Flow Biocatalysis: A Challenging Alternative for the Synthesis of APIs and Natural Compounds

 ,
,  ,
,  ,
, 
 , ,
, ,  and
and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
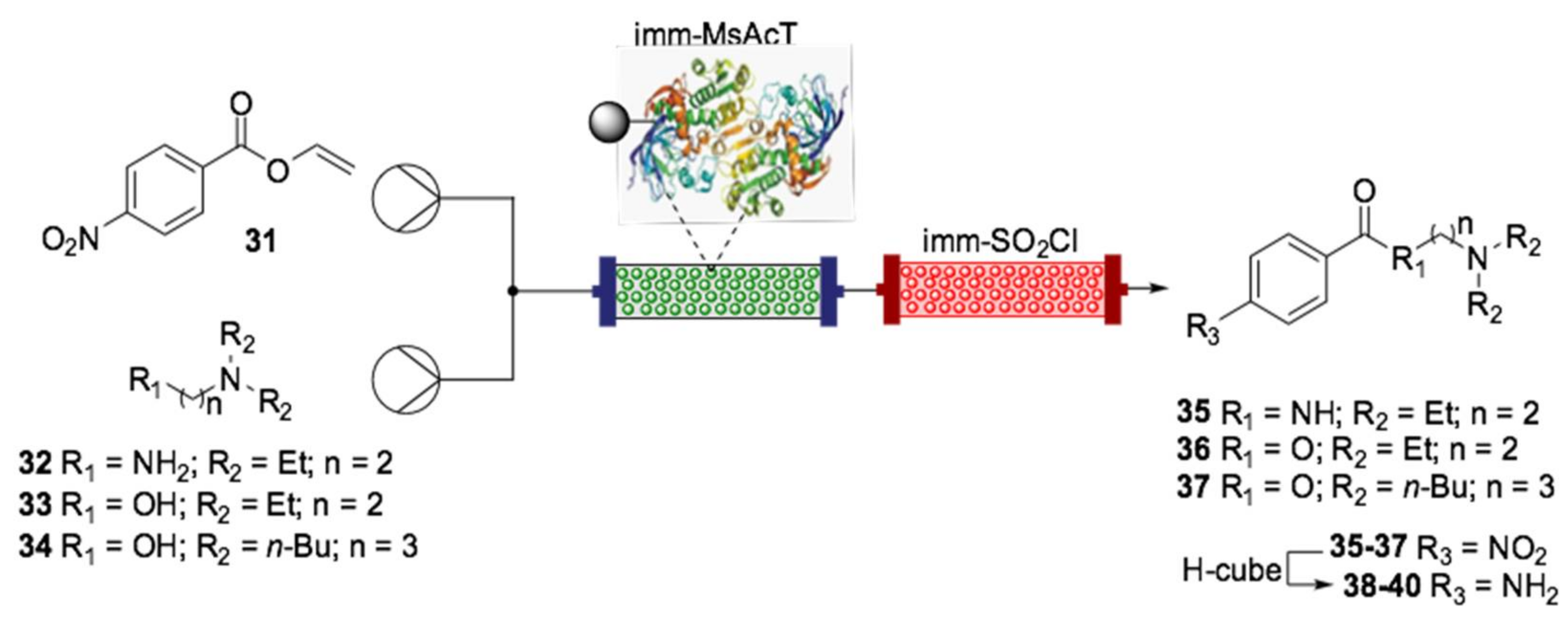{kind=link}
{kind=link}
{kind=link}
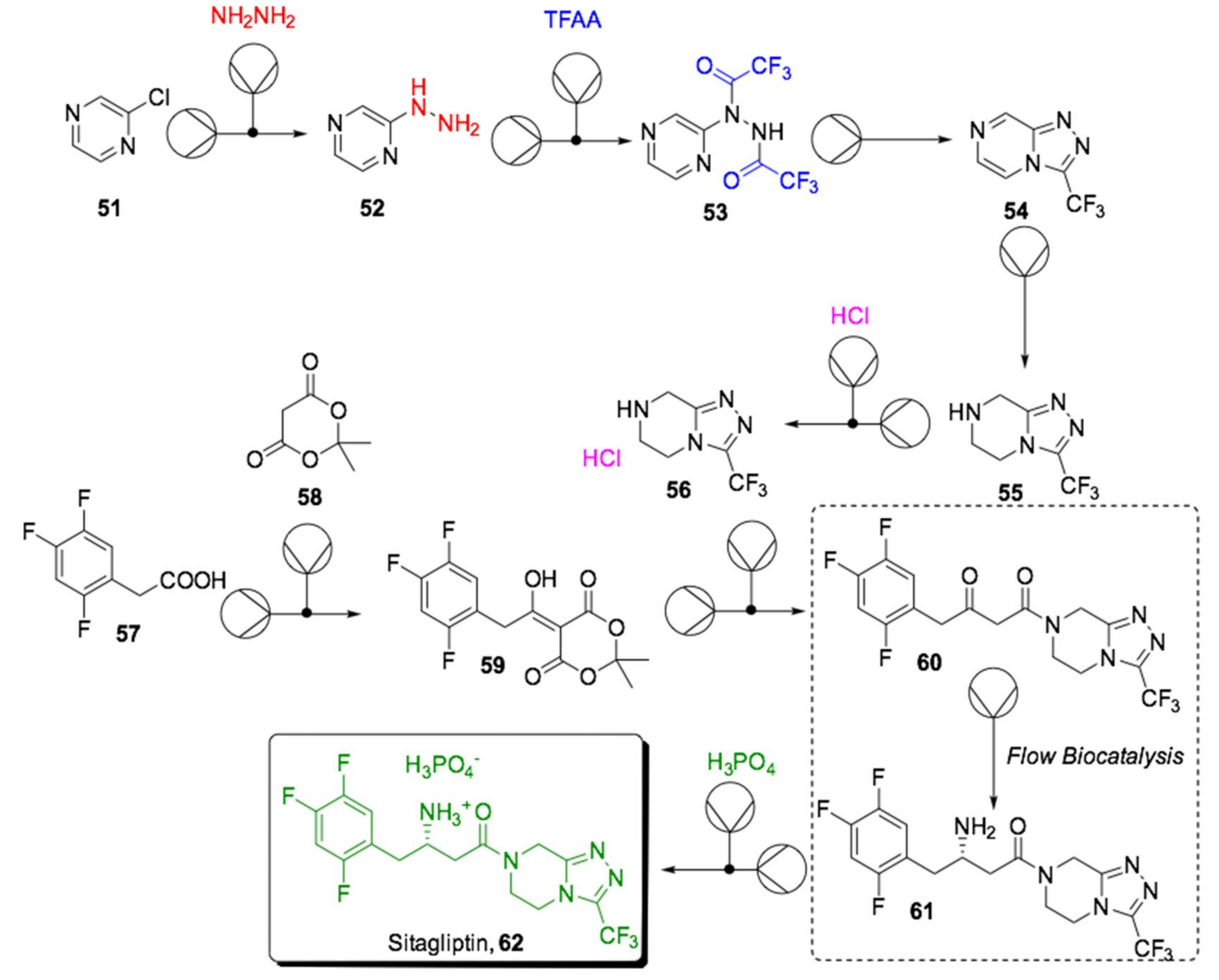{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Biocatalysis in Industry
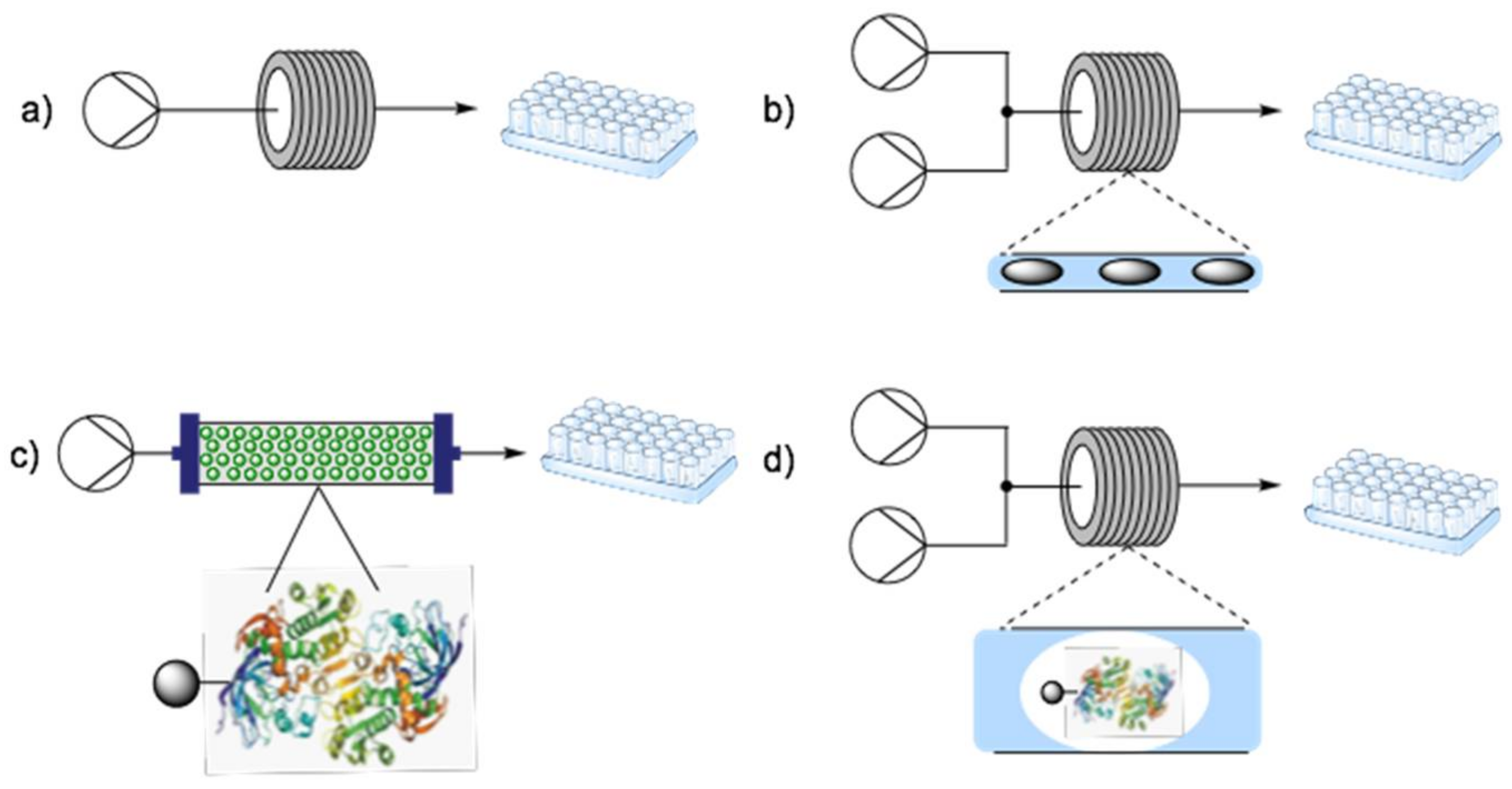
3. Fundamentals of Flow Biocatalysis
4. Flow Biocatalysis Applied to the Synthesis of Active Pharmaceutical Ingredients (APIs) and Their Precursors
4.1. Single-Step Flow Biocatalytic Systems
4.2. Flow Biocatalysis Applied to Multi-Step Synthesis
5. Flow Biocatalysis Applied to the Synthesis of Natural Products and Aromas
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE | Angiotensin-converting enzyme |
| AceKMs | Mycobacterium smegmatis acetate kinase |
| ADS | Amorphadiene synthases |
| AhPNP | Aeromonas hydrophila purine nucleoside phosphorylase |
| API | Active pharmaceutical ingredient |
| araA | Arabinofuranosyl adenine |
| araU | Arabinofuranosyl uracil |
| AS | Aristolochene synthase |
| BPR | Back pressure regulator |
| BS2m | Bacillus subtilis esterase |
| CCC | Counter current chromatography |
| CpUP | Clostridium perfrigens uridine phosphorylase |
| CLEASs | Crosslinked enzyme aggregates |
| CLECs | Crosslinked enzyme crystals |
| CSTR | Continuously stirred tank reactor |
| DHAA | Dihydroartemisinic aldehyde |
| DHAP | Dihydroxyacetone phosphate |
| DoE | Design of experiments |
| dUrd | Deoxyuridine |
| EMA | European Medicines Agency |
| FDA | Food and Drug Administration |
| FDP | Farnesyl diphosphate |
| FdUrd | 5-fluoro-2′-deoxyuridine |
| FEP | Fluorinated ethylene propylene |
| FruA | Fructose aldolase |
| G3PD Ec | Escherichia coli glycerol-3-phosphate dehydrogenase |
| Gd4olS | Germacradien-4-ol synthase |
| Gd11olS | Germacradien-11-ol synthase |
| GlpKTk | Thermococcus kodakarensis glycerol kinase |
| Gs-Lys6DH | Geobacillus stearothermophilus dehydrogenase |
| HEWT | Halomonas Elongata amine transaminase |
| ID | Internal diameter |
| IdUrd | 5-Iodo-2′-deoxyuridine |
| IMERs | Immobilized enzyme reactors |
| HCCC | High-performance countercurrent chromatography |
| HTS | High-throughput screening |
| LrNDT | Lactobacillus reuteri nucleoside 2′deoxyribosyltransferase |
| MsAcT | Mycobacterium smegmatis acyltransferase enzyme |
| MTBE | Methyl tert-butyl ether |
| NOXCa | NADH oxidase Clostridium aminovalericum |
| NSAID | Nonsteroidal anti-inflammatory drug |
| P5C | Pyrroline-5-carboxylate reductase |
| PBR | Packed-bed reactor |
| PTFE | Polytetrafluoroethylene |
| QbD | Quality by design |
| TTL | Thermomyces lanuginosus lipase |
| TTN | Total turnover number |
References
- Sheldon, R.A.; Brady, D.; Bode, M.L. The Hitchhiker’s guide to biocatalysis: Recent advances in the use of enzymes in organic synthesis. Chem. Sci. 2020, 11, 2587–2605. [Google Scholar] [CrossRef]
- Pasteur, L.C. Mémoire de L. Pasteur sur la fermentation de l’acide tartrique. C. R. Acad. Sci. Paris 1958, 46, 615–618. [Google Scholar]
- Bilal, M.; Iqbal, H.M.N. State-of-the-art strategies and applied perspectives of enzyme biocatalysis in food sector—current status and future trends. Crit. Rev. Food Sci. 2020, 60, 2052–2066. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; Brady, D. The limits to biocatalysis: Pushing the envelope. Chem. Commun. 2018, 54, 6088–6104. [Google Scholar] [CrossRef]
- Sheldon, R.A. Why green chemistry and sustainability of resources are essential to our future. J. Environ. Monit. 2008, 10, 406–407. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Green and sustainable manufacture of chemicals from biomass: State of the art. Green Chem. 2014, 16, 950–963. [Google Scholar] [CrossRef]
- The World Commission on Environmental Development. Our Common Future; Oxford University Press: Oxford, UK, 1987. [Google Scholar]
- The US National Research Council. Our Common Journey: A Transition Toward Sustainability; National Academy Press: Washington, DC, USA, 1999. [Google Scholar]
- Anastas, P.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Sheldon, R.A.; Woodley, J.M. Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 2018, 118, 801–838. [Google Scholar] [CrossRef] [PubMed]
- Krüger, A.; Schäfers, C.; Schröder, C.; Antranikian, G. Towards a sustainable biobased industry—Highlighting the impact of extremophiles. New Biotechnol. 2018, 40, 144–153. [Google Scholar] [CrossRef]
