
USP2-Related Cellular Signaling and Consequent Pathophysiological Outcomes


Abstract
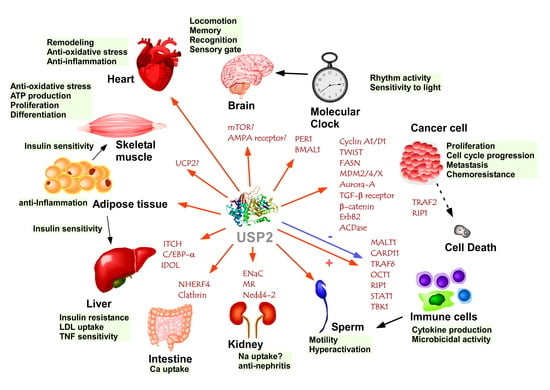
1. Introduction
2. Tumorigenesis
3. Apoptosis and Autophagy
4. Circadian Clock
5. Renal System
6. Energy Metabolism and Metabolic Disorders
7. Nervous System
8. Skeletal and Cardiac Muscles
9. Immune and Inflammatory Responses
10. Male Genital Tract
11. Perspectives
12. Short Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-PBA | 4-phenylbutyric acid |
| ACDase | Acid ceramidase |
| ActD | Actinomycin D |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| ANO1 | Anoctamin 1 |
| aP2 | Adipocyte protein 2 |
| APC | Adenomatous polyposis coli |
| ATS | Anti-thymocyte serum |
| BCL10 | B-cell lymphoma 10 |
| BMAL1 | Brain and muscle Arnt-like protein 1 |
| C/EBP | CCAAT-enhancer-binding protein |
| CARD1 | Caspase recruitment domain family member 11 |
| Cav1.2 | Calcium channel, voltage-dependent, L-type, α1C subunit |
| CCL | C-C motif ligand |
| CLOCK | Clock circadian regulator |
| cFLIP | Cellular FLICE-like inhibitory protein |
| cGAS | Cyclic GMP-AMP synthase |
| cIAP2 | Cellular inhibitor of apoptosis protein 2 |
| CRY | Cryptochromes |
| DIM | 3,3’-diinodolylmethane |
| DUB | Deubiquitinating enzyme |
| E4BP4 | E4 promoter-binding protein 4 |
| EGF | Epidermal growth factor |
| EGFR | EGF receptor |
| EMT | Epithelial-mesenchymal transition |
| ENaC | Epithelial sodium channel |
| ERBB2 | Erythroblastic oncogene B2 |
| ERK | Extracellular signal regulate kinase |
| ERS | Endoplasmic reticulum stress |
| FASN | Fatty acid synthase |
| G-6-Pase | Glucose-6-phosphatase |
| GBB10 | Growth factor receptor-bound protein 10 |
| GM-CSF | Granulocyte macrophage-colony stimulating factor |
| GSK | Glycogen synthase kinase |
| HCC | Hepatocellular carcinoma |
| HER2 | Human epidermal growth factor receptor 2 |
| HFD | High fat diet |
| HMGA2 | High mobility group protein A2 |
| HSD1 | 11β-hydroxysteroid dehydrogenase 1 |
| HSP | Heat shock protein |
| IDOL | Inducible degrader of the LDLR |
| IFN | interferon |
| IκB | NF-κB inhibitor |
| IKK | IκB kinase |
| IL | Interleukin |
| Imd | Immune deficiency |
| IRF3 | Interferon regulatory factor 3 |
| ITCH | Itchy homolog |
| JAK | Janus kinase |
| JNK | c-Jun N-terminal kinase |
| KO | Knockout |
| LPS | Lipopolysaccharide |
| LV | Left ventricular |
| MALT1 | Mucosa-associated lymphoid translocation gene 1 |
| MAV5 | Mitochondrial antiviral signaling protein |
| MDA5 | Melanoma differentiation-associated gene 5 |
| MDM | Murine double minute |
| MEF | Mouse embryo fibroblast |
| Mφ | Macrophage |
| MMP | Matrix metalloprotease |
| msUsp2KO | Macrophage-selective Usp2KO |
| mTOR | Mammalian target of rapamycin |
| Nedd | Neural precursor cell expressed developmentally down-regulated protein |
| NF-κB | Nuclear factor-κB |
| NHERF4 | Na+/H+ exchange regulatory cofactor 4 |
| NOX | NADPH oxidase |
| OCT | Octamer binding protein |
| OXPHOS | Oxidative phosphorylation |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PARP | Poly ADP-ribose polymerase |
| PBS | Phosphate-buffered saline |
| PCNA | Proliferating cell nuclear antigen |
| PEPCK | Phosphoenol pyruvate carboxykinase |
| PER | Period |
| PGC | PPARγ coactivator |
| PKA | Protein kinase A |
| PMA | Phorbol 12-myristate 13-acetate |
| PPAR | Peroxisome proliferator-activated receptor |
| PTS | Peroxisomal targeting signal 1 |
| pY701 | Phosphorylation at tyrosine 701 |
| RA | Rheumatoid arthritis |
| RIG-I | Retinoic acid-inducible gene-I |
| RIP1 | Receptor-interacting serine/threonine-protein kinase 1 |
| ROR | REV-ERB and retinoic acid receptor-related orphan receptor |
| SAPS | Scale for the assessment of positive symptoms |
| SCN | Suprachiasmatic nucleus |
| SeV | Sendai virus |
| siRNA | Small interfering RNA |
| SUMO | Small ubiquitin-related modifier |
| TAB | TAK1-binding protein |
| TAC | Transverse aortic construction |
| TAK1 | TGF-β-activated kinase 1 |
| TBK1 | TANK binding kinase 1 |
| TCR | T cell receptor |
| TGF | Transforming growth factor |
| TMBIM4 | Transmembrane BAX inhibitor motif containing 4 |
| TNF | Tumor necrosis factor |
| TNFR1 | TNF receptor 1 |
| TRADD | TNF receptor associated death domain protein |
| TRAF | TNF receptor-associated factor |
| TRAPV6 | Transient receptor potential vanilloid subfamily member 6 |
| TSG | Tumor suppressor gene |
| Tyk2 | Tyrosine kinase 2 |
| UCP2 | Uncoupling protein 2 |
| USP | Ubiquitin-specific protease |
| VSV | Vascular stomatitis virus |
| ZT | Zeitgeber time |
References
- Celebi, G.; Kesim, H.; Ozer, E.; Kutlu, O. The effect of dysfunctional ubiquitin enzymes in the pathogenesis of most common diseases. Int. J. Mol. Sci. 2020, 21, 6335. [Google Scholar] [CrossRef]
- Park, J.; Cho, J.; Song, E.J. Ubiquitin–proteasome system (UPS) as a target for anticancer treatment. Arch. Pharm. Res. 2020. [Google Scholar] [CrossRef]
- Yuan, T.; Yan, F.; Ying, M.; Cao, J.; He, Q.; Zhu, H.; Yang, B. Inhibition of Ubiquitin-Specific Proteases as a Novel Anticancer Therapeutic Strategy. Front. Pharmacol. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Baek, S.H.; Choi, K.S.; Yoo, Y.J.; Cho, J.M.; Baker, R.T.; Tanaka, K.; Chung, C.H. Molecular cloning of a novel ubiquitin-specific protease, UBP41, with isopeptidase activity in chick skeletal muscle. J. Biol. Chem. 1997, 272, 25560–25565. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Keriel, A.; Morales, C.R.; Bedard, N.; Zhao, Q.; Hingamp, P.; Lefrançois, S.; Combaret, L.; Wing, S.S. Divergent N-Terminal Sequences Target an Inducible Testis Deubiquitinating Enzyme to Distinct Subcellular Structures. Mol. Cell. Biol. 2000, 20, 6568–6578. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reglinski, K.; Keil, M.; Altendorf, S.; Waithe, D.; Eggeling, C.; Schliebs, W.; Erdmann, R. Peroxisomal import reduces the proapoptotic activity of deubiquitinating enzyme USP2. PLoS ONE 2015, 10, e0140685. [Google Scholar] [CrossRef]
- Gewies, A.; Grimm, S. UBP41 is a proapoptotic ubiquitin-specific protease. Cancer Res. 2003, 63, 682–688. [Google Scholar] [PubMed]
- Zhu, H.Q.; Gao, F.H. The molecular mechanisms of regulation on USP2’s alternative splicing and the significance of its products. Int. J. Biol. Sci. 2017, 13, 1489–1496. [Google Scholar] [CrossRef]
- Kitamura, H.; Kimura, S.; Shimamoto, Y.; Okabe, J.; Ito, M.; Miyamoto, T.; Naoe, Y.; Kikuguchi, C.; Meek, B.; Toda, C.; et al. Ubiquitin-specific protease 2-69 in macrophages potentially modulates metainflammation. FASEB J. 2013, 27, 4940–4953. [Google Scholar] [CrossRef]
