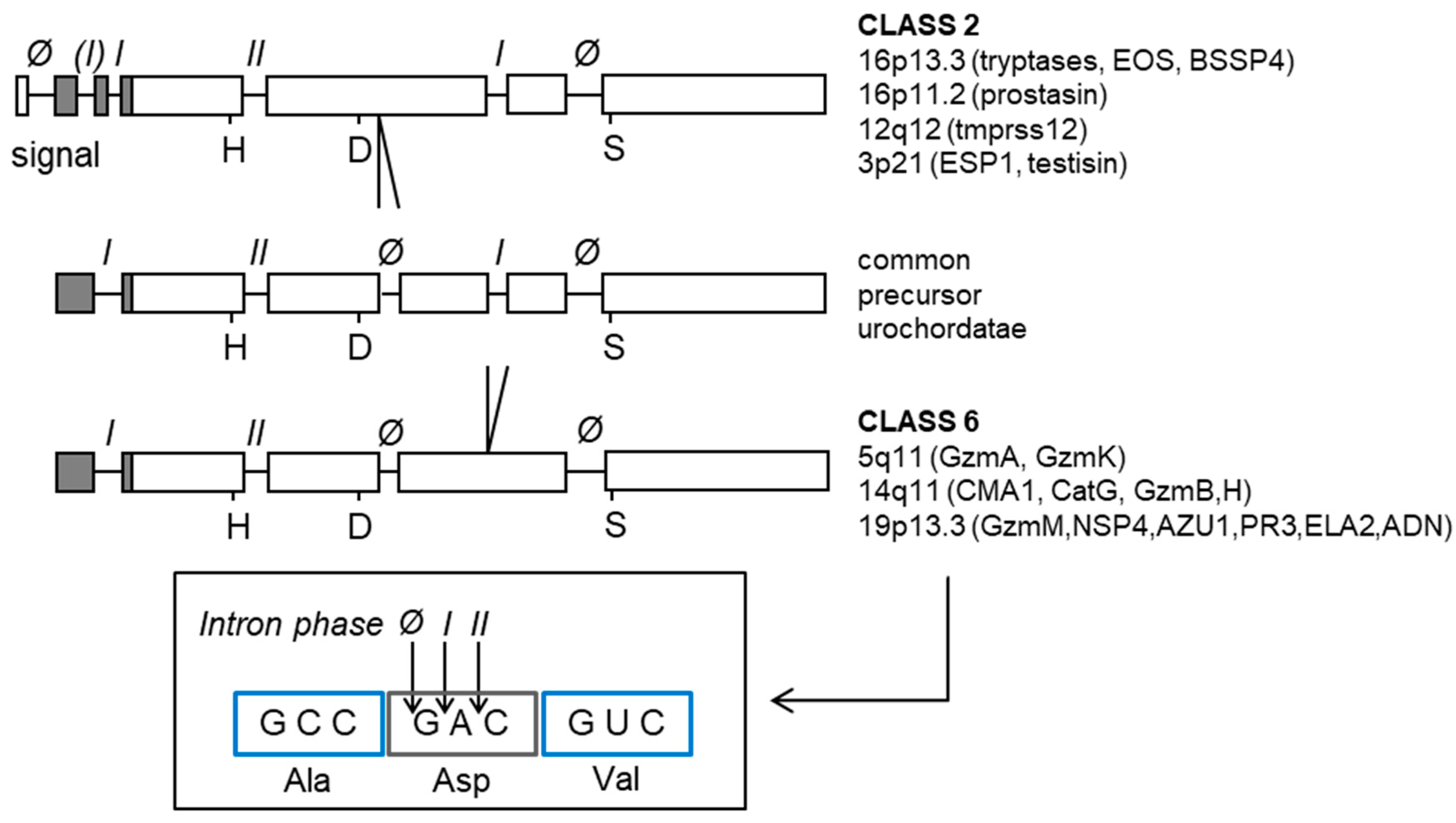
2. Simple Serine Proteases of Vertebrates are Trypsin Descendants
3. Repurposing of the Early Trypsin Ancestor, Complement Factor D
4. The Structural Basis for New S1 Specificities
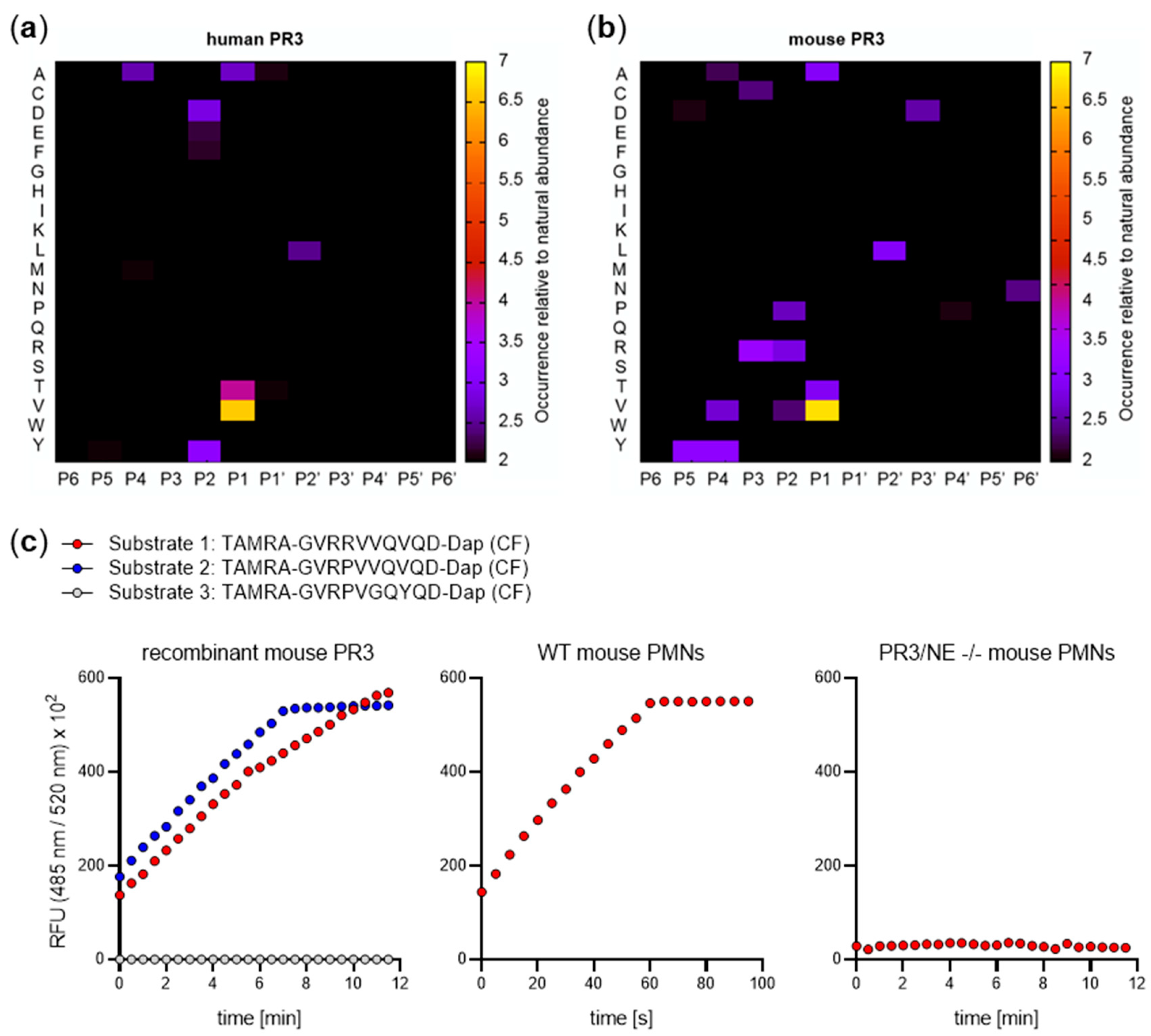
5. Substrate Profiling of Neutrophil Serine Proteases
6. Conversion of the Trypsin Ancestor into Chymotrypsin/Elastase-Like Enzymes
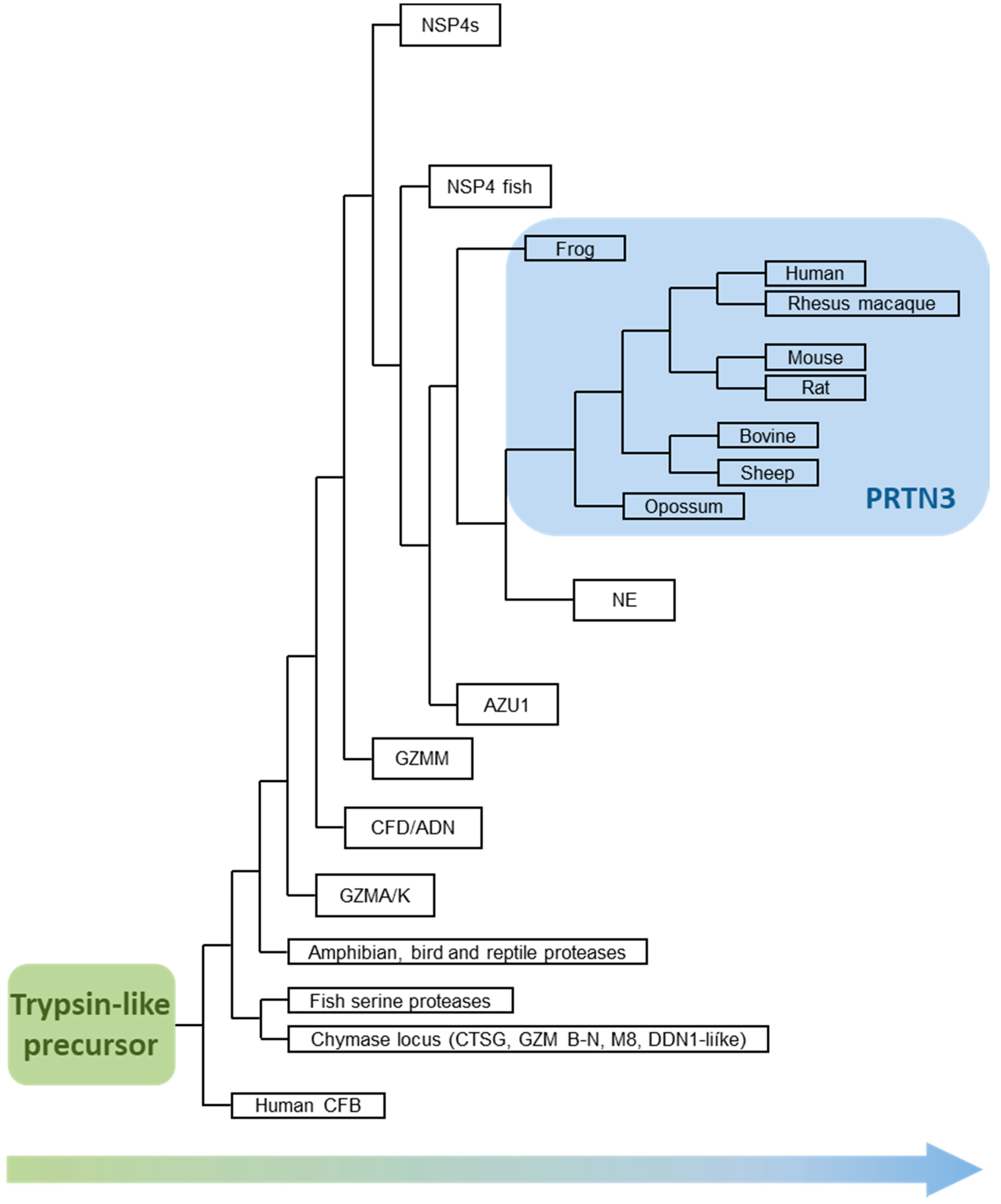
7. PR3 Preceded Leukocyte Elastase (ELANE)
8. Functional Divergence of PR3 in Mouse and Man
9. Regulation of Proteinase 3 Activity
10. Impact of Proteinase 3 on Human Disease
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADN | Adipsin |
| ANCA | Anti-neutrophil cytoplasmic antibody |
| AZU1 | Azurocidin 1 |
| BSSP4 | Brain-specific serine protease 4 |
| C1r-LP | C1r-like serine protease |
| C5a | Complement component 5a |
| C5aR | Complement component 5a receptor |
| CatG | Cathepsin G |
| CF | Cystic fibrosis |
| CFB | Complement factor B |
| CFD | Complement factor D |
| Cit | Citrullin |
| CMA1 | Chymase 1 |
| COPD | Chronic obstructive pulmonary disease |
| CTSG | Cathepsin G gene |
| DDN1 | Duodenase 1 |
| ECM | Extracellular matrix |
| ELA2 | Neutrophil elastase |
| ER | Endoplasmatic reticulum |
| ESP1 | Separin |
| FRET | Förster resonance energy transfer |
| GPA | Granulomatosis with polyangiitis |
| Gzm | Granzyme |
| GZMA/K | Granzyme A/K gene |
| GZMM | Granzyme M gene |
| IL-1β | Interleukin-1-beta |
| MNEI | Monocyte neutrophil elastase inhibitor |
| mPR3 | Membrane-bound proteinase 3 |
| NE | Neutrophil elastase |
| NET | Neutrophil extracellular trap |
| NSP4 | Neutrophil serine protease 4 |
| Orn | Ornithine |
| PGRN | Progranulin |
| PR3 | Proteinase 3 |
| PRTN3 | Proteinase 3 |
| RCL | Reactive center loop |
| TAMRA | Carboxytetramethylrhodamine |
| Tmprss12 | Transmembrane protease serine 12 |
| TNF-α | Tumor necrosis factor alpha |
| α1PI | Alpha-1 protease inhibitor |
References
- Perona, J.J.; Craik, C.S. Structural basis of substrate specificity in the serine proteases. Protein Sci. 1995, 4, 337–360. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Kopetzki, E.; Kresse, G.B.; Bode, W.; Huber, R.; Engh, R.A. New enzyme lineages by subdomain shuffling. Proc. Natl. Acad. Sci. USA 1998, 95, 9813–9818. [Google Scholar] [CrossRef]
- Jenne, D.E.; Kuhl, A. Production and applications of recombinant proteinase 3, Wegener’s autoantigen: Problems and perspectives. Clin. Nephrol. 2006, 66, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J. Evolutionary families of peptidases. Biochem. J. 1993, 290, 205–218. [Google Scholar] [CrossRef]
- Rojas, A.; Doolittle, R.F. The occurrence of type S1A serine proteases in sponge and jellyfish. J. Mol. Evol. 2002, 55, 790–794. [Google Scholar] [CrossRef]
- Ponczek, M.B.; Bijak, M.Z.; Nowak, P.Z. Evolution of thrombin and other hemostatic proteases by survey of protochordate, hemichordate, and echinoderm genomes. J. Mol. Evol. 2012, 74, 319–331. [Google Scholar] [CrossRef]
- Rogers, J. Exon shuffling and intron insertion in serine protease genes. Nature 1985, 315, 458–459. [Google Scholar] [CrossRef]
- Doolittle, R.F. Coagulation in vertebrates with a focus on evolution and inflammation. J. Innate Immun. 2011, 3, 9–16. [Google Scholar] [CrossRef]
- Irwin, D.M.; Robertson, K.A.; MacGillivray, R.T. Structure and evolution of the bovine prothrombin gene. J. Mol. Biol. 1988, 200, 31–45. [Google Scholar] [CrossRef]
