
Dual Role of Interleukin-10 in Murine NZB/W F1 Lupus

Abstract
1. Introduction
2. Results
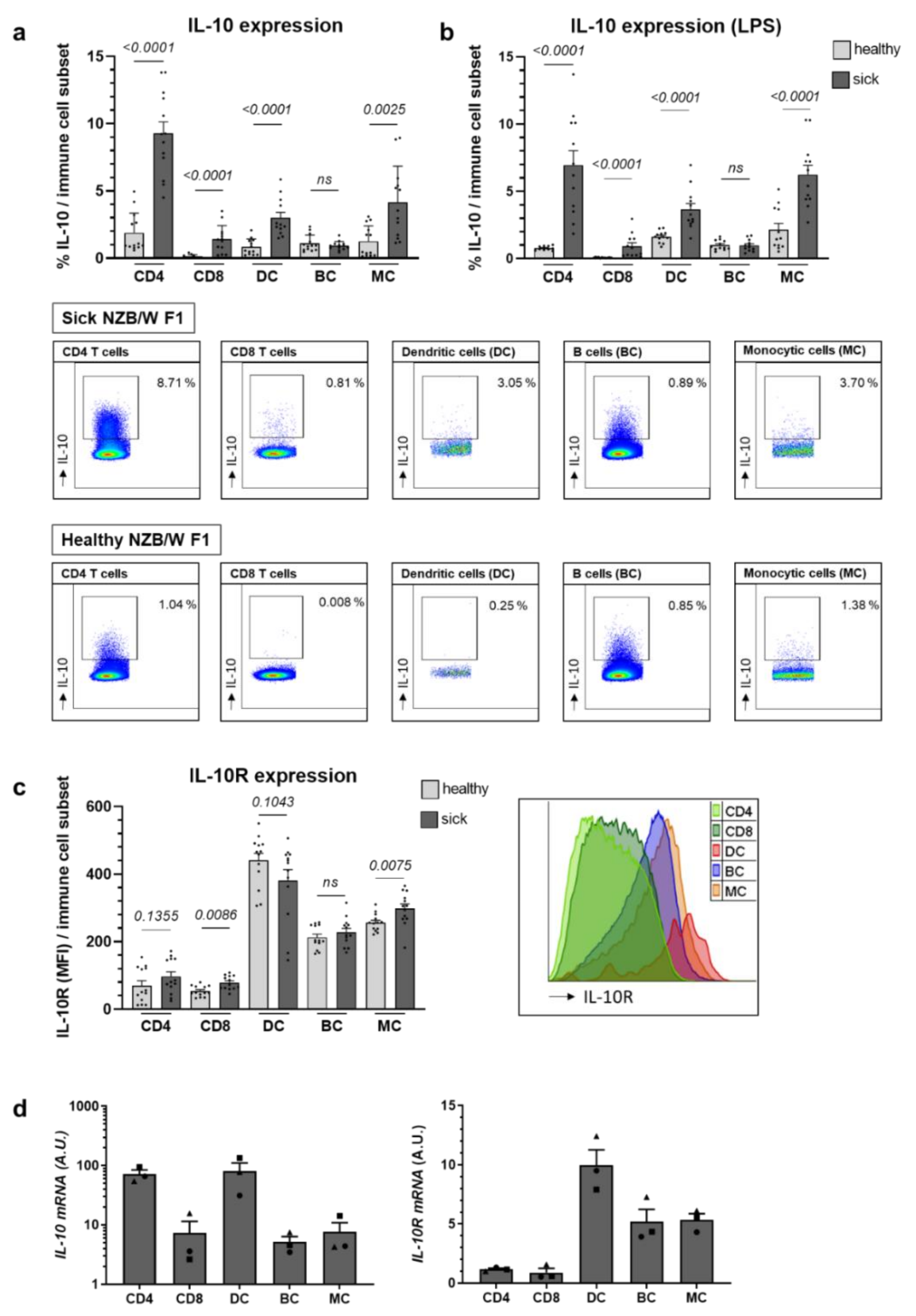
2.1. Il-10 Expression Largely Increases with Lupus Progression in NZB/W F1 Animals, While IL-10R Levels Remain Relatively Stable
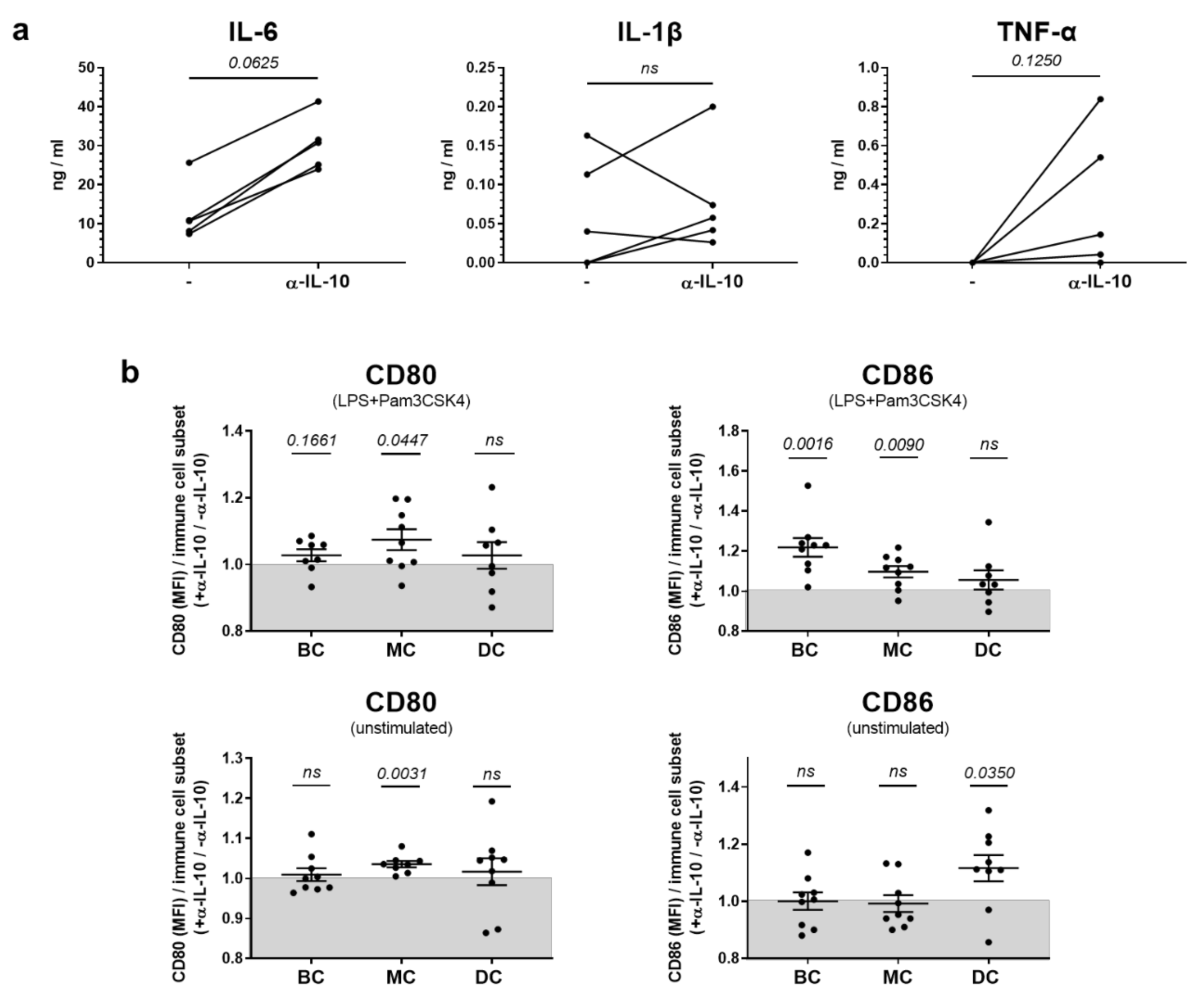
2.2. In Vitro, IL-10 Predominantly Influences the Production of Pro-Inflammatory Cytokines but Has Only Sporadic Effects on Co-Stimulatory Molecule Expression in Innate Immune Cells
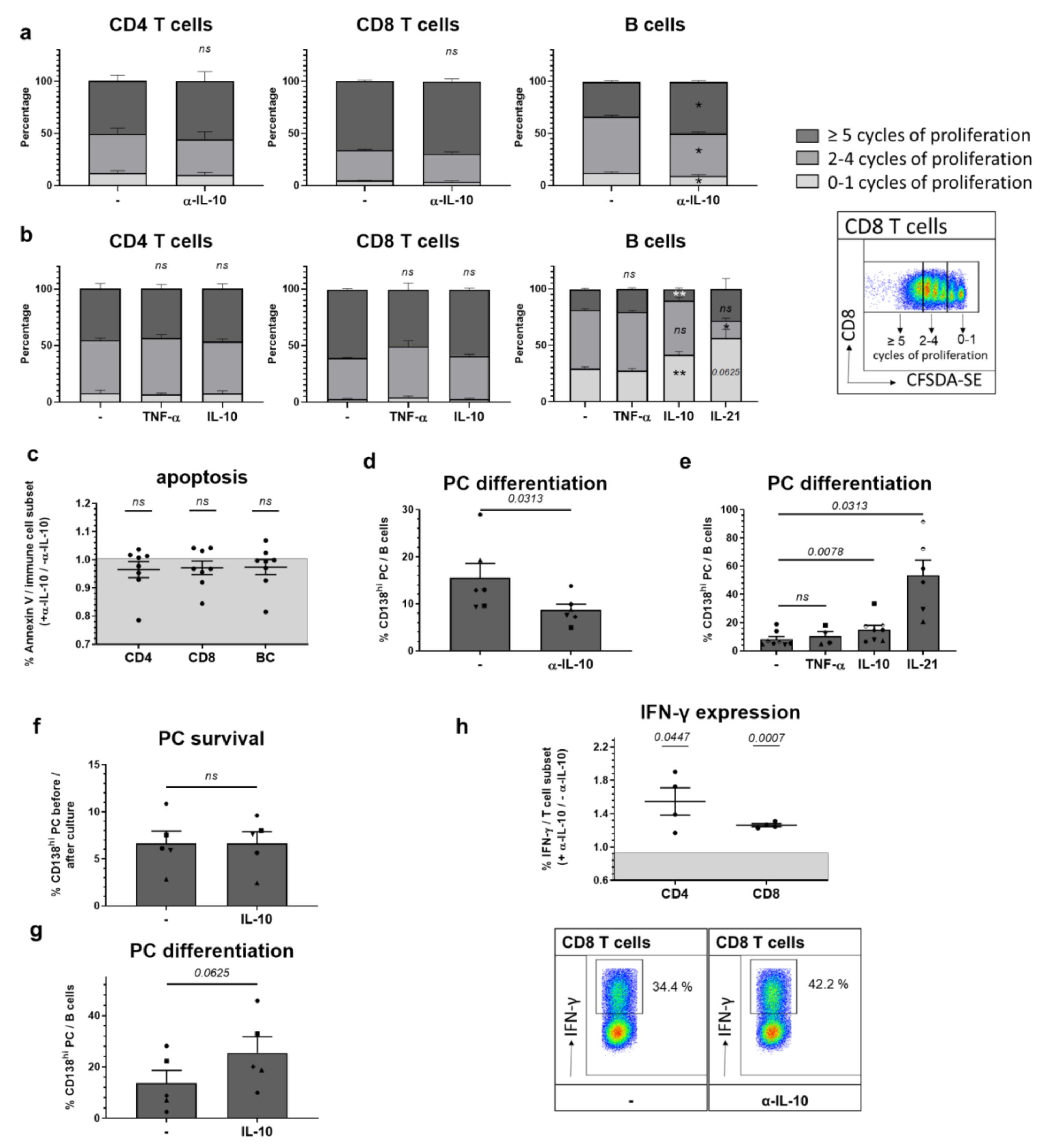
2.3. Among In Vitro Effects on Adaptive Immune Cells, IL-10 Most Prominently Slows B Cell Proliferation Triggering Plasma Cell Differentiation
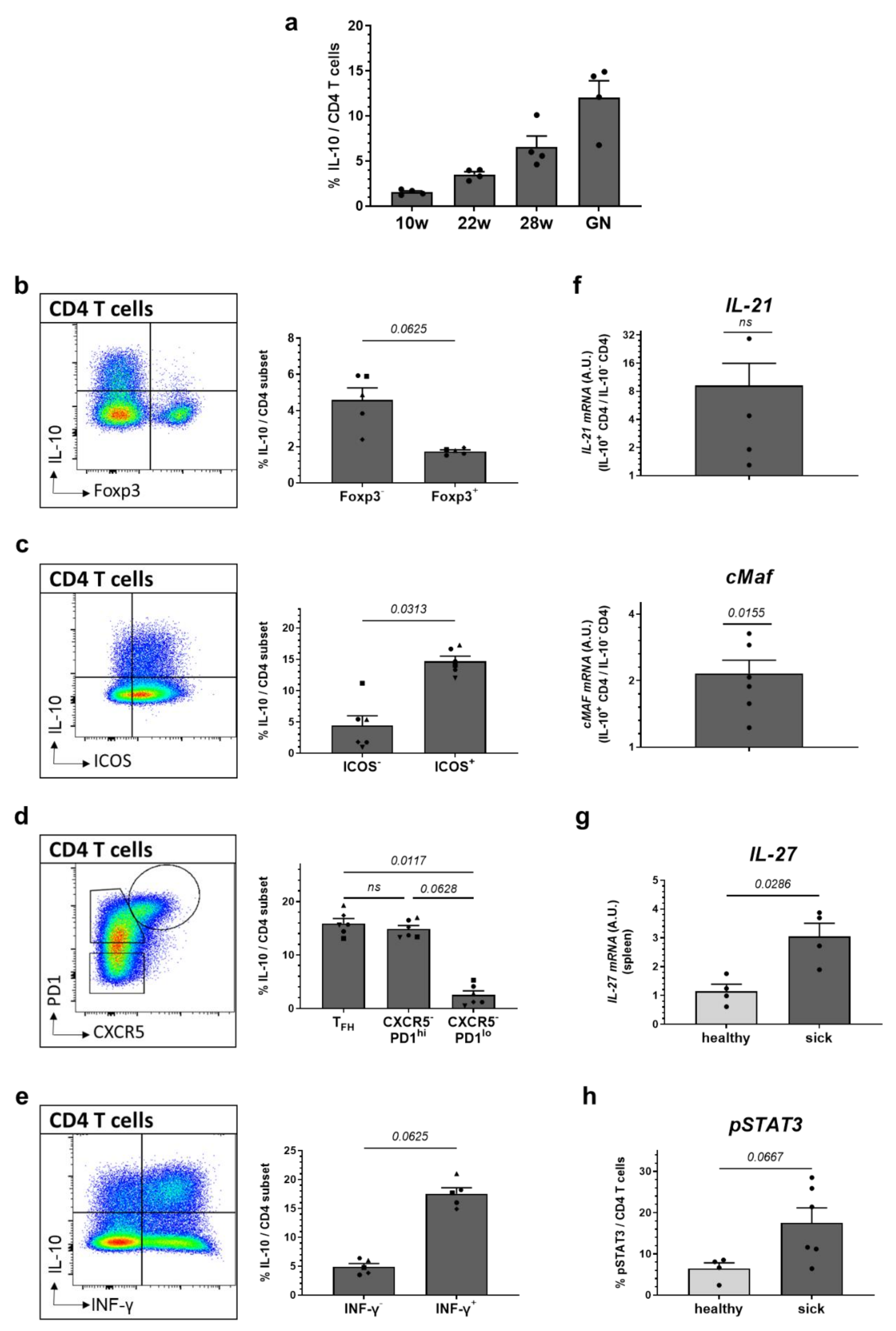
2.4. High Expression of IL-10 Is Found in ICOS+ and PD1+ Effector CD4 T Cells and Co-Incites with Increased Levels of IL-21, cMAF and IFN-γ
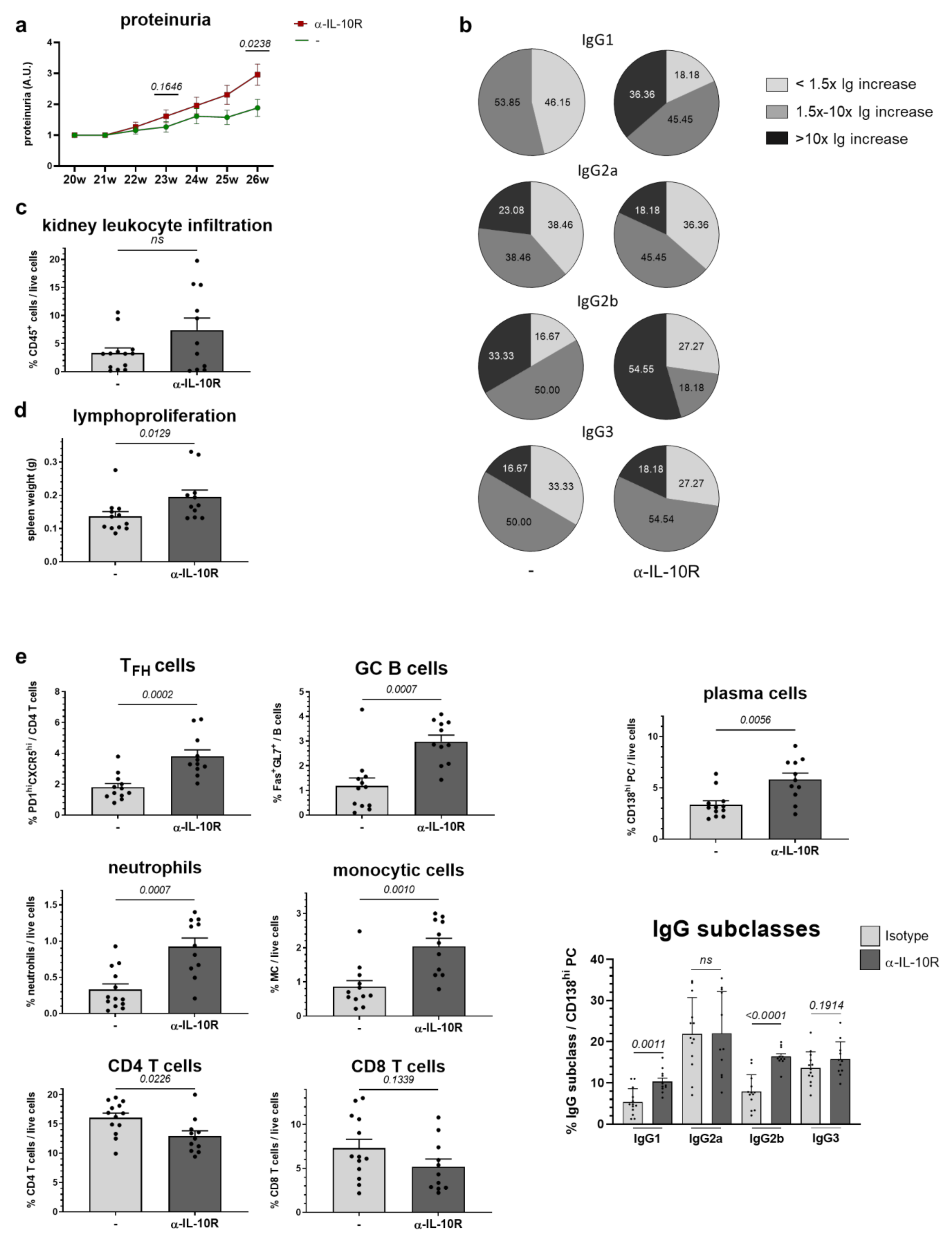
2.5. In Vivo Administration of α-IL-10R to NZB/W F1 Animals with Beginning Lupus Goes along with Moderate Immunologic Changes and Slightly Accelerates Disease Progression
3. Discussion
4. Materials and Methods
4.1. Mice and Models
4.2. Assessment of Proteinuria
4.3. Assessment of α-dsDNA Autoantibodies and IgG Subclasses (ELISA)
4.4. Cell Purification
4.5. Cell Culture
4.6. Flow Cytometry
4.7. Antibodies Used for Flow Cytometry
4.8. Immune Cell Phenotyping
4.9. Real-Time Quantitative PCR (RT-PCR)
4.10. Assessment of Cytokines (ELISA)
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| α-dsDNA | Anti-double stranded DNA autoantibodies |
| α-IL-10α-IL-10R | Anti-IL-10 antibodiesAnti-IL-10 receptor antibodies |
| A.U. | Arbitrary units |
| BC | B cells |
| Breg | regulatory B cells |
| BrdU | bromdesoxyuridin |
| CFDA-SE | carboxyfluorescein diacetate succinimidyl ester |
| DC | dendritic cells |
| ELISA | enzyme-linked immunosorbent assay |
| FACS | fluorescence-activated cell sorting |
| GC | germinal center |
| IFN-γ | interferon-γ |
| IgG | immunoglobulin G |
| IL-1β | interleukin-1β |
| IL-6 | interleukin-6 |
| IL-10 | interleukin-10 |
| IL-10R | interleukin-10 receptor |
| IL-21 | interleukin-21 |
| IL-27 | interleukin-27 |
| LPS | lipopolysaccharide |
| MC | monocytic cells |
| MFI | mean fluorescence intensity |
| NZB/W F1 | New Zealand black x New Zealand white F1 |
| PC | plasma cells |
| PMA | phorbol 12-myristate-13-acetate |
| RT-PCR | real-time quantitative PCR |
| TFH | follicular B-helper T cells |
| Th1 | T helper cell 1 |
| Th17 | T helper cell 17 |
| TLR | toll-like receptor |
| TNF-α | tumor necrosis factor-α |
| Treg | regulatory T cells |
| TR1 | inducible T regulatory type 1 |
| W | weeks |
References
- Saraiva, M.; Vieira, P.; Ogarra, A. Biology and therapeutic potential of interleukin-10. J. Exp. Med. 2020, 217, 1. [Google Scholar] [CrossRef]
- Glocker, E.-O.; Kotlarz, D.; Boztug, K.; Gertz, E.M.; Schäffer, A.A.; Noyan, F.; Perro, M.; Diestelhorst, J.; Allroth, A.; Murugan, D.; et al. Inflammatory Bowel Disease and Mutations Affecting the Interleukin-10 Receptor. N. Engl. J. Med. 2009, 361, 2033–2045. [Google Scholar] [CrossRef] [PubMed]
- Glocker, E.O.; Frede, N.; Perro, M.; Sebire, N.; Elawad, M.; Shah, N.; Grimbacher, B. Infant colitis—It’s in the genes. Lancet 2010, 376, 1272. [Google Scholar] [CrossRef]
- Moran, C.J.; Walters, T.D.; Guo, C.-H.; Kugathasan, S.; Klein, C.; Turner, D.; Wolters, V.M.; Bandsma, R.H.; Mouzaki, M.; Zachos, M.; et al. IL-10R Polymorphisms Are Associated with Very-early-onset Ulcerative Colitis. Inflamm. Bowel Dis. 2013, 19, 115–123. [Google Scholar] [CrossRef]
- Cardoso, A.; Gil Castro, A.; Martins, A.C.; Carriche, G.M.; Murigneux, V.; Castro, I.; Cumano, A.; Vieira, P.; Saraiva, M. The Dynamics of Interleukin-10-Afforded Protection during Dextran Sulfate Sodium-Induced Colitis. Front. Immunol. 2018, 9, 400. [Google Scholar] [PubMed]
- McInnes, I.; Illei, G.G.; Danning, C.L.; Yarboro, C.H.; Crane, M.; Kuroiwa, T.; Schlimgen, R.; Lee, E.; Foster, B.; Flemming, D.; et al. IL-10 improves skin disease and modulates endothelial activation and leukocyte effector function in patients with psoriatic arthritis. J. Immunol. 2001, 167, 4075–4082. [Google Scholar] [CrossRef]