- Hammer, S.C.; Knight, A.M.; Arnold, F.H. Design and evolution of enzymes for non-natural chemistry. Curr. Opin. Green Sustain. Chem. 2017, 7, 23–30. [Google Scholar] [CrossRef]
- Kim, J.Y.; Yoo, H.-W.; Lee, P.-G.; Lee, S.-G.; Seo, J.-H.; Kim, B.-G. In vivo Protein Evolution, Next Generation Protein Engineering Strategy: From Random Approach to Target-specific Approach. Biotechnol. Bioprocess Eng. 2019, 24, 85–94. [Google Scholar] [CrossRef]
- Eijsink, V.G.H.; Gåseidnes, S.; Borchert, T.V.; Burg, B.V.D. Directed evolution of enzyme stability. Biomol. Eng. 2005, 22, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, X.; Zhao, H. Biosystems design by directed evolution. AIChE J. 2019, 66, e16716. [Google Scholar] [CrossRef]
- Woodley, J.M. New opportunities for biocatalysis: Making pharmaceutical processes greener. Trends Biotechnol. 2008, 26, 321–327. [Google Scholar] [CrossRef]
- Ran, N.; Zhao, L.; Chen, Z.; Tao, J. Recent applications of biocatalysis in developing green chemistry for chemical synthesis at the industrial scale. Green Chem. 2008, 10, 361–372. [Google Scholar] [CrossRef]
- Omori, A.T.; Lobo, F.G.; Carolina, A.; Amaral, G.; Oliveira, C.d.S. Purple carrots: Better biocatalysts for the enantioselective reduction of acetophenones than common orange carrots (D. carota). J. Mol. Catal. B Enzym. 2016, 127, 93–97. [Google Scholar] [CrossRef]
- Carvalho, C.C.C.R. Whole cell biocatalysts: Essential workers from Nature to the industry. Microb. Biotechnol. 2017, 10, 250–263. [Google Scholar] [CrossRef]
- Patel, R.N. Biocatalysis for synthesis of pharmaceuticals. Bioorg. Med. Chem. 2018, 26, 1252–1274. [Google Scholar] [CrossRef]
- Thompson, M.P.; Peñafiel, I.; Cosgrove, S.C.; Turner, N.J. Biocatalysis Using Immobilized Enzymes in Continuous Flow for the Synthesis of Fine Chemicals. Org. Process Res. Dev. 2019, 23, 9–18. [Google Scholar] [CrossRef]
- Abdelraheem, E.M.M.; Busch, H.; Hanefeld, U.; Tonin, F. Biocatalysis explained: From pharmaceutical to bulk chemical production. React. Chem. Eng. 2019, 4, 1878–1894. [Google Scholar] [CrossRef]
- Adams, J.P.; Brown, M.J.B.; Diaz-Rodriguez, A.; Lloyd, R.C.; Roiban, G.-D. Biocatalysis: A Pharma Perspective. Adv. Synth. Catal. 2019, 361, 2421–2432. [Google Scholar] [CrossRef]
- Baumann, M.; Moody, T.S.; Smyth, M.; Wharry, S. A Perspective on Continuous Flow Chemistry in the Pharmaceutical Industry. Org. Process Res. Dev. 2020, 24, 1802–1813. [Google Scholar] [CrossRef]
- Wenda, S.; Illner, S.; Kragl, U. Industrial biotechnology—the future of green chemistry? Green Chem. 2011, 13, 3007–3047. [Google Scholar] [CrossRef]
- Liu, L.; Yang, H.; Shin, H.D.; Chen, R.R.; Li, J.; Du, G.; Chen, J. How to achieve high-level expression of microbial enzymes: Strategies and perspectives. Bioengineered 2013, 4, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [PubMed]
- Netto, C.G.C.M.; Toma, H.E.; Andrade, L.H. Superparamagnetic nanoparticles as versatile carriers and supporting materials for enzymes. J. Mol. Catal. B Enzym. 2013, 85–86, 71–92. [Google Scholar] [CrossRef]
- Diefenbach, X.W.; Farasat, I.; Guetschow, E.D.; Welch, C.J.; Kennedy, R.T.; Sun, S.; Moore, J.C. Enabling Biocatalysis by High-Throughput Protein Engineering Using Droplet Microfluidics Coupled to Mass Spectrometry. ACS Omega 2018, 3, 1498–1508. [Google Scholar] [CrossRef]
- Foley, A.M.; Maguire, A.R. The impact of recent developments in technologies which enable the increased use of biocatalysts. Eur. J. Org. Chem. 2019, 3713–3734. [Google Scholar] [CrossRef]
- Wang, W.; Nema, S.; Teagarden, D. Protein aggregation—Pathways and influencing factors. Int. J. Pharm. 2010, 390, 89–99. [Google Scholar] [CrossRef]
- Plutschack, M.B.; Pieber, B.; Gilmore, K.; Seeberger, P.H. The Hitchhiker’s Guide to Flow Chemistry. Chem. Rev. 2017, 117, 11796–11893. [Google Scholar] [CrossRef]
- Ley, S.V.; Fitzpatrick, D.E.; Myers, R.M.; Battilocchio, C.; Ingham, R.J. Machine-Assisted Organic Synthesis. Angew. Chem. Int. Ed. 2015, 54, 10122–10137. [Google Scholar] [CrossRef]
- De Santis, P.; Meyer, L.-E.; Kara, S. The rise of continuous flow biocatalysis–Fundamentals, very recent developments and future perspectives. React. Chem. Eng. 2020, 5, 2155–2184. [Google Scholar] [CrossRef]
- Guajardo, N.; Domínguez de María, P. Continuous Biocatalysis in Environmentally-Friendly Media: A Triple Synergy for Future Sustainable Processes. ChemCatChem 2019, 11, 3128–3137. [Google Scholar] [CrossRef]
- Britton, J.; Majumdar, S.; Weiss, G.A. Continuous flow biocatalysis. Chem. Soc. Rev. 2018, 47, 5891–5918. [Google Scholar] [CrossRef] [PubMed]