- Kitamura, H.; Ishino, T.; Shimamoto, Y.; Okabe, J.; Miyamoto, T.; Takahashi, E.; Miyoshi, I. Ubiquitin-specific protease 2 modulates the lipopolysaccharide-elicited expression of proinflammatory cytokines in macrophage-like HL-60 cells. Mediat. Inflamm. 2017, 2017, 6909415. [Google Scholar] [CrossRef]
- Molusky, M.M.; Li, S.; Ma, D.; Yu, L.; Lin, J.D. Ubiquitin-Specific Protease 2 Regulates Hepatic Gluconeogenesis and Diurnal Glucose Metabolism Through 11β-Hydroxysteroid Dehydrogenase 1. Diabetes 2012, 61, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Gousseva, N.; Baker, R.T. Gene Structure, Alternate Splicing, Tissue Distribution, Cellular Localization, and Developmental Expression Pattern of Mouse Deubiquitinating Enzyme Isoforms Usp2-45 and Usp2-69. Gene Expr. 2003, 11, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Haimerl, F.; Erhardt, A.; Sass, G.; Tiegs, G. Down-regulation of the de-ubiquitinating enzyme ubiquitin-specific protease 2 contributes to tumor necrosis factor-α-induced hepatocyte survival. J. Biol. Chem. 2009, 284, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Sacco, J.J.; Coulson, J.M.; Clague, M.J.; Urbé, S. Emerging roles of deubiquitinases in cancer-associated pathways. IUBMB Life 2010, 62, 140–157. [Google Scholar] [CrossRef]
- Bonacci, T.; Emanuele, M.J. Dissenting degradation: Deubiquitinases in cell cycle and cancer. Semin. Cancer Biol. 2020, 1–14. [Google Scholar] [CrossRef]
- Wing, S.S. Deubiquitinases in skeletal muscle atrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2130–2135. [Google Scholar] [CrossRef][Green Version]
- Verrey, F.; Fakitsas, P.; Adam, G.; Staub, O. Early transcriptional control of ENaC (de)ubiquitylation by aldosterone. Kidney Int. 2008, 73, 691–696. [Google Scholar] [CrossRef]
- Graner, E.; Tang, D.; Rossi, S.; Baron, A.; Migita, T.; Weinstein, L.J.; Lechpammer, M.; Huesken, D.; Zimmermann, J.; Signoretti, S.; et al. The isopeptidase USP2a regulates the stability of fatty acid synthase in prostate cancer. Cancer Cell 2004, 5, 253–261. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083.e5. [Google Scholar] [CrossRef]
- Tao, B.B.; He, H.; Shi, X.H.; Wang, C.L.; Li, W.Q.; Li, B.; Dong, Y.; Hu, G.H.; Hou, L.J.; Luo, C.; et al. Up-regulation of USP2a and FASN in gliomas correlates strongly with glioma grade. J. Clin. Neurosci. 2013, 20, 717–720. [Google Scholar] [CrossRef]
- Boustani, M.R.; Khoshnood, R.J.; Nikpasand, F.; Taleshi, Z.; Ahmadi, K.; Yahaghi, E.; Goudarzi, P.K. Overexpression of ubiquitin-specific protease 2a (USP2a) and nuclear factor erythroid 2-related factor 2 (Nrf2) in human gliomas. J. Neurol. Sci. 2016, 363, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Jeong, P.; Ha, Y.S.; Yun, S.J.; Yoon, H.Y.; Freeman, M.R.; Kim, J.; Kim, W.J. Assess the expression of ubiquitin specific protease USP2a for bladder cancer diagnosis. BMC Urol. 2015, 15, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Mao, Y.; Xiao, G.; Fei, X.; Wang, J.; Zhang, Y.; Liu, J.; Cheng, G.; Chen, X.; Wang, J.; et al. USP2 promotes cell migration and invasion in triple negative breast cancer cell lines. Tumor Biol. 2015, 36, 5415–5423. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, W.J.; Liu, Z.; Loda, M.F.; Freeman, M.R. The ubiquitin-specific protease USP2a enhances tumor progression by targeting cyclin A1 in bladder cancer. Cell Cycle 2012, 11, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Lee, H.J.; Saha, S.; Ruan, D.; Guo, H.; Chan, C.H. Inhibition of USP2 eliminates cancer stem cells and enhances TNBC responsiveness to chemotherapy. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef]
- Gelebart, P.; Zak, Z.; Anand, M.; Belch, A.; Lai, R. Blockade of fatty acid synthase triggers significant apoptosis in mantle cell lymphoma. PLoS ONE 2012, 7, e33738. [Google Scholar] [CrossRef]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Carrà, G.; Lingua, M.F.; Maffeo, B.; Taulli, R.; Morotti, A. P53 vs NF-κB: The role of nuclear factor-kappa B in the regulation of p53 activity and vice versa. Cell. Mol. Life Sci. 2020, 77, 4449–4458. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 1–14. [Google Scholar] [CrossRef]
- Dobbelstein, M.; Levine, A.J. Mdm2: Open questions. Cancer Sci. 2020, 111, 2203–2211. [Google Scholar] [CrossRef]
- Stevenson, L.F.; Sparks, A.; Allende-Vega, N.; Xirodimas, D.P.; Lane, D.P.; Saville, M.K. The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO J. 2007, 26, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Biskup, E.; Gjerdrum, L.M.R.; Niazi, O.; Ødum, N.; Gniadecki, R. Ubiquitin-specific protease 2 decreases p53-dependent apoptosis in cutaneous T-cell lymphoma. Oncotarget 2016, 7, 48391–48400. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X.; Wang, G.; Yang, Y.; Yuan, Y.; Ouyang, L. The past, present and future of potential small-molecule drugs targeting p53-MDM2/MDMX for cancer therapy. Eur. J. Med. Chem. 2019, 176, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Allende-Vega, N.; Sparks, A.; Lane, D.P.; Saville, M.K. MdmX is a substrate for the deubiquitinating enzyme USP2a. Oncogene 2010, 29, 432–441. [Google Scholar] [CrossRef][Green Version]
- Wang, C.L.; Wang, J.Y.; Liu, Z.Y.; Ma, X.M.; Wang, X.W.; Jin, H.; Zhang, X.P.; Fu, D.; Hou, L.J.; Lu, Y.C. Ubiquitin-specific protease 2a stabilizes MDM4 and facilitates the p53-mediated intrinsic apoptotic pathway in glioblastoma. Carcinogenesis 2014, 35, 1500–1509. [Google Scholar] [CrossRef]
- Shrestha, M.; Park, P.H. P53 Signaling Is Involved in Leptin-Induced Growth of Hepatic and Breast Cancer Cells. Korean J. Physiol. Pharmacol. 2016, 20, 487–498. [Google Scholar] [CrossRef][Green Version]
- Navarro, A.; Beà, S.; Jares, P.; Campo, E. Molecular Pathogenesis of Mantle Cell Lymphoma. Hematol. Oncol. Clin. North Am. 2020, 34, 795–807. [Google Scholar] [CrossRef]
- Qie, S.; Diehl, J.A. Cyclin D degradation by E3 ligases in cancer progression and treatment. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Shan, J.; Zhao, W.; Gu, W. Suppression of Cancer Cell Growth by Promoting Cyclin D1 Degradation. Mol. Cell 2009, 36, 469–476. [Google Scholar] [CrossRef]
- Magiera, K.; Tomala, M.; Kubica, K.; De Cesare, V.; Trost, M.; Zieba, B.J.; Kachamakova-Trojanowska, N.; Les, M.; Dubin, G.; Holak, T.A.; et al. Lithocholic Acid Hydroxyamide Destabilizes Cyclin D1 and Induces G0/G1 Arrest by Inhibiting Deubiquitinase USP2a. Cell Chem. Biol. 2017, 24, 458–470.e18. [Google Scholar] [CrossRef]
- Nepal, S.; Shrestha, A.; Park, P.H. Ubiquitin specific protease 2 acts as a key modulator for the regulation of cell cycle by adiponectin and leptin in cancer cells. Mol. Cell. Endocrinol. 2015, 412, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Pragani, R.; Fox, J.T.; Shen, M.; Parmar, K.; Gaudiano, E.F.; Liu, L.; Tanega, C.; McGee, L.; Hall, M.D.; et al. Small molecule inhibition of the ubiquitin-specific protease USP2 accelerates cyclin D1 degradation and leads to cell cycle arrest in colorectal cancer and mantle cell lymphoma models. J. Biol. Chem. 2016, 291, 24628–24640. [Google Scholar] [CrossRef] [PubMed]
