
DIscBIO: A User-Friendly Pipeline for Biomarker Discovery in Single-Cell Transcriptomics

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
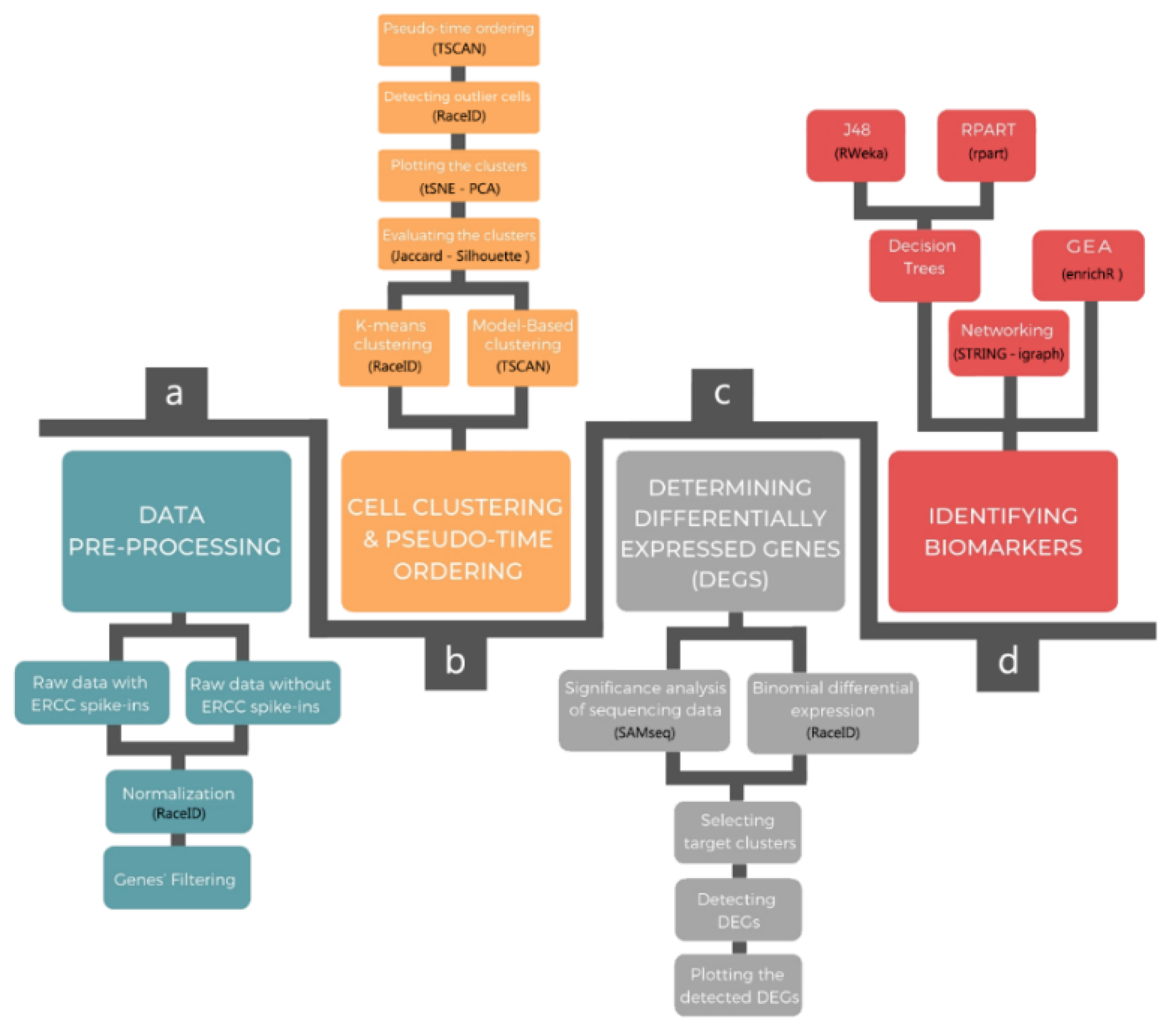
2. Pipeline Description
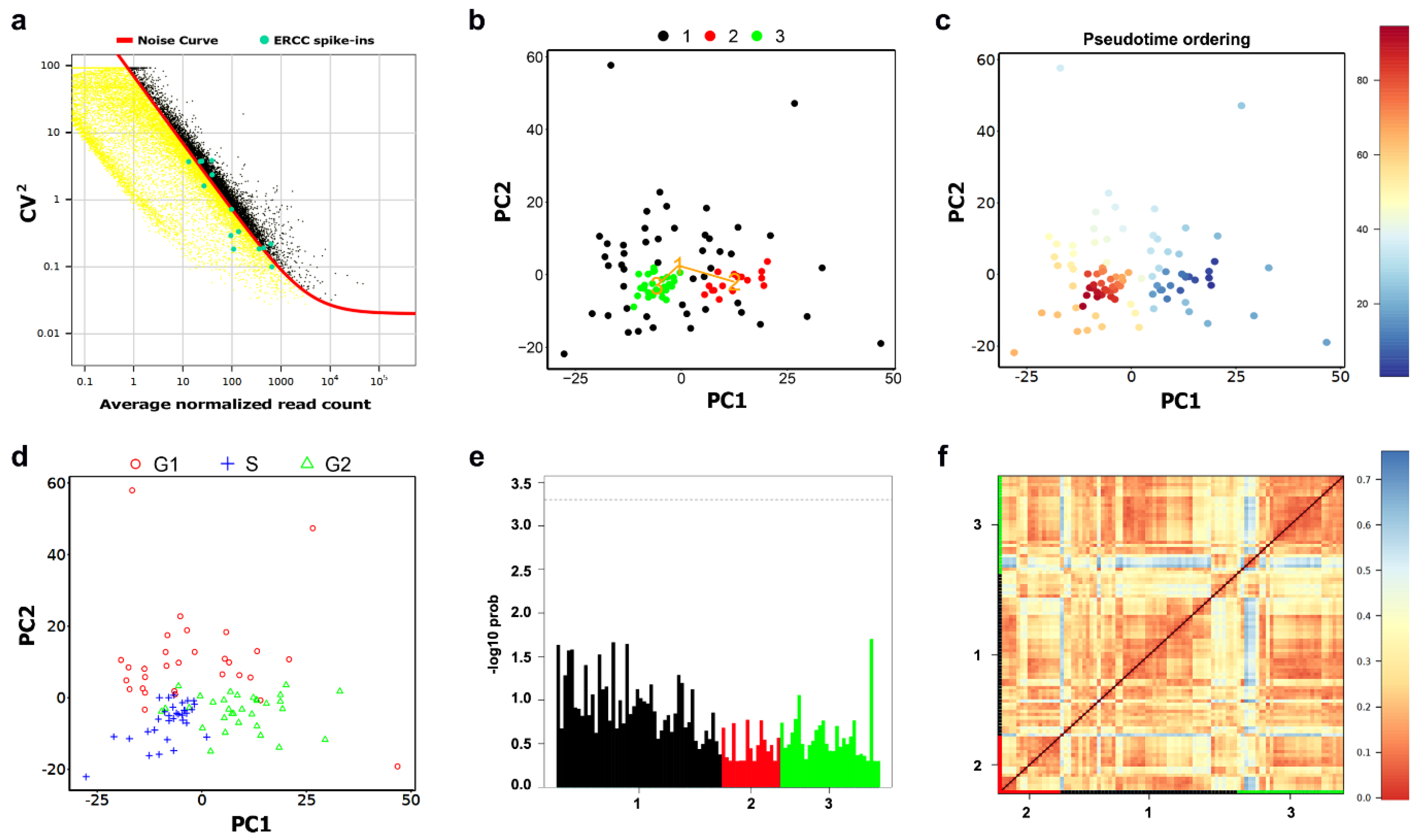
2.1. Data Pre-Processing
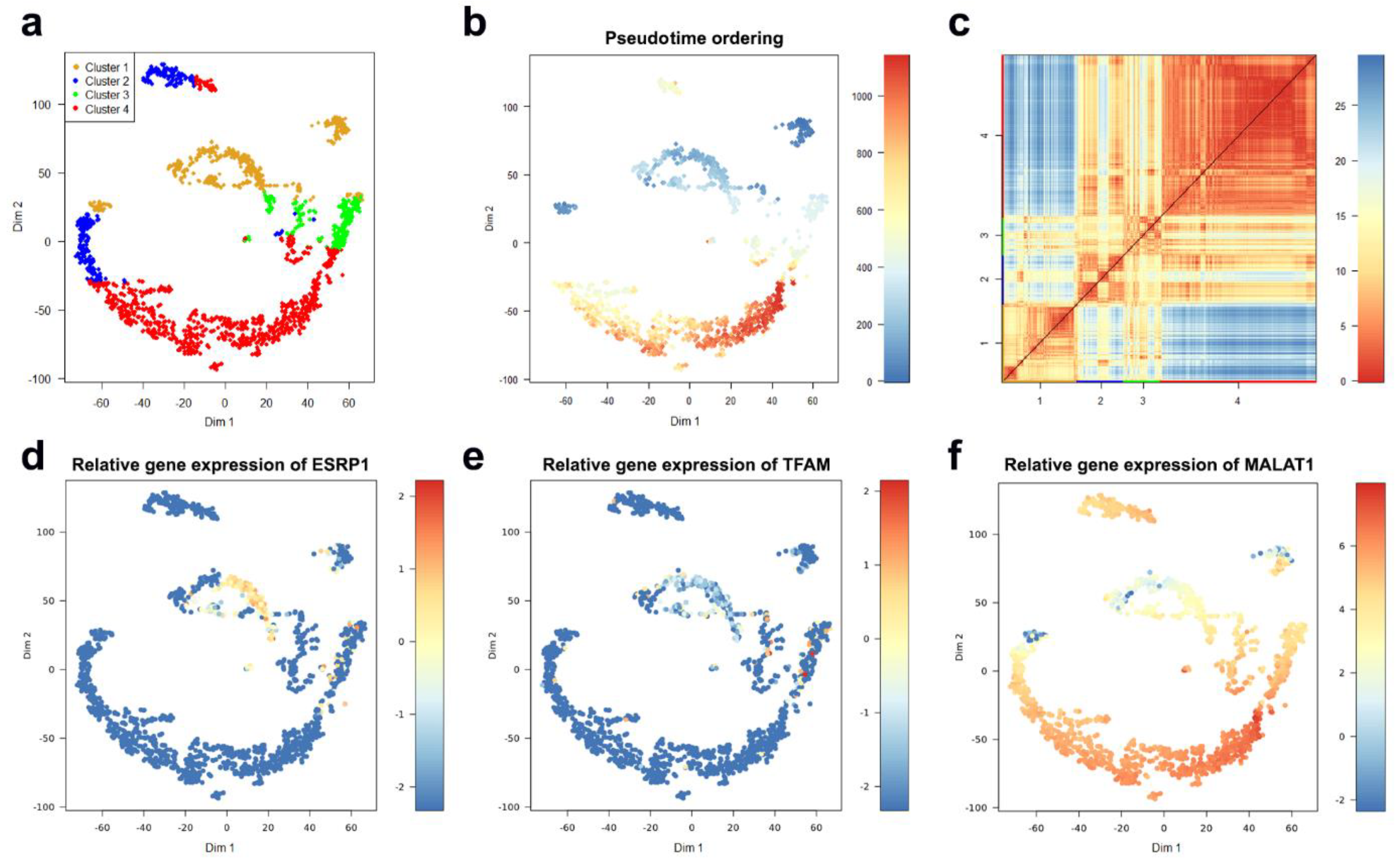
2.2. Cellular Clustering and Pseudo-Temporal Ordering
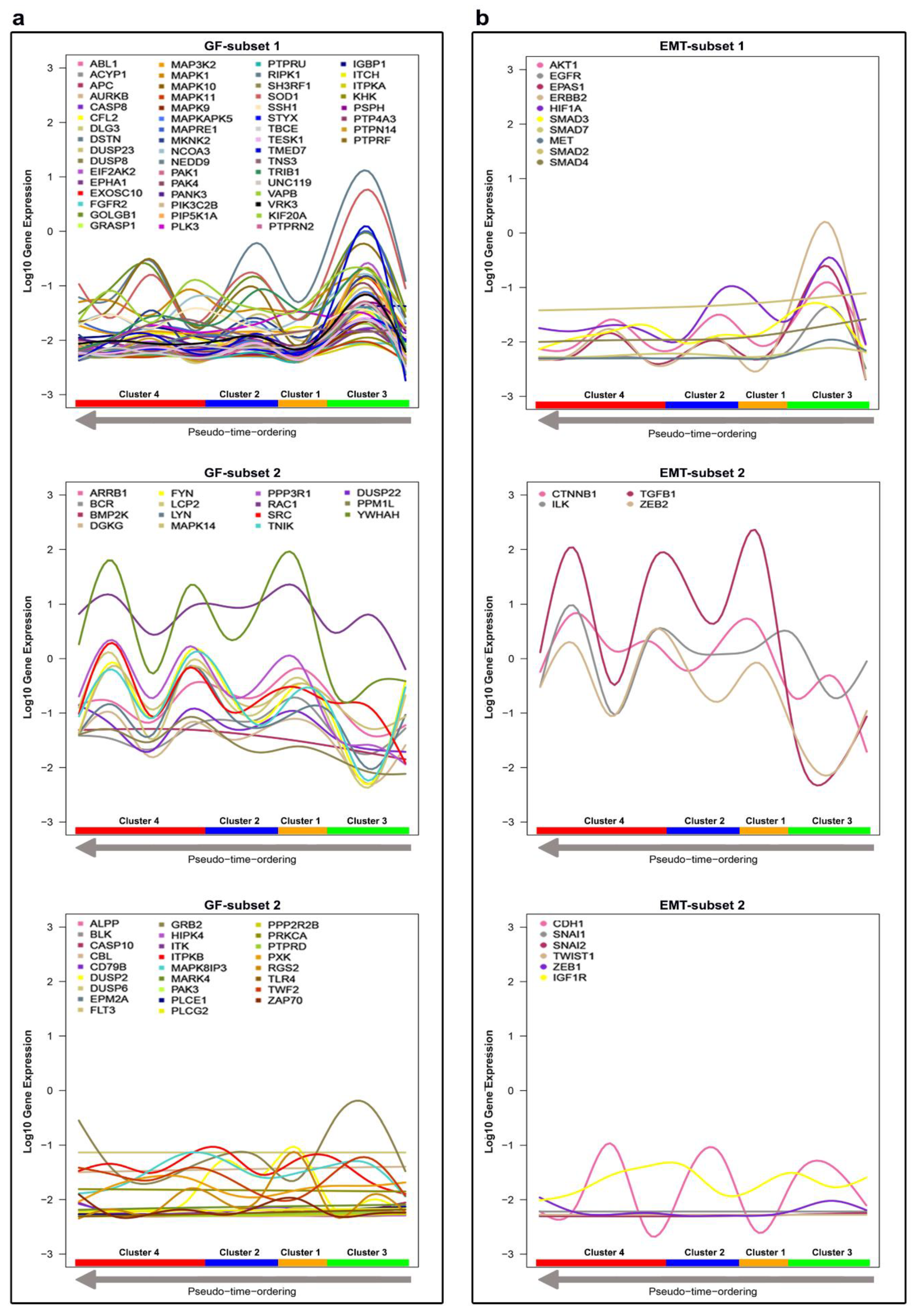
2.3. Determining DEGs
2.4. Identifying Biomarkers
3. Pipeline Extension
4. Case Studies
4.1. CTC Case Study
4.1.1. Characterization of CTC Subpopulations
4.1.2. Linking Alterations of the Golgi Apparatus with Cancer Progression
4.2. MLS Case Study
5. A Comparative Analysis of DIscBIO against Similar scRNAseq Pipelines
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stegle, O.; Teichmann, S.A.; Marioni, J.C. Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 2015, 16, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Grün, D.; van Oudenaarden, A. Design and analysis of single-cell sequencing experiments. Cell 2015, 163, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.H.; Lukowski, S.W.; Chiu, H.S.; Senabouth, A.; Bruxner, T.J.; Christ, A.N.; Palpant, N.J.; Powell, J.E. Single-cell RNA-seq of human induced pluripotent stem cells reveals cellular heterogeneity and cell state transitions between subpopulations. Genome Res. 2018, 28, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Leigh, N.D.; Dunlap, G.S.; Johnson, K.; Mariano, R.; Oshiro, R.; Wong, A.Y.; Bryant, D.M.; Miller, B.M.; Ratner, A.; Chen, A. Transcriptomic landscape of the blastema niche in regenerating adult axolotl limbs at single-cell resolution. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.; Weinreb, C.; Wagner, D.E.; Megason, S.; Peshkin, L.; Kirschner, M.W.; Klein, A.M. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science 2018, 360. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.E.; Weinreb, C.; Collins, Z.M.; Briggs, J.A.; Megason, S.G.; Klein, A.M. Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science 2018, 360, 981–987. [Google Scholar] [CrossRef]
