
The Cholinergic System, the Adrenergic System and the Neuropathology of Alzheimer’s Disease

Abstract
1. Introduction
2. Acetylcholine, Cholinergic System and Alzheimer’s Disease
2.1. Acetylcholine and Cholinergic Neurons
2.2. Acetylcholine Receptors and Cholinergic Signaling
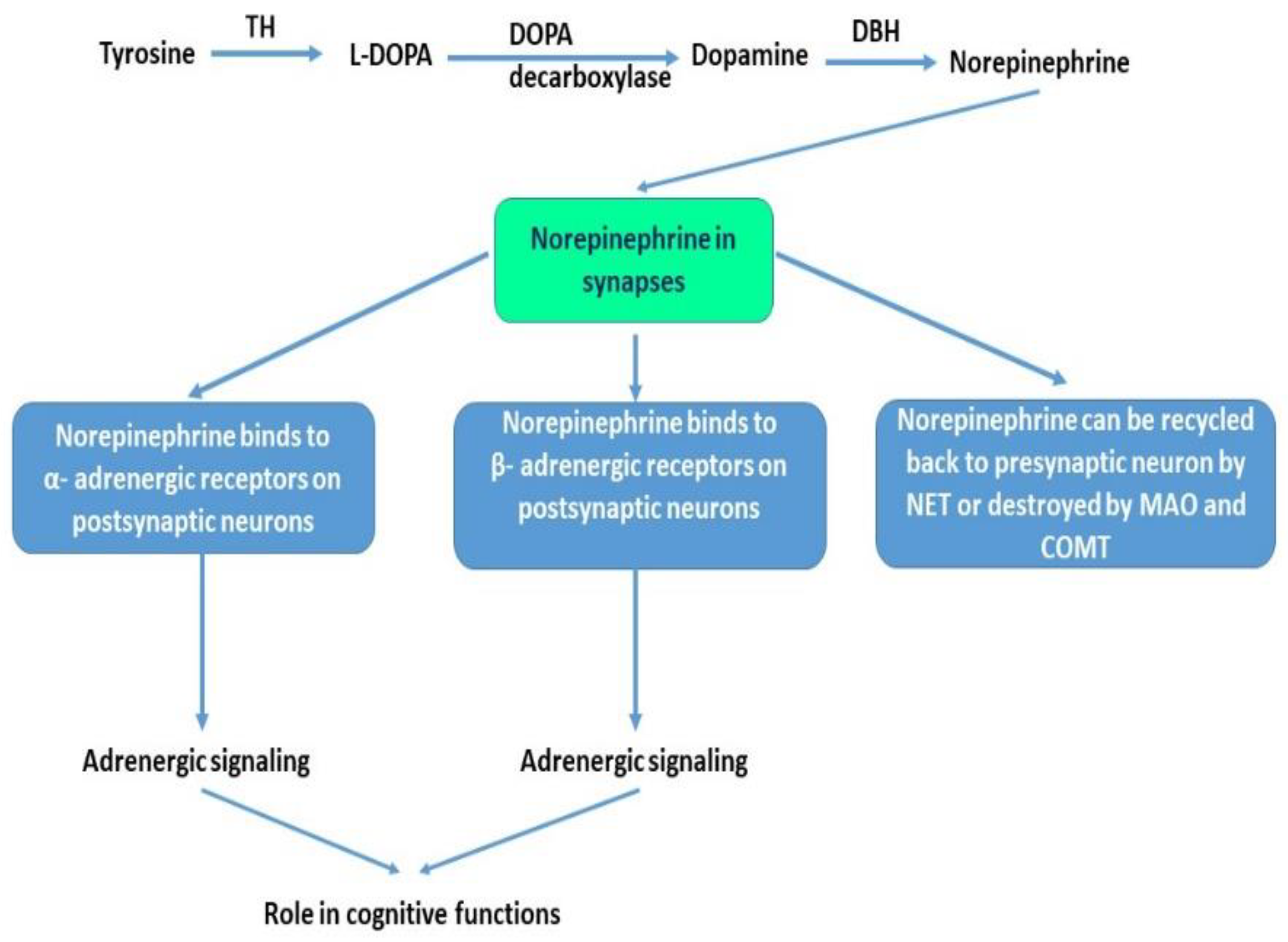
3. Norepinephrine, Adrenergic System and Alzheimer’s Disease
3.1. Norepinephine and LC-NE Neurons
3.2. Adrenergic Receptors and Adrenergic Signaling
4. Therapeutic Strategies for Alzheimer’s Disease and the Role of Epigenetics
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | Amyloid-beta Peptide |
| Ach | Acetylcholine |
| AchE | Acetylcholine Esterase |
| AchRs | Acetylcholine Receptors |
| AD | Alzheimer’s Disease |
| APP | Amyloid Precursor Protein |
| ARs | Adrenergic receptors |
| CBP | CREB-Binding Protein |
| CHT | Choline Transporter |
| ChAT | Choline Acetyltransferase |
| COMT | Catechol-O-methyltransferase |
| CSF | Cerebrospinal Fluid |
| DNMT | DNA Methyltransferase |
| DBH | Dopamine-β-Hydroxylase |
| GPCR | G-Protein-Coupled Receptor |
| H3K14 | Histone H3 Lysine 14 |
| H3S10 | Histoen H3 Serine 10 |
| nAchR | Nicotinic Acetylcholine Receptor |
| mAchR | Muscarinic Acetylcholine Receptor |
| LC | Locus Coeruleus |
| LTD | Long Term Depression |
| LTP | Long Term Potentiation |
| MAO | Monoamine Oxidase |
| NBM | Nucleus Basalis of Meynert |
| NE | Norepinephrine |
| NET | Norepinephrine Transporter |
| NGF | Nerve Growth Factor |
| NL | Neurofilament Light |
| PFC | Prefrontal Cortex |
| PS | Presenilin |
| TH | Tyrosine Hydroxylase |
| vAchT | Vesicular Acetylcholine Transporter |
References
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer Disease in the United States (2010–2050) Estimated Using the 2010 Census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Products—Data Briefs—Number 355—January 2020. Available online: https://www.cdc.gov/nchs/products/databriefs/db355.htm (accessed on 7 October 2020).