- Fu, Z.; Thorpe, M.; Akula, S.; Chahal, G.; Hellman, L.T. Extended Cleavage Specificity of Human Neutrophil Elastase, Human Proteinase 3, and Their Distant Ortholog Clawed Frog PR3-Three Elastases with Similar Primary but Different Extended Specificities and Stability. Front. Immunol. 2018, 9, 2387. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, L.; Szilagyi, L.; Rutter, W.J. Converting trypsin to chymotrypsin: The role of surface loops. Science 1992, 255, 1249–1253. [Google Scholar] [CrossRef]
- Perona, J.J.; Hedstrom, L.; Rutter, W.J.; Fletterick, R.J. Structural origins of substrate discrimination in trypsin and chymotrypsin. Biochemistry 1995, 34, 1489–1499. [Google Scholar] [CrossRef]
- Schulze, R.J.; Schott, M.B.; Casey, C.A.; Tuma, P.L.; McNiven, M.A. The cell biology of the hepatocyte: A membrane trafficking machine. J. Cell. Biol. 2019, 218, 2096–2112. [Google Scholar] [CrossRef]
- Fankboner, P.V. Digestive System of Invertebrates. In Encyclopedia of Life Sciences; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Neurath, H.; Walsh, K.A. Role of proteolytic enzymes in biological regulation (a review). Proc. Natl. Acad. Sci. USA 1976, 73, 3825–3832. [Google Scholar] [CrossRef]
- Chen, J.M.; Montier, T.; Ferec, C. Molecular pathology and evolutionary and physiological implications of pancreatitis-associated cationic trypsinogen mutations. Hum Genet 2001, 109, 245–252. [Google Scholar] [CrossRef]
- Le Marechal, C.; Masson, E.; Chen, J.M.; Morel, F.; Ruszniewski, P.; Levy, P.; Ferec, C. Hereditary pancreatitis caused by triplication of the trypsinogen locus. Nat. Genet. 2006, 38, 1372–1374. [Google Scholar] [CrossRef] [PubMed]
- Nemoda, Z.; Sahin-Toth, M. The tetra-aspartate motif in the activation peptide of human cationic trypsinogen is essential for autoactivation control but not for enteropeptidase recognition. J. Biol. Chem. 2005, 280, 29645–29652. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, S.; Ogasawara, M. Compartmentalized expression patterns of pancreatic- and gastric-related genes in the alimentary canal of the ascidian Ciona intestinalis: Evolutionary insights into the functional regionality of the gastrointestinal tract in Olfactores. Cell Tissue Res. 2017, 370, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Jenne, D.E. Structure of the azurocidin, proteinase 3, and neutrophil elastase genes. Implications for inflammation and vasculitis. Am. J. Respir. Crit. Care Med. 1994, 150, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, C.E. Hepatic ontogenesis. Hepatology 1985, 5, 1213–1221. [Google Scholar] [CrossRef]
- Zaret, K.S.; Grompe, M. Generation and regeneration of cells of the liver and pancreas. Science 2008, 322, 1490–1494. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.S.; Cook, K.S.; Yaglom, J.; Groves, D.L.; Volanakis, J.E.; Damm, D.; White, T.; Spiegelman, B.M. Adipsin and complement factor D activity: An immune-related defect in obesity. Science 1989, 244, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, A.; Lowell, B.B.; Damm, D.; Leibel, R.L.; Ravussin, E.; Jimerson, D.C.; Lesem, M.D.; Van Dyke, D.C.; Daly, P.A.; Chatis, P.; et al. Concentrations of adipsin in blood and rates of adipsin secretion by adipose tissue in humans with normal, elevated and diminished adipose tissue mass. Int. J. Obes. Relat. Metab. Disord. 1994, 18, 213–218. [Google Scholar]
- Volanakis, J.E.; Narayana, S.V. Complement factor D, a novel serine protease. Protein Sci. 1996, 5, 553–564. [Google Scholar] [CrossRef]
- Forneris, F.; Ricklin, D.; Wu, J.; Tzekou, A.; Wallace, R.S.; Lambris, J.D.; Gros, P. Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science 2010, 330, 1816–1820. [Google Scholar] [CrossRef] [PubMed]
