
Connexins during 500 Million Years—From Cyclostomes to Mammals


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
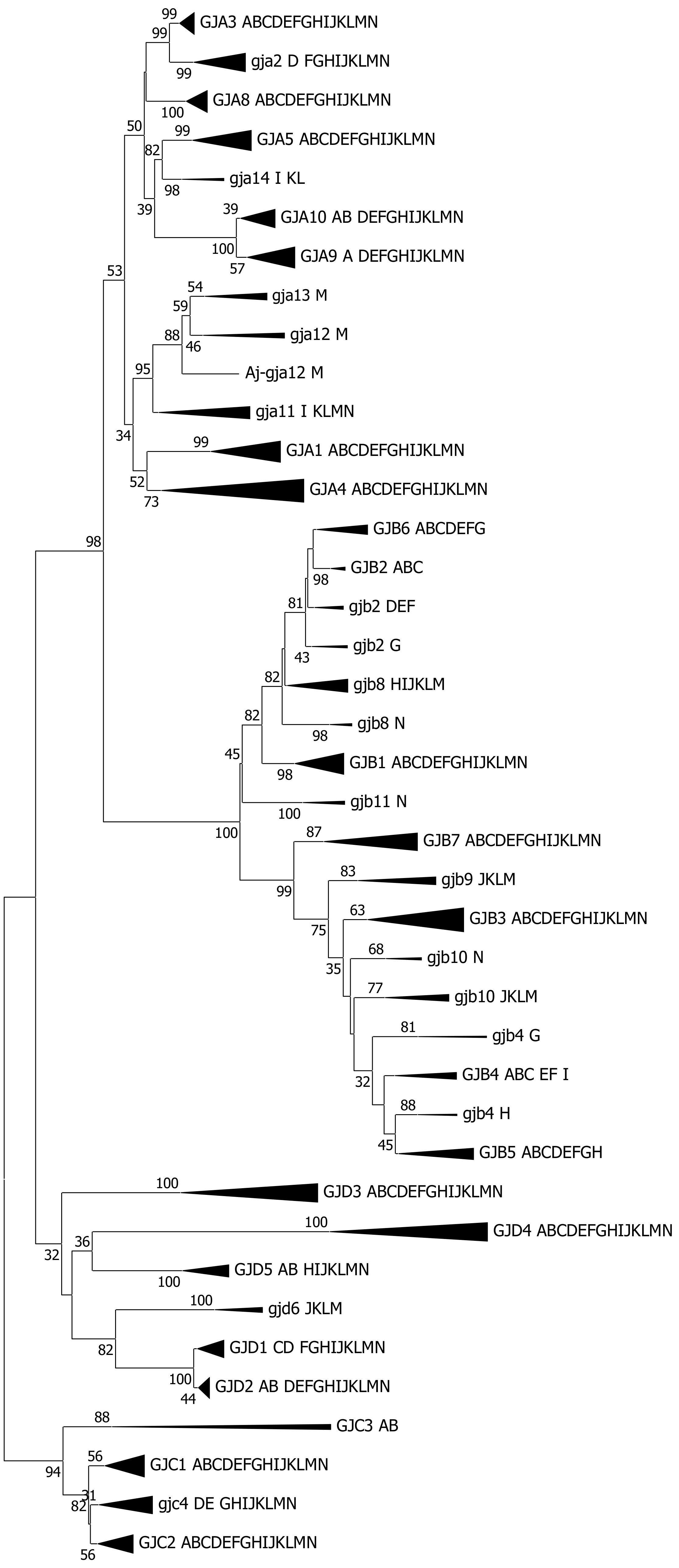
2. Results
2.1. On Genome Duplications and Ohnology
2.2. Chondrichthyes, Actinopterygii (Chondrostei, Holostei and Teleostei) and Sarcopterygii
2.3. Nonmammalian Tetrapods: Amphibia and the Greater Group of Reptilia
2.4. Mammalia
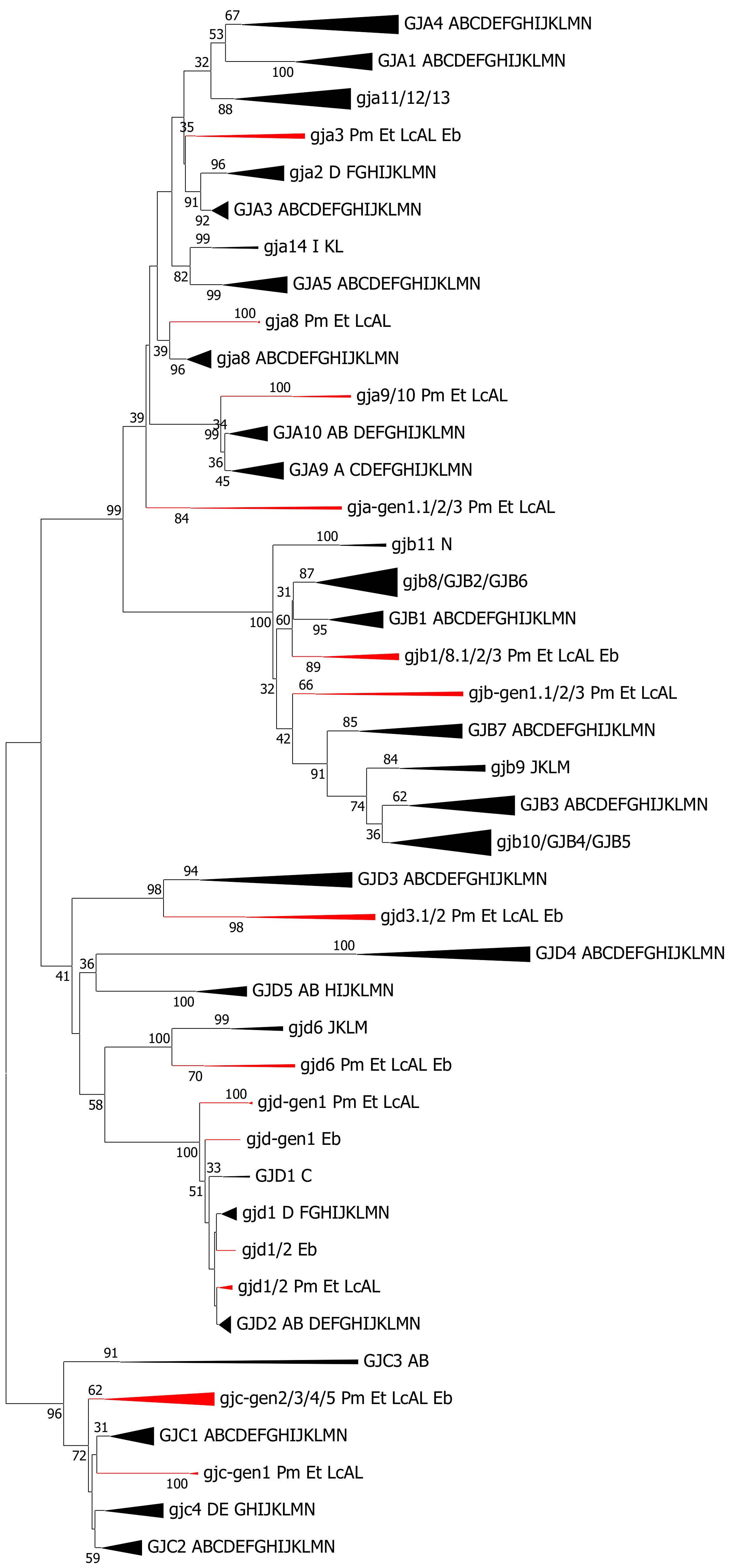
2.5. Cyclostomata: Lampreys and Hagfish
2.6. Comments on the Relationships between Gjb8 and GJB2/GJB6, and Gjb10 and GJB4/GJB5
3. Discussion
3.1. On Genome/Gene Duplication and Naming
3.2. The Number of Connexin Genes in Gnathostomata
3.3. The Connexin Gene Family Substructure
3.4. Cyclostomata vs. Gnathostomata Connexin Gene Families
4. Materials and Methods
4.1. Databanks and Collection of Sequences
- if an expected sequence was not found in a species,
- if only a partial sequence was found, we tried to find the corresponding sequence in a closely related species and
- if a single sequence from a certain species phylogenetically was located in an unexpected way.