- Sladitschek, H.L.; Fiuza, U.-M.; Pavlinic, D.; Benes, V.; Hufnagel, L.; Neveu, P.A. MorphoSeq: Full Single-Cell Transcriptome Dynamics Up to Gastrulation in a Chordate. Cell 2020. [Google Scholar] [CrossRef]
- Han, X.; Zhou, Z.; Fei, L.; Sun, H.; Wang, R.; Chen, Y.; Chen, H.; Wang, J.; Tang, H.; Ge, W. Construction of a human cell landscape at single-cell level. Nature 2020, 581, 303–309. [Google Scholar] [CrossRef]
- Libault, M.; Pingault, L.; Zogli, P.; Schiefelbein, J. Plant systems biology at the single-cell level. Trends Plant Sci. 2017, 22, 949–960. [Google Scholar] [CrossRef]
- Jean-Baptiste, K.; McFaline-Figueroa, J.L.; Alexandre, C.M.; Dorrity, M.W.; Saunders, L.; Bubb, K.L.; Trapnell, C.; Fields, S.; Queitsch, C.; Cuperus, J.T. Dynamics of gene expression in single root cells of Arabidopsis thaliana. Plant Cell 2019, 31, 993–1011. [Google Scholar] [CrossRef]
- Soneson, C.; Robinson, M.D. Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods 2018, 15, 255. [Google Scholar] [CrossRef] [PubMed]
- Duo, A.; Robinson, M.D.; Soneson, C. A systematic performance evaluation of clustering methods for single-cell RNA-seq data. F1000Res 2018, 7, 1141. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, V.Y.; Kirschner, K.; Schaub, M.T.; Andrews, T.; Yiu, A.; Chandra, T.; Natarajan, K.N.; Reik, W.; Barahona, M.; Green, A.R. SC3: Consensus clustering of single-cell RNA-seq data. Nat. Methods 2017, 14, 483–486. [Google Scholar] [CrossRef]
- Gardeux, V.; David, F.P.; Shajkofci, A.; Schwalie, P.C.; Deplancke, B. ASAP: A web-based platform for the analysis and interactive visualization of single-cell RNA-seq data. Bioinformatics 2017, 33, 3123–3125. [Google Scholar] [CrossRef]
- Zhu, X.; Wolfgruber, T.K.; Tasato, A.; Arisdakessian, C.; Garmire, D.G.; Garmire, L.X. Granatum: A graphical single-cell RNA-Seq analysis pipeline for genomics scientists. Genome Med. 2017, 9, 1–12. [Google Scholar] [CrossRef]
- Moussa, M.; Măndoiu, I.I. SC1: A web-based single cell RNA-seq analysis pipeline. In Proceedings of the 2018 IEEE 8th International Conference on Computational Advances in Bio and Medical Sciences (ICCABS), Las Vegas, NV, USA, 18–20 October 2018. [Google Scholar]
- Bacher, R.; Kendziorski, C. Design and computational analysis of single-cell RNA-sequencing experiments. Genome Biol 2016, 17, 63. [Google Scholar] [CrossRef]