- Abrera-Abeleda, M.A.; Xu, Y.; Pickering, M.C.; Smith, R.J.; Sethi, S. Mesangial immune complex glomerulonephritis due to complement factor D deficiency. Kidney Int. 2007, 71, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Novel mechanisms and functions of complement. Nat. Immunol. 2017, 18, 1288–1298. [Google Scholar] [CrossRef] [PubMed]
- Stahl, G.L.; Xu, Y.; Hao, L.; Miller, M.; Buras, J.A.; Fung, M.; Zhao, H. Role for the alternative complement pathway in ischemia/reperfusion injury. Am. J. Pathol. 2003, 162, 449–455. [Google Scholar] [CrossRef][Green Version]
- Turnberg, D.; Lewis, M.; Moss, J.; Xu, Y.; Botto, M.; Cook, H.T. Complement activation contributes to both glomerular and tubulointerstitial damage in adriamycin nephropathy in mice. J. Immunol. 2006, 177, 4094–4102. [Google Scholar] [CrossRef]
- Hiemstra, P.S.; Langeler, E.; Compier, B.; Keepers, Y.; Leijh, P.C.; van den Barselaar, M.T.; Overbosch, D.; Daha, M.R. Complete and partial deficiencies of complement factor D in a Dutch family. J. Clin. Investig. 1989, 84, 1957–1961. [Google Scholar] [CrossRef]
- Biesma, D.H.; Hannema, A.J.; van Velzen-Blad, H.; Mulder, L.; van Zwieten, R.; Kluijt, I.; Roos, D. A family with complement factor D deficiency. J. Clin. Investig. 2001, 108, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ma, M.; Ippolito, G.C.; Schroeder, H.W., Jr.; Carroll, M.C.; Volanakis, J.E. Complement activation in factor D-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 14577–14582. [Google Scholar] [CrossRef] [PubMed]
- Sprong, T.; Roos, D.; Weemaes, C.; Neeleman, C.; Geesing, C.L.; Mollnes, T.E.; van Deuren, M. Deficient alternative complement pathway activation due to factor D deficiency by 2 novel mutations in the complement factor D gene in a family with meningococcal infections. Blood 2006, 107, 4865–4870. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; McCrory, M.A.; Pass, C.; Bullard, D.C.; Ballantyne, C.M.; Xu, Y.; Briles, D.E.; Szalai, A.J. The virulence function of Streptococcus pneumoniae surface protein A involves inhibition of complement activation and impairment of complement receptor-mediated protection. J. Immunol. 2004, 173, 7506–7512. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.S.; Min, H.Y.; Johnson, D.; Chaplinsky, R.J.; Flier, J.S.; Hunt, C.R.; Spiegelman, B.M. Adipsin: A circulating serine protease homolog secreted by adipose tissue and sciatic nerve. Science 1987, 237, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zou, W.; Brestoff, J.R.; Rohatgi, N.; Wu, X.; Atkinson, J.P.; Harris, C.A.; Teitelbaum, S.L. Fat-Produced Adipsin Regulates Inflammatory Arthritis. Cell Rep. 2019, 27, 2809–2816. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Banoy, N.; Guseh, J.S.; Li, G.; Rubio-Navarro, A.; Chen, T.; Poirier, B.; Putzel, G.; Rosselot, C.; Pabon, M.A.; Camporez, J.P.; et al. Adipsin preserves beta cells in diabetic mice and associates with protection from type 2 diabetes in humans. Nat. Med. 2019, 25, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Schneider, D.S.; Soares, M.P. Disease tolerance as a defense strategy. Science 2012, 335, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J. MEROPS: The peptidase database. Nucleic Acids Res. 1999, 27, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Sichler, K.; Hopfner, K.P.; Kopetzki, E.; Huber, R.; Bode, W.; Brandstetter, H. The influence of residue 190 in the S1 site of trypsin-like serine proteases on substrate selectivity is universally conserved. FEBS Lett. 2002, 530, 220–224. [Google Scholar] [CrossRef]
- Pozzi, N.; Vogt, A.D.; Gohara, D.W.; Di Cera, E. Conformational selection in trypsin-like proteases. Curr. Opin. Struct. Biol. 2012, 22, 421–431. [Google Scholar] [CrossRef]
