
Genetic and Environmental Factors Influence the Pleomorphy of LRRK2 Parkinsonism



Abstract
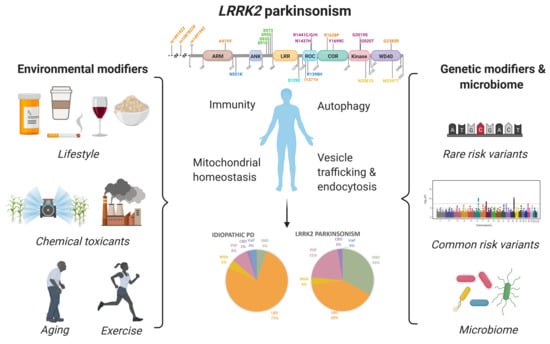
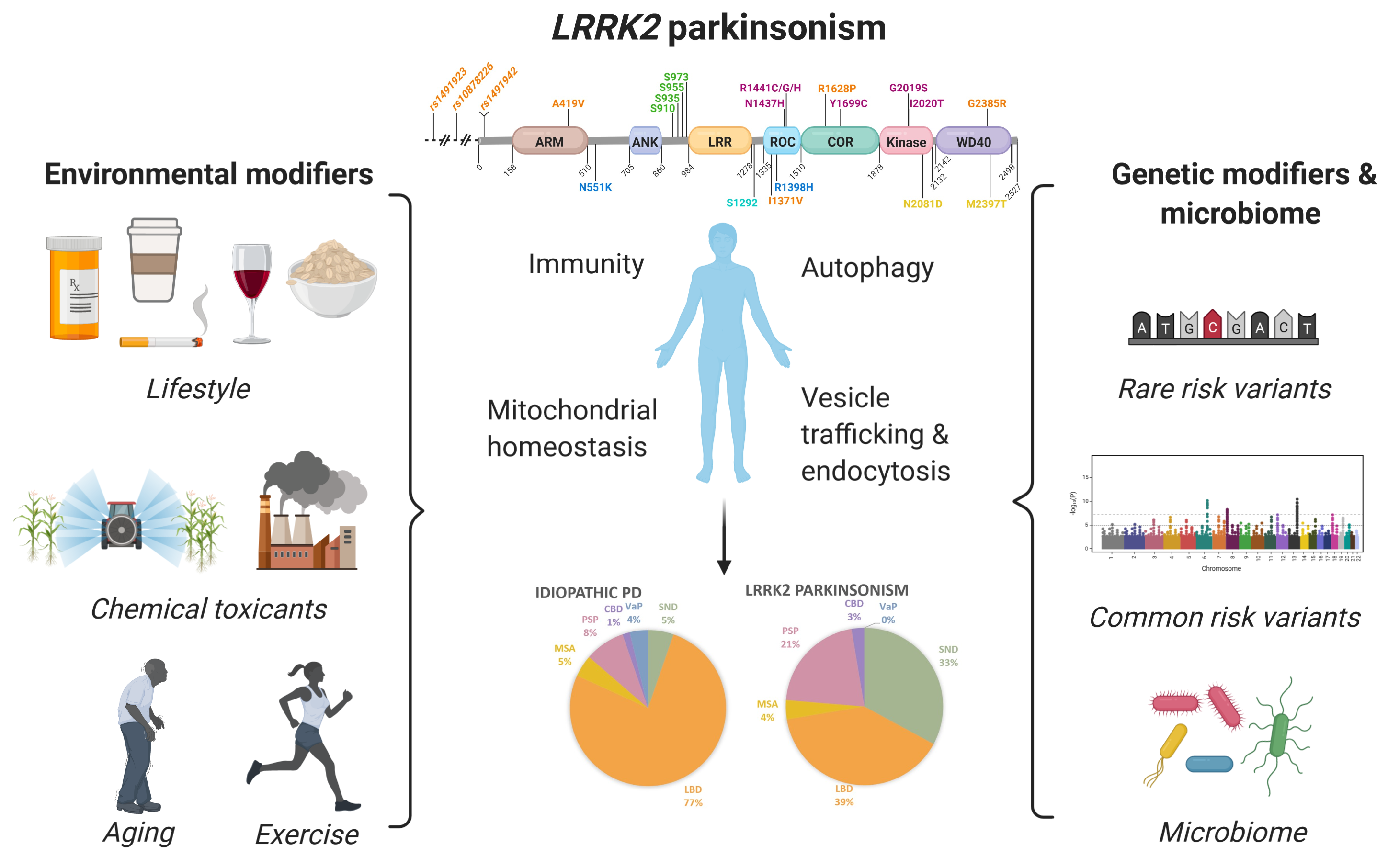
1. Introduction
1.1. Clinical Presentation and Incidence of PD
1.2. LRRK2 Variants, Haplotypes, and Penetrance PD
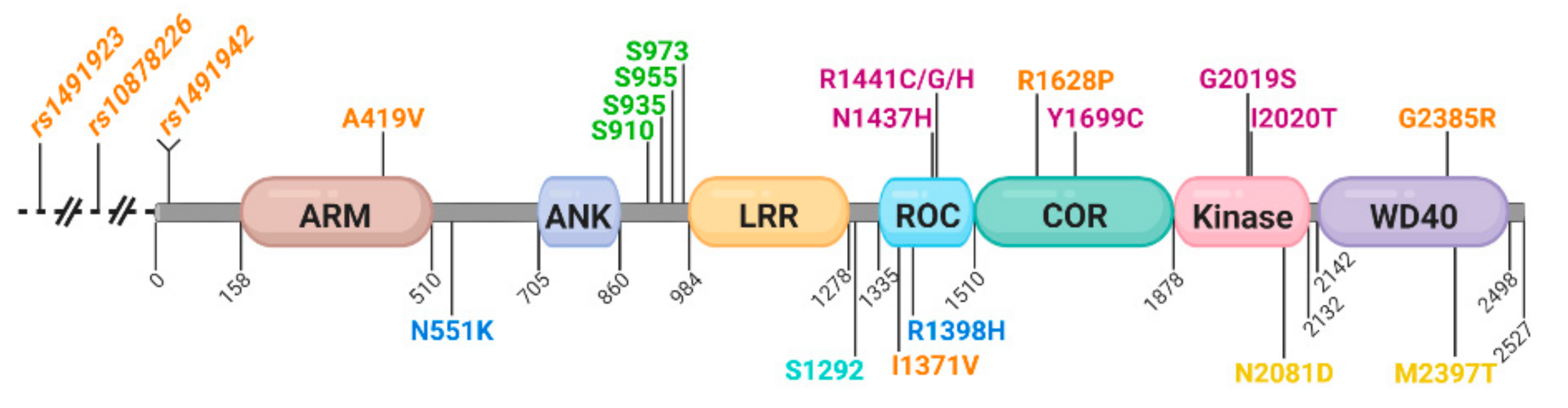
1.3. LRRK2 Domain Structure and the Impact of Inherited Mutations
1.4. LRRK2 Parkinsonism: Similarities and Differences to iPD
1.4.1. Motor and Nonmotor Features in LRRK2-PD
1.4.2. Cortico-Striato-Nigral Connectivity Alterations in Asymptomatic LRRK2 p.G2019S Mutation Carriers
1.5. Pleomorphic LRRK2 Neuropathology
2. Environmental Modifiers of LRRK2 Parkinsonism
2.1. Environmental and Lifestyle Factors Influence Penetrance and Age at Onset of LRRK2 Parkinsonism
2.2. Increased Susceptibility to Synthetic Toxicants in LRRK2 Animal Models
3. LRRK2 Risk and Protective Variants in Neurodegeneration and Genetic Modifiers of AAO and Penetrance in LRRK2 Parkinsonism
3.1. LRRK2 Modifier Risk Variants
3.2. Protective LRRK2 Variants in PD and MSA, but not for Essential Tremor or AD
3.3. LRRK2 Variants in Other Neurodegenerative Diseases
4. Summary and Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| α-syn | Alpha-synuclein |
| Aβ | Amyloid-beta |
| AAO | Age at onset |
| ANK | Ankyrin domain |
| ARM | Armadillo domain |
| BDNF | Brain-derived neurotrophic factor |
| BST1 | Bone marrow stromal cell antigen 1 |
| CBD | Corticobasal degeneration |
| CD | Crohn’s disease |
| COR | C-terminal of ROC domain |
| DLB | Dementia with Lewy bodies |
| DNM3 | Dynamin-3 |
| ET | Essential Tremor |
| GAK | Cyclin-G-associated kinase |
| GBA | Glucosidase, beta, acid, or glucocerebrosidase |
| GCI | Glia cytoplasmic inclusions |
| GTP | Guanosine triphosphate |
| GWAS | Genome-wide association study |
| HRV | Heart-rate variability |
| IBD | Inflammatory bowel disease |
| iPD | Idiopathic Parkinson’s disease |
| iPSC | Induced pluripotent stem cells |
| LB | Lewy body |
| LBD | Lewy body disorder |
| LRR | Leucine-rich repeat domain |
| LRRK2 | Leucine-rich repeat kinase 2 |
| MAPT | Microtubule-associated protein tau |
| MRI | Magnetic resonance imaging |
| MSA | Multiple system atrophy |
| PD | Parkinson’s disease |
| PDD | Parkinson’s disease dementia |
| PSP | Progressive supranuclear palsy |
| PSP-P | Progressive supranuclear palsy parkinsonism |
| PRS | Polygenic risk score |
| REM | Rapid eye movement |
| Roc | Ras of complex proteins |
| SND | Substantia nigra degeneration |
| SNP | Single nucleotide polymorphism |
| TDP-43 | Transactive response DNA-binding protein 43 |
| VAMP4 | Vesicle-associated membrane protein 4 |
| VaP | Vascular parkinsonism |
References
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Paisan-Ruiz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; Lopez de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Neuroanatomy and pathology of sporadic Parkinson’s disease. Adv. Anat. Embryol. Cell Biol. 2009, 201, 1–119. [Google Scholar] [PubMed]
- Clarke, C.E. Parkinson’s disease. BMJ 2007, 335, 441–445. [Google Scholar] [CrossRef]
- Marras, C.; Beck, J.C.; Bower, J.H.; Roberts, E.; Ritz, B.; Ross, G.W.; Abbott, R.D.; Savica, R.; Van Den Eeden, S.K.; Willis, A.W.; et al. Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis. 2018, 4, 21. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Postuma, R.B.; Poewe, W.; Litvan, I.; Lewis, S.; Lang, A.E.; Halliday, G.; Goetz, C.G.; Chan, P.; Slow, E.; Seppi, K.; et al. Validation of the MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2018, 33, 1601–1608. [Google Scholar] [CrossRef]