- Wenk, G.L. Neuropathologic Changes in Alzheimer’s Disease. J. Clin. Psychiatry 2003, 64 (Suppl. 9), 7–10. [Google Scholar] [PubMed]
- Calderon-Garcidueñas, A.L.; Duyckaerts, C. Alzheimer Disease. Handb. Clin. Neurol. 2017, 145, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The Cholinergic System in the Pathophysiology and Treatment of Alzheimer’s Disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.A.; McGonigle, P.; Van Bockstaele, E.J. Locus Coeruleus, Norepinephrine and Aβ Peptides in Alzheimer’s Disease. Neurobiol Stress 2015, 2, 73–84. [Google Scholar] [CrossRef]
- Woolf, N.J.; Butcher, L.L. Cholinergic Systems Mediate Action from Movement to Higher Consciousness. Behav. Brain Res. 2011, 221, 488–498. [Google Scholar] [CrossRef]
- Weinshenker, D. Long Road to Ruin: Noradrenergic Dysfunction in Neurodegenerative Disease. Trends Neurosci. 2018, 41, 211–223. [Google Scholar] [CrossRef]
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a Neuromodulator: Cholinergic Signaling Shapes Nervous System Function and Behavior. Neuron 2012, 76, 116–129. [Google Scholar] [CrossRef]
- Hasselmo, M.E.; Sarter, M. Modes and Models of Forebrain Cholinergic Neuromodulation of Cognition. Neuropsychopharmacology 2011, 36, 52–73. [Google Scholar] [CrossRef]
- Gill, S.K.; Ishak, M.; Dobransky, T.; Haroutunian, V.; Davis, K.L.; Rylett, R.J. 82-KDa Choline Acetyltransferase Is in Nuclei of Cholinergic Neurons in Human CNS and Altered in Aging and Alzheimer Disease. Neurobiol. Aging 2007, 28, 1028–1040. [Google Scholar] [CrossRef]
- Wurtman, R.J. Choline Metabolism as a Basis for the Selective Vulnerability of Cholinergic Neurons. Trends Neurosci. 1992, 15, 117–122. [Google Scholar] [CrossRef]
- Bartus, R.T.; Dean, R.L.; Beer, B.; Lippa, A.S. The Cholinergic Hypothesis of Geriatric Memory Dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef]
- D’Alton, S.; George, D.R. Changing Perspectives on Alzheimer’s Disease: Thinking Outside the Amyloid Box. J. Alzheimers Dis. 2011, 25, 571–581. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Khachaturian, A.S.; Vergallo, A.; Farlow, M.R.; Snyder, P.J.; Giacobini, E.; Khachaturian, Z.S. Revisiting the Cholinergic Hypothesis in Alzheimer’s Disease: Emerging Evidence from Translational and Clinical Research. J. Prev. Alzheimers Dis. 2019, 6, 2–15. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The Amyloid Cascade Hypothesis for Alzheimer’s Disease: An Appraisal for the Development of Therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Contestabile, A. The History of the Cholinergic Hypothesis. Behav. Brain Res. 2011, 221, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, P.J.; Price, D.L.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer Disease: Evidence for Selective Loss of Cholinergic Neurons in the Nucleus Basalis. Ann. Neurol. 1981, 10, 122–126. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s Disease and Senile Dementia: Loss of Neurons in the Basal Forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s Disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef]
- Parnetti, L.; Amici, S.; Lanari, A.; Romani, C.; Antognelli, C.; Andreasen, N.; Minthon, L.; Davidsson, P.; Pottel, H.; Blennow, K.; et al. Cerebrospinal Fluid Levels of Biomarkers and Activity of Acetylcholinesterase (AChE) and Butyrylcholinesterase in AD Patients before and after Treatment with Different AChE Inhibitors. Neurol. Sci. 2002, 23 (Suppl. 2), S95–S96. [Google Scholar] [CrossRef]
- Sharma, K. Cholinesterase Inhibitors as Alzheimer’s Therapeutics (Review). Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.V.; McKinney, M.; Coyle, J.T. Evidence for a Cholinergic Projection to Neocortex from Neurons in Basal Forebrain. Proc. Natl. Acad. Sci. USA 1979, 76, 5392–5396. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-Q.; Mobley, W.C. Exploring the Pathogenesis of Alzheimer Disease in Basal Forebrain Cholinergic Neurons: Converging Insights from Alternative Hypotheses. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, E.C.; Ananth, M.; Talmage, D.A.; Role, L.W. Basal Forebrain Cholinergic Circuits and Signaling in Cognition and Cognitive Decline. Neuron 2016, 91, 1199–1218. [Google Scholar] [CrossRef]
- Kamkwalala, A.R.; Newhouse, P.A. Beyond Acetylcholinesterase Inhibitors: Novel Cholinergic Treatments for Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 377–392. [Google Scholar] [CrossRef]
- Nyakas, C.; Granic, I.; Halmy, L.G.; Banerjee, P.; Luiten, P.G.M. The Basal Forebrain Cholinergic System in Aging and Dementia. Rescuing Cholinergic Neurons from Neurotoxic Amyloid-Β42 with Memantine. Behav. Brain Res. 2011, 221, 594–603. [Google Scholar] [CrossRef]
- Thiele, A. Muscarinic Signaling in the Brain. Ann. Rev. Neurosci. 2013, 36, 271–294. [Google Scholar] [CrossRef]
- Dani, J.A.; Bertrand, D. Nicotinic Acetylcholine Receptors and Nicotinic Cholinergic Mechanisms of the Central Nervous System. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 699–729. [Google Scholar] [CrossRef]