- Trachsel, E.; Bootz, F.; Silacci, M.; Kaspar, M.; Kosmehl, H.; Neri, D. Antibody-mediated delivery of IL-10 inhibits the progression of established collagen-induced arthritis. Arthritis Res. Ther. 2007, 9, R9. [Google Scholar] [CrossRef]
- Galeazzi, M.; Bazzichi, L.; Sebastiani, G.D.; Neri, D.; Garcia, E.; Ravenni, N.; Giovannoni, L.; Wilton, J.; Bardelli, M.; Baldi, C.; et al. A phase IB clinical trial with Dekavil (F8-IL10), an immunoregulatory ’armed antibody’ for the treatment of rheumatoid arthritis, used in combination wiIh methotrexate. Isr. Med. Assoc. J. 2014, 16, 666. [Google Scholar]
- Autio, K.A.; Oft, M. Pegylated Interleukin-10: Clinical Development of an Immunoregulatory Cytokine for Use in Cancer Therapeutics. Curr. Oncol. Rep. 2019, 21, 19. [Google Scholar] [CrossRef]
- Ouyang, W.; Ogarra, A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 2019, 50, 871–891. [Google Scholar]
- Wang, X.; Wong, K.; Ouyang, W.; Rutz, S. Targeting IL-10 Family Cytokines for the Treatment of Human Diseases. Cold Spring Harb. Perspect. Biol. 2019, 11, a028548. [Google Scholar] [CrossRef] [PubMed]
- Sabat, R.; Grütz, G.; Warszawska, K.; Kirsch, S.; Witte, E.; Wolk, K.; Geginat, J. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010, 21, 331–344. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Krause, C.D.; Izotova, L.S.; Pollack, B.P.; Wu, W.; Pestka, S. Identification and functional characterization of a second chain of the interleukin-10 receptor complex. EMBO J. 1997, 16, 5894–5903. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Rutz, S.; Crellin, N.K.; Valdez, P.; Hymowitz, S.G. Regulation and Functions of the IL-10 Family of Cytokines in Inflammation and Disease. Annu. Rev. Immunol. 2011, 29, 71–109. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Akdis, M.; Joss, A.; Akkoç, T.; Wenig, R.; Colonna, M.; Daigle, I.; Flory, E.; Blaser, K.; Akdis, C. IL-10 inhibits CD28 and ICOS costimulations of T cells via src homology 2 domain—containing protein tyrosine phosphatase 1. J. Allergy Clin. Immunol. 2007, 120, 76–83. [Google Scholar] [PubMed]
- Walter, M.R. The Molecular Basis of IL-10 Function: From Receptor Structure to the Onset of Signaling. Curr. Top. Microbiol. Immunol. 2014, 380, 191–212. [Google Scholar]
- Yoon, S.I.; Logsdon, N.J.; Sheikh, F.; Donnelly, R.P.; Walter, M.R. Conformational Changes Mediate Interleukin-10 Receptor 2 (IL-10R2) Binding to IL-10 and Assembly of the Signaling Complex. J. Biol. Chem. 2006, 281, 35088–35096. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, C.; Vodovotz, Y.; Nathan, C. Macrophage deactivation by interleukin 10. J. Exp. Med. 1991, 174, 1549–1555. [Google Scholar]
- Malefyt, R.D.W.; Abrams, J.; Bennett, B.; Figdor, C.G.; De Vries, J.E. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: An autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 1991, 174, 1209–1220. [Google Scholar] [CrossRef]
- Fiorentino, D.F.; Zlotnik, A.; Mosmann, T.R.; Howard, M.; O’Garra, A. IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 1991, 147, 3815–3822. [Google Scholar]
- Fiorentino, D.F.; Zlotnik, A.; Vieira, P.; Mosmann, T.R.; Howard, M.; Moore, K.W.; O’Garra, A. IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J. Immunol. 1991, 146, 3444–3451. [Google Scholar]
- Macatonia, S.E.; Doherty, T.M.; Knight, S.C.; O’Garra, A. Differential effect of IL-10 on dendritic cell-induced T cell proliferation and IFN-gamma production. J. Immunol. 1993, 150, 3755–3765. [Google Scholar]
- Groux, H.; Bigler, M.; De Vries, J.E.; Roncarolo, M.G. Interleukin-10 induces a long-term antigen-specific anergic state in human CD4+ T cells. J. Exp. Med. 1996, 184, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Kamanaka, M.; Huber, S.; Zenewicz, L.A.; Gagliani, N.; Rathinam, C.; O’Connor, W., Jr.; Wan, Y.Y.; Nakae, S.; Iwakura, Y.; Hao, L.; et al. Memory/effector (CD45RB(lo)) CD4 T cells are controlled directly by IL-10 and cause IL-22-dependent intestinal pathology. J. Exp. Med. 2011, 208, 1027–1040. [Google Scholar] [PubMed]