- Grün, D.; Lyubimova, A.; Kester, L.; Wiebrands, K.; Basak, O.; Sasaki, N.; Clevers, H.; Van Oudenaarden, A. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 2015, 525, 251–255. [Google Scholar] [CrossRef]
- Grün, D.; Muraro, M.J.; Boisset, J.-C.; Wiebrands, K.; Lyubimova, A.; Dharmadhikari, G.; van den Born, M.; Van Es, J.; Jansen, E.; Clevers, H. De novo prediction of stem cell identity using single-cell transcriptome data. Cell Stem Cell 2016, 19, 266–277. [Google Scholar] [CrossRef]
- Ji, Z.; Ji, H. TSCAN: Pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis. Nucleic Acids Res. 2016, 44, e117. [Google Scholar] [CrossRef]
- Brennecke, P.; Anders, S.; Kim, J.K.; Kolodziejczyk, A.A.; Zhang, X.; Proserpio, V.; Baying, B.; Benes, V.; Teichmann, S.A.; Marioni, J.C.; et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods 2013, 10, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Hu, Y.; Kelly, D.; Kim, J.; Li, M.; Zhang, N.R. Accounting for technical noise in differential expression analysis of single-cell RNA sequencing data. Nucleic Acids Res. 2017, 45, 10978–10988. [Google Scholar] [CrossRef] [PubMed]
- Goossens, N.; Nakagawa, S.; Sun, X.; Hoshida, Y. Cancer biomarker discovery and validation. Transl. Cancer Res. 2015, 4, 256. [Google Scholar] [PubMed]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M. STRING 8—A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, D412–D416. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Tung, C.-W.; Wu, M.-T.; Chen, Y.-K.; Wu, C.-C.; Chen, W.-C.; Li, H.-P.; Chou, S.-H.; Wu, D.-C.; Wu, I. Identification of biomarkers for esophageal squamous cell carcinoma using feature selection and decision tree methods. Sci. World J. 2013, 2013. [Google Scholar] [CrossRef]
- Huang, S.-H.; Tung, C.-W. Identification of consensus biomarkers for predicting non-genotoxic hepatocarcinogens. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Floares, A.; Birlutiu, A. Decision tree models for developing molecular classifiers for cancer diagnosis. In Proceedings of the 2012 International Joint Conference on Neural Networks (IJCNN), Brisbane, Australia, 10–15 June 2012; pp. 1–7. [Google Scholar]