- Beach, T.G.; Adler, C.H. Importance of low diagnostic Accuracy for early Parkinson’s disease. Mov. Disord. 2018, 33, 1551–1554. [Google Scholar] [CrossRef]
- Aarsland, D.; Creese, B.; Politis, M.; Chaudhuri, K.R.; Ffytche, D.H.; Weintraub, D.; Ballard, C. Cognitive decline in Parkinson disease. Nat. Rev. Neurol. 2017, 13, 217–231. [Google Scholar] [CrossRef]
- Pont-Sunyer, C.; Hotter, A.; Gaig, C.; Seppi, K.; Compta, Y.; Katzenschlager, R.; Mas, N.; Hofeneder, D.; Brucke, T.; Bayes, A.; et al. The onset of nonmotor symptoms in Parkinson’s disease (the ONSET PD study). Mov. Disord. 2015, 30, 229–237. [Google Scholar] [CrossRef]
- Poewe, W. Non-motor symptoms in Parkinson’s disease. Eur. J. Neurol. 2008, 15 (Suppl. 1), 14–20. [Google Scholar] [CrossRef]
- Moisan, F.; Kab, S.; Mohamed, F.; Canonico, M.; Le Guern, M.; Quintin, C.; Carcaillon, L.; Nicolau, J.; Duport, N.; Singh-Manoux, A.; et al. Parkinson disease male-to-female ratios increase with age: French nationwide study and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2016, 87, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Sellbach, A.N.; Boyle, R.S.; Silburn, P.A.; Mellick, G.D. Parkinson’s disease and family history. Parkinsonism Relat. Disord. 2006, 12, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Kay, D.M.; Zabetian, C.P.; Factor, S.A.; Nutt, J.G.; Samii, A.; Griffith, A.; Bird, T.D.; Kramer, P.; Higgins, D.S.; Payami, H. Parkinson’s disease and LRRK2: Frequency of a common mutation in U.S. movement disorder clinics. Mov. Disord. 2006, 21, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Benamer, H.T.; de Silva, R. LRRK2 G2019S in the North African population: A review. Eur. Neurol. 2010, 63, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Ozelius, L.J.; Senthil, G.; Saunders-Pullman, R.; Ohmann, E.; Deligtisch, A.; Tagliati, M.; Hunt, A.L.; Klein, C.; Henick, B.; Hailpern, S.M.; et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 2006, 354, 424–425. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shira, A.; Hutter, C.M.; Giladi, N.; Zabetian, C.P.; Orr-Urtreger, A. Ashkenazi Parkinson’s disease patients with the LRRK2 G2019S mutation share a common founder dating from the second to fifth centuries. Neurogenetics 2009, 10, 355–358. [Google Scholar] [CrossRef]
- Zabetian, C.P.; Hutter, C.M.; Yearout, D.; Lopez, A.N.; Factor, S.A.; Griffith, A.; Leis, B.C.; Bird, T.D.; Nutt, J.G.; Higgins, D.S.; et al. LRRK2 G2019S in families with Parkinson disease who originated from Europe and the Middle East: Evidence of two distinct founding events beginning two millennia ago. Am. J. Hum. Genet. 2006, 79, 752–758. [Google Scholar] [CrossRef]
- Monfrini, E.; Di Fonzo, A. Leucine-Rich Repeat Kinase (LRRK2) Genetics and Parkinson’s Disease. Adv. Neurobiol. 2017, 14, 3–30. [Google Scholar] [CrossRef]
- Marder, K.; Wang, Y.; Alcalay, R.N.; Mejia-Santana, H.; Tang, M.X.; Lee, A.; Raymond, D.; Mirelman, A.; Saunders-Pullman, R.; Clark, L.; et al. Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology 2015, 85, 89–95. [Google Scholar] [CrossRef]
- Trinh, J.; Guella, I.; Farrer, M.J. Disease Penetrance of Late-Onset Parkinsonism: A Meta-analysis. JAMA Neurol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Wang, Y.; Alcalay, R.N.; Mejia-Santana, H.; Saunders-Pullman, R.; Bressman, S.; Corvol, J.C.; Brice, A.; Lesage, S.; Mangone, G.; et al. Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov. Disord. 2017, 32, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Biskup, S.; West, A.B. Zeroing in on LRRK2-linked pathogenic mechanisms in Parkinson’s disease. Biochim. Biophys. Acta 2009, 1792, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Reed, X.; Kia, D.A.; Gan-Or, Z.; Lesage, S.; Pihlstrom, L.; Guerreiro, R.; Gibbs, J.R.; Sabir, M.; Ahmed, S.; et al. Frequency of Loss of Function Variants in LRRK2 in Parkinson Disease. JAMA Neurol. 2018, 75, 1416–1422. [Google Scholar] [CrossRef]