- They were very similar to the sequences already collected, with only a few nucleotide differences. The reasons for not including all sequences are (A) the sequences may come from different individuals of the same species, thus having small individual genetic variations, and do not reflect unique genes, (B) there could be errors in the genome assemblies, predicting gene duplications where there are, in reality, no gene duplications and (C) a gene duplication may have occurred in a single species, but in the present work, we are focusing on the larger picture. Examples of such excluded sequences are the zebra finch (Taeniopygia guttata) XM_002198742, XM_002186636 and XM_012571095 and the Chinese softshell turtle (Pelodiscus sinensis) XM_006125970.
- We were not able to define the proper conserved domains, including the cysteine codon signatures. Several predicted sequences did not contain the proper cysteine codon patterns in one or both of the conserved domains or the translation into a protein showed that the expected transmembrane domains deviated too much from the expected nonpolar sequences. An example of such excluded sequences is chicken (Gallus gallus) XM_015299660.
4.2. Alignment of Conserved Domains
4.3. Nomenclature Harmonization by Orthology Consistency
- If the sequence corresponded to a predicted or confirmed sequence in GenBank: (species abbreviation)-(potentially amended “Greek” name)-(accession number)-(database entry name if different from the amended “Greek” name). The species abbreviations are given in Figure 1. Example: for a species with the Latin name “Latin name” (abbreviated Ln), a predicted gene found in GenBank with the accession number “XM_number” may have the name gjd2like, and we find in our phylogenetic analyses that this sequence is a gjd5 sequence. In the phylogenetic trees, the sequence will be called Ln-gjd5-XM_number-gjd2like, while, in the text, the same sequence will be called Ln-gjd5.
- If the sequence corresponded to a sequence predicted in Ensembl (but not in GenBank): (species abbreviation)-(potentially amended “Greek” name)-(abbreviated Ensembl gene number)-(database entry name if different from the amended “Greek” name).
- If the sequence was not predicted in any database, but a transcript with the name could be found in the GenBank Transcriptome Shotgun Assembly (TSA) database: (species abbreviation)-(amended “Greek” name)-(scaffold accession number:start position direction)-(TSA name).
- If the sequence was not predicted in any database, nor found in the TSA database, but could be found in the genome assemblies: (species abbreviation)-(correct “Greek” name)-(scaffold accession number:start position direction).
4.4. Model Selection and Phylogenetic Analyses
5. Conclusions
- Cyclostomes do have a subfamily structure with alpha, beta, gamma and delta genes, but only a few of the genes show a phylogenetic affinity to genes that are found in the higher vertebrates. Thus, at the cyclostome–gnathostome split, an early version of the subfamily structure was established. The subfamily structure in cyclostomes is evidently more advanced than in tunicates [17].
- The connexin subfamily structures (alpha, beta, gamma, delta and epsilon) were established in cartilaginous fishes, and these subfamilies can be followed throughout the vertebrates. Most of the connexin genes in cartilaginous fishes can be classified into orthogroups together with genes from teleosts and other ray-finned fishes.
- It is not obviously evident how the gene family evolved from the “immature” version now found in cyclostomes to the “mature” version found in cartilaginous fishes. As it is assumed a whole genome duplication did not occur after the cyclostome–gnathostome split, the most likely possibility is a combination of gene duplications (that normally generates closely spaced duplicates) and regional duplications followed by rearrangements (that are more likely to generate similar genes on different chromosomes).
- There is no major change in the gene family in the transition from fishes to tetrapods.
- Birds have approximately the same number of connexin genes as other vertebrates (when genes generated from genome duplications or species hybridization are not considered).
- Due to the gene duplications and gene losses in certain vertebrate groups, the exact complement of the genes varies between the vertebrate groups. For example, gjd5 was lost in the Reptilia early after the divergence, as none of the four vertebrate groups Squamata, Testudines, Crocodylia or Aves possess this gene, but it is found in all fish groups and in other tetrapods, including placental mammals and marsupials.