- Hornik, K.; Buchta, C.; Zeileis, A. Open-source machine learning: R meets Weka. Comput. Stat. 2009, 24, 225–232. [Google Scholar] [CrossRef]
- Therneau, T.M.; Atkinson, B.; Ripley, M.B. The Rpart Package; R Foundation for Statistical Computing: Oxford, UK, 2010. [Google Scholar]
- Peng, R.D. Reproducible research in computational science. Science 2011, 334, 1226–1227. [Google Scholar] [CrossRef]
- Sandve, G.K.; Nekrutenko, A.; Taylor, J.; Hovig, E. Ten simple rules for reproducible computational research. PLoS Comput. Biol. 2013, 9, e1003285. [Google Scholar] [CrossRef]
- Ghannoum Salim, K.-L.A.; Waldir, L.; Damiano, F.; Min, R.K. ocbe-uio/DIscBIO: DIscBIO universe 1.0.1 (Version v1.0.1). Zenodo 2020. [Google Scholar] [CrossRef]
- Rule, A.; Birmingham, A.; Zuniga, C.; Altintas, I.; Huang, S.-C.; Knight, R.; Moshiri, N.; Nguyen, M.H.; Rosenthal, S.B.; Pérez, F. Ten Simple Rules for Writing and Sharing Computational Analyses in Jupyter Notebooks. PLoS Comput. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jupyter, P.; Bussonnier, M.; Forde, J.; Freeman, J.; Granger, B.; Head, T.; Holdgraf, C.; Kelley, K.; Nalvarte, G.; Osheroff, A. Binder 2.0-Reproducible, interactive, sharable environments for science at scale. In Proceedings of the 17th Python in Science Conference, Austin, TX, USA, 9–15 July 2018; p. 120. [Google Scholar]
- Shalek, A.K.; Satija, R.; Adiconis, X.; Gertner, R.S.; Gaublomme, J.T.; Raychowdhury, R.; Schwartz, S.; Yosef, N.; Malboeuf, C.; Lu, D. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 2013, 498, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Tarpey, T. A parametric k-means algorithm. Comput. Stat. 2007, 22, 71. [Google Scholar] [CrossRef] [PubMed]
- Fraley, C.; Raftery, A.E. Model-Based Clustering, Discriminant Analysis, and Density Estimation. J. Am. Stat. Assoc. 2002, 97, 611–631. [Google Scholar] [CrossRef]
- Tibshirani, R.; Walther, G.; Hastie, T. Estimating the number of clusters in a data set via the gap statistic. J. R. Stat. Soc. Ser. B Stat. Methodol. 2001, 63, 411–423. [Google Scholar] [CrossRef]