- Whiffin, N.; Armean, I.M.; Kleinman, A.; Marshall, J.L.; Minikel, E.V.; Goodrich, J.K.; Quaife, N.M.; Cole, J.B.; Wang, Q.; Karczewski, K.J.; et al. The effect of LRRK2 loss-of-function variants in humans. Nat. Med. 2020, 26, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Zhang, Y.; Sun, Q.; Pan, H.; Tang, B. A Comprehensive Analysis of Population Differences in LRRK2 Variant Distribution in Parkinson’s Disease. Front. Aging Neurosci. 2019, 11, 13. [Google Scholar] [CrossRef]
- Jaleel, M.; Nichols, R.J.; Deak, M.; Campbell, D.G.; Gillardon, F.; Knebel, A.; Alessi, D.R. LRRK2 phosphorylates moesin at threonine-558: Characterization of how Parkinson’s disease mutants affect kinase activity. Biochem. J. 2007, 405, 307–317. [Google Scholar] [CrossRef]
- Nichols, R.J.; Dzamko, N.; Morrice, N.A.; Campbell, D.G.; Deak, M.; Ordureau, A.; Macartney, T.; Tong, Y.; Shen, J.; Prescott, A.R.; et al. 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem. J. 2010, 430, 393–404. [Google Scholar] [CrossRef]
- Li, X.; Wang, Q.J.; Pan, N.; Lee, S.; Zhao, Y.; Chait, B.T.; Yue, Z. Phosphorylation-dependent 14-3-3 binding to LRRK2 is impaired by common mutations of familial Parkinson’s disease. PLoS ONE 2011, 6, e17153. [Google Scholar] [CrossRef]
- Zhang, P.; Fan, Y.; Ru, H.; Wang, L.; Magupalli, V.G.; Taylor, S.S.; Alessi, D.R.; Wu, H. Crystal structure of the WD40 domain dimer of LRRK2. Proc. Natl. Acad. Sci. USA 2019, 116, 1579–1584. [Google Scholar] [CrossRef]
- Marin, I.; van Egmond, W.N.; van Haastert, P.J. The Roco protein family: A functional perspective. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 3103–3110. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.A. The function of ROCO proteins in health and disease. Biol. Cell 2009, 101, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Wu, C.X.; Burlak, C.; Zhang, S.; Sahm, H.; Wang, M.; Zhang, Z.Y.; Vogel, K.W.; Federici, M.; Riddle, S.M.; et al. Parkinson disease-associated mutation R1441H in LRRK2 prolongs the “active state” of its GTPase domain. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Lewis, P.A.; Greggio, E.; Sluch, E.; Beilina, A.; Cookson, M.R. Structure of the ROC domain from the Parkinson’s disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc. Natl. Acad. Sci. USA 2008, 105, 1499–1504. [Google Scholar] [CrossRef]
- Rudi, K.; Ho, F.Y.; Gilsbach, B.K.; Pots, H.; Wittinghofer, A.; Kortholt, A.; Klare, J.P. Conformational heterogeneity of the Roc domains in C. tepidum Roc-COR and implications for human LRRK2 Parkinson mutations. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef]
- Gotthardt, K.; Weyand, M.; Kortholt, A.; Van Haastert, P.J.; Wittinghofer, A. Structure of the Roc-COR domain tandem of C. tepidum, a prokaryotic homologue of the human LRRK2 Parkinson kinase. EMBO J. 2008, 27, 2352. [Google Scholar] [CrossRef][Green Version]
- Jorgensen, N.D.; Peng, Y.; Ho, C.C.; Rideout, H.J.; Petrey, D.; Liu, P.; Dauer, W.T. The WD40 domain is required for LRRK2 neurotoxicity. PLoS ONE 2009, 4, e8463. [Google Scholar] [CrossRef]
- Greggio, E.; Zambrano, I.; Kaganovich, A.; Beilina, A.; Taymans, J.M.; Daniels, V.; Lewis, P.; Jain, S.; Ding, J.; Syed, A.; et al. The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J. Biol. Chem. 2008, 283, 16906–16914. [Google Scholar] [CrossRef]