- Tamborini, L.; Fernandes, P.; Paradisi, F.; Molinari, F. Flow Bioreactors as Complementary Tools for Biocatalytic Process Intensification. Trends Biotechnol. 2018, 36, 73–88. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, Q.; Shao, L.; Jia, Y.; Zhang, X. Microfluidic immobilized enzyme reactors for continuous biocatalysis. React. Chem. Eng. 2020, 5, 9–32. [Google Scholar] [CrossRef]
- Paradisi, F. Flow Biocatalysis. Catalysts 2020, 10, 645. [Google Scholar] [CrossRef]
- Martin, L.L.; Peschke, T.; Venturoni, F.; Mostarda, S. Pharmaceutical industry perspectives on flow chemocatalysis and biocatalysis. Curr. Opin. Green Sustain. Chem. 2020, 25, 100350. [Google Scholar] [CrossRef]
- Drauz, K.; Gröger, H.; May, O. Enzyme Catalysis. In Organic Synthesis, 3rd ed.; Drauz, K., Gröger, H., May, O., Eds.; Wiley-VCH: Weinheim, Germany, 2012; Volume 1–3. [Google Scholar]
- Liese, A.; Seelbach, K.; Wandrey, C. Industrial Biotransformations, 2nd ed.; Liese, A., Seelbach, K., Wandrey, C., Eds.; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Schäfer, B. Naturstoffe der Chemischen Industrie; Elsevier: München, Germany, 2007; p. 389. [Google Scholar]
- Arnold, F.H. Innovation by Evolution: Bringing New Chemistry to Life (Nobel Lecture). Angew. Chem. Int. Ed. 2019, 58, 14420–14426. [Google Scholar] [CrossRef]
- Riesenberg, D.; Guthke, R. High-cell-density cultivation of microorganisms. Appl. Microbiol. Biotechnol. 1999, 51, 422–430. [Google Scholar] [CrossRef]
- Grayson, J.I.; Roos, J.; Osswald, S. Development of a Commercial Process for (S)-β-Phenylalanine(1). Org. Process Res. Dev. 2012, 15, 1201–1205. [Google Scholar] [CrossRef]
- Gröger, H.; Chamouleau, F.; Orologas, N.; Rollmann, C.; Drauz, K.; Hummel, W.; Weckbecker, A.; May, O. Enantioselective Reduction of Ketones with “Designer Cells” at High Substrate Concentrations: Highly Efficient Access to Functionalized Optically Active Alcohols. Angew. Chem. Int. Ed. 2006, 45, 5677–5681. [Google Scholar] [CrossRef] [PubMed]
- Savile, C.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Gröger, H.; Drauz, K. Methods for the Enantioselective Biocatalytic Production of L-Amino Acids on an Industrial Scale. In Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions; Schmidt, E., Blaser, H.U., Eds.; Wiley-VCH: Weinheim, Germany, 2004; Chapter 1.8; p. 131. [Google Scholar] [CrossRef]
- Adebar, N.; Choi, J.E.; Schober, L.; Miyake, R.; Iura, T.; Kawabata, H.; Gröger, H. Overcoming Work-Up Limitations of Biphasic Biocatalytic Reaction Mixtures Through Liquid-Liquid Segmented Flow Processes. ChemCatChem 2019, 11, 5788–5793. [Google Scholar] [CrossRef]
- Lee, S.L.; O’Connor, T.F.; Yang, X.; Cruz, C.N.; Chatterjee, S.; Madurawe, R.D.; Moore, C.M.V.; Yu, L.X.; Woodcock, J. Modernizing Pharmaceutical Manufacturing: From Batch to Continuous Production. J. Pharm. Innov. 2015, 10, 191–199. [Google Scholar] [CrossRef]
- Chatterjee, S. FDA Perspective on Continuous Manufacturing. In Proceedings of the Annual International Foundation Process Analytical Chemistry (IFPAC) Meeting, Baltimore, MD, USA, 22–25 January 2012. [Google Scholar]
- Hernan, D. Continuous Manufacturing: Challenges and Opportunities. EMA Perspective. In Proceedings of the 3rd FDA/PQRI Conference on Advancing Product Quality, Rockville, MD, USA, 22–24 March 2017. [Google Scholar]
- Hoeks, F.W.J.M.M.; Kulla, H.; Meyer, H.P. Continuous cell-recycle process for l-carnitine production: Performance, engineering and downstream processing aspects compared with discontinuous processes. J. Biotechnol. 1992, 22, 117–128. [Google Scholar] [CrossRef]
- Rohner, M.; Meyer, H.P. Applications of modelling for bioprocess design and control in industrial production. Bioprocess. Eng. 1995, 13, 69–78. [Google Scholar] [CrossRef]
- Akwi, F.M.; Watts, P. Continuous flow chemistry: Where are we now? Recent applications, challenges and limitations. Chem. Commun. 2018, 54, 13894–13928. [Google Scholar] [CrossRef]
- Hartman, R.L. Flow chemistry remains an opportunity for chemists and chemical engineers. Curr. Opin. Chem. Eng. 2020, 29, 1–9. [Google Scholar] [CrossRef]
- Pastre, J.C.; Browne, D.L.; Ley, S.T. Flow chemistry syntheses of natural products. Chem. Soc. Rev. 2013, 42, 8849–8869. [Google Scholar] [CrossRef]
- Porta, R.; Benaglia, M.; Puglisi, A. Flow Chemistry: Recent Developments in the Synthesis of Pharmaceutical Products. Org. Process Res. Dev. 2016, 20, 2–25. [Google Scholar] [CrossRef]
- Hartman, R.L.; McMullen, J.P.; Jensen, K.F. Deciding Whether To Go with the Flow: Evaluating the Merits of Flow Reactors for Synthesis. Angew. Chem. Int. Ed. 2011, 50, 7502–7519. [Google Scholar] [CrossRef]