- Hennig, C. Cluster-wise assessment of cluster stability. Comput. Stat. Data Anal. 2007, 52, 258–271. [Google Scholar] [CrossRef]
- Lletı, R.; Ortiz, M.C.; Sarabia, L.A.; Sánchez, M.S. Selecting variables for k-means cluster analysis by using a genetic algorithm that optimises the silhouettes. Anal. Chim. Acta 2004, 515, 87–100. [Google Scholar] [CrossRef]
- Platzer, A. Visualization of SNPs with t-SNE. PLoS ONE 2013, 8, e56883. [Google Scholar] [CrossRef]
- Krijthe, J.H. Rtsne: T-Distributed Stochastic Neighbor Embedding using Barnes-Hut Implementation, R package version 0. 10; R Foundation for Statistical Computing: Oxford, UK, 2015. [Google Scholar]
- De Vienne, D.M.; Ollier, S.; Aguileta, G. Phylo-MCOA: A fast and efficient method to detect outlier genes and species in phylogenomics using multiple co-inertia analysis. Mol. Biol. Evol. 2012, 29, 1587–1598. [Google Scholar] [CrossRef]
- Li, J.; Tibshirani, R. Finding consistent patterns: A nonparametric approach for identifying differential expression in RNA-Seq data. Stat. Methods Med Res. 2013, 22, 519–536. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R.C.G.; Balasubramanian, N.; Li, J. Package ‘Samr’; R Foundation for Statistical Computing: Oxford, UK, 2015. [Google Scholar]
- Praktiknjo, S.D.; Obermayer, B.; Zhu, Q.; Fang, L.; Liu, H.; Quinn, H.; Stoeckius, M.; Kocks, C.; Birchmeier, W.; Rajewsky, N. Tracing tumorigenesis in a solid tumor model at single-cell resolution. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Xin, J.; Ren, X.; Chen, L.; Wang, Y. Identifying network biomarkers based on protein-protein interactions and expression data. BMC Med. Genom. 2015, 8, S11. [Google Scholar] [CrossRef]
- Xiao, H.; Yang, L.; Liu, J.; Jiao, Y.; Lu, L.; Zhao, H. Protein‑protein interaction analysis to identify biomarker networks for endometriosis. Exp. Ther. Med. 2017, 14, 4647–4654. [Google Scholar] [CrossRef]
- Traag, V.A.; Waltman, L.; van Eck, N.J. From Louvain to Leiden: Guaranteeing well-connected communities. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Kelly, S.T. Leiden: R Implementation of the Leiden Algorithm. R Package Version 0.3.6. 2020. Available online: https://github.com/TomKellyGenetics/leiden (accessed on 29 January 2021).