- Coomes, S.M.; Kannan, Y.; Pelly, V.; Entwistle, L.J.; Guidi, R.; Perez-Lloret, J.; Nikolov, N.; Muller, W.; Wilson, M.S. CD4+ Th2 cells are directly regulated by IL-10 during allergic airway inflammation. Mucosal Immunol. 2017, 10, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.; Samstein, R.M.; Treuting, P.; Liang, Y.; Pils, M.C.; Heinrich, J.-M.; Jack, R.S.; Wunderlich, F.T.; Brüning, J.C.; Müller, W.; et al. Interleukin-10 Signaling in Regulatory T Cells Is Required for Suppression of Th17 Cell-Mediated Inflammation. Immunity 2011, 34, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Murai, M.; Turovskaya, O.; Kim, G.; Madan, R.; Karp, C.L.; Cheroutre, H.; Kronenberg, M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 2009, 10, 1178–1184. [Google Scholar] [CrossRef]
- Groux, H.; Bigler, M.; De Vries, J.E.; Roncarolo, M.G. Inhibitory and stimulatory effects of IL-10 on human CD8+ T cells. J. Immunol. 1998, 160, 3188–3193. [Google Scholar]
- Mumm, J.B.; Emmerich, J.; Zhang, X.; Chan, I.; Wu, L.; Mauze, S.; Blaisdell, S.; Basham, B.; Dai, J.; Grein, J.; et al. IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell 2011, 20, 781–796. [Google Scholar] [CrossRef]
- Naing, A.; Infante, J.R.; Papadopoulos, K.P.; Chan, I.H.; Shen, C.; Ratti, N.P.; Rojo, B.; Autio, K.A.; Wong, D.J.; Patel, M.R.; et al. PEGylated IL-10 (Pegilodecakin) Induces Systemic Immune Activation, CD8(+) T Cell Invigoration and Polyclonal T Cell Expansion in Cancer Patients. Cancer Cell 2018, 34, 775–791.e3. [Google Scholar] [CrossRef]
- Chen, W.F.; Zlotnik, A. IL-10: A novel cytotoxic T cell differentiation factor. J. Immunol. 1991, 147, 528–534. [Google Scholar] [PubMed]
- Nizzoli, G.; Larghi, P.; Paroni, M.; Crosti, M.C.; Moro, M.; Neddermann, P.; Caprioli, F.; Pagani, M.; De Francesco, R.; Abrignani, S.; et al. IL-10 promotes homeostatic proliferation of human CD8+memory T cells and, when produced by CD1c+DCs, shapes naive CD8+T-cell priming. Eur. J. Immunol. 2016, 46, 1622–1632. [Google Scholar] [PubMed]
- Rousset, F.; Garcia, E.; Defrance, T.; Peronne, C.; Vezzio, N.; Hsu, D.H.; Kastelein, R.; Moore, K.W.; Banchereau, J. Interleukin 10 is a potent growth and differentiation factor for activated human B lymphocytes. Proc. Natl. Acad. Sci. USA 1992, 89, 1890–1893. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Hirohata, S. The role of IL-10 in human B cell activation, proliferation, and differentiation. J. Immunol. 1995, 154, 4341–4350. [Google Scholar] [PubMed]
- Fornek, J.L.; Tygrett, L.T.; Waldschmidt, T.J.; Poli, V.; Rickert, R.C.; Kansas, G.S. Critical role for Stat3 in T-dependent terminal differentiation of IgG B cells. Blood 2006, 107, 1085–1091. [Google Scholar] [CrossRef]
- Ettinger, R.; Sims, G.P.; Fairhurst, A.-M.; Robbins, R.; Da Silva, Y.S.; Spolski, R.; Leonard, W.J.; Lipsky, P.E. IL-21 Induces Differentiation of Human Naive and Memory B Cells into Antibody-Secreting Plasma Cells. J. Immunol. 2005, 175, 7867–7879. [Google Scholar] [CrossRef]
- Rekvig, O.P. Systemic Lupus Erythematosus: Definitions, Contexts, Conflicts, Enigmas. Front. Immunol. 2018, 9, 387. [Google Scholar] [CrossRef]
- Tsokos, G.C.; Lo, M.S.; Reis, P.C.; Sullivan, K.E. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2016, 12, 716–730. [Google Scholar] [CrossRef]
- Geginat, J.; Vasco, M.; Gerosa, M.; Tas, S.; Pagani, M.; Grassi, F.; Flavell, R.; Meroni, P.; Abrignani, S. IL-10 producing regulatory and helper T-cells in systemic lupus erythematosus. Semin. Immunol. 2019, 44, 101330. [Google Scholar]
- Peng, H.; Wang, W.; Zhou, M.; Li, R.; Pan, H.-F.; Ye, D. Role of interleukin-10 and interleukin-10 receptor in systemic lupus erythematosus. Clin. Rheumatol. 2013, 32, 1255–1266. [Google Scholar] [CrossRef]