- There are few connexin genes that are specific to a single taxonomic group of vertebrates. Gjb11 is specific to cartilaginous fishes. Gja12 and gja13 in teleosts are most likely generated by gene duplications from gja11, which, on the other hand, is found in most ray-finned fishes and cartilaginous fishes. For being more consistent with the nomenclature conventions, gja12 and gja13 should be fused into a single gene name, gja12, and hence, the genes should be detailed as gja12.1, gja12.2, gja12.1.1, gja12.1.2, etc. As a further consequence, the gene group that we call here gja14 (found in non-teleost ray-finned fishes and Latimeria) could then be called gja13.
- We suggest that GJB2 and GJB6 were generated by gene duplication from gjb8 shortly before the divergence of the Reptilia. Similarly, GJB4 and GJB5 were generated by gene duplication from gjb10 shortly before the divergence of the Amphibia.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harris, A.L. Emerging issues of connexin channels: Biophysics fills the gap. Q. Rev. Biophys. 2001, 34, 325–472. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, V.; Mikalsen, S.O. Connexins, gap junctional intercellular communication and kinases. Biol. Cell 2002, 94, 433–443. [Google Scholar] [CrossRef]
- Srinivas, M.; Verselis, V.K.; White, T.W. Human diseases associated with connexin mutations. Biochim. Biophys. Acta Biomembr. 2018, 1860, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Zefferino, R.; Piccoli, C.; Gioia, S.D.; Capitanio, N.; Conese, M. Gap junction intercellular communication in the carcinogenesis hallmarks: Is this a phenomenon or epiphenomenon? Cells 2019, 8, 896. [Google Scholar] [CrossRef]
- Laird, D.W.; Lampe, P.D. Therapeutic strategies targeting connexins. Nat. Rev. Drug Discov. 2018, 17, 905–921. [Google Scholar] [CrossRef]
- Delmar, M.; Laird, D.W.; Naus, C.C.; Nielsen, M.S.; Verselis, V.K.; White, T.W. Connexins and disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a029348. [Google Scholar] [CrossRef]
- Sonntag, S.; Sohl, G.; Dobrowolski, R.; Zhang, J.; Theis, M.; Winterhager, E.; Bukauskas, F.F.; Willecke, K. Mouse lens connexin23 (gje1) does not form functional gap junction channels but causes enhanced ATP release from HeLa cells. Eur. J. Cell Biol. 2009, 88, 65–77. [Google Scholar] [CrossRef]
- Cruciani, V.; Mikalsen, S.O. The connexin gene family in mammals. Biol. Chem. 2005, 386, 325–332. [Google Scholar] [CrossRef]
- Mikalsen, S.O.; Tausen, M.; í Kongsstovu, S. Phylogeny of teleost connexins reveals highly inconsistent intra- and interspecies use of nomenclature and misassemblies in recent teleost chromosome assemblies. BMC Genom. 2020, 21, 223. [Google Scholar]
- Human Gene Nomenclature Committee. Available online: https://www.genenames.org/about/guidelines/ (accessed on 11 February 2019).
- Zebrafish Information Network. Available online: https://wiki.zfin.org/display/general/ZFIN+Zebrafish+Nomenclature+Conventions (accessed on 25 March 2019).