- Kharchenko, P.V.; Silberstein, L.; Scadden, D.T. Bayesian approach to single-cell differential expression analysis. Nat. Methods 2014, 11, 740–742. [Google Scholar] [CrossRef]
- Peyvandipour, A.; Shafi, A.; Saberian, N.; Draghici, S. Identification of cell types from single cell data using stable clustering. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Choi, J.-H.; Kim, H.I.; Woo, H.G. scTyper: A comprehensive pipeline for the cell typing analysis of single-cell RNA-seq data. BMC Bioinform. 2020, 21, 1–8. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Aceto, N.; Bersani, F.; Madden, M.W.; Donaldson, M.C.; Desai, R.; Zhu, H.; Comaills, V.; Zheng, Z. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 2014, 345, 216–220. [Google Scholar] [CrossRef]
- Sarioglu, A.F.; Aceto, N.; Kojic, N.; Donaldson, M.C.; Zeinali, M.; Hamza, B.; Engstrom, A.; Zhu, H.; Sundaresan, T.K.; Miyamoto, D.T. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat. Methods 2015, 12, 685–691. [Google Scholar] [CrossRef]
- Jordan, N.V.; Bardia, A.; Wittner, B.S.; Benes, C.; Ligorio, M.; Zheng, Y.; Yu, M.; Sundaresan, T.K.; Licausi, J.A.; Desai, R. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature 2016, 537, 102–106. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell 2019, 176, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N.; Bardia, A.; Wittner, B.S.; Donaldson, M.C.; O'Keefe, R.; Engstrom, A.; Bersani, F.; Zheng, Y.; Comaills, V.; Niederhoffer, K. AR expression in breast cancer CTCs associates with bone metastases. Mol. Cancer Res. 2018, 16, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.; Gupta, K.; Sharma, S.; Hari, K.; Lee, Y.F.; Ramalingam, N.; Yap, Y.S.; West, J.; Bhagat, A.A.; Subramani, B.V. Integrative analysis and machine learning based characterization of single circulating tumor cells. J. Clin. Med. 2020, 9, 1206. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-H.; Chen, Y.-C.; Lin, E.; Brien, R.; Jung, S.; Chen, Y.-T.; Lee, W.; Hao, Z.; Sahoo, S.; Kang, H.M. Hydro-Seq enables contamination-free high-throughput single-cell RNA-sequencing for circulating tumor cells. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Yang, C.; Xia, B.-R.; Jin, W.-L.; Lou, G. Circulating tumor cells in precision oncology: Clinical applications in liquid biopsy and 3D organoid model. Cancer Cell Int. 2019, 19, 341. [Google Scholar] [CrossRef] [PubMed]
- Jie, X.-X.; Zhang, X.-Y.; Xu, C.-J. Epithelial-to-mesenchymal transition, circulating tumor cells and cancer metastasis: Mechanisms and clinical applications. Oncotarget 2017, 8, 81558. [Google Scholar] [CrossRef] [PubMed]
- Calaminus, S.D.; Guitart, A.V.; Sinclair, A.; Schachtner, H.; Watson, S.P.; Holyoake, T.L.; Kranc, K.R.; Machesky, L.M. Lineage tracing of Pf4-Cre marks hematopoietic stem cells and their progeny. PLoS ONE 2012, 7, e51361. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Berk, R.; Kosir, M.A. CXCL7-mediated stimulation of lymphangiogenic factors VEGF-C, VEGF-D in human breast cancer cells. J. Oncol. 2010, 2010, 939407. [Google Scholar] [CrossRef] [PubMed]
- Kozlova, N.; Morozevich, G.; Ushakova, N.; Berman, A. Implication of integrin α2β1 in proliferation and invasion of human breast carcinoma and melanoma cells: Noncanonical function of akt protein kinase. Biochemistry 2018, 83, 738–745. [Google Scholar] [CrossRef]
- Sossey-Alaoui, K.; Pluskota, E.; Davuluri, G.; Bialkowska, K.; Das, M.; Szpak, D.; Lindner, D.J.; Downs-Kelly, E.; Thompson, C.L.; Plow, E.F. Kindlin-3 enhances breast cancer progression and metastasis by activating Twist-mediated angiogenesis. FASEB J. 2014, 28, 2260–2271. [Google Scholar] [CrossRef]
- Coupland, L.A.; Chong, B.H.; Parish, C.R. Platelets and P-selectin control tumor cell metastasis in an organ-specific manner and independently of NK cells. Cancer Res. 2012, 72, 4662–4671. [Google Scholar] [CrossRef]
- Kim, J.; Piao, H.-L.; Kim, B.-J.; Yao, F.; Han, Z.; Wang, Y.; Xiao, Z.; Siverly, A.N.; Lawhon, S.E.; Ton, B.N. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet. 2018, 50, 1705–1715. [Google Scholar] [CrossRef]
- Farhan, H.; Rabouille, C. Signalling to and from the secretory pathway. J. Cell Sci. 2011, 124, 171–180. [Google Scholar] [CrossRef]