- Llorente, L.; Zou, W.; Levy, Y.; Richaud-Patin, Y.; Wijdenes, J.; Alcocer-Varela, J.; Morel-Fourrier, B.; Brouet, J.C.; Alarcon-Segovia, D.; Galanaud, P.; et al. Role of interleukin 10 in the B lymphocyte hyperactivity and autoantibody production of human systemic lupus erythematosus. J. Exp. Med. 1995, 181, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.B.; Lee, S.K.; Kim, D.S.; Lee, J.; Lee, C.H.; Song, C.H. Elevated interleukin-10 levels correlated with disease activity in systemic lupus erythematosus. Clin. Exp. Rheumatol. 1998, 16, 283–288. [Google Scholar] [PubMed]
- Uchida, M.; Ooka, S.; Goto, Y.; Suzuki, K.; Fujimoto, H.; Ishimori, K.; Matsushita, H.; Takakuwa, Y.; Kawahata, K. Anti-IL-10 antibody in systemic lupus erythematosus. Open Access Rheumatol. Res. Rev. 2019, 11, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Ravirajan, C.T.; Wang, Y.; Matis, L.A.; Papadaki, L.; Griffiths, M.H.; Latchman, D.S.; Isenberg, D.A. Effect of neutralizing antibodies to IL-10 and C5 on the renal damage caused by a pathogenic human anti-dsDNA antibody. Rheumatology 2004, 43, 442–447. [Google Scholar] [CrossRef]
- Llorente, L.; Richaud-Patin, Y.; García-Padilla, C.; Claret, E.; Jakez-Ocampo, J.; Cardiel, M.H.; Alcocer-Varela, J.; Grangeot-Keros, L.; Alarcón-Segovia, D.; Wijdenes, J.; et al. Clinical and biologic effects of anti-interleukin-10 monoclonal antibody administration in systemic lupus erythematosus. Arthritis Rheum. 2000, 43, 1790–1800. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, R.; Wang, H.; Jiang, P.; Zhang, J.; Zhang, M.; Gu, L.; Yang, X.; Zhang, M.; Ji, X. Serum IL-10 from systemic lupus erythematosus patients suppresses the differentiation and function of monocyte-derived dendritic cells. J. Biomed. Res. 2012, 26, 456–466. [Google Scholar] [CrossRef]
- Yin, Z.; Bahtiyar, G.; Zhang, N.; Liu, L.; Zhu, P.; Robert, M.E.; McNiff, J.; Madaio, M.P.; Craft, J. IL-10 Regulates Murine Lupus. J. Immunol. 2002, 169, 2148–2155. [Google Scholar] [CrossRef]
- Ishida, H.; Muchamuel, T.; Sakaguchi, S.; Andrade, S.; Menon, S.; Howard, M. Continuous administration of anti-interleukin 10 antibodies delays onset of autoimmunity in NZB/W F1 mice. J. Exp. Med. 1994, 179, 305–310. [Google Scholar] [CrossRef]
- Joo, H.; Coquery, C.; Xue, Y.; Gayet, I.; Dillon, S.R.; Punaro, M.; Zurawski, G.; Banchereau, J.; Pascual, V.; Oh, S. Serum from patients with SLE instructs monocytes to promote IgG and IgA plasmablast differentiation. J. Exp. Med. 2012, 209, 1335–1348. [Google Scholar] [CrossRef]
- Heine, G.; Drozdenko, G.; Grun, J.R.; Chang, H.D.; Radbruch, A.; Worm, M. Autocrine IL-10 promotes human B-cell differentiation into IgM- or IgG-secreting plasmablasts. Eur. J. Immunol. 2014, 44, 1615–1621. [Google Scholar] [CrossRef]
- Aringer, M.; Smolen, J.S. The role of tumor necrosis factor-alpha in systemic lupus erythematosus. Arthritis Res. Ther. 2008, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Aringer, M.; Smolen, J.S. SLE—Complex cytokine effects in a complex autoimmune disease: Tumor necrosis factor in systemic lupus erythematosus. Arthritis Res. Ther. 2003, 5, 172–177. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chevalier, N.; Macia, L.; Tan, J.; Mason, L.J.; Robert, R.; Thorburn, A.N.; Wong, C.H.Y.; Tsai, L.M.; Bourne, K.; Brink, R.; et al. The Role of Follicular Helper T Cell Molecules and Environmental Influences in Autoantibody Production and Progression to Inflammatory Arthritis in Mice. Arthritis Rheumatol. 2016, 68, 1026–1038. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.-L.E.A.; Hentze, C.; Amend, A.; Sprenger, D.T.L.; Finzel, S.; Daniel, C.; Salzer, U.; Rizzi, M.; Voll, R.E.; Chevalier, N. Low dietary fiber intake links development of obesity and lupus pathogenesis. Submitt. Publ. 2020, in press. [Google Scholar]
- Kulkarni, U.; Karsten, C.M.; Kohler, T.; Hammerschmidt, S.; Bommert, K.; Tiburzy, B.; Meng, L.; Thieme, L.; Recke, A.; Ludwig, R.J.; et al. IL-10 mediates plasmacytosis-associated immunodeficiency by inhibiting complement-mediated neutrophil migration. J. Allergy Clin. Immunol. 2016, 137, 1487–1497.e6. [Google Scholar] [CrossRef] [PubMed]