- Langston, J.W.; Schüle, B.; Rees, L.; Nichols, R.J.; Barlow, C. Multisystem Lewy body disease and the other parkinsonian disorders. Nat. Genet. 2015, 47, 1378–1384. [Google Scholar] [CrossRef]
- Lin, M.K.; Farrer, M.J. Genetics and genomics of Parkinson’s disease. Genome Med. 2014, 6, 48. [Google Scholar] [CrossRef]
- Mata, I.F.; Wedemeyer, W.J.; Farrer, M.J.; Taylor, J.P.; Gallo, K.A. LRRK2 in Parkinson’s disease: Protein domains and functional insights. Trends Neurosci. 2006, 29, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 2016, 5, 12813. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.K.; Hunter, T. Protein kinases 6. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1995, 9, 576–596. [Google Scholar]
- Hanks, S.K.; Quinn, A.M. Protein kinase catalytic domain sequence database: Identification of conserved features of primary structure and classification of family members. Methods Enzymol. 1991, 200, 38–62. [Google Scholar] [PubMed]
- Hanks, S.K.; Quinn, A.M.; Hunter, T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science 1988, 241, 42–52. [Google Scholar] [CrossRef]
- Greggio, E.; Cookson, M.R. Leucine-rich repeat kinase 2 mutations and Parkinson’s disease: Three questions. ASN Neuro 2009, 1. [Google Scholar] [CrossRef]
- Schmidt, S.H.; Knape, M.J.; Boassa, D.; Mumdey, N.; Kornev, A.P.; Ellisman, M.H.; Taylor, S.S.; Herberg, F.W. The dynamic switch mechanism that leads to activation of LRRK2 is embedded in the DFGpsi motif in the kinase domain. Proc. Natl. Acad. Sci. USA 2019, 116, 14979–14988. [Google Scholar] [CrossRef]
- Gilsbach, B.K.; Ho, F.Y.; Vetter, I.R.; van Haastert, P.J.; Wittinghofer, A.; Kortholt, A. Roco kinase structures give insights into the mechanism of Parkinson disease-related leucine-rich-repeat kinase 2 mutations. Proc. Natl. Acad. Sci. USA 2012, 109, 10322–10327. [Google Scholar] [CrossRef]
- Gilsbach, B.K.; Kortholt, A. Structural biology of the LRRK2 GTPase and kinase domains: Implications for regulation. Front. Mol. Neurosci. 2014, 7, 32. [Google Scholar] [CrossRef]
- Gloeckner, C.J.; Kinkl, N.; Schumacher, A.; Braun, R.J.; O’Neill, E.; Meitinger, T.; Kolch, W.; Prokisch, H.; Ueffing, M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum. Mol. Genet. 2006, 15, 223–232. [Google Scholar] [CrossRef]
- Nichols, R.J.; Dzamko, N.; Hutti, J.E.; Cantley, L.C.; Deak, M.; Moran, J.; Bamborough, P.; Reith, A.D.; Alessi, D.R. Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson’s disease. Biochem. J. 2009, 424, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Bender, S.; Kang, S.; Lin, R.; Glicksman, M.A.; Liu, M. The Parkinson disease-linked LRRK2 protein mutation I2020T stabilizes an active state conformation leading to increased kinase activity. J. Biol. Chem. 2014, 289, 13042–13053. [Google Scholar] [CrossRef] [PubMed]
- Rudenko, I.N.; Kaganovich, A.; Hauser, D.N.; Beylina, A.; Chia, R.; Ding, J.; Maric, D.; Jaffe, H.; Cookson, M.R. The G2385R variant of leucine-rich repeat kinase 2 associated with Parkinson’s disease is a partial loss-of-function mutation. Biochem. J. 2012, 446, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Leandrou, E.; Markidi, E.; Memou, A.; Melachroinou, K.; Greggio, E.; Rideout, H.J. Kinase activity of mutant LRRK2 manifests differently in hetero-dimeric vs. homo-dimeric complexes. Biochem. J. 2019, 476, 559–579. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E.; Hazrati, L.N.; Fujioka, S.; Wszolek, Z.K.; Dickson, D.W.; Ross, O.A.; Van Deerlin, V.M.; Trojanowski, J.Q.; Hurtig, H.I.; et al. Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol. 2015, 72, 100–105. [Google Scholar] [CrossRef]