- Gotti, C.; Zoli, M.; Clementi, F. Brain Nicotinic Acetylcholine Receptors: Native Subtypes and Their Relevance. Trends Pharmacol. Sci. 2006, 27, 482–491. [Google Scholar] [CrossRef]
- Wu, J.; Ishikawa, M.; Zhang, J.; Hashimoto, K. Brain Imaging of Nicotinic Receptors in Alzheimer’s Disease. Available online: https://www.hindawi.com/journals/ijad/2010/548913/ (accessed on 13 November 2020). [CrossRef]
- Teipel, S.J.; Meindl, T.; Grinberg, L.; Grothe, M.; Cantero, J.L.; Reiser, M.F.; Möller, H.-J.; Heinsen, H.; Hampel, H. The Cholinergic System in Mild Cognitive Impairment and Alzheimer’s Disease: An in Vivo MRI and DTI Study. Hum. Brain Mapp. 2011, 32, 1349–1362. [Google Scholar] [CrossRef]
- Egea, J.; Buendia, I.; Parada, E.; Navarro, E.; León, R.; Lopez, M.G. Anti-Inflammatory Role of Microglial Alpha7 NAChRs and Its Role in Neuroprotection. Biochem. Pharmacol. 2015, 97, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Sabri, O.; Meyer, P.M.; Gräf, S.; Hesse, S.; Wilke, S.; Becker, G.-A.; Rullmann, M.; Patt, M.; Luthardt, J.; Wagenknecht, G.; et al. Cognitive Correlates of Α4β2 Nicotinic Acetylcholine Receptors in Mild Alzheimer’s Dementia. Brain 2018, 141, 1840–1854. [Google Scholar] [CrossRef] [PubMed]
- Guillem, K.; Bloem, B.; Poorthuis, R.B.; Loos, M.; Smit, A.B.; Maskos, U.; Spijker, S.; Mansvelder, H.D. Nicotinic Acetylcholine Receptor Β2 Subunits in the Medial Prefrontal Cortex Control Attention. Science 2011, 333, 888–891. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Lee, D.H.S.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. β-Amyloid1–42 Binds to Α7 Nicotinic Acetylcholine Receptor with High Affinity Implications for Alzheimer’s disease pathology. J. Biol. Chem. 2000, 275, 5626–5632. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.-G.; Qian, Y.-H. Alpha 7 Nicotinic Acetylcholine Receptor and Its Effects on Alzheimer’s Disease. Neuropeptides 2019, 73, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Huang, Y.; Xue, F.; Simard, A.; DeChon, J.; Li, G.; Zhang, J.; Lucero, L.; Wang, M.; Sierks, M.; et al. A Novel Nicotinic Acetylcholine Receptor Subtype in Basal Forebrain Cholinergic Neurons with High Sensitivity to Amyloid Peptides. J. Neurosci. 2009, 29, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Huang, Y.; Shen, J.; Steffensen, S.; Wu, J. Functional Α7β2 Nicotinic Acetylcholine Receptors Expressed in Hippocampal Interneurons Exhibit High Sensitivity to Pathological Level of Amyloid β Peptides. BMC Neurosci. 2012, 13, 155. [Google Scholar] [CrossRef]
- Picomolar Amyloid-β Positively Modulates Synaptic Plasticity and Memory in Hippocampus | Journal of Neuroscience. Available online: https://www.jneurosci.org/content/28/53/14537 (accessed on 1 November 2020).
- Plant, L.D.; Boyle, J.P.; Smith, I.F.; Peers, C.; Pearson, H.A. The Production of Amyloid β Peptide Is a Critical Requirement for the Viability of Central Neurons. J. Neurosci. 2003, 23, 5531–5535. [Google Scholar] [CrossRef]
- Lombardo, S.; Maskos, U. Role of the Nicotinic Acetylcholine Receptor in Alzheimer’s Disease Pathology and Treatment. Neuropharmacology 2015, 96, 255–262. [Google Scholar] [CrossRef]
- Chu, L.W.; Ma, E.S.K.; Lam, K.K.Y.; Chan, M.F.; Lee, D.H.S. Increased Alpha 7 Nicotinic Acetylcholine Receptor Protein Levels in Alzheimer’s Disease Patients. Dement. Geriatr. Cogn. Disord. 2005, 19, 106–112. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Stucky, A.; Liu, J.; Shen, C.; Trocme-Thibierge, C.; Morain, P. Dissociating Beta-Amyloid from Alpha 7 Nicotinic Acetylcholine Receptor by a Novel Therapeutic Agent, S 24795, Normalizes Alpha 7 Nicotinic Acetylcholine and NMDA Receptor Function in Alzheimer’s Disease Brain. J. Neurosci. 2009, 29, 10961–10973. [Google Scholar] [CrossRef] [PubMed]
- Hoskin, J.L.; Al-Hasan, Y.; Sabbagh, M.N. Nicotinic Acetylcholine Receptor Agonists for the Treatment of Alzheimer’s Dementia: An Update. Nicotine Tob. Res. 2018, 21, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Yakel, J.L. The Effect of Α7 Nicotinic Receptor Activation on Glutamatergic Transmission in the Hippocampus. Biochem. Pharmacol. 2015, 97, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Janak, P.H.; Tye, K.M. From Circuits to Behaviour in the Amygdala. Nature 2015, 517, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Hascup, K.N.; Hascup, E.R. Soluble Amyloid-Β42 Stimulates Glutamate Release through Activation of the Α7 Nicotinic Acetylcholine Receptor. J. Alzheimers Dis. 2016, 53, 337–347. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Green, K.N.; Liang, K.; Tran, L.; Chen, Y.; Leslie, F.M.; LaFerla, F.M. Chronic Nicotine Administration Exacerbates Tau Pathology in a Transgenic Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2005, 102, 3046–3051. [Google Scholar] [CrossRef]