- Tomala, M.D.; Magiera-Mularz, K.; Kubica, K.; Krzanik, S.; Zieba, B.; Musielak, B.; Pustula, M.; Popowicz, G.M.; Sattler, M.; Dubin, G.; et al. Identification of small-molecule inhibitors of USP2a. Eur. J. Med. Chem. 2018, 150, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Xiang, X.; Hao, L.; Wang, T.; Lai, Y.; Abudoureyimu, M.; Zhou, H.; Feng, B.; Chu, X.; Wang, R. The role of Aurora-A in human cancers and future therapeutics. Am. J. Cancer Res. 2020, 10, 2705–2729. [Google Scholar] [PubMed]
- Vangenderen, C.; Harkness, T.A.A.; Arnason, T.G. The role of Anaphase Promoting Complex activation, inhibition and substrates in cancer development and progression. Aging (Albany. NY). 2020, 12, 15818–15855. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Solomon, L.R.; Pereda-Lopez, A.; Giranda, V.L.; Luo, Y.; Johnson, E.F.; Shoemaker, A.R.; Leverson, J.; Liu, X. Ubiquitin-specific cysteine protease 2a (USP2a) regulates the stability of Aurora-A. J. Biol. Chem. 2011, 286, 38960–38968. [Google Scholar] [CrossRef] [PubMed]
- Calabrò, M.L.; Lazzari, N.; Rigotto, G.; Tonello, M.; Sommariva, A. Role of epithelial–mesenchymal plasticity in pseudomyxoma peritonei: Implications for locoregional treatments. Int. J. Mol. Sci. 2020, 21, 9120. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Luo, W.; Sun, J.; Yang, N.; Zhang, L.W.; Zhao, Z.; Zhang, Z.; Wu, H. Usp2-69 overexpression slows down the progression of rat anti-Thy1.1 nephritis. Exp. Mol. Pathol. 2016, 101, 249–258. [Google Scholar] [CrossRef]
- Moses, H.L.; Roberts, A.B.; Derynck, R. The discovery and early days of TGF-b: A historical perspective. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Ahmadi, A.; Najafi, M.; Farhood, B.; Mortezaee, K. Transforming growth factor-β signaling: Tumorigenesis and targeting for cancer therapy. J. Cell. Physiol. 2019, 234, 12173–12187. [Google Scholar] [CrossRef]
- Dardare, J.; Witz, A.; Merlin, J.L.; Gilson, P.; Harlé, A. SMAD4 and the TGFΒ pathway in patients with pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 3534. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, X.; Wang, Q.; Deng, Y.; Li, K.; Zhang, M.; Zhang, Q.; Zhou, J.; Wang, H.Y.; Bai, P.; et al. USP2a Supports Metastasis by Tuning TGF-β Signaling. Cell Rep. 2018, 22, 2442–2454. [Google Scholar] [CrossRef] [PubMed]
- Gurzu, S.; Kobori, L.; Fodor, D.; Jung, I. Epithelial Mesenchymal and Endothelial Mesenchymal Transitions in Hepatocellular Carcinoma: A Review. Biomed. Res. Int. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Teeuwssen; Fodde Wnt Signaling in Ovarian Cancer Stemness, EMT, and Therapy Resistance. J. Clin. Med. 2019, 8, 1658. [CrossRef] [PubMed]
- Chiu, H.-C.; Li, C.-J.; Yiang, G.-T.; Tsai, A.; Wu, M.-Y. Epithelial to Mesenchymal Transition and Cell Biology of Molecular Regulation in Endometrial Carcinogenesis. J. Clin. Med. 2019, 8, 439. [Google Scholar] [CrossRef] [PubMed]
- van der Wal, T.; van Amerongen, R. Walking the tight wire between cell adhesion and WNT signalling: A balancing act for β -catenin. Open Biol. 2020, 10, 200267. [Google Scholar] [CrossRef]
- Park, H.B.; Kim, J.W.; Baek, K.H. Regulation of wnt signaling through ubiquitination and deubiquitination in cancers. Int. J. Mol. Sci. 2020, 21, 3904. [Google Scholar] [CrossRef]
- Kim, J.; Alavi Naini, F.; Sun, Y.; Ma, L. Ubiquitin-specific peptidase 2a (USP2a) deubiquitinates and stabilizes β-catenin. Am. J. Cancer Res. 2018, 8, 1823–1836. [Google Scholar]
- Rimawi, M.F.; Schiff, R.; Osborne, C.K. Targeting HER2 for the treatment of breast cancer. Annu. Rev. Med. 2015, 66, 111–128. [Google Scholar] [CrossRef]
- Castagnola, P.; Bellese, G.; Birocchi, F.; Gagliani, M.C.; Tacchetti, C.; Cortese, K. Identification of an HSP90 modulated multi-step process for ERBB2 degradation in breast cancer cells. Oncotarget 2016, 7, 85411–85429. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Liu, C.; Du, S.; Feng, L.; Luan, X.; Zhang, Y.; Shi, Y.; Wang, T.; Wu, Y.; et al. Neratinib induces ErbB2 ubiquitylation and endocytic degradation via HSP90 dissociation in breast cancer cells. Cancer Lett. 2016, 382, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, S.; Li, Q.; Shi, Y.; Wu, Y.; Liu, F.; Wang, S.; Zaky, M.Y.; Yousuf, W.; Sun, Q.; et al. The deubiquitylase USP2 maintains ErbB2 abundance via counteracting endocytic degradation and represents a therapeutic target in ErbB2-positive breast cancer. Cell Death Differ. 2020, 2710–2725. [Google Scholar] [CrossRef] [PubMed]
- Kataria, H.; Alizadeh, A.; Karimi-Abdolrezaee, S. Neuregulin-1/ErbB network: An emerging modulator of nervous system injury and repair. Prog. Neurobiol. 2019, 180, 101643. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Huang, C.; Chen, H.; Liu, K.; Xiang, P.; Yao, N.; Yang, L.; Zhou, L.; Wu, Q.; Zheng, Y.; et al. SDCBP/MDA-9/syntenin phosphorylation by AURKA promotes esophageal squamous cell carcinoma progression through the EGFR-PI3K-Akt signaling pathway. Oncogene 2020, 39, 5405–5419. [Google Scholar] [CrossRef]
- Kim, B.; Park, Y.S.; Kim, Y.H. Clathrin-mediated EGFR endocytosis as a potential therapeutic strategy for overcoming primary resistance of EGFR TKI in wild-type EGFR non-small cell lung cancer. Cancer Med. 2020, 1–14. [Google Scholar] [CrossRef]
- Liu, Z.; Zanata, S.M.; Kim, J.; Peterson, M.A.; Di Vizio, D.; Chirieac, L.R.; Pyne, S.; Agostini, M.; Freeman, M.R.; Loda, M. The ubiquitin-specific protease USP2a prevents endocytosis-mediated EGFR degradation. Oncogene 2013, 32, 1660–1669. [Google Scholar] [CrossRef]
- Duarte, C.; Akkaoui, J.; Yamada, C.; Ho, A.; Mao, C.; Movila, A. Elusive Roles of the Different Ceramidases in Human Health, Pathophysiology, and Tissue Regeneration. Cells 2020, 9, 1379. [Google Scholar] [CrossRef]
- Grbčić, P.; Sedić, M. Sphingosine 1-Phosphate Signaling and Metabolism in Chemoprevention and Chemoresistance in Colon Cancer. Molecules 2020, 25, 2436. [Google Scholar] [CrossRef]
- Govindarajah, N.; Clifford, R.; Bowden, D.; Sutton, P.A.; Parsons, J.L.; Vimalachandran, D. Sphingolipids and acid ceramidase as therapeutic targets in cancer therapy. Crit. Rev. Oncol. Hematol. 2019, 138, 104–111. [Google Scholar] [CrossRef]
- Mizutani, N.; Inoue, M.; Omori, Y.; Ito, H.; Tamiya-Koizumi, K.; Takagi, A.; Kojima, T.; Nakamura, M.; Iwaki, S.; Nakatochi, M.; et al. Increased acid ceramidase expression depends on upregulation of androgen-dependent deubiquitinases, USP2, in a human prostate cancer cell line, LNCaP. J. Biochem. 2015, 158, 309–319. [Google Scholar] [CrossRef]