- Henderson, M.X.; Sengupta, M.; Trojanowski, J.Q.; Lee, V.M.Y. Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol. Commun. 2019, 7, 183. [Google Scholar] [CrossRef]
- Takanashi, M.; Funayama, M.; Matsuura, E.; Yoshino, H.; Li, Y.; Tsuyama, S.; Takashima, H.; Nishioka, K.; Hattori, N. Isolated nigral degeneration without pathological protein aggregation in autopsied brains with LRRK2 p.R1441H homozygous and heterozygous mutations. Acta Neuropathol. Commun. 2018, 6, 105. [Google Scholar] [CrossRef]
- Lee, K.; Nguyen, K.D.; Sun, C.; Liu, M.; Zafar, F.; Saetern, J.; Flierl, A.; Tetrud, J.W.; Langston, J.W.; Dickson, D.; et al. LRRK2 p.Ile1371Val Mutation in a Case with Neuropathologically Confirmed Multi-System Atrophy. J. Parkinsons Dis. 2018, 8, 93–100. [Google Scholar] [CrossRef]
- Schneider, S.A.; Alcalay, R.N. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov. Disord. 2017, 32, 1504–1523. [Google Scholar] [CrossRef]
- Ysselstein, D.; Nguyen, M.; Young, T.J.; Severino, A.; Schwake, M.; Merchant, K.; Krainc, D. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat. Commun. 2019, 10, 5570. [Google Scholar] [CrossRef]
- Bonet-Ponce, L.; Beilina, A.; Williamson, C.D.; Lindberg, E.; Kluss, J.H.; Saez-Atienzar, S.; Landeck, N.; Kumaran, R.; Mamais, A.; Bleck, C.K.E.; et al. LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C.; Lewis, P.A. LRRK2 and Autophagy. Adv. Neurobiol. 2017, 14, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Beilina, A.; Smith, N.; Li, Y.; Kim, M.; Kumaran, R.; Kaganovich, A.; Mamais, A.; Adame, A.; Iba, M.; et al. LRRK2 mediates microglial neurotoxicity via NFATc2 in rodent models of synucleinopathies. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Wallings, R.L.; Herrick, M.K.; Tansey, M.G. LRRK2 at the Interface Between Peripheral and Central Immune Function in Parkinson’s. Front. Neurosci. 2020, 14, 443. [Google Scholar] [CrossRef]
- Ahmadi Rastegar, D.; Dzamko, N. Leucine Rich Repeat Kinase 2 and Innate Immunity. Front. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- Howlett, E.H.; Jensen, N.; Belmonte, F.; Zafar, F.; Hu, X.; Kluss, J.; Schüle, B.; Kaufman, B.A.; Greenamyre, J.T.; Sanders, L.H. LRRK2 G2019S-induced mitochondrial DNA damage is LRRK2 kinase dependent and inhibition restores mtDNA integrity in Parkinson’s disease. Hum. Mol. Genet. 2017, 26, 4340–4351. [Google Scholar] [CrossRef]
- Singh, A.; Zhi, L.; Zhang, H. LRRK2 and mitochondria: Recent advances and current views. Brain Res. 2019, 1702, 96–104. [Google Scholar] [CrossRef]
- Wang, S.; Kelly, K.; Brotchie, J.M.; Koprich, J.B.; West, A.B. Exosome markers of LRRK2 kinase inhibition. NPJ Parkinson’s Dis. 2020, 6, 32. [Google Scholar] [CrossRef]
- Candelario, K.M.; Balaj, L.; Zheng, T.; Skog, J.; Scheffler, B.; Breakefield, X.; Schüle, B.; Steindler, D.A. Exosome/Microvesicle Content is Altered in LRRK2 Mutant iPSC-derived Neural Cells. J. Comp. Neurol. 2019. [Google Scholar] [CrossRef]
- Christensen, K.V.; Hentzer, M.; Oppermann, F.S.; Elschenbroich, S.; Dossang, P.; Thirstrup, K.; Egebjerg, J.; Williamson, D.S.; Smith, G.P. LRRK2 exonic variants associated with Parkinson’s disease augment phosphorylation levels for LRRK2-Ser1292 and Rab10-Thr73. bioRxiv 2018, 447946. [Google Scholar] [CrossRef]
- Hui, K.Y.; Fernandez-Hernandez, H.; Hu, J.; Schaffner, A.; Pankratz, N.; Hsu, N.Y.; Chuang, L.S.; Carmi, S.; Villaverde, N.; Li, X.; et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- West, A.B.; Moore, D.J.; Choi, C.; Andrabi, S.A.; Li, X.; Dikeman, D.; Biskup, S.; Zhang, Z.; Lim, K.L.; Dawson, V.L.; et al. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 2007, 16, 223–232. [Google Scholar] [CrossRef]