- Llorente, L.; Richaud-Patin, Y.; Wijdenes, J.; Alcocer-Varela, J.; Maillot, M.C.; Durand-Gasselin, I.; Fourrier, B.M.; Galanaud, P.; Emilie, D. Spontaneous production of interleukin-10 by B lymphocytes and monocytes in systemic lupus erythematosus. Eur. Cytokine Netw. 1993, 4, 421–427. [Google Scholar]
- Llorente, L.; Richaud-Patin, Y.; Fior, R.; Alcocer-Varela, J.; Wijdenes, J.; Fourrier, B.M.; Galanaud, P.; Emilie, D. In vivo production of interleukin-10 by non-T cells in rheumatoid arthritis, Sjogren’s syndrome, and systemic lupus erythematosus. A potential mechanism of B lymphocyte hyperactivity and autoimmunity. Arthritis Rheum. 1994, 37, 1647–1655. [Google Scholar] [CrossRef]
- Gelati, M.; Lamperti, E.; Dufour, A.; Corsini, E.; Venegoni, E.; Milanese, C.; Nespolo, A.; Salmaggi, A. IL-10 production in multiple sclerosis patients, SLE patients and healthy controls: Preliminary findings. Ital. J. Neurol. Sci. 1997, 18, 191–194. [Google Scholar] [CrossRef]
- Kristjansdottir, H.; Gunnlaugsdottir, B.; Arnason, A.; Lundberg, I.; Klareskog, L.; Steinsson, K. Increased number of interleukin-10-producing cells in systemic lupus erythematosus patients and their first-degree relatives and spouses in Icelandic multicase families. Arthritis Rheum. 1999, 42, 1649–1654. [Google Scholar]
- Hagiwara, E.; Gourley, M.F.; Lee, S.; Klinman, D.K. Disease severity in patients with systemic lupus erythematosus correlates with an increased ratio of interleukin-10:interferon-gamma-secreting cells in the peripheral blood. Arthritis Rheum. 1996, 39, 379–385. [Google Scholar] [CrossRef]
- Houssiau, F.A.; Lefebvre, C.; Berghe, M.V.; Lambert, M.; Devogelaer, J.P.; Renauld, J.C. Serum interleukin 10 titers in systemic lupus erythematosus reflect disease activity. Lupus 1995, 4, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Miret, C.; Font, J.; Molina, R.; Garcia-Carrasco, M.; Filella, X.; Ramos, M.; Cervera, R.; Ballesta, A.; Ingelmo, M. Relationship of oncogenes (sFas, Bcl-2) and cytokines (IL-10, alfa-TNF) with the activity of systemic lupus erythematosus. Anticancer. Res. 2001, 21, 3053–3059. [Google Scholar] [PubMed]
- Blenman, K.R.M.; Duan, B.; Xu, Z.; Wan, S.; Atkinson, M.A.; Flotte, T.R.; Croker, B.P.; Morel, L. IL-10 regulation of lupus in the NZM2410 murine model. Lab. Investig. 2006, 86, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Emilie, D.; Zou, W.; Fior, R.; Llorente, L.; Durandy, A.; Crevon, M.-C.; Maillot, M.-C.; Durand-Gasselin, I.; Raphael, M.; Peuchmaur, M.; et al. Production and Roles of IL-6, IL-10, and IL-13 in B-Lymphocyte Malignancies and in B-Lymphocyte Hyperactivity of HIV Infection and Autoimmunity. Methods 1997, 11, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Batten, M.; Kljavin, N.M.; Li, J.; Walter, M.J.; De Sauvage, F.J.; Ghilardi, N. Cutting Edge: IL-27 Is a Potent Inducer of IL-10 but Not FoxP3 in Murine T Cells. J. Immunol. 2008, 180, 2752–2756. [Google Scholar] [CrossRef] [PubMed]
- Alarcon-Riquelme, M.E.; Lindqvist, A.K.; Jonasson, I.; Johanneson, B.; Sandino, S.; Alcocer-Varela, J.; Granados, J.; Kristjansdottir, H.; Grondal, G.; Svenungsson, E.; et al. Genetic analysis of the contribution of IL10 to systemic lupus erythematosus. J. Rheumatol. 1999, 26, 2148–2152. [Google Scholar] [PubMed]
- Gibson, A.W.; Edberg, J.C.; Wu, J.; Westendorp, R.G.J.; Huizinga, T.W.; Kimberly, R.P. Novel single nucleotide polymorphisms in the distal IL-10 promoter affect IL-10 production and enhance the risk of systemic lupus erythematosus. J. Immunol. 2001, 166, 3915–3922. [Google Scholar] [CrossRef] [PubMed]
- Johanneson, B.; Lima, G.; von Salome, J.; Alarcon-Segovia, D.; Alarcon-Riquelme, M.E. A major susceptibility locus for systemic lupus erythemathosus maps to chromosome 1q31. Am. J. Hum. Genet. 2002, 71, 1060–1071. [Google Scholar] [CrossRef][Green Version]
- Zhao, M.; Tang, J.; Gao, F.; Wu, X.; Liang, Y.; Yin, H.; Lu, Q. Hypomethylation ofIL10andIL13Promoters in CD4+T Cells of Patients with Systemic Lupus Erythematosus. J. Biomed. Biotechnol. 2010, 2010, 931018. [Google Scholar] [CrossRef]