- Eyjolfsdottir, H.; Eriksdotter, M.; Linderoth, B.; Lind, G.; Juliusson, B.; Kusk, P.; Almkvist, O.; Andreasen, N.; Blennow, K.; Ferreira, D.; et al. Targeted Delivery of Nerve Growth Factor to the Cholinergic Basal Forebrain of Alzheimer’s Disease Patients: Application of a Second-Generation Encapsulated Cell Biodelivery Device. Alzheimers Res. Ther. 2016, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Wallenstein, G.V.; Vago, D.R. Intrahippocampal Scopolamine Impairs Both Acquisition and Consolidation of Contextual Fear Conditioning. Neurobiol. Learn. Memory 2001, 75, 245–252. [Google Scholar] [CrossRef]
- Avila, J. Muscarinic Receptors and Alzheimer’s Disease. Neurodegener. Dis. Manag. 2011, 1, 267–269. [Google Scholar] [CrossRef]
- Jiang, S.; Li, Y.; Zhang, C.; Zhao, Y.; Bu, G.; Xu, H.; Zhang, Y.-W. M1 Muscarinic Acetylcholine Receptor in Alzheimer’s Disease. Neurosci. Bull. 2014, 30, 295–307. [Google Scholar] [CrossRef]
- Caccamo, A.; Oddo, S.; Billings, L.M.; Green, K.N.; Martinez-Coria, H.; Fisher, A.; LaFerla, F.M. M1 Receptors Play a Central Role in Modulating AD-like Pathology in Transgenic Mice. Neuron 2006, 49, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.C.; Goldsmith, P.J.; Jackson, K.; Sanger, H.E.; Evans, D.A.; Mogg, A.J.; Broad, L.M. Current Status of Muscarinic M1 and M4 Receptors as Drug Targets for Neurodegenerative Diseases. Neuropharmacology 2018, 136, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.; Sørensen, G.; Dencker, D. Physiological Roles of CNS Muscarinic Receptors Gained from Knockout Mice. Neuropharmacology 2018, 136 Pt C, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Dennis, S.H.; Pasqui, F.; Colvin, E.M.; Sanger, H.; Mogg, A.J.; Felder, C.C.; Broad, L.M.; Fitzjohn, S.M.; Isaac, J.T.R.; Mellor, J.R. Activation of Muscarinic M1 Acetylcholine Receptors Induces Long-Term Potentiation in the Hippocampus. Cereb. Cortex 2016, 26, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Gulledge, A.T. M1 and M4 Receptors Modulate Hippocampal Pyramidal Neurons. J. Neurophysiol. 2010, 105, 779–792. [Google Scholar] [CrossRef]
- Gulledge, A.T.; Bucci, D.J.; Zhang, S.S.; Matsui, M.; Yeh, H.H. M1 Receptors Mediate Cholinergic Modulation of Excitability in Neocortical Pyramidal Neurons. J. Neurosci. 2009, 29, 9888–9902. [Google Scholar] [CrossRef]
- Medeiros, R.; Kitazawa, M.; Caccamo, A.; Baglietto-Vargas, D.; Estrada-Hernandez, T.; Cribbs, D.H.; Fisher, A.; LaFerla, F.M. Loss of Muscarinic M1 Receptor Exacerbates Alzheimer’s Disease–Like Pathology and Cognitive Decline. Am. J. Pathol. 2011, 179, 980–991. [Google Scholar] [CrossRef]
- Development of M1 mAChR Allosteric and Bitopic Ligands: Prospective Therapeutics for the Treatment of Cognitive Deficits | ACS Chemical Neuroscience. Available online: https://pubs.acs.org/doi/10.1021/cn400086m (accessed on 11 November 2020).
- Bainbridge, N.K.; Koselke, L.R.; Jeon, J.; Bailey, K.R.; Wess, J.; Crawley, J.N.; Wrenn, C.C. Learning and Memory Impairments in a Congenic C57BL/6 Strain of Mice That Lacks the M2 Muscarinic Acetylcholine Receptor Subtype. Behav. Brain Res. 2008, 190, 50–58. [Google Scholar] [CrossRef]
- Hagena, H.; Hansen, N.; Manahan-Vaughan, D. β-Adrenergic Control of Hippocampal Function: Subserving the Choreography of Synaptic Information Storage and Memory. Cereb. Cortex 2016, 26, 1349–1364. [Google Scholar] [CrossRef]
- Hansen, N.; Manahan-Vaughan, D. Hippocampal Long-Term Potentiation That Is Elicited by Perforant Path Stimulation or That Occurs in Conjunction with Spatial Learning Is Tightly Controlled by Beta-Adrenoreceptors and the Locus Coeruleus. Hippocampus 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Song, S.; Jiang, L.; Oyarzabal, E.A.; Wilson, B.; Li, Z.; Shih, Y.-Y.I.; Wang, Q.; Hong, J.-S. Loss of Brain Norepinephrine Elicits Neuroinflammation-Mediated Oxidative Injury and Selective Caudo-Rostral Neurodegeneration. Mol. Neurobiol. 2019, 56, 2653–2669. [Google Scholar] [CrossRef] [PubMed]
- Leanza, G.; Gulino, R.; Zorec, R. Noradrenergic Hypothesis Linking Neurodegeneration-Based Cognitive Decline and Astroglia. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Borodovitsyna, O.; Flamini, M.; Chandler, D. Noradrenergic Modulation of Cognition in Health and Disease. Neural Plast. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Weinshenker, D. Functional Consequences of Locus Coeruleus Degeneration in Alzheimer’s Disease. Curr. Alzheimer Res. 2008, 5, 342–345. [Google Scholar] [CrossRef]
- Hammerschmidt, T.; Kummer, M.P.; Terwel, D.; Martinez, A.; Gorji, A.; Pape, H.-C.; Rommelfanger, K.S.; Schroeder, J.P.; Stoll, M.; Schultze, J.; et al. Selective Loss of Noradrenaline Exacerbates Early Cognitive Dysfunction and Synaptic Deficits in APP/PS1 Mice. Biol. Psychiatry 2013, 73, 454–463. [Google Scholar] [CrossRef]
- Perez, D.M. Α1-Adrenergic Receptors in Neurotransmission, Synaptic Plasticity, and Cognition. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Betts, M.J.; Kirilina, E.; Otaduy, M.C.G.; Ivanov, D.; Acosta-Cabronero, J.; Callaghan, M.F.; Lambert, C.; Cardenas-Blanco, A.; Pine, K.; Passamonti, L.; et al. Locus Coeruleus Imaging as a Biomarker for Noradrenergic Dysfunction in Neurodegenerative Diseases. Brain 2019, 142, 2558–2571. [Google Scholar] [CrossRef]