- Fitch, W.M. Distinguishing homologous from analogous proteins. Syst. Zool. 1970, 19, 99–113. [Google Scholar] [CrossRef]
- Fitch, W.M. Homology. A personal view on some of the problems. Trends Genet. 2000, 16, 227–231. [Google Scholar] [CrossRef]
- Premzl, M. Comparative genomic analysis of eutherian connexin genes. Sci. Rep. 2019, 9, 16938. [Google Scholar] [CrossRef] [PubMed]
- de Boer, T.P.; van der Heyden, M.A. Xenopus connexins: How frogs bridge the gap. Differentiation 2005, 73, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, V.; Mikalsen, S.O. The vertebrate connexin family. Cell. Mol. Life Sci. 2006, 63, 1125–1140. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, V.; Mikalsen, S.O. Evolutionary selection pressure and family relationships among connexin genes. Biol. Chem. 2007, 388, 253–264. [Google Scholar] [CrossRef]
- Eastman, S.D.; Chen, T.H.; Falk, M.M.; Mendelson, T.C.; Iovine, M.K. Phylogenetic analysis of three complete gap junction gene families reveals lineage-specific duplications and highly supported gene classes. Genomics 2006, 87, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Douzery, E.J.; Snell, E.A.; Bapteste, E.; Delsuc, F.; Philippe, H. The timing of eukaryotic evolution: Does a relaxed molecular clock reconcile proteins and fossils? Proc. Natl. Acad. Sci. USA 2004, 101, 15386–15391. [Google Scholar] [CrossRef]
- Vandepoele, K.; De Vos, W.; Taylor, J.S.; Meyer, A.; Van de Peer, Y. Major events in the genome evolution of vertebrates: Paranome age and size differ considerably between ray-finned fishes and land vertebrates. Proc. Natl. Acad. Sci. USA 2004, 101, 1638–1643. [Google Scholar] [CrossRef]
- Hurley, I.A.; Mueller, R.L.; Dunn, K.A.; Schmidt, E.J.; Friedman, M.; Ho, R.K.; Prince, V.E.; Yang, Z.; Thomas, M.G.; Coates, M.I. A new time-scale for ray-finned fish evolution. Proc. Biol. Sci. 2007, 274, 489–498. [Google Scholar] [CrossRef]
- Bininda-Emonds, O.R.; Cardillo, M.; Jones, K.E.; MacPhee, R.D.; Beck, R.M.D.; Grenyer, R.; Price, S.A.; Vos, R.A.; Gittleman, J.L.; Purvis, A. The delayed rise of present-day mammals. Nature 2007, 446, 507–512. [Google Scholar] [CrossRef]
- Zhang, G.; Li, C.; Li, Q.; Li, B.; Larkin, D.M.; Lee, C.; Storz, J.F.; Antunes, A.; Greenwold, M.J.; Meredith, R.W.; et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 2014, 346, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Lovell, P.V.; Wirthlin, M.; Wilhelm, L.; Minx, P.; Lazar, N.H.; Carbone, L.; Warren, W.C.; Mello, C.V. Conserved syntenic clusters of protein coding genes are missing in birds. Genome Biol. 2014, 15, 565. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Stiller, J.; Deng, Y.; Armstrong, J.; Fang, Q.; Reeve, A.H.; Xie, D.; Chen, G.; Guo, C.; Faircloth, B.C.; et al. Dense sampling of bird diversity increases power of comparative genomics. Nature 2020, 587, 252–257. [Google Scholar] [CrossRef] [PubMed]
- White, T.W.; Wang, H.; Mui, R.; Litteral, J.; Brink, P.R. Cloning and functional expression of invertebrate connexins from Halocynthia pyriformis. FEBS Lett. 2004, 577, 42–48. [Google Scholar] [CrossRef]
- Okamura, Y.; Nishino, A.; Murata, Y.; Nakajo, K.; Iwasaki, H.; Ohtsuka, Y.; Tanaka-Kunishima, M.; Takahashi, N.; Hara, Y.; Yoshida, T.; et al. Comprehensive analysis of the ascidian genome reveals novel insights into the molecular evolution of ion channel genes. Physiol. Genom. 2005, 22, 269–282. [Google Scholar] [CrossRef]
- O’Brien, J.; al-Ubaidi, M.R.; Ripps, H. Connexin 35: A gap-junctional protein expressed preferentially in the skate retina. Mol. Biol. Cell 1996, 7, 233–243. [Google Scholar] [CrossRef][Green Version]