- Facciotti, F.; Larghi, P.; Bosotti, R.; Vasco, C.; Gagliani, N.; Cordiglieri, C.; Mazzara, S.; Ranzani, V.; Rottoli, E.; Curti, S.; et al. Evidence for a pathogenic role of extrafollicular, IL-10–producing CCR6+B helper T cells in systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 2020, 117, 7305–7316. [Google Scholar] [CrossRef]
- Rivino, L.; Gruarin, P.; Häringer, B.; Steinfelder, S.; Lozza, L.; Steckel, B.; Weick, A.; Sugliano, E.; Jarrossay, D.; Kühl, A.A.; et al. CCR6 is expressed on an IL-10–producing, autoreactive memory T cell population with context-dependent regulatory function. J. Exp. Med. 2010, 207, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Leiss, H.; Niederreiter, B.; Bandur, T.; Schwarzecker, B.; Blüml, S.; Steiner, G.; Ulrich, W.; Smolen, J.S.; Stummvoll, G.H. Pristane-induced lupus as a model of human lupus arthritis: Evolvement of autoantibodies, internal organ and joint inflammation. Lupus 2013, 22, 778–792. [Google Scholar] [CrossRef] [PubMed]
- Caielli, S.; Veiga, D.T.; Balasubramanian, P.; Athale, S.; Domic, B.; Murat, E.; Banchereau, R.; Xu, Z.; Chandra, M.; Chung, C.-H.; et al. A CD4+ T cell population expanded in lupus blood provides B cell help through interleukin-10 and succinate. Nat. Med. 2019, 25, 75–81. [Google Scholar] [PubMed]
- Gregori, S.; Roncarolo, M.G. Engineered T Regulatory Type 1 Cells for Clinical Application. Front. Immunol. 2018, 9, 233. [Google Scholar] [PubMed]
- Undeutsch, R.; Humrich, J.Y.; Papendieck, A.; Riemekasten, G. CD4 T cells producing IL-10 have a beneficial effect in murine lupus. Ann. Rheum. Dis. 2011, 70, A73. [Google Scholar] [CrossRef]
- Ding, Y.; Qin, L.; Zamarin, D.; Kotenko, S.V.; Pestka, S.; Moore, K.W.; Bromberg, J.S. Differential IL-10R1 Expression Plays a Critical Role in IL-10-Mediated Immune Regulation. J. Immunol. 2001, 167, 6884–6892. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.-M.; Wang, J.; Sun, Z.-R.; Ma, F.-M.; Zhang, Q.-R.; Hirose, S.; Jiang, Y. Polymorphism of the mouse gene for the interleukin 10 receptor alpha chain (Il10ra) and its association with the autoimmune phenotype. Immunogenetics 2005, 57, 697–702. [Google Scholar] [CrossRef]
- Cairns, A.P.; Crockard, A.D.; Bell, A.L. Interleukin-10 receptor expression in systemic lupus erythematosus and rheumatoid arthritis. Clin. Exp. Rheumatol. 2003, 21, 83–86. [Google Scholar]
- Valencia-Pacheco, G.; Layseca-Espinosa, E.; Niño-Moreno, P.; Portales-Pérez, D.P.; Baranda, L.; Rosenstein, Y.; Abud-Mendoza, C.; González-Amaro, R. Expression and function of IL?10R in mononuclear cells from patients with systemic lupus erythematosus. Scand. J. Rheumatol. 2006, 35, 368–378. [Google Scholar] [CrossRef]
- Jacob, C.O.; McDevitt, H.O. Tumour necrosis factor-α in murine autoimmune ’lupus’ nephritis. Nat. Cell Biol. 1988, 331, 356–358. [Google Scholar] [CrossRef]
- Gordon, C.; Ranges, G.E.; Greenspan, J.S.; Wofsy, D. Chronic therapy with recombinant tumor necrosis factor-alpha in autoimmune NZB/NZW F1 mice. Clin. Immunol. Immunopathol. 1989, 52, 421–434. [Google Scholar] [CrossRef]
- Jacob, C.O.; Hwang, F.; Lewis, G.D.; Stall, A.M. Tumor necrosis factor alpha in murine systemic lupus erythematosus disease models: Implications for genetic predisposition and immune regulation. Cytokine 1991, 3, 551–561. [Google Scholar] [CrossRef]
- Watanabe, Y.; Jacob, C.O. Regulation of MHC class II antigen expression. Opposing effects of tumor necrosis factor-alpha on IFN-gamma-induced HLA-DR and Ia expression depends on the maturation and differentiation stage of the cell. J. Immunol. 1991, 146, 899–905. [Google Scholar]
- Lopez, P.; Gutierrez, C.; Suarez, A. IL-10 and TNFalpha genotypes in SLE. J. Biomed. Biotechnol. 2010, 2010, 838390. [Google Scholar] [CrossRef]
- Ban, L.; Zhang, J.; Wang, L.; Kuhtreiber, W.; Burger, D.; Faustman, D.L. Selective death of autoreactive T cells in human diabetes by TNF or TNF receptor 2 agonism. Proc. Natl. Acad. Sci. USA 2008, 105, 13644–13649. [Google Scholar] [CrossRef]