- Benassi, B.; Flavin, R.; Marchionni, L.; Zanata, S.; Pan, Y.; Chowdhury, D.; Marani, M.; Strano, S.; Muti, P.; Blandino, G.; et al. MYC is activated by USP2a-mediated modulation of MicroRNAs in prostate cancer. Cancer Discov. 2012, 2, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Benassi, B.; Marani, M.; Loda, M.; Blandino, G. USP2a alters chemotherapeutic response by modulating redox. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef]
- Li, M.; Sun, Q.; Wang, X. Transcriptional landscape of human cancers. Oncotarget 2017, 8, 34534–34551. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Xiong, Z.; Xiao, W.; Yuan, C.; Wang, C.; Huang, Y.; Tong, J.; Shi, J.; Chen, Z.; Liu, C.; et al. Downregulation of ubiquitin-specific protease 2 possesses prognostic and diagnostic value and promotes the clear cell renal cell carcinoma progression. Ann. Transl. Med. 2020, 8, 319. [Google Scholar] [CrossRef]
- Priolo, C.; Tang, D.; Brahamandan, M.; Benassi, B.; Sicinska, E.; Ogino, S.; Farsetti, A.; Porrello, A.; Finn, S.; Zimmermann, J.; et al. The isopeptidase USP2a protects human prostate cancer from apoptosis. Cancer Res. 2006, 66, 8625–8632. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Pazarentzos, E.; Datler, C.; Iwasawa, R.; Abuali, G.; Lin, B.; Grimm, S. De-ubiquitinating protease USP2a targets RIP1 and TRAF2 to mediate cell death by TNF. Cell Death Differ. 2012, 19, 891–899. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Datler, C.; Pazarentzos, E.; Lin, B.; Chaisaklert, W.; Abuali, G.; Grimm, S. De-ubiquitinating proteases USP2a and USP2c cause apoptosis by stabilising RIP1. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1353–1365. [Google Scholar] [CrossRef]
- da Fonseca, L.G.; Reig, M.; Bruix, J. Tyrosine Kinase Inhibitors and Hepatocellular Carcinoma. Clin. Liver Dis. 2020, 24, 719–737. [Google Scholar] [CrossRef]
- Cabral, L.K.D.; Tiribelli, C.; Sukowati, C.H.C. Sorafenib resistance in hepatocellular carcinoma: The relevance of genetic heterogeneity. Cancers (Basel) 2020, 12, 1576. [Google Scholar] [CrossRef]
- Liu, D.; Fan, Y.; Li, J.; Cheng, B.; Lin, W.; Li, X.; Du, J.; Ling, C. Inhibition of cFLIP overcomes acquired resistance to sorafenib via reducing ER stress-related autophagy in hepatocellular carcinoma. Oncol. Rep. 2018, 40, 2206–2214. [Google Scholar] [CrossRef] [PubMed]
- Molusky, M.M.; Ma, D.; Buelow, K.; Yin, L.; Lin, J.D. Peroxisomal Localization and Circadian Regulation of Ubiquitin-Specific Protease 2. PLoS ONE 2012, 7, e47970. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Huang, L.; Zhao, J.; Chen, S.; Liu, J.; Li, G. The circadian clock and inflammation: A new insight. Clin. Chim. Acta 2021, 512, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Dierickx, P.; Van Laake, L.W.; Geijsen, N. Circadian clocks: From stem cells to tissue homeostasis and regeneration. EMBO Rep. 2018, 19, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Lahens, N.F.; Ballance, H.I.; Hughes, M.E.; Hogenesch, J.B. A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. USA 2014, 111, 16219–16224. [Google Scholar] [CrossRef]
- Yang, Y.; Duguay, D.; Bédard, N.; Rachalski, A.; Baquiran, G.; Na, C.H.; Fahrenkrug, J.; Storch, K.F.; Peng, J.; Wing, S.S.; et al. Regulation of behavioral circadian rhythms and clock protein PER1 by the deubiquitinating enzyme USP2. Biol. Open 2012, 1, 789–801. [Google Scholar] [CrossRef]
- Scoma, H.D.; Humby, M.; Yadav, G.; Zhang, Q.; Fogerty, J.; Besharse, J.C. The de-ubiquitinylating enzyme, USP2, is associated with the circadian clockwork and regulates its sensitivity to light. PLoS ONE 2011, 6, e25382. [Google Scholar] [CrossRef]
- Lee, J.; Lee, Y.; Lee, M.J.; Park, E.; Kang, S.H.; Chung, C.H.; Lee, K.H.; Kim, K. Dual Modification of BMAL1 by SUMO2/3 and Ubiquitin Promotes Circadian Activation of the CLOCK/BMAL1 Complex. Mol. Cell. Biol. 2008, 28, 6056–6065. [Google Scholar] [CrossRef]
- Yang, Y.; Duguay, D.; Fahrenkrug, J.; Cermakian, N.; Wing, S.S. USP2 regulates the intracellular localization of PER1 and circadian gene expression. J. Biol. Rhythm. 2014, 29, 243–256. [Google Scholar] [CrossRef]
- Pouly, D.; Chenaux, S.; Martin, V.; Babis, M.; Koch, R.; Nagoshi, E.; Katanaev, V.L.; Gachon, F.; Staub, O. USP2-45 is a circadian clock output effector regulating calcium absorption at the post- Translational level. PLoS ONE 2016, 11, e0145155. [Google Scholar] [CrossRef]
- Fecher-Trost, C.; Wissenbach, U.; Weissgerber, P. TRPV6: From identification to function. Cell Calcium 2017, 67, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Manning, J.A.; Kumar, S. Physiological Functions of Nedd4-2: Lessons from Knockout Mouse Models. Trends Biochem. Sci. 2018, 43, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Fakitsas, P.; Adam, G.; Daidié, D.; Van Bemmelen, M.X.; Fouladkou, F.; Patrignani, A.; Wagner, U.; Warth, R.; Camargo, S.M.R.; Staub, O.; et al. Early aldosterone-induced gene product regulates the epithelial sodium channel by deubiquitylation. J. Am. Soc. Nephrol. 2007, 18, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Oberfeld, B.; Ruffieux-Daidié, D.; Vitagliano, J.J.; Pos, K.M.; Verrey, F.; Staub, O. Ubiquitin-specific protease 2-45 (Usp2-45) binds to epithelial Na+ channel (ENaC)-ubiquitylating enzyme Nedd4-2. Am. J. Physiol. Ren. Physiol. 2011, 301, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Ruffieux-Daidié, D.; Poirot, O.; Boulkroun, S.; Verrey, F.; Kellenberger, S.; Staub, O. Deubiquitylation regulates activation and proteolytic cleavage of ENaC. J. Am. Soc. Nephrol. 2008, 19, 2170–2180. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tomkovicz, V.R.; Butler, P.L.; Ochoa, L.A.; Peterson, Z.J.; Snyder, P.M. Ubiquitin-specific peptidase 8 (USP8) regulates endosomal trafficking of the epithelial Na+ channel. J. Biol. Chem. 2013, 288, 5389–5397. [Google Scholar] [CrossRef]
- Ruffieux-Daidié, D.; Staub, O. Intracellular ubiquitylation of the epithelial Na+ channel controls extracellular proteolytic channel activation via conformational change. J. Biol. Chem. 2011, 286, 2416–2424. [Google Scholar] [CrossRef]
- Cannavo, A.; Bencivenga, L.; Liccardo, D.; Elia, A.; Marzano, F.; Gambino, G.; D’Amico, M.L.; Perna, C.; Ferrara, N.; Rengo, G.; et al. Aldosterone and mineralocorticoid receptor system in cardiovascular physiology and pathophysiology. Oxid. Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef]
- Faresse, N.; Vitagliano, J.J.; Staub, O. Differential ubiquitylation of the mineralocorticoid receptor is regulated by phosphorylation. FASEB J. 2012, 26, 4373–4382. [Google Scholar] [CrossRef]