- Nixon-Abell, J.; Berwick, D.C.; Granno, S.; Spain, V.A.; Blackstone, C.; Harvey, K. Protective LRRK2 R1398H Variant Enhances GTPase and Wnt Signaling Activity. Front. Mol. Neurosci. 2016, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Aasly, J.O.; Vilariño-Güell, C.; Dachsel, J.C.; Webber, P.J.; West, A.B.; Haugarvoll, K.; Johansen, K.K.; Toft, M.; Nutt, J.G.; Payami, H.; et al. Novel pathogenic LRRK2 p.Asn1437His substitution in familial Parkinson’s disease. Mov. Disord. 2010, 25, 2156–2163. [Google Scholar] [CrossRef] [PubMed]
- West, A.B.; Moore, D.J.; Biskup, S.; Bugayenko, A.; Smith, W.W.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 16842–16847. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.A.; Greggio, E.; Beilina, A.; Jain, S.; Baker, A.; Cookson, M.R. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem. Biophys. Res. Commun. 2007, 357, 668–671. [Google Scholar] [CrossRef]
- Shu, Y.; Ming, J.; Zhang, P.; Wang, Q.; Jiao, F.; Tian, B. Parkinson-Related LRRK2 Mutation R1628P Enables Cdk5 Phosphorylation of LRRK2 and Upregulates Its Kinase Activity. PLoS ONE 2016, 11, e0149739. [Google Scholar] [CrossRef]
- Fava, V.M.; Xu, Y.Z.; Lettre, G.; Van Thuc, N.; Orlova, M.; Thai, V.H.; Tao, S.; Croteau, N.; Eldeeb, M.A.; MacDougall, E.J.; et al. Pleiotropic effects for Parkin and LRRK2 in leprosy type-1 reactions and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 15616–15624. [Google Scholar] [CrossRef]
- Daniëls, V.; Vancraenenbroeck, R.; Law, B.M.; Greggio, E.; Lobbestael, E.; Gao, F.; De Maeyer, M.; Cookson, M.R.; Harvey, K.; Baekelandt, V.; et al. Insight into the mode of action of the LRRK2 Y1699C pathogenic mutant. J. Neurochem. 2011, 116, 304–315. [Google Scholar] [CrossRef]
- Ikezu, T.; Koro, L.; Wolozin, B.; Farraye, F.A.; Strongosky, A.J.; Wszolek, Z.K. Crohn’s and Parkinson’s Disease-Associated LRRK2 Mutations Alter Type II Interferon Responses in Human CD14(+) Blood Monocytes Ex Vivo. J. Neuroimmune Pharmacol. 2020, 15, 794–800. [Google Scholar] [CrossRef]
- Fava, V.M.; Manry, J.; Cobat, A.; Orlova, M.; Van Thuc, N.; Ba, N.N.; Thai, V.H.; Abel, L.; Alcais, A.; Schurr, E.; et al. A Missense LRRK2 Variant Is a Risk Factor for Excessive Inflammatory Responses in Leprosy. PLoS Negl. Trop. Dis. 2016, 10, e0004412. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lee, J.; Krummey, S.; Lu, W.; Cai, H.; Lenardo, M.J. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat. Immunol. 2011, 12, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Schüle, B.; Munhoz, R.P.; Rogaeva, E.; Langston, J.W.; Kasten, M.; Meaney, C.; Klein, C.; Wadia, P.M.; Lim, S.Y.; et al. Phenotype in parkinsonian and nonparkinsonian LRRK2 G2019S mutation carriers. Neurology 2011, 77, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Zhang, Y.; Pan, H.; Xu, Q.; Guo, J.; Tang, B.; Sun, Q. Clinical Heterogeneity Among LRRK2 Variants in Parkinson’s Disease: A Meta-Analysis. Front. Aging Neurosci. 2018, 10, 283. [Google Scholar] [CrossRef]
- Orr-Urtreger, A.; Shifrin, C.; Rozovski, U.; Rosner, S.; Bercovich, D.; Gurevich, T.; Yagev-More, H.; Bar-Shira, A.; Giladi, N. The LRRK2 G2019S mutation in Ashkenazi Jews with Parkinson disease: Is there a gender effect? Neurology 2007, 69, 1595–1602. [Google Scholar] [CrossRef]
- Saunders-Pullman, R.; Mirelman, A.; Alcalay, R.N.; Wang, C.; Ortega, R.A.; Raymond, D.; Mejia-Santana, H.; Orbe-Reilly, M.; Johannes, B.A.; Thaler, A.; et al. Progression in the LRRK2-Asssociated Parkinson Disease Population. JAMA Neurol. 2018, 75, 312–319. [Google Scholar] [CrossRef]