- Müller, S.T.R.; Wirth, T. Diazo Compounds in Continuous-Flow Technology. ChemSusChem 2015, 8, 245–250. [Google Scholar] [CrossRef]
- Hock, K.J.; Koenigs, R.M. The Generation of Diazo Compounds in Continuous-Flow. Chem. Eur. J. 2018, 24, 10571–10583. [Google Scholar] [CrossRef]
- O’Mahony, R.M.; Lynch, D.; Hayes, H.L.D.; Thuama, E.N.; Donnellan, P.; Jones, R.C.; Glennon, B.; Collins, S.G.; Maguire, A.R. Exploiting the Continuous in situ Generation of Mesyl Azide for Use in a Telescoped Process. Eur. J. Org. Chem. 2017, 6533–6539. [Google Scholar] [CrossRef]
- Mallia, C.J.; Baxendale, I.R. The Use of Gases in Flow Synthesis. Org. Process Res. Dev. 2016, 20, 327–360. [Google Scholar] [CrossRef]
- Shukla, C.A.; Kulkarni, A.A. Automating multistep flow synthesis: Approach and challenges in integrating chemistry, machines and logic. Beilstein J. Org. Chem. 2017, 13, 960–987. [Google Scholar] [CrossRef]
- Reizman, B.J.; Jensen, K.F. Feedback in Flow for Accelerated Reaction Development. Acc. Chem. Res. 2016, 49, 1786–1796. [Google Scholar] [CrossRef]
- Clayton, A.D.; Manson, J.A.; Taylor, C.J.; Chamberlain, T.W.; Taylor, B.A.; Clemens, G.; Bourne, R.A. Algorithms for the self-optimisation of chemical reactions. React. Chem. Eng. 2019, 4, 1545–1554. [Google Scholar] [CrossRef]
- McQuade, D.T.; Seeberger, P.H. Applying Flow Chemistry: Methods, Materials, and Multistep Synthesis. J. Org. Chem. 2013, 78, 6384–6389. [Google Scholar] [CrossRef]
- Bloemendal, V.R.L.J.; Janssen, M.A.C.H.; Hest, J.C.M.v.; Rutjes, F.P.J.T. Continuous one-flow multi-step synthesis of active pharmaceutical ingredients. React. Chem. Eng. 2020, 5, 1186–1197. [Google Scholar] [CrossRef]
- Cambié, D.; Bottecchia, C.; Straathof, N.J.W.; Hessel, V.; Noël, T. Applications of Continuous-Flow Photochemistry in Organic Synthesis. Chem. Rev. 2016, 116, 10276–10341. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.C.; Berenguer-Murcia, O.A.; Torres, R.; Fernandez-Lafuente, R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013, 42, 6290–6307. [Google Scholar] [CrossRef] [PubMed]
- Fassouane, A.; Laval, J.M.; Moiroux, J.; Bourdillon, C. Electrochemical regeneration of NAD in a plug-flow reactor. Biotechnol. Bioeng. 1990, 35, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.K.; Choban, E.R.; Kane, C.; Tzedakis, T.; Kenis, P.J. Laminar flow-based electrochemical microreactor for efficient regeneration of nicotinamide cofactors for biocatalysis. J. Am. Chem. Soc. 2005, 127, 10466–10467. [Google Scholar] [CrossRef] [PubMed]
- Lindeque, R.M.; Woodley, J.M. The Effect of Dissolved Oxygen on Kinetics during Continuous Biocatalytic Oxidations. Org. Process Res. Dev. 2020, 24, 2055–2063. [Google Scholar] [CrossRef]
- Rudroff, F.; Mihovilovic, M.D.; Gröger, H.; Snajdrova, R.; Iding, H.; Bornscheuer, U.T. Opportunities and challenges for combining chemo- and biocatalysis. Nature Cat. 2018, 1, 12–22. [Google Scholar] [CrossRef]
- Yuryev, R.; Strompen, S.; Liese, A. Coupled chemo(enzymatic) reactions in continuous flow. Beilstein J. Org. Chem. 2011, 7, 1449–1467. [Google Scholar] [CrossRef]
- Fink, M.J.; Schön, M.; Rudroff, F.; Schnürch, M.; Mihovilovic, M.D. Single operation stereoselective synthesis of aerangis lactones: Combining continuous flow hydrogenation and biocatalysts in a chemoenzymatic sequence. ChemCatChem 2013, 5, 724–727. [Google Scholar] [CrossRef]
- Wohlgemuth, R.; Plazl, I.; Žnidaršič-Plazl, P.; Gernaey, K.V.; Woodley, J.M. Microscale technology and bio-catalytic processes: Opportunities and challenges for synthesis. Trends Biotechnol. 2015, 33, 302–314. [Google Scholar] [CrossRef]
- Yao, X.; Zhang, Y.; Du, L.; Liu, J.; Yao, J. Review of the applications of microreactors. Renew. Sustain. Energy Rev. 2015, 47, 519–539. [Google Scholar] [CrossRef]
- Tomaszewski, B.; Schmid, A.; Buehler, K. Biocatalytic production of catechols using a high pressure tube-in-tube segmented flow microreactor. Org. Process Res. Dev. 2014, 18, 1516–1526. [Google Scholar] [CrossRef]
- Ringborg, R.H.; Pedersen, A.T.; Woodley, J.M. Automated Determination of Oxygen-Dependent Enzyme Kinetics in a Tube-in-Tube Flow Reactor. ChemCatChem 2017, 9, 3273. [Google Scholar] [CrossRef]
- Peris, E.; Okafor, O.; Kulcinskaja, E.; Goodridge, R.; Luis, S.V.; Garcia-Verdugo, E.; O’Reilly, E.; Sans, V. Tuneable 3D-printed bioreactors for transaminations under continuous-flow. Green Chem. 2017, 19, 5345–5349. [Google Scholar] [CrossRef]