- Faresse, N.; Debonneville, A.; Staub, O. USP2-45 represses aldosterone mediated responses by decreasing mineralocorticoid receptor availability. Cell. Physiol. Biochem. 2013, 31, 462–472. [Google Scholar] [CrossRef]
- Jin, H.S.; Hong, K.W.; Lim, J.E.; Hwang, S.Y.; Lee, S.H.; Shin, C.; Park, H.K.; Oh, B. Genetic variations in the sodium balance-regulating genes ENaC, NEDD4L, NDFIP2 and USP2 influence blood pressure and hypertension. Kidney Blood Press. Res. 2010, 33, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Pouly, D.; Debonneville, A.; Ruffieux-Daidié, D.; Maillard, M.; Abriel, H.; Loffing, J.; Staub, O. Mice carrying ubiquitin-specific protease 2 (Usp2) gene inactivation maintain normal sodium balance and blood pressure. Am. J. Physiol. Ren. Physiol. 2013, 305, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Barthel, A.; Schmoll, D. Novel concepts in insulin regulation of hepatic gluconeogenesis. Am. J. Physiol. Endocrinol. Metab. 2003, 285. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.J.; Suzuki, S.; Segars, J.H.; Kino, T. CRTC2 is a coactivator of GR and couples GR and CREB in the regulation of hepatic gluconeogenesis. Mol. Endocrinol. 2016, 30, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, T.; Ohta, Y.; Taguchi, A.; Hiroshige, S.; Kajimura, Y.; Fukuda, N.; Yamamoto, K.; Nakabayashi, H.; Fujimoto, R.; Yanai, A.; et al. Liver-specific dysregulation of clock-controlled output signal impairs energy metabolism in liver and muscle. Biochem. Biophys. Res. Commun. 2020. [Google Scholar] [CrossRef]
- Piccinin, E.; Villani, G.; Moschetta, A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: The role of PGC1 coactivators. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 160–174. [Google Scholar] [CrossRef]
- Hirano, T. Pathophysiology of Diabetic Dyslipidemia. J. Atheroscler. Thromb. 2018, 25, 771–782. [Google Scholar] [CrossRef]
- McCormick, S.P.A.; Schneider, W.J. Lipoprotein(a) catabolism: A case of multiple receptors. Pathology 2019, 51, 155–164. [Google Scholar] [CrossRef]
- Yang, H.X.; Zhang, M.; Long, S.Y.; Tuo, Q.H.; Tian, Y.; Chen, J.X.; Zhang, C.P.; Liao, D.F. Cholesterol in LDL receptor recycling and degradation. Clin. Chim. Acta 2020, 500, 81–86. [Google Scholar] [CrossRef]
- Nelson, J.K.; Sorrentino, V.; Trezza, R.A.; Heride, C.; Urbe, S.; Distel, B.; Zelcer, N. The deubiquitylase USP2 regulates the ldlr pathway by counteracting the E3-Ubiquitin Ligase IDOL. Circ. Res. 2016, 118, 410–419. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Orliaguet, L.; Dalmas, E.; Drareni, K.; Venteclef, N.; Alzaid, F. Mechanisms of Macrophage Polarization in Insulin Signaling and Sensitivity. Front. Endocrinol. (Lausanne) 2020, 11, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yun, K.; Mu, R. A review on the biology and properties of adipose tissue macrophages involved in adipose tissue physiological and pathophysiological processes. Lipids Health Dis. 2020, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kaji, H. Adipose tissue-derived plasminogen activator inhibitor-1 function and regulation. Compr. Physiol. 2016, 6, 1873–1896. [Google Scholar] [CrossRef] [PubMed]
- Virdis, A.; Colucci, R.; Bernardini, N.; Blandizzi, C.; Taddei, S.; Masi, S. Microvascular Endothelial Dysfunction in Human Obesity: Role of TNF- α. J. Clin. Endocrinol. Metab. 2018, 104, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K. Anti-inflammatory effects of exercise: Role in diabetes and cardiovascular disease. Eur. J. Clin. Invest. 2017, 47, 600–611. [Google Scholar] [CrossRef]
- Saito, N.; Kimura, S.; Miyamoto, T.; Fukushima, S.; Amagasa, M.; Shimamoto, Y.; Nishioka, C.; Okamoto, S.; Toda, C.; Washio, K.; et al. Macrophage ubiquitin-specific protease 2 modifies insulin sensitivity in obese mice. Biochem. Biophys. Rep. 2017. [Google Scholar] [CrossRef]
- Lee, M.K.; Kim, T.S. Histone H4-Specific Deacetylation at Active Coding Regions by Hda1C. Mol. Cells 2020, 43, 841–847. [Google Scholar] [CrossRef]
- Hughes, A.L.; Kelley, J.R.; Klose, R.J. Understanding the interplay between CpG island-associated gene promoters and H3K4 methylation. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194567. [Google Scholar] [CrossRef]
- Li, C.; Zhang, J.; Xu, H.; Chang, M.; Lv, C.; Xue, W.; Song, Z.; Zhang, L.; Zhang, X.; Tian, X. Retigabine ameliorates acute stress-induced impairment of spatial memory retrieval through regulating USP2 signaling pathways in hippocampal CA1 area. Neuropharmacology 2018, 135, 151–162. [Google Scholar] [CrossRef]
- Mastaitis, J.W.; Wurmbach, E.; Cheng, H.; Sealfon, S.C.; Mobbs, C. V Acute Induction of Gene Expression in Brain and Liver by Insulin-Induced Hypoglycemia. Diabetes 2005, 54, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Kapogiannis, D.; Avgerinos, K.I. Brain Glucose and Ketone Utilization in Brain Aging and Neurodegenerative Diseases, 1st ed.; Academic Press: San Diego, CA, USA; Volume 154, 2020; ISBN 9780128200766. [Google Scholar]
- Shi, M.M.; Fan, K.M.; Qiao, Y.N.; Xu, J.H.; Qiu, L.J.; Li, X.; Liu, Y.; Qian, Z.Q.; Wei, C.L.; Han, J.; et al. Hippocampal µ-opioid receptors on GABAergic neurons mediate stress-induced impairment of memory retrieval. Mol. Psychiatry 2020, 25, 977–992. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.S.; Zhong, G.; Cao, H.X.; Hu, Y.; Hong, X.Y.; Li, T.; Li, X.; Liu, Q.; Wang, Q.; Ke, D.; et al. Repeated Restraint Stress Led to Cognitive Dysfunction by NMDA Receptor-Mediated Hippocampal CA3 Dendritic Spine Impairments in Juvenile Sprague-Dawley Rats. Front. Mol. Neurosci. 2020, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bousman, C.A.; Chana, G.; Glatt, S.J.; Chandler, S.D.; May, T.; Lohr, J.; Kremen, W.S.; Tsuang, M.T.; Everall, I.P. Positive symptoms of psychosis correlate with expression of ubiquitin proteasome genes in peripheral blood. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Anazi, S.; Maddirevula, S.; Faqeih, E.; Alsedairy, H.; Alzahrani, F.; Shamseldin, H.E.; Patel, N.; Hashem, M.; Ibrahim, N.; Abdulwahab, F.; et al. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol. Psychiatry 2017, 22, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Srikanta, S.B.; Stojkovic, K.; Cermakian, N. Behavioral phenotyping of mice lacking the deubiquitinase USP2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Park, K.C.; Kim, J.H.; Choi, E.J.; Min, S.W.; Rhee, S.; Baek, S.H.; Chung, S.S.; Bang, O.; Park, D.; Chiba, T.; et al. Antagonistic regulation of myogenesis by two deubiquitinating enzymes, UBP45 and UBP69. Proc. Natl. Acad. Sci. USA 2002, 99, 9733–9738. [Google Scholar] [CrossRef]