- Mestre, T.A.; Pont-Sunyer, C.; Kausar, F.; Visanji, N.P.; Ghate, T.; Connolly, B.S.; Gasca-Salas, C.; Kern, D.S.; Jain, J.; Slow, E.J.; et al. Clustering of motor and nonmotor traits in leucine-rich repeat kinase 2 G2019S Parkinson’s disease nonparkinsonian relatives: A multicenter family study. Mov. Disord. 2018, 33, 960–965. [Google Scholar] [CrossRef]
- Joers, V.; Emborg, M.E. Modeling and imaging cardiac sympathetic neurodegeneration in Parkinson’s disease. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 125–159. [Google Scholar]
- Alonso, A.; Huang, X.; Mosley, T.H.; Heiss, G.; Chen, H. Heart rate variability and the risk of Parkinson disease: The Atherosclerosis Risk in Communities study. Ann. Neurol. 2015, 77, 877–883. [Google Scholar] [CrossRef]
- Visanji, N.P.; Bhudhikanok, G.S.; Mestre, T.A.; Ghate, T.; Udupa, K.; AlDakheel, A.; Connolly, B.S.; Gasca-Salas, C.; Kern, D.S.; Jain, J.; et al. Heart rate variability in leucine-rich repeat kinase 2-associated Parkinson’s disease. Mov. Disord. 2017, 32, 610–614. [Google Scholar] [CrossRef]
- Carricarte Naranjo, C.; Marras, C.; Visanji, N.P.; Cornforth, D.J.; Sanchez-Rodriguez, L.; Schule, B.; Goldman, S.M.; Estevez, M.; Stein, P.K.; Lang, A.E.; et al. Increased markers of cardiac vagal activity in leucine-rich repeat kinase 2-associated Parkinson’s disease. Clin. Auton Res. 2019, 29, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Helmich, R.C.; Thaler, A.; van Nuenen, B.F.; Gurevich, T.; Mirelman, A.; Marder, K.S.; Bressman, S.; Orr-Urtreger, A.; Giladi, N.; Bloem, B.R.; et al. Reorganization of corticostriatal circuits in healthy G2019S LRRK2 carriers. Neurology 2015, 84, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Jacob, Y.; Rosenberg-Katz, K.; Gurevich, T.; Helmich, R.C.; Bloem, B.R.; Orr-Urtreger, A.; Giladi, N.; Mirelman, A.; Hendler, T.; Thaler, A. Network abnormalities among non-manifesting Parkinson disease related LRRK2 mutation carriers. Hum. Brain Mapp. 2019, 40, 2546–2555. [Google Scholar] [CrossRef] [PubMed]
- Vilas, D.; Segura, B.; Baggio, H.C.; Pont-Sunyer, C.; Compta, Y.; Valldeoriola, F.; Jose Marti, M.; Quintana, M.; Bayes, A.; Hernandez-Vara, J.; et al. Nigral and striatal connectivity alterations in asymptomatic LRRK2 mutation carriers: A magnetic resonance imaging study. Mov. Disord. 2016, 31, 1820–1828. [Google Scholar] [CrossRef]
- Thaler, A.; Mirelman, A.; Helmich, R.C.; van Nuenen, B.F.; Rosenberg-Katz, K.; Gurevich, T.; Orr-Urtreger, A.; Marder, K.; Bressman, S.; Bloem, B.R.; et al. Neural correlates of executive functions in healthy G2019S LRRK2 mutation carriers. Cortex 2013, 49, 2501–2511. [Google Scholar] [CrossRef]
- Thaler, A.; Gonen, T.; Mirelman, A.; Helmich, R.C.; Gurevich, T.; Orr-Urtreger, A.; Bloem, B.R.; Giladi, N.; Hendler, T.; Consortium, L.A.J. Altered reward-related neural responses in non-manifesting carriers of the Parkinson disease related LRRK2 mutation. Brain Imaging Behav. 2019, 13, 1009–1020. [Google Scholar] [CrossRef]
- Liu, S.Y.; Wile, D.J.; Fu, J.F.; Valerio, J.; Shahinfard, E.; McCormick, S.; Mabrouk, R.; Vafai, N.; McKenzie, J.; Neilson, N.; et al. The effect of LRRK2 mutations on the cholinergic system in manifest and premanifest stages of Parkinson’s disease: A cross-sectional PET study. Lancet Neurol. 2018, 17, 309–316. [Google Scholar] [CrossRef]
- Dickson, D.W. Neuropathology of Parkinson disease. Parkinsonism Relat. Disord. 2018, 46 (Suppl. 1), S30–S33. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Neuropathological Staging of Brain Pathology in Sporadic Parkinson’s disease: Separating the Wheat from the Chaff. J. Parkinsons Dis. 2017, 7, S71–S85. [Google Scholar] [CrossRef]