- Jing, H.; Xu, Y.; Carson, M.; Moore, D.; Macon, K.J.; Volanakis, J.E.; Narayana, S.V. New structural motifs on the chymotrypsin fold and their potential roles in complement factor B. EMBO J. 2000, 19, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Ligoudistianou, C.; Xu, Y.; Garnier, G.; Circolo, A.; Volanakis, J.E. A novel human complement-related protein, C1r-like protease (C1r-LP), specifically cleaves pro-C1s. Biochem. J. 2005, 387, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Circolo, A.; Jing, H.; Wang, Y.; Narayana, S.V.; Volanakis, J.E. Mutational analysis of the primary substrate specificity pocket of complement factor B. Asp(226) is a major structural determinant for p(1)-Arg binding. J. Biol. Chem. 2000, 275, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Wicher, K.B.; Fries, E. Prohaptoglobin is proteolytically cleaved in the endoplasmic reticulum by the complement C1r-like protein. Proc. Natl. Acad. Sci. USA 2004, 101, 14390–14395. [Google Scholar] [CrossRef] [PubMed]
- Perona, J.J.; Tsu, C.A.; McGrath, M.E.; Craik, C.S.; Fletterick, R.J. Relocating a negative charge in the binding pocket of trypsin. J. Mol. Biol. 1993, 230, 934–949. [Google Scholar] [CrossRef]
- Chen, S.; Yim, J.J.; Bogyo, M. Synthetic and biological approaches to map substrate specificities of proteases. Biol. Chem. 2019, 401, 165–182. [Google Scholar] [CrossRef]
- Ivry, S.L.; Meyer, N.O.; Winter, M.B.; Bohn, M.F.; Knudsen, G.M.; O’Donoghue, A.J.; Craik, C.S. Global substrate specificity profiling of post-translational modifying enzymes. Protein Sci. 2018, 27, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Overall, C.M.; Blobel, C.P. In search of partners: Linking extracellular proteases to substrates. Nat. Rev. Mol. Cell. Biol. 2007, 8, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Sabino, F.; Hermes, O.; Egli, F.E.; Kockmann, T.; Schlage, P.; Croizat, P.; Kizhakkedathu, J.N.; Smola, H.; auf dem Keller, U. In vivo assessment of protease dynamics in cutaneous wound healing by degradomics analysis of porcine wound exudates. Mol. Cell Proteom. 2015, 14, 354–370. [Google Scholar] [CrossRef] [PubMed]
- Kam, C.M.; Kerrigan, J.E.; Dolman, K.M.; Goldschmeding, R.; Born, A.E.V.D.; Powers, J.C. Substrate and inhibitor studies on proteinase 3. FEBS Lett. 1992, 297, 119–123. [Google Scholar] [CrossRef]
- Kasperkiewicz, P.; Poreba, M.; Snipas, S.J.; Parker, H.; Winterbourn, C.C.; Salvesen, G.S.; Drag, M. Design of ultrasensitive probes for human neutrophil elastase through hybrid combinatorial substrate library profiling. Proc. Natl. Acad. Sci. USA 2014, 111, 2518–2523. [Google Scholar] [CrossRef]
- Wysocka, M.; Lesner, A.; Gruba, N.; Korkmaz, B.; Gauthier, F.; Kitamatsu, M.; Anna, Ł.; Rolka, K. Three Wavelength Substrate System of Neutrophil Serine Proteinases. Anal. Chem. 2012, 84, 7241–7248. [Google Scholar] [CrossRef] [PubMed]
- Poreba, M.; Szalek, A.; Rut, W.; Kasperkiewicz, P.; Rutkowska-Wlodarczyk, I.; Snipas, S.J.; Itoh, Y.; Turk, D.; Turk, B.; Overall, C.M.; et al. Highly sensitive and adaptable fluorescence-quenched pair discloses the substrate specificity profiles in diverse protease families. Sci. Rep. 2017, 7, 43135. [Google Scholar] [CrossRef]
- Thorpe, M.; Fu, Z.; Chahal, G.; Akula, S.; Kervinen, J.; de Garavilla, L.; Hellman, L. Extended cleavage specificity of human neutrophil cathepsin G: A low activity protease with dual chymase and tryptase-type specificities. PLoS ONE 2018, 13, e0195077. [Google Scholar] [CrossRef]