- Grudzien, A.; Shaw, P.; Weintraub, S.; Bigio, E.; Mash, D.C.; Mesulam, M.M. Locus Coeruleus Neurofibrillary Degeneration in Aging, Mild Cognitive Impairment and Early Alzheimer’s Disease. Neurobiol. Aging 2007, 28, 327–335. [Google Scholar] [CrossRef]
- Theofilas, P.; Dunlop, S.; Heinsen, H.; Grinberg, L.T. Turning on the Light Within: Subcortical Nuclei of the Isodentritic Core and Their Role in Alzheimer’s Disease Pathogenesis. J. Alzheimers Dis. 2015, 46, 17–34. [Google Scholar] [CrossRef]
- Arendt, T.; Brückner, M.K.; Morawski, M.; Jäger, C.; Gertz, H.-J. Early Neurone Loss in Alzheimer’s Disease: Cortical or Subcortical? Acta Neuropathol. Commun. 2015, 3, 10. [Google Scholar] [CrossRef]
- Chan-Palay, V.; Asan, E. Alterations in Catecholamine Neurons of the Locus Coeruleus in Senile Dementia of the Alzheimer Type and in Parkinson’s Disease with and without Dementia and Depression. J. Comp. Neurol. 1989, 287, 373–392. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.S.; Boyle, P.A.; Yu, L.; Barnes, L.L.; Schneider, J.A.; Bennett, D.A. Life-Span Cognitive Activity, Neuropathologic Burden, and Cognitive Aging. Neurology 2013, 81, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Szot, P.; White, S.S.; Greenup, J.L.; Leverenz, J.B.; Peskind, E.R.; Raskind, M.A. Compensatory Changes in the Noradrenergic Nervous System in the Locus Ceruleus and Hippocampus of Postmortem Subjects with Alzheimer’s Disease and Dementia with Lewy Bodies. J. Neurosci. 2006, 26, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.N.; Alter, S.P.; Wang, M.; Goldstein, D.S.; Miller, G.W. Reduced Vesicular Storage of Catecholamines Causes Progressive Degeneration in the Locus Ceruleus. Neuropharmacology 2014, 76, 97–105. [Google Scholar] [CrossRef] [PubMed]
- McMillan, P.J.; White, S.S.; Franklin, A.; Greenup, J.L.; Leverenz, J.B.; Raskind, M.A.; Szot, P. Differential Response of the Central Noradrenergic Nervous System to the Loss of Locus Coeruleus Neurons in Parkinson’s Disease and Alzheimer’s Disease. Brain Res. 2011, 1373, 240–252. [Google Scholar] [CrossRef]
- Szabadi, E. Functional Neuroanatomy of the Central Noradrenergic System. J. Psychopharmacol. 2013, 27, 659–693. [Google Scholar] [CrossRef]
- Zweig, R.M.; Ross, C.A.; Hedreen, J.C.; Steele, C.; Cardillo, J.E.; Whitehouse, P.J.; Folstein, M.F.; Price, D.L. Neuropathology of Aminergic Nuclei in Alzheimer’s Disease. Progress Clin. Biol. Res. 1989, 317, 353–365. [Google Scholar] [CrossRef]
- Mustapic, M.; Presecki, P.; Pivac, N.; Mimica, N.; Hof, P.R.; Simic, G.; Folnegovic-Smalc, V.; Muck-Seler, D. Genotype-Independent Decrease in Plasma Dopamine Beta-Hydroxylase Activity in Alzheimer’s Disease. Progress Neuro-Psychopharmacol. Biol. Psychiatry 2013, 44, 94–99. [Google Scholar] [CrossRef]
- Stefani, A.; Olivola, E.; Liguori, C.; Hainsworth, A.H.; Saviozzi, V.; Angileri, G.; D’Angelo, V.; Galati, S.; Pierantozzi, M. Catecholamine-Based Treatment in AD Patients: Expectations and Delusions. Front. Aging Neurosci. 2015, 7, 67. [Google Scholar] [CrossRef]
- Guzmán-Ramos, K.; Moreno-Castilla, P.; Castro-Cruz, M.; McGaugh, J.L.; Martínez-Coria, H.; LaFerla, F.M.; Bermúdez-Rattoni, F. Restoration of Dopamine Release Deficits during Object Recognition Memory Acquisition Attenuates Cognitive Impairment in a Triple Transgenic Mice Model of Alzheimer’s Disease. Learn. Mem. 2012, 19, 453–460. [Google Scholar] [CrossRef]
- Perkovic, M.N.; Strac, D.S.; Tudor, L.; Konjevod, M.; Erjavec, G.N.; Pivac, N. Catechol-O-Methyltransferase, Cognition and Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Gannon, M.; Wang, Q. Complex Noradrenergic Dysfunction in Alzheimer’s Disease: Low Norepinephrine Input Is Not Always to Blame. Brain Res. 2019, 1702, 12–16. [Google Scholar] [CrossRef] [PubMed]
- McBurney-Lin, J.; Lu, J.; Zuo, Y.; Yang, H. Locus Coeruleus-Norepinephrine Modulation of Sensory Processing and Perception: A Focused Review. Neurosci. Biobehav. Rev. 2019, 105, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Gannon, M.; Che, P.; Chen, Y.; Jiao, K.; Roberson, E.D.; Wang, Q. Noradrenergic Dysfunction in Alzheimer’s Disease. Front. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Leverenz, J.B.; Miller, M.A.; Dobie, D.J.; Peskind, E.R.; Raskind, M.A. Increased Alpha 2-Adrenergic Receptor Binding in Locus Coeruleus Projection Areas in Dementia with Lewy Bodies. Neurobiol. Aging 2001, 22, 555–561. [Google Scholar] [CrossRef]
- Hashimoto, E.; Ozawa, H.; Saito, T.; Gsell, W.; Takahata, N.; Riederer, P.; Frölich, L. Impairment of G(Salpha) Function in Human Brain Cortex of Alzheimer’s Disease: Comparison with Normal Aging. J. Neural Transm. 2004, 111, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto-Sasaki, M.; Ozawa, H.; Saito, T.; Rösler, M.; Riederer, P. Impaired Phosphorylation of Cyclic AMP Response Element Binding Protein in the Hippocampus of Dementia of the Alzheimer Type. Brain Res. 1999, 824, 300–303. [Google Scholar] [CrossRef]
- Cowburn, R.F.; O’Neill, C.; Bonkale, W.L.; Ohm, T.G.; Fastbom, J. Receptor-G-Protein Signalling in Alzheimer’s Disease. Biochem. Soc. Symp. 2001, 67, 163–175. [Google Scholar] [CrossRef]