- Bershadsky, A.D.; Futerman, A.H. Disruption of the Golgi apparatus by brefeldin A blocks cell polarization and inhibits directed cell migration. Proc. Natl. Acad. Sci. USA 1994, 91, 5686–5689. [Google Scholar] [CrossRef]
- Yadav, S.; Puri, S.; Linstedt, A.D. A primary role for Golgi positioning in directed secretion, cell polarity, and wound healing. Mol. Biol. Cell 2009, 20, 1728–1736. [Google Scholar] [CrossRef] [PubMed]
- Petrosyan, A. Onco-Golgi: Is fragmentation a gate to cancer progression? Biochem. Mol. Biol. J. 2015, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Farhan, H.; Wendeler, M.W.; Mitrovic, S.; Fava, E.; Silberberg, Y.; Sharan, R.; Zerial, M.; Hauri, H.P. MAPK signaling to the early secretory pathway revealed by kinase/phosphatase functional screening. J. Cell Biol. 2010, 189, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Chia, J.; Goh, G.; Racine, V.; Ng, S.; Kumar, P.; Bard, F. RNAi screening reveals a large signaling network controlling the Golgi apparatus in human cells. Mol. Syst. Biol. 2012, 8, 629. [Google Scholar] [CrossRef]
- Millarte, V.; Boncompain, G.; Tillmann, K.; Perez, F.; Sztul, E.; Farhan, H. Phospholipase C γ1 regulates early secretory trafficking and cell migration via interaction with p115. Mol. Biol. Cell 2015, 26, 2263–2278. [Google Scholar] [CrossRef] [PubMed]
- Joshi, G.; Chi, Y.; Huang, Z.; Wang, Y. Aβ-induced Golgi fragmentation in Alzheimer’s disease enhances Aβ production. Proc. Natl. Acad. Sci. USA 2014, 111, E1230–E1239. [Google Scholar] [CrossRef]
- Haase, G.; Rabouille, C. Golgi fragmentation in ALS motor neurons. New mechanisms targeting microtubules, tethers, and transport vesicles. Front. Neurosci. 2015, 9, 448. [Google Scholar] [CrossRef]
- Kim, S.B.; Zhang, L.; Yoon, J.; Lee, J.; Min, J.; Li, W.; Grishin, N.V.; Moon, Y.-A.; Wright, W.E.; Shay, J.W. Truncated adenomatous polyposis coli mutation induces Asef-activated Golgi fragmentation. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef]
- Khoshbakht, S.; Jamalkandi, S.A.; Masudi-Nejad, A. Metastasis progression through the interplay between the immune system and Epithelial-Mesenchymal-Transition in circulating breast tumor cells. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Joosse, S.A.; Hannemann, J.; Spötter, J.; Bauche, A.; Andreas, A.; Müller, V.; Pantel, K. Changes in keratin expression during metastatic progression of breast cancer: Impact on the detection of circulating tumor cells. Clin. Cancer Res. 2012, 18, 993–1003. [Google Scholar] [CrossRef]
- Jung, H.; Kim, B.; Moon, B.I.; Oh, E.-S. Cytokeratin 18 is necessary for initiation of TGF-β1-induced epithelial–mesenchymal transition in breast epithelial cells. Mol. Cell. Biochem. 2016, 423, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Wang, C.; Fu, N.; Liu, L.; Zhu, D.; Wei, Z.; Zhang, H.; Xing, J.; Wang, Y. Downregulation of cytokeratin 18 enhances BCRP-mediated multidrug resistance through induction of epithelial-mesenchymal transition and predicts poor prognosis in breast cancer. Oncol. Rep. 2019, 41, 3015–3026. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Marengo, M.S.; Oltean, S.; Kemeny, G.; Bitting, R.L.; Turnbull, J.D.; Herold, C.I.; Marcom, P.K.; George, D.J.; Garcia-Blanco, M.A. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol. Cancer Res. 2011, 9, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Kong, L.; Liu, Y.; Qu, H. dbEMT: An epithelial-mesenchymal transition associated gene resource. Sci. Rep. 2015, 5, 11459. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGFbeta signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef]
- Tan, X.; Banerjee, P.; Guo, H.-F.; Ireland, S.; Pankova, D.; Ahn, Y.-h.; Nikolaidis, I.M.; Liu, X.; Zhao, Y.; Xue, Y. Epithelial-to-mesenchymal transition drives a pro-metastatic Golgi compaction process through scaffolding protein PAQR11. J. Clin. Investig. 2017, 127, 117–131. [Google Scholar] [CrossRef]
- Karlsson, J.; Kroneis, T.; Jonasson, E.; Larsson, E.; Ståhlberg, A. Transcriptomic characterization of the human cell cycle in individual unsynchronized cells. J. Mol. Biol. 2017, 429, 3909–3924. [Google Scholar] [CrossRef]
- Crozat, A.; Åman, P.; Mandahl, N.; Ron, D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 1993, 363, 640–644. [Google Scholar] [CrossRef]