- Kessel, A.; Rosner, I.; Rozenbaum, M.; Zisman, D.; Sagiv, A.; Shmuel, Z.; Sabo, E.; Toubi, E. Increased CD8+ T cell apoptosis in scleroderma is associated with low levels of NF-kappa B. J. Clin. Immunol. 2004, 24, 30–36. [Google Scholar] [CrossRef]
- Hayashi, T.; Faustman, D. Essential role of human leukocyte antigen-encoded proteasome subunits in NF-kappaB activation and prevention of tumor necrosis factor-alpha-induced apoptosis. J. Biol. Chem. 2000, 275, 5238–5247. [Google Scholar] [CrossRef]
- Qin, H.Y.; Chaturvedi, P.; Singh, B. In vivo apoptosis of diabetogenic T cells in NOD mice by IFN-gamma/TNF-alpha. Int. Immunol. 2004, 16, 1723–1732. [Google Scholar] [CrossRef]
- Christen, U.; Wolfe, T.; Mohrle, U.; Hughes, A.C.; Rodrigo, E.; Green, E.A.; Flavell, R.A.; von Herrath, M.G. A dual role for TNF-alpha in type 1 diabetes: Islet-specific expression abrogates the ongoing autoimmune process when induced late but not early during pathogenesis. J. Immunol. 2001, 166, 7023–7032. [Google Scholar]
- Boswell, J.M.; Yui, M.A.; Burt, D.W.; Kelley, V.E. Increased tumor necrosis factor and IL-1 beta gene expression in the kidneys of mice with lupus nephritis. J. Immunol. 1988, 141, 3050–3054. [Google Scholar]
- Yokoyama, H.; Kreft, B.; Kelley, V.R. Biphasic increase in circulating and renal TNF-α in MRL-lpr mice with differing regulatory mechanisms. Kidney Int. 1995, 47, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Brennan, D.C.; Yui, M.A.; Wuthrich, R.P.; Kelley, V.E. Tumor necrosis factor and IL-1 in New Zealand Black/White mice. Enhanced gene expression and acceleration of renal injury. J. Immunol. 1989, 143, 3470–3475. [Google Scholar]
- Nakamura, T.; Ebihara, I.; Fukui, M.; Osada, S.; Tomino, Y.; Masaki, T.; Goto, K.; Furuichi, Y.; Koide, H. Renal Expression of mRNAs for Endothelin-1, Endothelin-3 and Endothelin Receptors in NZB/W F1 Mice. Kidney Blood Press. Res. 1993, 16, 233–243. [Google Scholar] [CrossRef]
- Emmerich, J.; Mumm, J.B.; Chan, I.H.; LaFace, D.; Truong, H.; McClanahan, T.K.; Gorman, D.M.; Oft, M. IL-10 Directly Activates and Expands Tumor-Resident CD8+ T Cells without De Novo Infiltration from Secondary Lymphoid Organs. Cancer Res. 2012, 72, 3570–3581. [Google Scholar] [CrossRef]
- Xi, J.; Xu, M.; Song, Z.; Li, H.; Xu, S.; Wang, C.; Song, H.; Bai, J. Stimulatory role of interleukin 10 in CD8+ T cells through STATs in gastric cancer. Tumor Biol. 2017, 39, 1010428317706209. [Google Scholar] [CrossRef]
- Lin, X.; Wang, X.; Xiao, F.; Ma, K.; Liu, L.; Wang, X.; Xu, D.; Wang, F.; Shi, X.; Liu, D.; et al. IL-10-producing regulatory B cells restrain the T follicular helper cell response in primary Sjögren’s syndrome. Cell. Mol. Immunol. 2019, 16, 921–931. [Google Scholar] [CrossRef]
- Flores-Borja, F.; Bosma, A.; Ng, D.; Reddy, V.; Ehrenstein, M.R.; Isenberg, D.A.; Mauri, C. CD19+CD24hiCD38hi B Cells Maintain Regulatory T Cells While Limiting TH1 and TH17 Differentiation. Sci. Transl. Med. 2013, 5, 173ra23. [Google Scholar] [CrossRef]
- Yang, M.; Deng, J.; Liu, Y.; Ko, K.-H.; Wang, X.; Jiao, Z.; Wang, S.; Hua, Z.; Sun, L.; Srivastava, G.; et al. IL-10–Producing Regulatory B10 Cells Ameliorate Collagen-Induced Arthritis via Suppressing Th17 Cell Generation. Am. J. Pathol. 2012, 180, 2375–2385. [Google Scholar] [CrossRef]
- Yang, M.; Rui, K.; Wang, S.; Lu, L. Regulatory B cells in autoimmune diseases. Cell. Mol. Immunol. 2013, 10, 122–132. [Google Scholar]
- Dennis, K.L.; Saadalla, A.; Blatner, N.R.; Wang, S.; Venkateswaran, V.; Gounari, F.; Cheroutre, H.; Weaver, C.T.; Roers, A.; Egilmez, N.K.; et al. T-cell Expression of IL10 Is Essential for Tumor Immune Surveillance in the Small Intestine. Cancer Immunol. Res. 2015, 3, 806–814. [Google Scholar] [CrossRef][Green Version]
- Cope, A.; Le Friec, G.; Cardone, J.; Kemper, C. The Th1 life cycle: Molecular control of IFN-gamma to IL-10 switching. Trends Immunol. 2011, 32, 278–286. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Healthy | Sick | |||
|---|---|---|---|---|
| Untreated | Untreated | α-IL-10R-Treated | ||
| Spleen | Mean (±SEM) | Mean (±SEM) | Mean (±SEM) | p-Value |