- Peng, M.; Mittmann, E.; Wenger, L.; Hubbuch, J.; Engqvist, M.K.M.; Niemeyer, C.M.; Rabe, K.S. 3D-Printed Phenacrylate Decarboxylase Flow Reactors for the Chemoenzymatic Synthesis of 4-Hydroxystilben. Chem. Eur. J. 2019, 25, 15998–16001. [Google Scholar] [CrossRef] [PubMed]
- Schmieg, B.; Döbber, J.; Kirschhöfer, F.; Pohl, M.; Franzreb, M. Advantages of Hydrogel-Based 3D-Printed Enzyme Reactors and Their Limitations for Biocatalysis. Front. Bioeng. Biotechnol. 2019, 6, 211. [Google Scholar] [CrossRef]
- Spano, M.B.; Tran, B.H.; Majumdar, S.; Weiss, G.A. 3D-Printed Labware for High-Throughput Immobilization of Enzymes. J. Org. Chem. 2020, 85, 8480–8488. [Google Scholar] [CrossRef]
- Baneyx, F. Recombinant protein expression in Escherichia coli. Curr. Opin. Biotechnol. 1999, 10, 411–421. [Google Scholar] [CrossRef]
- Overton, T.W. Recombinant protein production in bacterial hosts. Drug Discov. Today 2014, 19, 590–601. [Google Scholar] [CrossRef]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in microbial systems. Front. Microbiol. 2014, 5, 341. [Google Scholar] [CrossRef]
- Lin, B.; Tao, Y. Whole-cell biocatalysts by design. Microb. Cell Fact. 2017, 16, 106. [Google Scholar] [CrossRef]
- Wachtmeister, J.; Rother, D. Recent advances in whole cell biocatalysis techniques bridging from investigative to industrial scale. Curr. Opin. Biotechnol. 2016, 42, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Rolf, J.; Rosenthal, K.; Lütz, S. Application of Cell-Free Protein Synthesis for Faster Biocatalyst Development. Catalysts 2019, 9, 190. [Google Scholar] [CrossRef]
- Ni, Y.; Chen, R.R. Accelerating whole-cell biocatalysis by reducing outer membrane permeability barrier. Biotechnol. Bioeng. 2004, 87, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.R. Permeability issues in whole-cell bioprocesses and cellular membrane engineering. Appl. Microbiol. Biotechnol. 2007, 74, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Bolivar, J.M.; Nidetzky, B. Multiphase biotransformations in microstructured reactors: Opportunities for biocatalytic process intensification and smart flow processing. Green Process. Synth. 2013, 2, 541–559. [Google Scholar] [CrossRef]
- Zdarta, J.; Meyer, A.S.; Jesionowski, T.; Pinelo, M. General Overview of Support Materials for Enzyme Immobilization: Characteristics, Properties, Practical Utility. Catalysts 2018, 8, 92. [Google Scholar] [CrossRef]
- Dos Santos, J.C.S.; Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Rodrigues, R.C.; Fernandez-Lafuente, R. Importance of the Support Properties for Immobilization or Purification of Enzymes. ChemCatChem 2015, 7, 2413–2432. [Google Scholar] [CrossRef]
- Lilly, M.D. Enzymes immobilized to cellulose. Method. Enzymol. 1976, 44, 46–53. [Google Scholar] [CrossRef]
- Cristóvão, R.O.; Silvério, S.C.; Tavares, A.P.M.; Brígida, A.I.S.; Loureiro, J.M.; Boaventura, R.A.R.; Macedo, E.A.; Coelho, M.A.Z. Green coconut fiber: A novel carrier for the immobilization of commercial laccase by covalent attachment for textile dyes decolourization. World J. Microbiol. Biotechnol. 2012, 28, 2827–2838. [Google Scholar] [CrossRef]
- De Souza Lima, J.; Costa, F.N.; Bastistella, M.A.; de Araújo, P.H.H.; De Oliveira, D. Functionalized kaolin as support for endoglucanase immobilization. Bioprocess Biosyst. Eng. 2019, 42, 1165–1173. [Google Scholar] [CrossRef]
- Vinu, A.; Murugesan, V.; Hartmann, M. Adsorption of Lysozyme over Mesoporous Molecular Sieves MCM-41 and SBA-15: Influence of pH and Aluminum Incorporation. J. Phys. Chem. B 2004, 108, 7323–7330. [Google Scholar] [CrossRef]
- Shi, Q.-H.; Tian, Y.; Dong, X.-Y.; Bai, S.; Sun, Y. Chitosan-coated silica beads as immobilized metal affinity support for protein adsorption. Biochem. Eng. J. 2003, 16, 317–322. [Google Scholar] [CrossRef]
- Zucca, P.; Fernandez-Lafuente, R.; Sanjust, E. Agarose and Its Derivatives as Supports for Enzyme Immobilization. Molecules 2016, 21, 1577. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Wang, G.; Han, W.; Shen, Y.; Uyama, H. An ideal enzyme immobilization carrier: A hierarchically porous cellulose monolith fabricated by phase separation method. Pure Appl. Chem. 2018, 90, 1055–1062. [Google Scholar] [CrossRef]
- Jackson, E.; Correa, S.; Betancor, L. Cellulose-Based Nanosupports for Enzyme Immobilization. In Cellulose-Based Superabsorbent Hydrogels. Polymers and Polymeric Composites: A Reference Series; Mondal, M., Ed.; Springer: Cham, Switzerland, 2019. [Google Scholar]