- Jo, V.Y.; Fletcher, C.D. WHO classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology 2014, 46, 95–104. [Google Scholar] [CrossRef]
- Ståhlberg, A.; Gustafsson, C.K.; Engtröm, K.; Thomsen, C.; Dolatabadi, S.; Jonasson, E.; Li, C.-Y.; Ruff, D.; Chen, S.-M.; Åman, P. Normal and functional TP53 in genetically stable myxoid/round cell liposarcoma. PLoS ONE 2014, 9, e113110. [Google Scholar] [CrossRef] [PubMed]
- Hofvander, J.; Viklund, B.; Isaksson, A.; Brosjö, O.; von Steyern, F.V.; Rissler, P.; Mandahl, N.; Mertens, F. Different patterns of clonal evolution among different sarcoma subtypes followed for up to 25 years. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shah, G.H.; Bhensdadia, C.; Ganatra, A.P. An empirical evaluation of density-based clustering techniques. Int. J. Soft Comput. Eng. IJSCE ISSN 2012, 22312307, 216–223. [Google Scholar]
- Bellas, A.; Bouveyron, C.; Cottrell, M.; Lacaille, J. Model-based clustering of high-dimensional data streams with online mixture of probabilistic PCA. Adv. Data Anal. Classif. 2013, 7, 281–300. [Google Scholar] [CrossRef]
- O’Dell, P. Midkine Is Associated with Poor Prognosis of Myxoid Liposarcoma; University of Eastern Finland: Eastern Finland, Finland, 2018. [Google Scholar]
- Yang, N.; Wang, C.; Wang, Z.; Zona, S.; Lin, S.; Wang, X.; Yan, M.; Zheng, F.; Li, S.; Xu, B. FOXM1 recruits nuclear Aurora kinase A to participate in a positive feedback loop essential for the self-renewal of breast cancer stem cells. Oncogene 2017, 36, 3428–3440. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Tan, X.; Lin, J.; Liu, R.-y.; Chen, S.; Geng, R.; Wu, J.; Huang, W. CDC27 induces metastasis and invasion in colorectal cancer via the promotion of epithelial-to-mesenchymal transition. J. Cancer 2017, 8, 2626. [Google Scholar] [CrossRef]
- Xin, Y.; Ning, S.; Zhang, L.; Cui, M. CDC27 facilitates gastric cancer cell proliferation, invasion and metastasis via twist-induced epithelial-mesenchymal transition. Cell. Physiol. Biochem. 2018, 50, 501–511. [Google Scholar] [CrossRef]
- Chen, W.; Dong, J.; Haiech, J.; Kilhoffer, M.C.; Zeniou, M. Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells Int. 2016, 2016, 1740936. [Google Scholar] [CrossRef]
- Spencer, S.L.; Cappell, S.D.; Tsai, F.-C.; Overton, K.W.; Wang, C.L.; Meyer, T. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 2013, 155, 369–383. [Google Scholar] [CrossRef]
- Naetar, N.; Soundarapandian, V.; Litovchick, L.; Goguen, K.L.; Sablina, A.A.; Bowman-Colin, C.; Sicinski, P.; Hahn, W.C.; DeCaprio, J.A.; Livingston, D.M. PP2A-mediated regulation of Ras signaling in G2 is essential for stable quiescence and normal G1 length. Mol. Cell 2014, 54, 932–945. [Google Scholar] [CrossRef]
- Sutcu, H.H.; Ricchetti, M. Loss of heterogeneity, quiescence, and differentiation in muscle stem cells. Stem Cell Investig. 2018, 5, 9. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghannoum, S.; Leoncio Netto, W.; Fantini, D.; Ragan-Kelley, B.; Parizadeh, A.; Jonasson, E.; Ståhlberg, A.; Farhan, H.; Köhn-Luque, A. DIscBIO: A User-Friendly Pipeline for Biomarker Discovery in Single-Cell Transcriptomics. Int. J. Mol. Sci. 2021, 22, 1399. https://doi.org/10.3390/ijms22031399
Ghannoum S, Leoncio Netto W, Fantini D, Ragan-Kelley B, Parizadeh A, Jonasson E, Ståhlberg A, Farhan H, Köhn-Luque A. DIscBIO: A User-Friendly Pipeline for Biomarker Discovery in Single-Cell Transcriptomics. International Journal of Molecular Sciences. 2021; 22(3):1399. https://doi.org/10.3390/ijms22031399
Chicago/Turabian StyleGhannoum, Salim, Waldir Leoncio Netto, Damiano Fantini, Benjamin Ragan-Kelley, Amirabbas Parizadeh, Emma Jonasson, Anders Ståhlberg, Hesso Farhan, and Alvaro Köhn-Luque. 2021. "DIscBIO: A User-Friendly Pipeline for Biomarker Discovery in Single-Cell Transcriptomics" International Journal of Molecular Sciences 22, no. 3: 1399. https://doi.org/10.3390/ijms22031399
APA StyleGhannoum, S., Leoncio Netto, W., Fantini, D., Ragan-Kelley, B., Parizadeh, A., Jonasson, E., Ståhlberg, A., Farhan, H., & Köhn-Luque, A. (2021). DIscBIO: A User-Friendly Pipeline for Biomarker Discovery in Single-Cell Transcriptomics. International Journal of Molecular Sciences, 22(3), 1399. https://doi.org/10.3390/ijms22031399