- Sohl, G.; Degen, J.; Teubner, B.; Willecke, K. The murine gap junction gene connexin36 is highly expressed in mouse retina and regulated during brain development. FEBS Lett. 1998, 428, 27–31. [Google Scholar] [CrossRef]
- Green, R.E.; Braun, E.L.; Armstrong, J.; Earl, D.; Nguyen, N.; Hickey, G.; Vandewege, M.W.; John, J.A.S.; Capella-Gutiérrez, S.; Castoe, T.A.; et al. Three crocodilian genomes reveal ancestral patterns of evolution among archosaurs. Science 2014, 346, 1254449. [Google Scholar] [CrossRef]
- Ghosh, A.; Johnson, M.G.; Osmanski, A.B.; Louha, S.; Bayona-Vásquez, N.J.; Glenn, T.C.; Gongora, J.; Green, R.E.; Isberg, S.; Stevens, R.D.; et al. A high-quality reference genome assembly of the saltwater crocodile, Crocodylus porosus, reveals patterns of selection in Crocodylidae. Genome Biol. Evol. 2020, 12, 3635–3646. [Google Scholar] [CrossRef]
- Wang, Z.; Pascual-Anaya, J.; Zadissa, A.; Li, W.; Niimura, Y.; Huang, Z.; Li, C.; White, S.; Xiong, Z.; Fang, D.; et al. The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan. Nat. Genet. 2013, 45, 701–706. [Google Scholar] [CrossRef]
- Alföldi, J.; Di Palma, F.; Grabherr, M.; Williams, C.; Kong, L.; Mauceli, E.; Russell, P.J.; Lowe, C.B.; Glor, R.E.; Jaffe, J.D.; et al. The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature 2011, 477, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Amemiya, C.; Alfoeldi, J.; Lee, A.P.; Fan, S.; Philippe, H.; Maccallum, I.; Braasch, I.; Manousaki, T.; Schneider, I.; Rohner, N.; et al. The African coelacanth genome provides insights into tetrapod evolution. Nature 2013, 496, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Braasch, I.; Gehrke, A.R.; Smith, J.J.; Kawasaki, K.; Manousaki, T.; Pasquier, J.; Amores, A.; Desvignes, T.; Batzel, P.; Catchen, J.; et al. The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat. Genet. 2016, 48, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Stöck, M.; Kneitz, S.; Klopp, C.; Woltering, J.M.; Adolfi, M.C.; Feron, R.; Prokopov, D.; Makunin, A.; Kichigin, I.; et al. The sterlet sturgeon genome sequence and the mechanisms of segmental rediploidization. Nat. Ecol. Evol. 2020, 4, 841–852. [Google Scholar] [CrossRef]
- Read, T.D.; Petit, R.A., 3rd; Joseph, S.J.; Alam, M.T.; Weil, M.R.; Ahmad, M.; Bhimani, R.; Vuong, J.S.; Haase, C.P.; Webb, D.H.; et al. Draft sequencing and assembly of the genome of the world’s largest fish, the whale shark: Rhincodon typus Smith 1828. BMC Genom. 2017, 18, 532. [Google Scholar]
- King, B.L.; Gillis, J.A.; Carlisle, H.R.; Dahn, R.D. A natural deletion of the HoxC cluster in elasmobranch fishes. Science 2011, 334, 1517. [Google Scholar] [CrossRef]
- Wyffels, J.; King, B.L.; Vincent, J.; Chen, C.; Wu, C.H.; Polson, S.W. SkateBase, an elasmobranch genome project and collection of molecular resources for chondrichthyan fishes. F1000Res 2014, 3, 191. [Google Scholar] [CrossRef]
- Venkatesh, B.; Lee, A.P.; Ravi, V.; Maurya, A.K.; Lian, M.M.; Swann, J.B.; Ohta, Y.; Flajnik, M.F.; Sutoh, Y.; Kasahara, M.; et al. Elephant shark genome provides unique insights into gnathostome evolution. Nature 2014, 505, 174–179. [Google Scholar] [CrossRef]
- Smith, J.J.; Kuraku, S.; Holt, C.; Sauka-Spengler, T.; Jiang, N.; Campbell, M.S.; Yandell, M.D.; Manousaki, T.; Meyer, A.; Bloom, O.E.; et al. Sequencing of the sea lamprey (Petromyzon marinus) genome provides insights into vertebrate evolution. Nat. Genet. 2013, 45, 415–421. [Google Scholar] [CrossRef]
- Hess, J.E.; Smith, J.J.; Timoshevskaya, N.; Baker, C.; Caudill, C.C.; Graves, D.; Keefer, M.L.; Kinziger, A.P.; Moser, M.L.; Porter, L.L.; et al. Genomic islands of divergence infer a phenotypic landscape in Pacific lamprey. Mol. Ecol. 2020, 29, 3841–3856. [Google Scholar] [CrossRef]