- Adkison, A.M.; Raptis, S.Z.; Kelley, D.G.; Pham, C.T. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J. Clin. Investig. 2002, 109, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Jenne, D.E.; Tschopp, J. Granzymes: A family of serine proteases in granules of cytolytic T lymphocytes. Curr. Top Microbiol. Immunol. 1989, 140, 33–47. [Google Scholar] [CrossRef]
- Perera, N.C.; Schilling, O.; Kittel, H.; Back, W.; Kremmer, E.; Jenne, D.E. NSP4, an elastase-related protease in human neutrophils with arginine specificity. Proc. Natl. Acad. Sci. USA 2012, 109, 6229–6234. [Google Scholar] [CrossRef] [PubMed]
- Perera, N.C.; Jenne, D.E. Perspectives and potential roles for the newly discovered NSP4 in the immune system. Expert Rev. Clin. Immunol. 2012, 8, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Bleackley, R.C. A molecular view of cytotoxic T lymphocyte induced killing. Biochem. Cell. Biol. 2005, 83, 747–751. [Google Scholar] [CrossRef]
- Han, M.V.; Demuth, J.P.; McGrath, C.L.; Casola, C.; Hahn, M.W. Adaptive evolution of young gene duplicates in mammals. Genome Res. 2009, 19, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Siddiq, M.A.; Hochberg, G.K.; Thornton, J.W. Evolution of protein specificity: Insights from ancestral protein reconstruction. Curr. Opin. Struct. Biol. 2017, 47, 113–122. [Google Scholar] [CrossRef]
- Wouters, M.A.; Liu, K.; Riek, P.; Husain, A. A despecialization step underlying evolution of a family of serine proteases. Mol. Cell 2003, 12, 343–354. [Google Scholar] [CrossRef]
- Tsu, C.A.; Perona, J.J.; Fletterick, R.J.; Craik, C.S. Structural basis for the broad substrate specificity of fiddler crab collagenolytic serine protease 1. Biochemistry 1997, 36, 5393–5401. [Google Scholar] [CrossRef]
- Grant, G.A.; Henderson, K.O.; Eisen, A.Z.; Bradshaw, R.A. Amino acid sequence of a collagenolytic protease from the hepatopancreas of the fiddler crab, Uca pugilator. Biochemistry 1980, 19, 4653–4659. [Google Scholar] [CrossRef] [PubMed]
- Perera, N.C.; Wiesmuller, K.H.; Larsen, M.T.; Schacher, B.; Eickholz, P.; Borregaard, N.; Jenne, D.E. NSP4 is stored in azurophil granules and released by activated neutrophils as active endoprotease with restricted specificity. J. Immunol. 2013, 191, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- AhYoung, A.P.; Lin, S.J.; Gerhardy, S.; van Lookeren Campagne, M.; Kirchhofer, D. An ancient mechanism of arginine-specific substrate cleavage: What’s ’up’ with NSP4? Biochimie 2019, 166, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.J.; Dong, K.C.; Eigenbrot, C.; van Lookeren Campagne, M.; Kirchhofer, D. Structures of neutrophil serine protease 4 reveal an unusual mechanism of substrate recognition by a trypsin-fold protease. Structure 2014, 22, 1333–1340. [Google Scholar] [CrossRef]
- Akula, S.; Thorpe, M.; Boinapally, V.; Hellman, L. Granule Associated Serine Proteases of Hematopoietic Cells—An Analysis of Their Appearance and Diversification during Vertebrate Evolution. PLoS ONE 2015, 10, e0143091. [Google Scholar] [CrossRef]
- De Poot, S.A.; Bovenschen, N. Granzyme M: Behind enemy lines. Cell Death Differ. 2014, 21, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Sacerdot, C.; Louis, A.; Bon, C.; Berthelot, C.; Roest Crollius, H. Chromosome evolution at the origin of the ancestral vertebrate genome. Genome Biol. 2018, 19, 166. [Google Scholar] [CrossRef]
- Puente, X.S.; Sánchez, L.M.; Overall, C.M.; López-Otín, C. Human and mouse proteases: A comparative genomic approach. Nat. Rev. Genet. 2003, 4, 544–558. [Google Scholar] [CrossRef]