| CD11b+Ly6Ghi neutrophils/live cells (%) | 0.1491 (0.05937) | 0.3301 (0.07797) | 0.9236 (0.1183) | 0.0007 |
| CD11chi DC/live cells (%) | 0.04564 (0.00500) | 0.2169 (0.02838) | 0.2435 (0.04614) | ns |
| CD80+/DC (MFI) | 264.0 (30.49) | 226.0 (15.13) | ns | |
| CD86+/DC (MFI) | 1486 (60.27) | 1582 (85.68) | ns | |
| I-A/I-E+/DC (MFI) | 11329 (1088) | 9366 (754.8) | 0.1500 | |
| ICOSL+/DC (MFI) | 378.1 (28.30) | 383.7 (58.30) | ns | |
| CD11b+ monocytic cells/live cells (%) | 0.4704 (0.1371) | 0.8572 (0.1792) | 2.034 (0.2418) | 0.0010 |
| CD80+/MC (MFI) | 381.2 (52.22) | 370.4 (48.49) | ns | |
| CD86+/MC (MFI) | 765.1 (20.96) | 803.2 (41.81) | ns | |
| I-A/I-E+/MC (MFI) | 1220 (40.97) | 1182 (37.34) | ns | |
| ICOSL+/MC (MFI) | 528.8 (17.39) | 557.6 (35.00) | ns | |
| TCRβ+CD4+ T cells/live cells (%) | 17.57 (0.2906) | 16.05 (0.7924) | 12.93 (0.9114) | 0.0226 |
| CD44hi/CD4+ T cells (%) | 12.42 (1.069) | 52.2 (4.080) | 61.09 (4.546) | 0.1226 |
| CXCR5hiPD1hi/CD4+ T cells (%) | 0.3583 (0.02892) | 1.802 (0.2429) | 3.820 (0.4114) | 0.0002 |
| IFN-γ+/CD4+ T cells (%) | 5.845 (0.5039) | 13.36 (1.763) | 12.95 (1.480) | ns |
| IL-17+/CD4+ T cells (%) | 0.0798 (0.00634) | 0.2590 (0.05745) | 0.3790 (0.03174) | 0.0973 |
| IL-10+/CD4+ T cells (%) | 1.122 (0.1058) | 5.058 (0.6076) | 6.345 (0.7977) | 0.1674 |
| FoxP3+/CD4+ T cells (%) | 14.58 (0.5437) | 2311 (1.654) | 20.24 (1.640) | 0.1911 |
| Annexin V+/CD4+ T cells (%) | 11.05 (1.202) | 20.59 (3.446) | 0.0548 | |
| BrdU+/CD4+ T cells (%) | 2.164 (0.2925) | 3.615 (0.4924) | 0.0184 | |
| TCRβ+CD8+ T cells/live cells (%) | 11.38 (0.3842) | 7.312 (0.9999) | 5.196 (0.8768) | 0.1339 |
| CD44hi/CD8+ T cells (%) | 8.280 (0.7897) | 16.92 (0.7688) | 17.70 (1.406) | ns |
| IFN-γ+/CD8+ T cells (%) | 14.02 (0.7795) | 10.16 (1.259) | 11.47 (1.047) | ns |
| Annexin V+/CD8+ T cells (%) | 9.525 (0.8064) | 16.09 (2.492) | 0.0352 | |
| BrdU+/CD8+ T cells (%) | 1.098 (0.2335) | 1.180 (0.2155) | ns | |
| TCRβ−B220+ B cells/live cells (%) | 31.10 (1.002) | 34.20 (0.8938) | 32.55 (1.1419) | ns |
| FashiGL7hi/B220+ B cells (%) | 0.3167 (0.03127) | 1.181 (0.3272) | 2.983 (0.2629) | 0.0007 |
| CD80+/B220+ B cells (MFI) | 141.8 (15.79) | 120.0 (6.43) | ns | |
| CD86+/B220+ B cells (MFI) | 631.0 (36.82) | 750.3 (27.73) | 0.0358 | |
| I-A/I-E+/B220+ B cells (MFI) | 13046 (665.9) | 13279 (397.8) | ns | |
| ICOSL+/B220+ B cells (MFI) | 189.2 (12.67) | 169.3 (22.63) | ns | |
| Annexin V+/B220+ B cells (%) | 26.80 (1.905) | 28.51 (1.689) | ns | |
| BrdU+/B220+ B cells (%) | 5.336 (0.6873) | 8.005 (0.6776) | 0.0020 | |
| CD138hi plasma cells/live cells (%) | 0.8245 (0.1023) | 3.359 (0.3818) | 5.826 (0.6163) | 0.0056 |
| IgG1+/CD138hi (%) | 8.362 (2.790) | 5.365 (0.9021) | 10.35 (0.8451) | 0.0011 |
| IgG2a+/CD138hi (%) | 6.1013 (0.7867) | 21.88 (2.451) | 22.04 (3.053) | ns |
| IgG2b+/CD138hi (%) | 8.2868 (0.9861) | 7.915 (1.132) | 16.39 (0.6913) | <0.0001 |
| IgG3+/CD138hi (%) | 6.318 (0.8690) | 13.58 (1.098) | 15.77 (1.274) | 0.1914 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amend, A.; Wickli, N.; Schäfer, A.-L.; Sprenger, D.T.L.; Manz, R.A.; Voll, R.E.; Chevalier, N. Dual Role of Interleukin-10 in Murine NZB/W F1 Lupus. Int. J. Mol. Sci. 2021, 22, 1347. https://doi.org/10.3390/ijms22031347
Amend A, Wickli N, Schäfer A-L, Sprenger DTL, Manz RA, Voll RE, Chevalier N. Dual Role of Interleukin-10 in Murine NZB/W F1 Lupus. International Journal of Molecular Sciences. 2021; 22(3):1347. https://doi.org/10.3390/ijms22031347
Chicago/Turabian StyleAmend, Anaïs, Natalie Wickli, Anna-Lena Schäfer, Dalina T. L. Sprenger, Rudolf A. Manz, Reinhard E. Voll, and Nina Chevalier. 2021. "Dual Role of Interleukin-10 in Murine NZB/W F1 Lupus" International Journal of Molecular Sciences 22, no. 3: 1347. https://doi.org/10.3390/ijms22031347
APA StyleAmend, A., Wickli, N., Schäfer, A.-L., Sprenger, D. T. L., Manz, R. A., Voll, R. E., & Chevalier, N. (2021). Dual Role of Interleukin-10 in Murine NZB/W F1 Lupus. International Journal of Molecular Sciences, 22(3), 1347. https://doi.org/10.3390/ijms22031347