- Bedard, N.; Yang, Y.; Gregory, M.; Cyr, D.G.; Suzuki, J.; Yu, X.; Chian, R.-C.; Hermo, L.; O’Flaherty, C.; Smith, C.E.; et al. Mice lacking the USP2 deubiquitinating enzyme have severe male subfertility associated with defects in fertilization and sperm motility. Biol. Reprod. 2011, 85, 594–604. [Google Scholar] [CrossRef]
- Wing, S.S. Deubiquitinating enzymes in skeletal muscle atrophy—An essential role for USP19. Int. J. Biochem. Cell Biol. 2016, 79, 462–468. [Google Scholar] [CrossRef]
- Gonçalves, T.M.; De Almeida Regitano, L.C.; Koltes, J.E.; Cesar, A.S.M.; Da Silva Andrade, S.C.; Mourão, G.B.; Gasparin, G.; Moreira, G.C.M.; Fritz-Waters, E.; Reecy, J.M.; et al. Gene co-expression analysis indicates potential pathways and regulators of beef tenderness in Nellore cattle. Front. Genet. 2018, 9, 1–18. [Google Scholar] [CrossRef]
- Hashimoto, M.; Saito, N.; Ohta, H.; Yamamoto, K.; Tashiro, A.; Nakazawa, K.; Inanami, O.; Kitamura, H. Inhibition of ubiquitin-specific protease 2 causes accumulation of reactive oxygen species, mitochondria dysfunction, and intracellular ATP decrement in C2C12 myoblasts. Physiol. Rep. 2019, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Li, P.; Hong, J.; Wang, M.; Liu, Y.; Gao, Y.; Dong, J.; Gu, H.; Li, L. Overexpression of Ubiquitin-Specific Protease 2 (USP2) in the Heart Suppressed Pressure Overload-Induced Cardiac Remodeling. Mediat. Inflamm. 2020, 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shuja, Z.; Colecraft, H.M. Regulation of microdomain voltage-gated L-type calcium channels in cardiac health and disease. Curr. Opin. Physiol. 2018, 2, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Rougier, J.-S.; Albesa, M.; Syam, N.; Halet, G.; Abriel, H.; Viard, P. Ubiquitin-specific protease USP2-45 acts as a molecular switch to promote α2δ-1-induced downregulation of Ca v1.2 channels. Pflug. Arch. 2015, 467, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Totzke, J.; Scarneo, S.A.; Yang, K.W.; Haystead, T.A.J. TAK1: A potent tumour necrosis factor inhibitor for the treatment of inflammatory diseases. Open Biol. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Fechtner, S.; Fox, D.A.; Ahmed, S. Transforming growth factor β activated kinase 1: A potential therapeutic target for rheumatic diseases. Rheumatol (United Kingdom) 2017, 56, 1060–1068. [Google Scholar] [CrossRef][Green Version]
- Jarosz-Griffiths, H.H.; Holbrook, J.; Lara-Reyna, S.; McDermott, M.F. TNF receptor signalling in autoinflammatory diseases. Int. Immunol. 2019, 31, 639–648. [Google Scholar] [CrossRef]
- Metzig, M.; Nickles, D.; Falschlehner, C.; Lehmann-Koch, J.; Straub, B.K.; Roth, W.; Boutros, M. An RNAi screen identifies USP2 as a factor required for TNF-α-induced NF-κB signaling. Int. J. Cancer 2011, 129, 607–618. [Google Scholar] [CrossRef]
- Zhang, C. Flare-up of cytokines in rheumatoid arthritis and their role in triggering depression: Shared common function and their possible applications in treatment (Review). Biomed. Rep. 2020, 14. [Google Scholar] [CrossRef]
- Dey, M.; Zhao, S.S.; Moots, R.J. Anti-TNF biosimilars in rheumatology: The end of an era? Expert Opin. Biol. Ther. 2020. [Google Scholar] [CrossRef]
- Akhtar, N.; Singh, A.K.; Ahmed, S. MicroRNA-17 Suppresses TNF-α Signaling by Interfering with TRAF2 and cIAP2 Association in Rheumatoid Arthritis Synovial Fibroblasts. J. Immunol. 2016, 197, 2219–2228. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Qin, Z.; Li, Q.; Wan, J.J.; Cheng, M.H.; Wang, P.Y.; Su, D.F.; Yu, J.G.; Liu, X. MicroRNA-124 negatively regulates LPS-induced TNF-α production in mouse macrophages by decreasing protein stability. Acta Pharmacol. Sin. 2016, 37, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Lork, M.; Staal, J.; Beyaert, R. Ubiquitination and phosphorylation of the CARD11-BCL10-MALT1 signalosome in T cells. Cell. Immunol. 2019, 340, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, X.; Wang, S.; Shu, H.B.; Liu, Y. USP2a positively regulates TCR-induced NF-κB activation by bridging MALT1-TRAF6. Protein Cell 2013, 4, 62–70. [Google Scholar] [CrossRef]
- He, X.; Li, Y.; Li, C.; Liu, L.-J.; Zhang, X.; Liu, Y.; Shu, H. USP2a negatively regulates IL-1β- and virus-induced NF-κB activation by deubiquitinating TRAF6. J. Mol. Cell Biol. 2013, 5, 39–47. [Google Scholar] [CrossRef]
- Ruiz-Lafuente, N.; Muro, M.; Minguela, A.; Parrado, A. The transcriptional response of mouse spleen B cells to IL-4: Comparison to the response of human peripheral blood B cells. Biochem. Biophys. Rep. 2018, 16, 56–61. [Google Scholar] [CrossRef]
- Nie, L.; Cai, S.-Y.; Shao, J.-Z.; Chen, J. Toll-Like Receptors, Associated Biological Roles, and Signaling Networks in Non-Mammals. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Tanji, T.; Ip, Y.T. Regulators of the Toll and Imd pathways in the Drosophila innate immune response. Trends Immunol. 2005, 26, 193–198. [Google Scholar] [CrossRef]
- Engel, E.; Viargues, P.; Mortier, M.; Taillebourg, E.; Couté, Y.; Thevenon, D.; Fauvarque, M.O. Identifying USPs regulating immune signals in Drosophila: USP2 deubiquitinates Imd and promotes its degradation by interacting with the proteasome. Cell Commun. Signal. 2014, 12, 1–14. [Google Scholar] [CrossRef]
- Walter, M.R. The Role of Structure in the Biology of Interferon Signaling. Front. Immunol. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Johnson, H.M.; Noon-Song, E.; Ahmed, C.M. Noncanonical IFN signaling, steroids, and stats: A probable role of V-ATPase. Mediat. Inflamm. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.M.; Ahmed, C.M. Noncanonical IFN Signaling: Mechanistic Linkage of Genetic and Epigenetic Events. Mediat. Inflamm. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhao, P.; Liu, J.; Yuan, Y.; Cheng, Q.; Zuo, Y.; Qian, L.; Liu, C.; Guo, T.; Zhang, L.; et al. Deubiquitinase USP2a Sustains Interferons Antiviral Activity by Restricting Ubiquitination of Activated STAT1 in the Nucleus. PLoS Pathog. 2016, 12, e1005764. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, X.; Zhang, M.; Zhao, W.; Gao, C. Ubiquitin-Specific Protease 2b Negatively Regulates IFN-β Production and Antiviral Activity by Targeting TANK-Binding Kinase 1. J. Immunol. 2014, 193, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, H.; Liu, Y.; Sun, J.; Zhao, Z.; Chen, Q.; Guo, M.; Ma, D.; Zhang, Z. Expression of USP2-69 in mesangial cells in vivo and in vitro. Pathol. Int. 2010, 60, 184–192. [Google Scholar] [CrossRef]
- Hashimoto, M.; Kimura, S.; Kanno, C.; Yanagawa, Y.; Watanabe, T.; Okabe, J.; Nagano, M. Macrophage ubiquitin-specific protease 2 contributes to motility, hyperactivation, capacitation, and in vitro fertilization activity of mouse sperm. Cell. Mol. Life Sci. 2020. [Google Scholar] [CrossRef]