- Warren, W.C.; Hillier, L.W.; Marshall Graves, J.A.; Birney, E.; Ponting, C.P.; Grutzner, F.; Belov, K.; Miller, W.; Clarke, L.; Chinwalla, A.T.; et al. Genome analysis of the platypus reveals unique signatures of evolution. Nature 2008, 453, 175–183. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kumar, A. Bayesian phylogeny analysis of vertebrate serpins illustrates evolutionary conservation of the intron and indels based six groups classification system from lampreys for ∼500 MY. PeerJ. 2015, 16, e1026. [Google Scholar] [CrossRef] [PubMed]
- Benarafa, C.; Simon, H.U. Role of granule proteases in the life and death of neutrophils. Biochem. Biophys. Res. Commun. 2017, 482, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Kalsheker, N.; Morley, S.; Morgan, K. Gene regulation of the serine proteinase inhibitors alpha1-antitrypsin and alpha1-antichymotrypsin. Biochem. Soc. Trans. 2002, 30, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, B.; Attucci, S.; Hazouard, E.; Ferrandiere, M.; Jourdan, M.L.; Brillard-Bourdet, M.; Juliano, L.; Gauthier, F. Discriminating between the activities of human neutrophil elastase and proteinase 3 using serpin-derived fluorogenic substrates. J. Biol. Chem. 2002, 277, 39074–39081. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, E.; Korkmaz, B.; Gauthier, F.; Brandsdal, B.O.; Witko-Sarsat, V.; Reuter, N. Inspection of the binding sites of proteinase3 for the design of a highly specific substrate. J. Med. Chem. 2006, 49, 1248–1260. [Google Scholar] [CrossRef]
- Biniossek, M.L.; Niemer, M.; Maksimchuk, K.; Mayer, B.; Fuchs, J.; Huesgen, P.F.; McCafferty, D.G.; Turk, B.; Fritz, G.; Mayer, J.; et al. Identification of Protease Specificity by Combining Proteome-Derived Peptide Libraries and Quantitative Proteomics. Mol. Cell Proteom. 2016, 15, 2515–2524. [Google Scholar] [CrossRef]
- Schilling, O.; Biniossek, M.L.; Mayer, B.; Elsasser, B.; Brandstetter, H.; Goettig, P.; Stenman, U.H.; Koistinen, H. Specificity profiling of human trypsin-isoenzymes. Biol. Chem. 2018, 399, 997–1007. [Google Scholar] [CrossRef]
- Halbwachs-Mecarelli, L.; Bessou, G.; Lesavre, P.; Lopez, S.; Witko-Sarsat, V. Bimodal distribution of proteinase 3 (PR3) surface expression reflects a constitutive heterogeneity in the polymorphonuclear neutrophil pool. FEBS Lett. 1995, 374, 29–33. [Google Scholar] [CrossRef]
- Korkmaz, B.; Kuhl, A.; Bayat, B.; Santoso, S.; Jenne, D.E. A hydrophobic patch on proteinase 3, the target of autoantibodies in Wegener granulomatosis, mediates membrane binding via NB1 receptors. J. Biol. Chem. 2008, 283, 35976–35982. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.; Eulenberg-Gustavus, C.; Bergmann, A.; Jerke, U.; Kettritz, R. Lessons from a double-transgenic neutrophil approach to induce antiproteinase 3 antibody-mediated vasculitis in mice. J. Leukoc. Biol. 2016, 100, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.J.; Campbell, M.A.; Owen, C.A. Bioactive proteinase 3 on the cell surface of human neutrophils: Quantification, catalytic activity, and susceptibility to inhibition. J. Immunol. 2000, 165, 3366–3374. [Google Scholar] [CrossRef]
- Schreiber, A.; Luft, F.C.; Kettritz, R. Membrane proteinase 3 expression and ANCA-induced neutrophil activation. Kidney Int. 2004, 65, 2172–2183. [Google Scholar] [CrossRef]
- Huntington, J.A.; Read, R.J.; Carrell, R.W. Structure of a serpin-protease complex shows inhibition by deformation. Nature 2000, 407, 923–926. [Google Scholar] [CrossRef]
- Poller, W.; Willnow, T.E.; Hilpert, J.; Herz, J. Differential recognition of alpha 1-antitrypsin-elastase and alpha 1-antichymotrypsin-cathepsin G complexes by the low density lipoprotein receptor-related protein. J. Biol. Chem. 1995, 270, 2841–2845. [Google Scholar] [CrossRef]