- Dyer-Reaves, K.; Goodman, A.M.; Nelson, A.R.; McMahon, L.L. Alpha1-Adrenergic Receptor Mediated Long-Term Depression at CA3-CA1 Synapses Can Be Induced via Accumulation of Endogenous Norepinephrine and Is Preserved Following Noradrenergic Denervation. Front. Synaptic Neurosci. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Katsouri, L.; Vizcaychipi, M.P.; McArthur, S.; Harrison, I.; Suárez-Calvet, M.; Lleo, A.; Lloyd, D.G.; Ma, D.; Sastre, M. Prazosin, an α(1)-Adrenoceptor Antagonist, Prevents Memory Deterioration in the APP23 Transgenic Mouse Model of Alzheimer’s Disease. Neurobiol. Aging 2013, 34, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Thathiah, A.; De Strooper, B. The Role of G Protein-Coupled Receptors in the Pathology of Alzheimer’s Disease. Nat. Rev. Neurosci. 2011, 12, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fu, Q.; Zhou, Y.; Xu, B.; Shi, Q.; Igwe, B.; Matt, L.; Hell, J.W.; Wisely, E.V.; Oddo, S.; et al. Β2 Adrenergic Receptor, Protein Kinase A (PKA) and c-Jun N-Terminal Kinase (JNK) Signaling Pathways Mediate Tau Pathology in Alzheimer Disease Models. J. Biol. Chem. 2013, 288, 10298–10307. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yuen, E.Y.; Zhou, Y.; Yan, Z.; Xiang, Y.K. Amyloid β Peptide-(1–42) Induces Internalization and Degradation of Β2 Adrenergic Receptors in Prefrontal Cortical Neurons. J. Biol. Chem. 2011, 286, 31852–31863. [Google Scholar] [CrossRef]
- Chen, Y.; Peng, Y.; Che, P.; Gannon, M.; Liu, Y.; Li, L.; Bu, G.; van Groen, T.; Jiao, K.; Wang, Q. Α2A Adrenergic Receptor Promotes Amyloidogenesis through Disrupting APP-SorLA Interaction. Proc. Natl. Acad. Sci. USA 2014, 111, 17296–17301. [Google Scholar] [CrossRef] [PubMed]
- Coradazzi, M.; Gulino, R.; Fieramosca, F.; Falzacappa, L.V.; Riggi, M.; Leanza, G. Selective Noradrenaline Depletion Impairs Working Memory and Hippocampal Neurogenesis. Neurobiol. Aging 2016, 48, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Murchison, C.F.; Schutsky, K.; Jin, S.-H.; Thomas, S.A. Norepinephrine and SS₁-Adrenergic Signaling Facilitate Activation of Hippocampal CA1 Pyramidal Neurons during Contextual Memory Retrieval. Neuroscience 2011, 181, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, M.; Young, M.B.; Lestini, M.M.; Schutsky, K.; Thomas, S.A. Redundant Catecholamine Signaling Consolidates Fear Memory via Phospholipase C. J. Neurosci. 2012, 32, 1932–1941. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ouyang, M.; Ganellin, C.R.; Thomas, S.A. The Slow Afterhyperpolarization: A Target of Β1-Adrenergic Signaling in Hippocampus-Dependent Memory Retrieval. J. Neurosci. 2013, 33, 5006–5016. [Google Scholar] [CrossRef]
- Evans, A.K.; Ardestani, P.M.; Yi, B.; Park, H.H.; Lam, R.K.; Shamloo, M. Beta-Adrenergic Receptor Antagonism Is Proinflammatory and Exacerbates Neuroinflammation in a Mouse Model of Alzheimer’s Disease. Neurobiol. Dis. 2020, 146, 105089. [Google Scholar] [CrossRef]
- Kelly, S.C.; He, B.; Perez, S.E.; Ginsberg, S.D.; Mufson, E.J.; Counts, S.E. Locus Coeruleus Cellular and Molecular Pathology during the Progression of Alzheimer’s Disease. Acta Neuropathol. Commun. 2017, 5. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal Phosphorylation of the Microtubule-Associated Protein Tau (Tau) in Alzheimer Cytoskeletal Pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Iba, M.; McBride, J.D.; Guo, J.L.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.-Y. Tau Pathology Spread in PS19 Tau Transgenic Mice Following Locus Coeruleus (LC) Injections of Synthetic Tau Fibrils Is Determined by the LC’s Afferent and Efferent Connections. Acta Neuropathol. 2015, 130, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.P.; Hammerschmidt, T.; Martinez, A.; Terwel, D.; Eichele, G.; Witten, A.; Figura, S.; Stoll, M.; Schwartz, S.; Pape, H.-C.; et al. Ear2 Deletion Causes Early Memory and Learning Deficits in APP/PS1 Mice. J. Neurosci. 2014, 34, 8845–8854. [Google Scholar] [CrossRef] [PubMed]
- Hansen, N. The Longevity of Hippocampus-Dependent Memory Is Orchestrated by the Locus Coeruleus-Noradrenergic System. Neural Plast. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Hou, J.; Ping, J.; Cai, D. Advances in Developing Novel Therapeutic Strategies for Alzheimer’s Disease. Mol. Neurodegener. 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Maity, S.; Jarome, T.J.; Blair, J.; Lubin, F.D.; Nguyen, P.V. Noradrenaline Goes Nuclear: Epigenetic Modifications during Long-Lasting Synaptic Potentiation Triggered by Activation of β-Adrenergic Receptors. J. Physiol. 2016, 594, 863–881. [Google Scholar] [CrossRef]
- Lonze, B.E.; Ginty, D.D. Function and Regulation of CREB Family Transcription Factors in the Nervous System. Neuron 2002, 35, 605–623. [Google Scholar] [CrossRef]
- Epigenetics of Brain Disorders: The Paradigm of Alzheimer’s Disease | OMICS International. Available online: https://www.omicsonline.org/open-access/epigenetics-of-brain-disorders-the-paradigm-of-alzheimer-s-disease-2161-0460-1000229.php?aid=72455&view=mobile (accessed on 17 January 2021).