- Abualsaud, D.; Hashem, M.; AlHashem, A.; Alkuraya, F.S. Survey of disorders of sex development in a large cohort of patients with diverse Mendelian phenotypes. Am. J. Med. Genet. Part A 2020, 1–12. [Google Scholar] [CrossRef]
- Ito, M.; Kitamura, H.; Kikuguchi, C.; Hase, K.; Ohno, H.; Ohara, O. SP600125 Inhibits Cap-dependent Translation Independently of the c-Jun N-terminal Kinase Pathway. Cell Struct. Funct. 2011, 36, 27–33. [Google Scholar] [CrossRef][Green Version]
- Mirza, M.U.; Ahmad, S.; Abdullah, I.; Froeyen, M.; Xing, J.; Li, P.; Hong, J.; Wang, M.; Liu, Y.; Gao, Y.; et al. Identification of novel human USP2 inhibitor and its putative role in treatment of COVID-19 by inhibiting SARS-CoV-2 papain-like (PLpro) protease. Comput. Biol. Chem. 2020, 89, 1–12. [Google Scholar] [CrossRef]
- Krzystanek, K.; Rasmussen, H.B.; Grunnet, M.; Staub, O.; Olesen, S.P.; Abriel, H.; Jespersen, T. Deubiquitylating enzyme USP2 counteracts Nedd4-2mediated downregulation of KCNQ1 potassium channels. Hear. Rhythm. 2012, 9, 440–448. [Google Scholar] [CrossRef]
- Alonso, V.; Magyar, C.E.; Wang, B.; Bisello, A.; Friedman, P.A. Ubiquitination-deubiquitination balance dictates ligand-stimulated PTHR sorting. J. Bone Miner. Res. 2011, 26, 2923–2934. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.H.; Yang, S.W.; Park, J.M.; Seol, J.H.; Iemura, S.; Natsume, T.; Murata, S.; Tanaka, K.; Jeon, Y.J.; Chung, C.H. Control of AIF-mediated cell death by antagonistic functions of CHIP ubiquitin E3 ligase and USP2 deubiquitinating enzyme. Cell Death Differ. 2011, 18, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Reyes-Turcu, F.; Licchesi, J.D.F.; Odenwaelder, P.; Wilkinson, K.D.; Barford, D. Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep. 2009, 10, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H. Recent Advances and Contradictions in the Study of the Individual Roles of Ubiquitin Ligases That Regulate RIG-I-Like Receptor-Mediated Antiviral Innate Immune Responses. Front. Immunol. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Wang, Y.S.; Wu, K.P.; Jiang, H.K.; Kurkute, P.; Chen, R.H. Branched Ubiquitination: Detection Methods, Biological Functions and Chemical Synthesis. Molecules 2020, 25, 5200. [Google Scholar] [CrossRef]
- McDowell, G.S.; Philpott, A. Non-canonical ubiquitylation: Mechanisms and consequences. Int. J. Biochem. Cell Biol. 2013, 45, 1833–1842. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Vere, G.; Kealy, R.; Kessler, B.M.; Pinto-Fernandez, A. Ubiquitomics: An overview and future. Biomolecules 2020, 10, 1453. [Google Scholar] [CrossRef]
- Back, S.; Gorman, A.W.; Vogel, C.; Silva, G.M. Site-Specific K63 Ubiquitinomics Provides Insights into Translation Regulation under Stress. J. Proteome Res. 2018, 18. [Google Scholar] [CrossRef]
- Lu, M.; Chen, W.; Zhuang, W.; Zhan, X. Label-free quantitative identification of abnormally ubiquitinated proteins as useful biomarkers for human lung squamous cell carcinomas. EPMA J. 2020, 11, 73–94. [Google Scholar] [CrossRef] [PubMed]
- Issaenko, O.A.; Amerik, A.Y. Chalcone-based small-molecule inhibitors attenuate malignant phenotype via targeting deubiquitinating enzymes. Cell Cycle 2012, 11, 1804–1817. [Google Scholar] [CrossRef] [PubMed]
- Shimrit, O.; Maya, R.; Adi, H.; Amir, A.; Ashraf, B. Harnessing the oxidation susceptibility of deubiquitinases for inhibition with small molecules. Angew. Chem. Int. Ed. 2015, 54, 599–603. [Google Scholar] [CrossRef]
- Chuang, S.J.; Cheng, S.C.; Tang, H.C.; Sun, C.Y.; Chou, C.Y. 6-Thioguanine is a noncompetitive and slow binding inhibitor of human deubiquitinating protease USP2. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Chojnacki, M.; Zhang, D.; Talarowska, M.; Gałecki, P.; Szemraj, J.; Fushman, D.; Nakasone, M.A. Characterizing polyubiquitinated forms of the neurodegenerative ubiquitin mutant UBB+1. FEBS Lett. 2016, 590, 4573–4585. [Google Scholar] [CrossRef]
- Hospenthal, M.K.; Mevissen, T.E.T.; Komander, D. Deubiquitinase–based analysis of ubiquitin chain architecture using Ubiquitin Chain Restriction ( UbiCRest ). Nat. Protoc. 2016, 10, 349–361. [Google Scholar] [CrossRef]
- Kitamura, H.; Ito, M.; Yuasa, T.; Kikuguchi, C.; Hijikata, A.; Takayama, M.; Kimura, Y.; Yokoyama, R.; Kaji, T.; Ohara, O. Genome-wide identification and characterization of transcripts translationally regulated by bacterial lipopolysaccharide in macrophage-like J774.1 cells. Physiol. Genom. 2008, 33, 121–132. [Google Scholar] [CrossRef]
- Faas, M.M.; de Vos, P. Mitochondrial function in immune cells in health and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165845. [Google Scholar] [CrossRef]
- Ayres, J.S. Immunometabolism of infections. Nat. Rev. Immunol. 2020, 20, 79–80. [Google Scholar] [CrossRef]
- Qian, X.; Yang, Z.; Mao, E.; Chen, E. Regulation of fatty acid synthesis in immune cells. Scand. J. Immunol. 2018, 88. [Google Scholar] [CrossRef]
- Sadiku, P.; Willson, J.A.; Ryan, E.M.; Sammut, D.; Coelho, P.; Watts, E.R.; Grecian, R.; Young, J.M.; Bewley, M.; Arienti, S.; et al. Neutrophils Fuel Effective Immune Responses through Gluconeogenesis and Glycogenesis. Cell Metab. 2020, 1–13. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| USP2 Species | USP2 Isoform | USP2-Affected Cell/Tissue/Animal | Experimental Manipulation | Direct Target | Function of USP2 | Reference |
|---|---|---|---|---|---|---|
| Human | USP2-1 | Cell line (Bladder cancer) | OE, KD | Cyclin A1 | Proliferation (+), Invasion (+), Migration (+), Chemoresistance (+) | [24] |
| Human | USP2-1 | Cell line (Breast cancer) | KD, Blocker | TWIST | EMT (+), Proliferation (+), Chemoresistance (+), Self-renewal (+) | [25] |
| Human | USP2-1 | Cell line (Prostate cancer, Hepatoma) | OE, KD | FASN | Apoptosis (-), Lipogenesis (+) | [18,19] |
| Human | USP2-1 | Cell line (Osteosarcoma, Lung carcinoma Embryonal carcinoma T-cell lymphoma) | OE, KD | MDM2 | p53 signaling (-), Apoptosis (-), Chemo resistance (+) | [31,32] |
| Human | USP2-1 | Cell line (Lung carcinoma Embryonal carcinoma) | OE, KD | MDMX | Chemo resistance (+) | [34] |
| Human | USP2-1 | Cell line (Glioblastoma) | OE, KD | MDM4 | p53 signaling (-), Apoptosis (-) | [35] |
| Human | USP2 | Cell line (Hepatoma, Breast cancer) | KD | - | p53 expression (+) | [36] |
| Human | USP2-1 | Cell line (Osteosarcoma, Kidney cells, Colorectal carcinoma, Breast cancer, Prostate cancer) | OE, KD | Cyclin D1 | Cell cycle (+) | [39] |
| Human | USP2 | Cell line (Hepatoma, Breast cancer) | OE, KD | Cyclin D1 | Cell cycle (+) | [41] |
| Human | USP2-1 | Cell line (Pancreatic carcinoma, Colorectal carcinoma, Kidney carcinoma) | OE, KD | Aurora-A | Proliferation (+), Mitosis (+) | [46] |