- Suo, H.; Xu, L.; Xue, Y.; Qiu, X.; Huang, H.; Hu, Y. Ionic liquids-modified cellulose coated magnetic nanoparticles for enzyme immobilization: Improvement of catalytic performance. Carbohydr. Polym. 2020, 234, 115914. [Google Scholar] [CrossRef]
- Peschke, T.; Bitterwolf, P.; Gallus, S.; Hu, Y.; Oelschlaeger, C.; Willenbacher, N.; Rabe, K.; Niemeyer, C.M. Self-Assembling All-Enzyme Hydrogels for Flow Biocatalysis. Angew. Chem. Int. Ed. 2018, 130, 17153–17514. [Google Scholar] [CrossRef]
- Menegatti, T.; Žnidaršič-Plazl, P. Copolymeric Hydrogel-Based Immobilization of Yeast Cells for Continuous Biotransformation of Fumaric Acid in a Microreactor. Micromachines 2019, 10, 867. [Google Scholar] [CrossRef]
- Pauly, H.J.; Gröger, H.; Patel, A.V. Developing Multicompartment Biopolymer Hydrogel Beads for Tandem Chemoenzymatic One-Pot Process. Catalysts 2019, 9, 547. [Google Scholar] [CrossRef]
- Valikhani, D.; Bolivar, J.M.; Nidetzky, B. Enzyme Immobilization in Wall-Coated Flow Microreactors. Methods Mol. Biol. 2020, 2100, 243–257. [Google Scholar] [CrossRef]
- Cen, Y.-K.; Liu, Y.-X.; Xue, Y.-P.; Zheng, Y.-G. Immobilization of enzymes in/on membranes and their applications. Adv. Synth. Catal. 2019, 361, 5500–5515. [Google Scholar] [CrossRef]
- Ranieri, G.; Mazzei, R.; Wu, Z.; Li, K.; Giorno, L. Use of a Ceramic Membrane to Improve the Performance of Two-Separate-Phase Biocatalytic Membrane Reactor. Molecules 2016, 21, 345. [Google Scholar] [CrossRef] [PubMed]
- Adebar, N.; Gröger, H. Heterogeneous Catalysts “on the Move”: Flow Chemistry with Fluid Immobilised (Bio)Catalysts. Eur. J. Org. Chem. 2020, 6062–6067. [Google Scholar] [CrossRef]
- Mohamad, N.R.; Marzuki, N.H.C.; Buang, N.A.; Huyop, F.; Wahab, R.A. An overview of technologies for immobilization of enzymes and surface analysis techniques for immobilized enzymes. Biotechnol. Biotechnol. Equip. 2015, 29, 205–220. [Google Scholar] [CrossRef]
- Lindeque, R.M.; Woodley, J.M. Reactor Selection for Effective Continuous Biocatalytic Production of Pharmaceuticals. Catalysts 2019, 9, 262. [Google Scholar] [CrossRef]
- Csajági, C.; Szatzker, G.; Tőke, E.R.; Ürge, L.; Darvasa, F.; Poppe, L. Enantiomer selective acylation of racemic alcohols by lipases in continuous-flow bioreactors. Tetrahedron Asymmetry 2008, 19, 237–246. [Google Scholar] [CrossRef]
- Weidenhamer, J.D.; Macias, F.A.; Fisher, N.H.; Williamson, B. Just how insoluble are monoterpenes? J. Chem. Ecol. 1993, 19, 1799–1807. [Google Scholar] [CrossRef] [PubMed]
- Breitmaier, E. Sesquiterpenes. In Terpenes: Flavours, Fragrances, Pharmaca, Pheromones; Breitmaier, E., Ed.; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Zhu, C.; Cook, S.P. A Concise Synthesis of (+)-Artemisinin. J. Am. Chem. Soc. 2012, 134, 13577–13579. [Google Scholar] [CrossRef] [PubMed]
- Yadav, J.S.; Thirupathaiah, B.; Srihari, P. A concise stereoselective total synthesis of (+)-artemisinin. Tetrahedron 2010, 66, 2005–2009. [Google Scholar] [CrossRef]
- Posthumus, M.A.; Schmidt, C.O.; De Kraker, J.W.; Konig, W.A.; Franssen, M.C. Amorpha-4,11-diene synthase catalyses the first probable step in artemisinin biosynthesis. Phytochemistry 1999, 52, 843–854. [Google Scholar] [CrossRef]
- Inprakhon, P.; Wongthongdee, N.; Amornsakchai, T.; Pongtharankul, T.; Sunintaboon, P.; Wiemann, L.O.; Durand, A.; Sieber, V. Lipase-catalyzed synthesis of sucrose monoester: Increased productivity by combining enzyme pretreatment and non-aqueous biphasic medium. J. Biotechnol. 2017, 259, 182–190. [Google Scholar] [CrossRef]
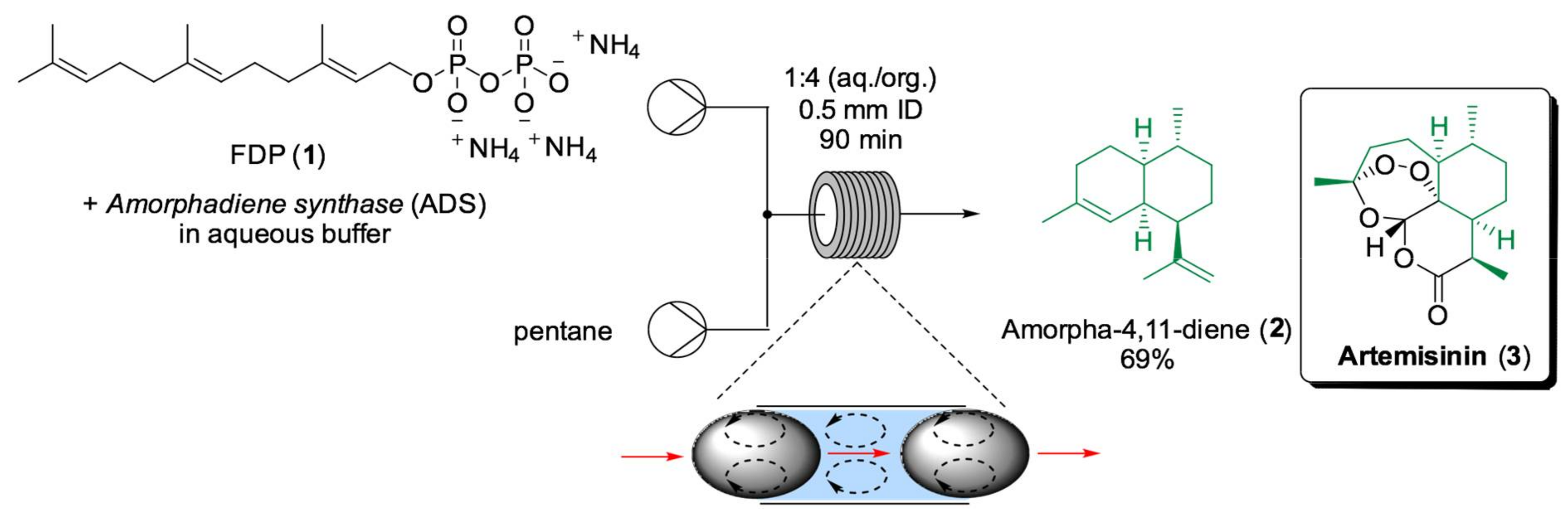
- Tang, X.; Allemann, R.K.; Wirth, T. Optimising Terpene Synthesis with Flow Biocatalysis. Eur. J. Org. Chem. 2017, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Kashid, M.N.; Agar, D.W. Hydrodynamics of liquid–liquid slug flow capillary microreactor: Flow regimes, slug size and pressure drop. Chem. Eng. J. 2007, 131, 1–13. [Google Scholar] [CrossRef]
- Kashid, M.N.; Agar, D.W.; Turek, S. CFD modelling of mass transfer with and without chemical reaction in the liquid–liquid slug flow microreactor. Chem. Eng. Sci. 2007, 62, 5102–5109. [Google Scholar] [CrossRef]
- Cascón, O.; Richter, G.; Allemann, R.K.; Wirth, T. Efficient Terpene Synthase Catalysis by Extraction in Flow. ChemPlusChem 2013, 78, 1334–1337. [Google Scholar] [CrossRef]
- Faraldos, J.A.; Miller, D.J.; Gonzalez, V.; Yoosuf-Aly, Z.; Cascon, O.; Li, A.; Allemann, R.K. A 1,6-Ring Closure Mechanism for (+)-δ-Cadinene Synthase? J. Am. Chem. Soc. 2012, 134, 5900–5908. [Google Scholar] [CrossRef]
- Huynh, F.; Tailby, M.; Finniear, A.; Stephens, K.; Allemann, R.K.; Wirth, T. Accelerating Biphasic Biocatalysis through New Process Windows. Angew. Chem. Int. Ed. 2020, 59, 16490–16495. [Google Scholar] [CrossRef]