- Meyer, P.-F.; Pichet Binette, A.; Gonneaud, J.; Breitner, J.C.S.; Villeneuve, S. Characterization of Alzheimer Disease Biomarker Discrepancies Using Cerebrospinal Fluid Phosphorylated Tau and AV1451 Positron Emission Tomography. JAMA Neurol. 2020, 77, 508. [Google Scholar] [CrossRef]
- Zetterberg, H.; Bendlin, B.B. Biomarkers for Alzheimer’s Disease—Preparing for a New Era of Disease-Modifying Therapies. Mol. Psychiatry 2020, 1–13. [Google Scholar] [CrossRef]
- Khoury, R.; Ghossoub, E. Diagnostic Biomarkers of Alzheimer’s Disease: A State-of-the-Art Review. Biomark. Neuropsychiatry 2019, 1, 100005. [Google Scholar] [CrossRef]
- Puranik, N.; Yadav, D.; Yadav, S.K.; Chavda, V.K.; Jin, J.-O. Proteomics and Neurodegenerative Disorders: Advancements in the Diagnostic Analysis. Curr. Protein Pept. Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lashley, T.; Schott, J.M.; Weston, P.; Murray, C.E.; Wellington, H.; Keshavan, A.; Foti, S.C.; Foiani, M.; Toombs, J.; Rohrer, J.D.; et al. Molecular Biomarkers of Alzheimer’s Disease: Progress and Prospects. Dis. Model Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Lao, K.; Ji, N.; Zhang, X.; Qiao, W.; Tang, Z.; Gou, X. Drug Development for Alzheimer’s Disease: Review. J. Drug Target 2019, 27, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Kumar, A.; Tripathi, T.; Kumar, A. Muscarinic and Nicotinic Acetylcholine Receptor Agonists: Current Scenario in Alzheimer’s Disease Therapy. J. Pharm. Pharmacol. 2018, 70, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Femminella, G.D.; Rengo, G.; Pagano, G.; de Lucia, C.; Komici, K.; Parisi, V.; Cannavo, A.; Liccardo, D.; Vigorito, C.; Filardi, P.P.; et al. β-Adrenergic Receptors and G Protein-Coupled Receptor Kinase-2 in Alzheimer’s Disease: A New Paradigm for Prognosis and Therapy? J. Alzheimers Dis. 2013, 34, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-T.; Wang, N.-D.; Ma, T.; Jiang, H.; Guan, J.; Tan, L. Roles of β-Adrenergic Receptors in Alzheimer’s Disease: Implications for Novel Therapeutics. Brain Res. Bull. 2011, 84, 111–117. [Google Scholar] [CrossRef]
- Huang, L.-K.; Chao, S.-P.; Hu, C.-J. Clinical Trials of New Drugs for Alzheimer Disease. J. Biomed. Sci. 2020, 27. [Google Scholar] [CrossRef]
- Wang, J.; Yu, J.-T.; Tan, M.-S.; Jiang, T.; Tan, L. Epigenetic Mechanisms in Alzheimer’s Disease: Implications for Pathogenesis and Therapy. Ageing Res. Rev. 2013, 12, 1024–1041. [Google Scholar] [CrossRef]
- Coppieters, N.; Dragunow, M. Epigenetics in Alzheimer’s Disease: A Focus on DNA Modifications. Curr. Pharm. Des. 2011, 17, 3398–3412. [Google Scholar] [CrossRef]
- Chouliaras, L.; Rutten, B.P.F.; Kenis, G.; Peerbooms, O.; Visser, P.J.; Verhey, F.; van Os, J.; Steinbusch, H.W.M.; van den Hove, D.L.A. Epigenetic Regulation in the Pathophysiology of Alzheimer’s Disease. Progress Neurobiol. 2010, 90, 498–510. [Google Scholar] [CrossRef]
- Sezgin, Z.; Dincer, Y. Alzheimer’s Disease and Epigenetic Diet. Neurochem. Int. 2014, 78, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-Y.; Aromolaran, K.A.; Zukin, R.S. The Emerging Field of Epigenetics in Neurodegeneration and Neuroprotection. Nat. Rev. Neurosci. 2017, 18, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wei, Q.; Liu, F.-F.; Hu, F.; Xie, A.-J.; Zhu, L.-Q.; Liu, D. Synaptic Dysfunction in Alzheimer’s Disease: Aβ, Tau, and Epigenetic Alterations. Mol. Neurobiol. 2018, 55, 3021–3032. [Google Scholar] [CrossRef] [PubMed]
- Prasad, G.R.; Jho, E.H. A Concise Review of Human Brain Methylome during Aging and Neurodegenerative Diseases. BMB Rep. 2019, 52, 577–588. [Google Scholar] [CrossRef] [PubMed]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s Disease: Early Alterations in Brain DNA Methylation at ANK1, BIN1, RHBDF2 and Other Loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef]
- West, R.L.; Lee, J.M.; Maroun, L.E. Hypomethylation of the Amyloid Precursor Protein Gene in the Brain of an Alzheimer’s Disease Patient. J. Mol. Neurosci. 1995, 6, 141–146. [Google Scholar] [CrossRef]
- Bakulski, K.M.; Dolinoy, D.C.; Sartor, M.A.; Paulson, H.L.; Konen, J.R.; Lieberman, A.P.; Albin, R.L.; Hu, H.; Rozek, L.S. Genome-Wide DNA Methylation Differences Between Late-Onset Alzheimer’s Disease and Cognitively Normal Controls in Human Frontal Cortex. J. Alzheimers Dis. 2012, 29, 571–588. [Google Scholar] [CrossRef]
- Zhang, K.; Schrag, M.; Crofton, A.; Trivedi, R.; Vinters, H.; Kirsch, W. Targeted Proteomics for Quantification of Histone Acetylation in Alzheimer’s Disease. Proteomics 2012, 12, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, D.; Delvaux, E.; Nolz, J.; Tan, Y.; Grover, A.; Oddo, S.; Coleman, P.D. Aberrant Intracellular Localization of H3k4me3 Demonstrates an Early Epigenetic Phenomenon in Alzheimer’s Disease. Neurobiol. Aging 2015, 36, 3121–3129. [Google Scholar] [CrossRef] [PubMed]