- Mehta, T.K.; Ravi, V.; Yamasaki, S.; Lee, A.P.; Lian, M.M.; Tay, B.-H.; Tohari, S.; Yanai, S.; Tay, A.; Brenner, S.; et al. Evidence for at least six Hox clusters in the Japanese lamprey (Lethenteron japonicum). Proc. Natl. Acad. Sci. USA 2013, 110, 16044–16049. [Google Scholar] [CrossRef] [PubMed]
- Bergsten, J. A review of long-branch attraction. Cladistics 2005, 21, 163–193. [Google Scholar] [CrossRef]
- Siddal, M.E.; Whiting, M.F. Long-branch abstractions. Cladistics 1999, 15, 9–24. [Google Scholar] [CrossRef]
- Postlethwait, J.H. The zebrafish genome in context: Ohnologs gone missing. J. Exp. Zool. B Mol. Dev. Evol. 2007, 308, 563–577. [Google Scholar] [CrossRef]
- Wolfe, K. Robustness—It’s not where you think it is. Nat. Genet. 2000, 25, 3–4. [Google Scholar] [CrossRef]
- Cheng, P.; Huang, Y.; Du, H.; Li, C.; Lv, Y.; Ruan, R.; Ye, H.; Bian, C.; You, X.; Xu, J.; et al. Draft genome and complete Hox-cluster characterization of the sterlet (Acipenser ruthenus). Front. Genet. 2019, 10, 776. [Google Scholar] [CrossRef]
- Havelka, M.; Bytyutskyy, D.; Symonová, R.; Ráb, P.; Flajshans, M. The second highest chromosome count among vertebrates is observed in cultured sturgeon and is associated with genome plasticity. Genet. Sel. Evol. 2016, 48, 12. [Google Scholar] [CrossRef]
- Yin, Z.-T.; Zhu, F.; Lin, F.-B.; Jia, T.; Wang, Z.; Sun, D.-T.; Li, G.-S.; Zhang, C.-L.; Smith, J.; Yang, N.; et al. Revisiting avian ‘missing’ genes from de novo assembled transcripts. BMC Genom. 2019, 20, 4. [Google Scholar] [CrossRef]
- Kuraku, S.; Kuratani, S. Time scale for cyclostome evolution inferred with a phylogenetic diagnosis of hagfish and lamprey cDNA sequences. Zool. Sci. 2006, 23, 1053–1064. [Google Scholar] [CrossRef]
- Heimberg, A.M.; Cowper-Sal-lari, R.; Semon, M.; Donoghue, P.C.; Peterson, K.J. microRNAs reveal the interrelationships of hagfish, lampreys, and gnathostomes and the nature of the ancestral vertebrate. Proc. Natl. Acad. Sci. USA 2010, 107, 19379–19383. [Google Scholar] [CrossRef]
- MUSCLE—MUltiple Sequence Comparison by Log-Expectation. Available online: https://www.ebi.ac.uk/Tools/msa/muscle/ (accessed on 9 December 2020).
- Clustal Omega—Multiple Sequence Alignment. Available online: https://www.ebi.ac.uk/Tools/msa/clustalo/ (accessed on 9 December 2020).
- Kohno, S.; Nakai, Y.; Satoh, S.; Yoshida, M.; Kobayashi, H. Chromosome elimination in the Japanese hagfish, Eptatretus burgeri (Agnatha, Cyclostomata). Cytogenet. Cell Genet 1986, 41, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.J.; Timoshevskaya, N.; Ye, C.; Holt, C.; Keinath, M.C.; Parker, H.J.; Cook, M.E.; Hess, J.E.; Narum, S.R.; Lamanna, F.; et al. The sea lamprey germline genome provides insights into programmed genome rearrangement and vertebrate evolution. Nat. Genet. 2018, 50, 270–277. [Google Scholar] [CrossRef]
- Koonin, E.V. Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet. 2005, 39, 309–338. [Google Scholar] [CrossRef]
- Lien, S.; Koop, B.F.; Sandve, S.R.; Miller, J.R.; Kent, M.P.; Nome, T.; Hvidsten, T.R.; Leong, J.S.; Minkley, D.R.; Zimin, A.; et al. The Atlantic salmon genome provides insights into rediploidization. Nature 2016, 533, 200–205. [Google Scholar] [CrossRef]
- Session, A.M.; Uno, Y.; Kwon, T.; Chapman, J.A.; Toyoda, A.; Takahashi, S.; Fukui, A.; Hikosaka, A.; Suzuki, A.; Kondo, M.; et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature 2016, 538, 336–343. [Google Scholar] [CrossRef]
- Guidelines for Nomenclature of Genes, Genetic Markers, Alleles, and Mutations in Mouse and Rat. Available online: http://www.informatics.jax.org/mgihome/nomen/gene.shtml (accessed on 11 December 2020).