- Berthod, A.; Faure, K. Separations with a Liquid Stationary Phase: Countercurrent Chromatography or Centrifugal Partition Chromatography. In Analytical Separation Science; Anderson, J., Berthod, A., Pino, V., Stalcup, A.M., Eds.; Wiley-VCH: Weinheim, Germany, 2015; Volume 4, pp. 1177–1206. [Google Scholar]
- Berthod, A. Countercurrent Chromatography. Compr. Anal. Chem. 2002, 38, 1–20. [Google Scholar] [CrossRef]
- Skalicka-Woźniak, K.; Garrard, I.K. Counter-current chromatography for the separation of terpenoids: A comprehensive review with respect to the solvent systems employed. Phytochem. Rev. 2014, 13, 547–572. [Google Scholar] [CrossRef]
- Valikhani, D.; Srivastava, P.L.; Allemann, R.K.; Wirth, T. Immobilised Enzymes for Sesquiterpene Synthesis in Batch and Flow Systems. ChemCatChem 2020, 12, 2194–2197. [Google Scholar] [CrossRef]
- Hugentobler, K.G.; Rasparini, M.; Thompson, L.A.; Jolley, K.E.; Blacker, A.J.; Turner, N.J. Comparison of a Batch and Flow Approach for the Lipase-Catalyzed Resolution of a Cyclopropanecarboxylate Ester, A Key Building Block for the Synthesis of Ticagrelor. Org. Process Res. Dev. 2017, 21, 195–199. [Google Scholar] [CrossRef]
- Wynne, S.; Djakiew, D. NSAID Inhibition of Prostate Cancer Cell Migration Is Mediated by Nag-1 Induction via the p38 MAPK-p75NTR Pathway. Mol. Cancer Res. 2010, 8, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- Tamborini, L.; Romano, D.; Pinto, A.; Bertolani, A.; Molinari, F.; Conti, P. An efficient method for the lipase-catalysed resolution and in-line purification of racemic flurbiprofen in a continuous-flow reactor. J. Mol. Catal. B Enzym. 2012, 84, 78–82. [Google Scholar] [CrossRef]
- Tamborini, L.; Romano, D.; Pinto, A.; Contente, M.; Iannuzzi, M.C.; Conti, P.; Molinari, F. Biotransformation with whole microbial systems in a continuous flow reactor: Resolution of (RS)-flurbiprofen using Aspergillus oryzae by direct esterification with ethanol in organic solvent. Tetrahedron Lett. 2013, 54, 6090–6093. [Google Scholar] [CrossRef]
- Roura Padrosa, D.; De Vitis, V.; Contente, M.; Molinari, F.; Paradisi, F. Overcoming Water Insolubility in Flow: Enantioselective Hydrolysis of Naproxen Ester. Catalysts 2019, 9, 232. [Google Scholar] [CrossRef]
- Andrade, L.H.; Kroutil, W.; Jamison, T.F. Continuous flow synthesis of chiral amines in organic solvents: Immobilization of E. coli cells containing both ω-transaminase and PLP. Org. Lett. 2014, 16, 6092–6095. [Google Scholar] [CrossRef]
- De Vitis, V.; Dall’Oglio, F.; Pinto, A.; De Micheli, C.; Molinari, F.; Conti, P.; Romano, D.; Tamborini, L. Chemoenzymatic Synthesis in Flow Reactors: A Rapid and Convenient Preparation of Captopril. ChemistryOpen 2017, 6, 668–673. [Google Scholar] [CrossRef]
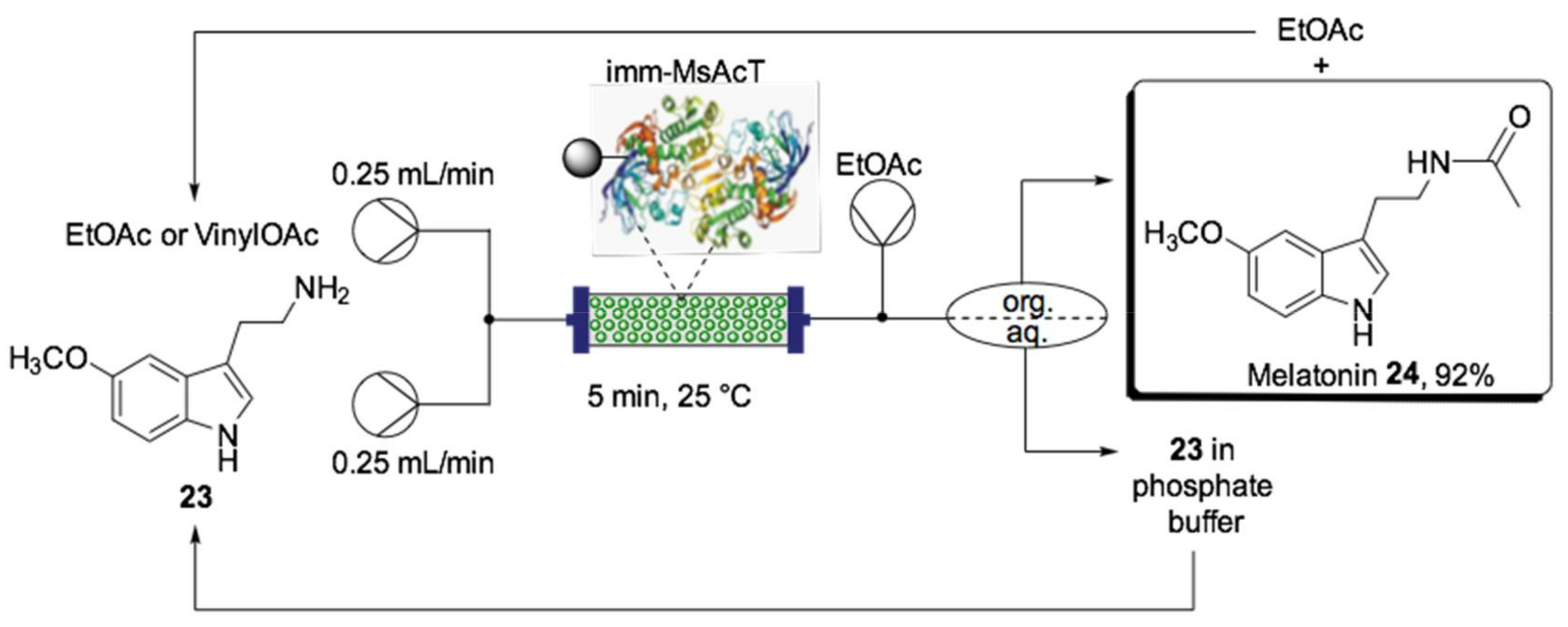
- Contente, M.L.; Farris, S.; Tamborini, L.; Molinari, F.; Paradisi, F. Flow-based enzymatic synthesis of melatonin and other high value tryptamine derivatives: A five-minute intensified process. Green Chem. 2019, 21, 3263–3266. [Google Scholar] [CrossRef]
- Tamborini, L.; Previtali, C.; Annunziata, F.; Bavaro, T.; Terreni, M.; Calleri, E.; Rinaldi, F.; Pinto, A.; Speranza, G.; Ubiali, D.; et al. An enzymatic flow-based preparative route to vidarabine. Molecules 2020, 25, 1223. [Google Scholar] [CrossRef]
- Rinaldi, F.; Fernández-Lucas, J.; de la Fuente, D.; Zheng, C.; Bavaro, T.; Peters, B.; Massolini, G.; Annunziata, F.; Conti, P.; de la Mata, I.; et al. Immobilized enzyme reactors based on nucleoside phosphorylases and 2′-deoxyribosyltransferase for the in-flow synthesis of pharmaceutically relevant nucleoside analogues. Bioresour. Technol. 2020, 307, 1–9. [Google Scholar] [CrossRef]
- Annunziata, F.; Letizia Contente, M.; Betti, D.; Pinna, C.; Molinari, F.; Tamborini, L.; Pinto, A. Efficient Chemo-Enzymatic Flow Synthesis of High Value Amides and Esters. Catalysts 2020, 10, 939. [Google Scholar] [CrossRef]
- Britton, J.; Raston, C.L. Multi-step continuous-flow synthesis. Chem. Soc. Rev. 2017, 46, 1250–1271. [Google Scholar] [CrossRef] [PubMed]
- Roura Padrosa, D.; Benítez-Mateos, A.I.; Calvey, L.; Paradisi, F. Cell-free biocatalytic syntheses of l-pipecolic acid: A dual strategy approach and process intensification in flow. Green Chem. 2020, 22, 5310–5316. [Google Scholar] [CrossRef]
- Hartley, C.J.; Williams, C.C.; Scoble, J.A.; Churches, Q.I.; North, A.; French, N.G.; Nebl, T.; Coia, G.; Warden, A.C.; Simpson, G.; et al. Engineered enzymes that retain and regenerate their cofactors enable continuous-flow biocatalysis. Nat. Catal. 2019, 2, 1006–1015. [Google Scholar] [CrossRef]