- Di Francesco, A.; Arosio, B.; Falconi, A.; Micioni Di Bonaventura, M.V.; Karimi, M.; Mari, D.; Casati, M.; Maccarrone, M.; D’Addario, C. Global Changes in DNA Methylation in Alzheimer’s Disease Peripheral Blood Mononuclear Cells. Brain Behav. Immun. 2015, 45, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Lardenoije, R.; Roubroeks, J.A.Y.; Pishva, E.; Leber, M.; Wagner, H.; Iatrou, A.; Smith, A.R.; Smith, R.G.; Eijssen, L.M.T.; Kleineidam, L.; et al. Alzheimer’s Disease-Associated (Hydroxy)Methylomic Changes in the Brain and Blood. Clin. Epigenetics 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, M.F.; Beal, M.F.; Bird, E.D.; Martin, J.B. Oxytocin in Alzheimer’s Disease: Postmortem Brain Levels. Neurology 1987, 37, 1001–1003. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.M.; Yates, P.O.; Marcyniuk, B. Changes in Alzheimer’s Disease in the Magnocellular Neurones of the Supraoptic and Paraventricular Nuclei of the Hypothalamus and Their Relationship to the Noradrenergic Deficit. Clin. Neuropathol. 1985, 4, 127–134. [Google Scholar] [PubMed]
- Cummings, J.; Scheltens, P.; McKeith, I.; Blesa, R.; Harrison, J.E.; Bertolucci, P.H.F.; Rockwood, K.; Wilkinson, D.; Wijker, W.; Bennett, D.A.; et al. Effect Size Analyses of Souvenaid in Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2017, 55, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Twisk, J.W.R.; Blesa, R.; Scarpini, E.; von Arnim, C.A.F.; Bongers, A.; Harrison, J.; Swinkels, S.H.N.; Stam, C.J.; de Waal, H.; et al. Efficacy of Souvenaid in Mild Alzheimer’s Disease: Results from a Randomized, Controlled Trial. J. Alzheimers Dis. 2012, 31, 225–236. [Google Scholar] [CrossRef] [PubMed]
- De Waal, H.; Stam, C.J.; Lansbergen, M.M.; Wieggers, R.L.; Kamphuis, P.J.G.H.; Scheltens, P.; Maestú, F.; van Straaten, E.C.W. The Effect of Souvenaid on Functional Brain Network Organisation in Patients with Mild Alzheimer’s Disease: A Randomised Controlled Study. PLoS ONE 2014, 9, e86558. [Google Scholar] [CrossRef]
- Van Wijk, N.; Broersen, L.M.; de Wilde, M.C.; Hageman, R.J.J.; Groenendijk, M.; Sijben, J.W.C.; Kamphuis, P.J.G.H. Targeting Synaptic Dysfunction in Alzheimer’s Disease by Administering a Specific Nutrient Combination. J. Alzheimers Dis. 2014, 38, 459–479. [Google Scholar] [CrossRef]
- Bekdash, R.A. Neuroprotective Effects of Choline and Other Methyl Donors. Nutrients 2019, 11, 2995. [Google Scholar] [CrossRef]
- Muñoz Fernández, S.S.; Lima Ribeiro, S.M. Nutrition and Alzheimer Disease. Clin. Geriatr. Med. 2018, 34, 677–697. [Google Scholar] [CrossRef]
- Athanasopoulos, D.; Karagiannis, G.; Tsolaki, M. Recent Findings in Alzheimer Disease and Nutrition Focusing on Epigenetics. Adv. Nutr. 2016, 7, 917–927. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Description | Outcomes | References |
|---|---|---|
| α7 nAchRs | Enhancement of LTP in the hippocampus and in the amygdala | [46,47] |
| 3xTg-AD mouse model | Reduction in α7 nAchRs levels in an age-dependent manner | [49] |
| Blockade of mAchR signaling by an antagonist in rats | Impairment in the acquisition and consolidation of contextual fear conditioning | [51] |
| Muscarinic agonist against M1 mAchRs in a 3xTg-AD mouse model | Decrease in β-amyloid plaques accumulation, decrease in tau hyperphosphorylation, improvement in cholinergic activity in the cortex and the hippocampus and improved some cognitive functions | [54] |
| Knockout of M1 and M4 mAchRs in mice | Impacted hippocampal circuitry and hippocampal release of Ach | [57,58] |
| Lack of M1 AchR in mouse PFC | Impairment of cholinergic signaling in pyramidal neurons and cue detection deficits | [59] |
| M2 AchR knockout mice | Deficit in specific learning and memory tests | [62] |
| Description | Outcomes or Role | References |
|---|---|---|
| Blockade of α1 ARs in APP23 transgenic mice model of AD | Impairment in cognitive functions | [94] |
| β2 AR in PFC of APP/PS1 transgenic mouse model | Alteration in Amyloid Precursor Protein (APP) processing and mediation of Aβ-induced tau pathology | [97] |
| α2A AR in cerebral cortex of AD transgenic mice model | Regulation of APP proteolytic processing and Aβ production and secretion | [98] |
| Blockade of β ARs in APP mouse model | Impairment in cognitive functions, and behavior. Inducing inflammation with chronic blockade of these receptors | [103] |
| β1 AR activation in hippocampal pyramidal neurons in mice | Role in contextual and spatial memory consolidation and retrieval | [100,101,102] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bekdash, R.A. The Cholinergic System, the Adrenergic System and the Neuropathology of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 1273. https://doi.org/10.3390/ijms22031273
Bekdash RA. The Cholinergic System, the Adrenergic System and the Neuropathology of Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(3):1273. https://doi.org/10.3390/ijms22031273
Chicago/Turabian StyleBekdash, Rola A. 2021. "The Cholinergic System, the Adrenergic System and the Neuropathology of Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 3: 1273. https://doi.org/10.3390/ijms22031273
APA StyleBekdash, R. A. (2021). The Cholinergic System, the Adrenergic System and the Neuropathology of Alzheimer’s Disease. International Journal of Molecular Sciences, 22(3), 1273. https://doi.org/10.3390/ijms22031273