| Human Mouse | USP2-1 | Mouse, Cell line (Cervix epithelioid carcinoma, Hepatoma, Colorectal carcinoma, Kidney cells), | OE, KD, KOBlocker | TGF-β receptor | TGF-β signaling (+), Metastasis (+), Tumor growth (+), Mortality (-) | [52] |
| Human | USP2-1 | Cell line (Kidney cell) | OE, KD, Blocker | β-catenin | Wnt/β-catenin signaling (+) | [58] |
| Human | USP2 | Cell line (Breast cancer), Mouse | OE, KD Blocker | ErbB2 | Tumor growth (+), Proliferation (+), Cell cycle (+) | [62] |
| Human | USP2-1 | Cell line (Prostate cancer) | OE, KD | ACDase | Proliferation (+)? | [70] |
| Human | USP2 | Cell line (Kidney carcinoma) | OE | Unidentified | Proliferation (-), Migration (-), Invasion (-) | [74] |
| Human | USP2-2 | Cell line (Cervix epithelioid carcinoma, Kidney cells) | OE | MDM2? | Apoptosis (+) | [7] |
| Human | USP2-1 | Cell line (Prostate epithelial cells, Prostate cancer, Colon cancer, Breast cancer, Sarcoma, Fibroblasts) Mouse | OE, KD | Unidentified | Apoptosis (-), Cell growth (+), p53 signaling(-) | [75] |
| Human | USP2-1 | Cell line (Hepatoma) | OE | ITCH? | Apoptosis (+) | [81] |
| Mouse | USP2-2 | Tissue (Liver), Hepatocyte | OE, KD | ITCH | Apoptosis (+), TNF-resistance (-) | [13] |
| Human | USP2-1 | Cell line (Cervix epithelioid carcinoma, Breast cancer, Kidney cell) | OE, KD | TRAF2, RIP1 | Apoptosis (+) or (-), TNF-signaling (+), NF-κB activation (-), p38 signaling (-), JNK signaling (-) | [77,78] |
| Human | USP2-2 | Cell line (Breast cancer) | OE, KD | TRAF2, RIP1 | Apoptosis (+) | [78] |
| Human | USP2-1 USP2-2 USP2-3 USP2-4 | Cell line (Kidney cell), | OE | Unidentified | Apoptosis (+) | [6] |
| Mouse | USP2-1 USP2-4 | Mouse, Tissue (Liver) Fibroblast, Cell line (Kidney cell) | OE, KO | PER1 | Control of circadian period, Circadian gene expression (+) | [86] |
| Mouse | USP2-4 | Mouse, Tissue (SCN), Cell line (Kidney cell) Fibroblast | OE, KO | BMAL1 | Control of circadian period, Circadian gene expression (+) | [87,88], |
| Mouse | USP2-4 | Mouse, Fruit fly, Cell line (Kidney cell) | OE, KD, KO | NHERF4, Clathrin heavy chain | Calcium absorption (+), Sodium balance (n.i.) | [90,102] |
| Mouse | USP2-4 | Cell line (Kidney cell) | OE | Cav1.2, α2δ-1 subunit | Calcium uptake (-), Surface calcium channel (-) | [135] |
| Mouse | USP2-4 | Tissue (Oocyte), Cell line (Kidney cell) | OE | ENaC, Nedd4-2 | Sodium uptake (+) Surface ENaC expression (+) ENaC activation (+) | [93,94,95] |
| No information | USP2-4 | Cell line (Kidney cell) | OE | Unidentified | Surface ENaC expression (+) | [96] |
| No information | USP2-4 | Cell line (Kidney cell) | OE, KD | MR | Aldosterone signaling (-) | [100] |
| Mouse | USP2-4 | Tissue (Liver), Cell line (Kidney cell) | OE, KD | C/EBPα | Gluconeogenesis (+) Glucose sensitivity (-) Insulin signaling (-) Glucocorticoid signaling (+) | [11] |
| Human | USP2-1 USP2-4 | Fibroblast, Cell line (Kidney cell, Hepatoma, Epidermoid carcinoma, Cervix epithelioid carcinoma) | OE, KD | IDOL | LDL uptake (+) | [110] |
| Human, Mouse | USP2-1 | Mouse, Tissue (Adipose tissue, Liver, Skeletal muscle), Cell line (Macrophage, Myocyte, Adipocyte) | OE, KD | Unidentified | Inflammation (-), Cytokine production (-), Insulin signaling (+) Adipocity (-) Histone modification (-) | [9,117] |
| Mouse | USP2 | Tissue (Hippocampus) | Down-regulation by stress | mTOR?, AMPA receptor? | Autophagy (-)?, Spatial memory (+)? | [120] |
| Human | USP2-1 | Human | Mutation | Unidentified | Development (+), Seizure (-), Muscle strength (+), Reproduction (+) | [126] |
| Mouse | USP2 | Mouse | KO | Unidentified | Locomotion (+) or (n.i.) Motor coordination (+) Recognition (+) Sensory response (+) Anxiety (+) | [86,87,127] |
| Rat | USP2-1 USP2-4 | Cell line (Myoblast) | OE, DN | Unidentified | Differentiation (+, USP2-1; -, USP2-4) | [128] |
| Mouse | USP2 | Cell line (Myoblast) | KO, Blocker | UCP2? | Oxidative stress (-) ATP production (+) Proliferation (+) Differentiation (+) | [132] |
| No information | USP2 | Tissue (Heart) | OE | Unidentified | Cardiac function (+), Fibrosis (-), Inflammation (-), Cytokine production (-), Oxidative stress (-), Akt signaling (+), NF-κB signaling (+), ERK signaling (+) | [133] |
| Human | USP2 | Cell line (Kidney cell, Hepatoma, Cervix epithelioid carcinoma) | KD | Unidentified | NF-κB signaling (+), Cytokine production (+) | [139] |
| Mouse | USP2 | Cell line (Macrophage) | KD | TNF-α? | Cytokine production (+) | [143] |
| Human | USP2-1 | Cell line (Kidney cell, T cell) | OE, KD | MALT1, CARD11, TRAF6 | TCR signaling (+), NF-κB signaling (+), Cytokine production (+) | [145] |
| Human, Mouse | USP2-1, USP2-4 | Macrophage, Cell line (Macrophage) | OE, KD | OCT1 | OCT1/2 signaling (-) Cytokine production (-) | [10] |
| Human | USP2-1 | Cell line (Colorectal carcinoma, Kidney cell) | OE, KD, KO | TRAF6 | NF-κB signaling (-), Cytokine production (-) | [146] |
| Fruit fly | USP2 | Fruit fly, Cell line (Macrophage) | OE, KD | Imd | Antimicrobial activity (-), NF-κB signaling (-) | [150] |
| Human | USP2-1 | Vascular endothelial cell, Cell line (Kidney cell, Fibrosarcoma, Lung carcinoma, Hepatoma, Epithelial carcinoma, Sarcoma) | OE, KD | STAT1 | Antiviral activity (+), IFN signaling (+) | [154] |
| Human | USP2-4 | Cell line (Kidney cell) | OE, KD | TBK1 | Antiviral activity (-), TRIF signaling (-), STING signaling (-), IFNβ signaling (-), Cytokine production (-) | [155] |
| No information | USP2-1 | Tissue (Kidney), Mesangial cell | OE | Unidentified | Inflammation (-), Fibrosis (-), Mesangial cell activation (-) | [48] |
| Mouse | USP2 | Tissue (Testis), Sperm | KO | Unidentified | Sperm motility (+), Sperm capacitation (+), Spermatogenesis (+) Fertilization (+), | [129] |
| Mouse | USP2 | Tissue (Testis), Sperm, Macrophage | KO | Unidentified | Sperm motility (+), Sperm capacitation (+), Fertilization (+), Cytokine production (+) | [157] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitamura, H.; Hashimoto, M. USP2-Related Cellular Signaling and Consequent Pathophysiological Outcomes. Int. J. Mol. Sci. 2021, 22, 1209. https://doi.org/10.3390/ijms22031209
Kitamura H, Hashimoto M. USP2-Related Cellular Signaling and Consequent Pathophysiological Outcomes. International Journal of Molecular Sciences. 2021; 22(3):1209. https://doi.org/10.3390/ijms22031209
Chicago/Turabian StyleKitamura, Hiroshi, and Mayuko Hashimoto. 2021. "USP2-Related Cellular Signaling and Consequent Pathophysiological Outcomes" International Journal of Molecular Sciences 22, no. 3: 1209. https://doi.org/10.3390/ijms22031209
APA StyleKitamura, H., & Hashimoto, M. (2021). USP2-Related Cellular Signaling and Consequent Pathophysiological Outcomes. International Journal of Molecular Sciences, 22(3), 1209. https://doi.org/10.3390/ijms22031209