- Cooley, J.; Takayama, T.K.; Shapiro, S.D.; Schechter, N.M.; Remold-O’Donnell, E. The serpin MNEI inhibits elastase-like and chymotrypsin-like serine proteases through efficient reactions at two active sites. Biochemistry 2001, 40, 15762–15770. [Google Scholar] [CrossRef]
- Loison, F.; Xu, Y.; Luo, H.R. Proteinase 3 and Serpin B1: A novel pathway in the regulation of caspase-3 activation, neutrophil spontaneous apoptosis, and inflammation. Inflamm. Cell Signal 2014, 1. [Google Scholar] [CrossRef]
- Barrett, A.J.; Starkey, P.M. The interaction of alpha 2-macroglobulin with proteinases. Characteristics and specificity of the reaction, and a hypothesis concerning its molecular mechanism. Biochem. J. 1973, 133, 709–724. [Google Scholar] [CrossRef]
- Wiedow, O.; Luademann, J.; Utecht, B. Elafin is a potent inhibitor of proteinase 3. Biochem. Biophys. Res. Commun. 1991, 174, 6–10. [Google Scholar] [CrossRef]
- Beatty, K.; Bieth, J.; Travis, J. Kinetics of association of serine proteinases with native and oxidized alpha-1-proteinase inhibitor and alpha-1-antichymotrypsin. J. Biol. Chem. 1980, 255, 3931–3934. [Google Scholar] [CrossRef]
- Korkmaz, B.; Kellenberger, C.; Viaud-Massuard, M.C.; Gauthier, F. Selective inhibitors of human neutrophil proteinase 3. Curr. Pharm. Des. 2013, 19, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Hinkofer, L.C.; Hummel, A.M.; Stone, J.H.; Hoffman, G.S.; Merkel, P.A.; Spiera, E.R.; St Clair, W.; McCune, J.W.; Davis, J.C.; Specks, U.; et al. Allosteric modulation of proteinase 3 activity by anti-neutrophil cytoplasmic antibodies in granulomatosis with polyangiitis. J. Autoimmun. 2015, 59, 43–52. [Google Scholar] [CrossRef]
- Hinkofer, L.C.; Seidel, S.A.; Korkmaz, B.; Silva, F.; Hummel, A.M.; Braun, D.; Jenne, D.E.; Specks, U. A monoclonal antibody (MCPR3-7) interfering with the activity of proteinase 3 by an allosteric mechanism. J. Biol. Chem. 2013, 288, 26635–26648. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.P.; Casal Moura, M.; Thompson, G.E.; Nelson, D.R.; Hummel, A.M.; Jenne, D.E.; Emerling, D.; Volkmuth, W.; Robinson, W.H.; Specks, U. Remote Activation of a Latent Epitope in an Autoantigen Decoded with Simulated B-Factors. Front. Immunol. 2019, 10, 2467. [Google Scholar] [CrossRef]
- Jenne, D.E.; Tschopp, J.; Ludemann, J.; Utecht, B.; Gross, W.L. Wegener’s autoantigen decoded. Nature 1990, 346, 520. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Dau, T.; Jenne, D.E. Tailor-made inflammation: How neutrophil serine proteases modulate the inflammatory response. J. Mol. Med. 2011, 89, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Kettritz, R. How anti-neutrophil cytoplasmic autoantibodies activate neutrophils. Clin. Exp. Immunol. 2012, 169, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C.; Falk, R.J.; Gasim, A.H. Pathogenesis of antineutrophil cytoplasmic autoantibody vasculitis. Curr. Opin. Nephrol. Hypertens. 2011, 20, 263–270. [Google Scholar] [CrossRef]
- Sinden, N.J.; Baker, M.J.; Smith, D.J.; Kreft, J.U.; Dafforn, T.R.; Stockley, R.A. alpha-1-antitrypsin variants and the proteinase/antiproteinase imbalance in chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L179–L190. [Google Scholar] [CrossRef] [PubMed]