- Goundry, W.R.F.; Dai, K.; Gonzalez, M.; Legg, D.; O’Kearney-McMullan, A.; Morrison, J.; Stark, A.; Siedlecki, P.; Tomlin, P.; Yang, J. Development and Scale-up of a Route to ATR Inhibitor AZD6738. Org. Process Res. Dev. 2019, 23, 1333–1342. [Google Scholar] [CrossRef]
- Ho, C.-H.; Yi, J.; Wang, X. Biocatalytic Continuous Manufacturing of Diabetes Drug: Plantwide Process Modeling, Optimization, and Environmental and Economic Analysis. ACS Sustain. Chem. Eng. 2019, 7, 1038–1051. [Google Scholar] [CrossRef]
- World Demand for Flavor, Fragrances to Exceed $26 Billion in 2020. 2016. Available online: http://www.preparedfoods.com/articles/118094-world-demand-for-flavor-fragrances-to-exceed-26-billion-in-2020 (accessed on 5 December 2020).
- Romero, M.D.; Calvo, L.; Alba, C.; Daneshfar, A.; Ghaziaskar, H.S. Enzymatic synthesis of isoamyl acetate with immobilized Candida antarctica lipase in n-hexane. Enzyme Microbial. Technol. 2005, 37, 42–48. [Google Scholar] [CrossRef]
- Sakakibara, I.; Katsuhara, T.; Ikeya, Y.; Hayashi, K.; Mitsuhashi, H. Cannabisin A, an arylnaphthalene lignanamide from fruits of Cannabis sativa. Phytochemistry 1991, 30, 3013–3016. [Google Scholar] [CrossRef]
- Baxendale, I.R.; Griffiths-Jones, C.M.; Ley, S.V.; Tranmer, G.K. Preparation of the Neolignan Natural Product Grossamide by a Continuous-Flow Process. Synlett 2006, 427–430. [Google Scholar] [CrossRef]
- Gumel, A.M.; Annuar, M.S.M. Thermomyces lanuginosus lipase-catalyzed synthesis of natural flavor esters in a continuous flow microreactor. 3 Biotech 2016, 6. [Google Scholar] [CrossRef]
- Contente, M.L.; Dall’Oglio, F.; Tamborini, L.; Molinari, F.; Pardisi, F. Highly Efficient Oxidation of Amines to Aldehydes with Flow-based Biocatalysis. ChemCatChem 2017, 9, 1–7. [Google Scholar] [CrossRef]
- Lozano, P.; Bernal, J.M.; Navarro, A. Clean enzymatic process for producing flavour esters by direct esterification in switchable ionic liquid/solid phases. Green Chem. 2012, 14, 3026–3033. [Google Scholar] [CrossRef]
- Todero, L.M.; Bassi, J.J.; Lage, F.A.P.; Corradini, M.C.A.C.; Barboza, J.C.S.; Hirata, D.B.; Mendes, A.A. Enzymatic synthesis of isoamyl butyrate catalyzed by immobilized lipase on poly-methacrylate particles: Optimization, reusability and mass transfer studies. Bioprocess Biosyst. Eng. 2015, 38, 1601–1613. [Google Scholar] [CrossRef] [PubMed]
- Salvi, H.M.; Kamble, M.P.; Yadav, G.D. Synthesis of Geraniol Esters in a Continuous-Flow Packed-Bed Reactor of Immobilized Lipase: Optimization of Process Parameters and Kinetic Modeling. Appl. Biochem. Biotechnol. 2018, 184, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Adarme, C.A.A.; Leão, R.A.C.; de Souza, S.P.; Itabaiana, I., Jr.; Souza, R.O.M.A.; Rezende, C.M. Continuous-Flow Chemo and Enzymatic Synthesis of Monoterpenic Esters with Integrated Purification. Mol. Catal. 2018, 453, 39–46. [Google Scholar] [CrossRef]
- Contente, M.L.; Tamborini, L.; Molinari, F.; Paradisi, F. Aromas flow: Eco-friendly, continuous, and scalable preparation of flavour esters. J. Flow. Chem. 2020, 10, 235–240. [Google Scholar] [CrossRef]




























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santi, M.; Sancineto, L.; Nascimento, V.; Braun Azeredo, J.; Orozco, E.V.M.; Andrade, L.H.; Gröger, H.; Santi, C. Flow Biocatalysis: A Challenging Alternative for the Synthesis of APIs and Natural Compounds. Int. J. Mol. Sci. 2021, 22, 990. https://doi.org/10.3390/ijms22030990
Santi M, Sancineto L, Nascimento V, Braun Azeredo J, Orozco EVM, Andrade LH, Gröger H, Santi C. Flow Biocatalysis: A Challenging Alternative for the Synthesis of APIs and Natural Compounds. International Journal of Molecular Sciences. 2021; 22(3):990. https://doi.org/10.3390/ijms22030990
Chicago/Turabian StyleSanti, Micol, Luca Sancineto, Vanessa Nascimento, Juliano Braun Azeredo, Erika V. M. Orozco, Leandro H. Andrade, Harald Gröger, and Claudio Santi. 2021. "Flow Biocatalysis: A Challenging Alternative for the Synthesis of APIs and Natural Compounds" International Journal of Molecular Sciences 22, no. 3: 990. https://doi.org/10.3390/ijms22030990
APA StyleSanti, M., Sancineto, L., Nascimento, V., Braun Azeredo, J., Orozco, E. V. M., Andrade, L. H., Gröger, H., & Santi, C. (2021). Flow Biocatalysis: A Challenging Alternative for the Synthesis of APIs and Natural Compounds. International Journal of Molecular Sciences, 22(3), 990. https://doi.org/10.3390/ijms22030990