- Delsuc, F.; Brinkmann, H.; Chourrout, D.; Philippe, H. Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature 2006, 439, 965–968. [Google Scholar] [CrossRef]
- Ohno, S. Evolution by Gene Duplication; Springer: Berlin, Germany, 1970. [Google Scholar]
- Dehal, P.; Boore, J.L. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 2005, 3, e314. [Google Scholar] [CrossRef]
- Panopoulou, G.; Hennig, S.; Groth, D.; Krause, A.; Poustka, A.J.; Herwig, R.; Vingron, M.; Lehrach, H. New evidence for genome-wide duplications at the origin of vertebrates using an amphioxus gene set and completed animal genomes. Genome Res. 2003, 13, 1056–1066. [Google Scholar] [CrossRef]
- Panopoulou, G.; Poustka, A.J. Timing and mechanism of ancient vertebrate genome duplications—The adventure of a hypothesis. Trends Genet. 2005, 21, 559–567. [Google Scholar] [CrossRef]
- Kuraku, S.; Meyer, A.; Kuratani, S. Timing of genome duplications relative to the origin of the vertebrates: Did cyclostomes diverge before or after? Mol. Biol. Evol. 2009, 26, 47–59. [Google Scholar] [CrossRef]
- Hufton, A.L.; Groth, D.; Vingron, M.; Lehrach, H.; Poustka, A.J.; Panopoulou, G. Early vertebrate whole genome duplications were predated by a period of intense genome rearrangement. Genome Res. 2008, 18, 1582–1591. [Google Scholar] [CrossRef]
- Escriva, H.; Manzon, L.; Youson, J.; Laudet, V. Analysis of lamprey and hagfish genes reveals a complex history of gene duplications during early vertebrate evolution. Mol. Biol. Evol. 2002, 19, 1440–1450. [Google Scholar] [CrossRef]
- Holland, L.Z.; Ocampo Daza, D. A new look at an old question: When did the second whole genome duplication occur in vertebrate evolution? Genome Biol. 2018, 19, 209. [Google Scholar] [CrossRef]
- Smith, J.J.; Keinath, M.C. The sea lamprey meiotic map improves resolution of ancient vertebrate genome duplications. Genome Res. 2015, 25, 1081–1090. [Google Scholar] [CrossRef]
- Bennett, M.V.; Zheng, X.; Sogin, M.L. The connexins and their family tree. Soc. Gen. Physiol. Ser. 1994, 49, 223–233. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikalsen, S.-O.; í Kongsstovu, S.; Tausen, M. Connexins during 500 Million Years—From Cyclostomes to Mammals. Int. J. Mol. Sci. 2021, 22, 1584. https://doi.org/10.3390/ijms22041584
Mikalsen S-O, í Kongsstovu S, Tausen M. Connexins during 500 Million Years—From Cyclostomes to Mammals. International Journal of Molecular Sciences. 2021; 22(4):1584. https://doi.org/10.3390/ijms22041584
Chicago/Turabian StyleMikalsen, Svein-Ole, Sunnvør í Kongsstovu, and Marni Tausen. 2021. "Connexins during 500 Million Years—From Cyclostomes to Mammals" International Journal of Molecular Sciences 22, no. 4: 1584. https://doi.org/10.3390/ijms22041584
APA StyleMikalsen, S.-O., í Kongsstovu, S., & Tausen, M. (2021). Connexins during 500 Million Years—From Cyclostomes to Mammals. International Journal of Molecular Sciences, 22(4), 1584. https://doi.org/10.3390/ijms22041584