- Gudmann, N.S.; Manon-Jensen, T.; Sand, J.M.B.; Diefenbach, C.; Sun, S.; Danielsen, A.; Karsdal, M.A.; Leeming, D.J. Lung tissue destruction by proteinase 3 and cathepsin G mediated elastin degradation is elevated in chronic obstructive pulmonary disease. Biochem. Biophys. Res. Commun. 2018, 503, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Sinden, N.J.; Stockley, R.A. Proteinase 3 activity in sputum from subjects with alpha-1-antitrypsin deficiency and COPD. Eur. Respir. J. 2013, 41, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Witko-Sarsat, V.; Halbwachs-Mecarelli, L.; Schuster, A.; Nusbaum, P.; Ueki, I.; Canteloup, S.; Lenoir, G.; Descamps-Latscha, B.; Nadel, J.A. Proteinase 3, a potent secretagogue in airways, is present in cystic fibrosis sputum. Am. J. Respir. Cell Mol. Biol. 1999, 20, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Coeshott, C.; Ohnemus, C.; Pilyavskaya, A.; Ross, S.; Wieczorek, M.; Kroona, H.; Leimer, A.H.; Cheronis, J. Converting enzyme-independent release of tumor necrosis factor alpha and IL-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc. Natl. Acad. Sci. USA 1999, 96, 6261–6266. [Google Scholar] [CrossRef]
- Padrines, M.; Wolf, M.; Walz, A.; Baggiolini, M. Interleukin-8 processing by neutrophil elastase, cathepsin G and proteinase-3. FEBS Lett. 1994, 352, 231–235. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Krumbholz, M.; Schonermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Grone, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef]
- Zhu, J.; Nathan, C.; Jin, W.; Sim, D.; Ashcroft, G.S.; Wahl, S.M.; Lacomis, L.; Erdjument-Bromage, H.; Tempst, P.; Wright, C.D.; et al. Conversion of proepithelin to epithelins: Roles of SLPI and elastase in host defense and wound repair. Cell 2002, 111, 867–878. [Google Scholar] [CrossRef]
- Rao, N.V.; Wehner, N.G.; Marshall, B.C.; Gray, W.R.; Gray, B.H.; Hoidal, J.R. Characterization of proteinase-3 (PR-3), a neutrophil serine proteinase. Structural and functional properties. J. Biol. Chem. 1991, 266, 9540–9548. [Google Scholar] [CrossRef]
- Van den Berg, C.W.; Tambourgi, D.V.; Clark, H.W.; Hoong, S.J.; Spiller, O.B.; McGreal, E.P. Mechanism of neutrophil dysfunction: Neutrophil serine proteases cleave and inactivate the C5a receptor. J. Immunol. 2014, 192, 1787–1795. [Google Scholar] [CrossRef]
- Schonermarck, U.; Lamprecht, P.; Csernok, E.; Gross, W.L. Prevalence and spectrum of rheumatic diseases associated with proteinase 3-antineutrophil cytoplasmic antibodies (ANCA) and myeloperoxidase-ANCA. Rheumatology 2001, 40, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Toonen, E.J.; Mirea, A.M.; Tack, C.J.; Stienstra, R.; Ballak, D.B.; van Diepen, J.A.; Hijmans, A.; Chavakis, T.; Dokter, W.H.; Pham, C.T.; et al. Activation of proteinase 3 contributes to Non-alcoholic Fatty Liver Disease (NAFLD) and insulin resistance. Mol. Med. 2016, 22. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiss, S.A.I.; Rehm, S.R.T.; Perera, N.C.; Biniossek, M.L.; Schilling, O.; Jenne, D.E. Origin and Expansion of the Serine Protease Repertoire in the Myelomonocyte Lineage. Int. J. Mol. Sci. 2021, 22, 1658. https://doi.org/10.3390/ijms22041658
Weiss SAI, Rehm SRT, Perera NC, Biniossek ML, Schilling O, Jenne DE. Origin and Expansion of the Serine Protease Repertoire in the Myelomonocyte Lineage. International Journal of Molecular Sciences. 2021; 22(4):1658. https://doi.org/10.3390/ijms22041658
Chicago/Turabian StyleWeiss, Stefanie A. I., Salome R. T. Rehm, Natascha C. Perera, Martin L. Biniossek, Oliver Schilling, and Dieter E. Jenne. 2021. "Origin and Expansion of the Serine Protease Repertoire in the Myelomonocyte Lineage" International Journal of Molecular Sciences 22, no. 4: 1658. https://doi.org/10.3390/ijms22041658
APA StyleWeiss, S. A. I., Rehm, S. R. T., Perera, N. C., Biniossek, M. L., Schilling, O., & Jenne, D. E. (2021). Origin and Expansion of the Serine Protease Repertoire in the Myelomonocyte Lineage. International Journal of Molecular Sciences, 22(4), 1658. https://doi.org/10.3390/ijms22041658