- Karanth, S.; Nelson, P.T.; Katsumata, Y.; Kryscio, R.J.; Schmitt, F.A.; Fardo, D.W.; Cykowski, M.D.; Jicha, G.A.; Van Eldik, L.J.; Abner, E.L. Prevalence and Clinical Phenotype of Quadruple Misfolded Proteins in Older Adults. JAMA Neurol. 2020. [Google Scholar] [CrossRef]
- Poulopoulos, M.; Levy, O.A.; Alcalay, R.N. The neuropathology of genetic Parkinson’s disease. Mov. Disord. 2012, 27, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Poulopoulos, M.; Cortes, E.; Vonsattel, J.P.; Fahn, S.; Waters, C.; Cote, L.J.; Moskowitz, C.; Honig, L.S.; Clark, L.N.; Marder, K.S.; et al. Clinical and pathological characteristics of LRRK2 G2019S patients with PD. J. Mol. Neurosci. 2012, 47, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Agin-Liebes, J.; Cortes, E.; Vonsattel, J.P.; Marder, K.; Alcalay, R.N. Movement disorders rounds: A case of missing pathology in a patient with LRRK2 Parkinson’s disease. Parkinsonism Relat. Disord. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Stoessl, A.J.; Yokoyama, T.; Kowa, H.; Wszolek, Z.K.; Yagishita, S. Familial parkinsonism: Study of original Sagamihara PARK8 (I2020T) kindred with variable clinicopathologic outcomes. Parkinsonism Relat. Disord. 2009, 15, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Riboldi, G.M.; Palma, J.A.; Cortes, E.; Iida, M.A.; Sikder, T.; Henderson, B.; Raj, T.; Walker, R.H.; Crary, J.F.; Kaufmann, H.; et al. Early-onset pathologically proven multiple system atrophy with LRRK2 G2019S mutation. Mov. Disord. 2019, 34, 1080–1082. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Song, Y.J.; Murphy, K.; Holton, J.L.; Lashley, T.; Revesz, T.; Gai, W.P.; Halliday, G.M. LRRK2 and parkin immunoreactivity in multiple system atrophy inclusions. Acta Neuropathol. 2008, 116, 639–646. [Google Scholar] [CrossRef]
- Ling, H.; Kara, E.; Bandopadhyay, R.; Hardy, J.; Holton, J.; Xiromerisiou, G.; Lees, A.; Houlden, H.; Revesz, T. TDP-43 pathology in a patient carrying G2019S LRRK2 mutation and a novel p.Q124E MAPT. Neurobiol. Aging 2013, 34, 2889.e5–2889.e9. [Google Scholar] [CrossRef]
- Vilas, D.; Sharp, M.; Gelpi, E.; Genis, D.; Marder, K.S.; Cortes, E.; Vonsattel, J.P.; Tolosa, E.; Alcalay, R.N. Clinical and neuropathological features of progressive supranuclear palsy in Leucine rich repeat kinase (LRRK2) G2019S mutation carriers. Mov. Disord. 2018, 33, 335–338. [Google Scholar] [CrossRef]
- Ujiie, S.; Hatano, T.; Kubo, S.; Imai, S.; Sato, S.; Uchihara, T.; Yagishita, S.; Hasegawa, K.; Kowa, H.; Sakai, F.; et al. LRRK2 I2020T mutation is associated with tau pathology. Parkinsonism Relat. Disord. 2012, 18, 819–823. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Pletnikova, O.; Geiger, J.T.; Murphy, N.A.; Abramzon, Y.; Rudow, G.; Mamais, A.; Sabir, M.S.; Crain, B.; Ahmed, S.; et al. Genetic analysis of neurodegenerative diseases in a pathology cohort. Neurobiol. Aging 2019, 76, 214.e1–214.e9. [Google Scholar] [CrossRef]
- Wszolek, Z.K.; Pfeiffer, R.F.; Tsuboi, Y.; Uitti, R.J.; McComb, R.D.; Stoessl, A.J.; Strongosky, A.J.; Zimprich, A.; Muller-Myhsok, B.; Farrer, M.J.; et al. Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology 2004, 62, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Contreras, M.; Heckman, M.G.; Tacik, P.; Diehl, N.; Brown, P.H.; Soto-Ortolaza, A.I.; Christopher, E.A.; Walton, R.L.; Ross, O.A.; Golbe, L.I.; et al. Study of LRRK2 variation in tauopathy: Progressive supranuclear palsy and corticobasal degeneration. Mov. Disord. 2017, 32, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Giordana, M.T.; D’Agostino, C.; Albani, G.; Mauro, A.; Di Fonzo, A.; Antonini, A.; Bonifati, V. Neuropathology of Parkinson’s disease associated with the LRRK2 Ile1371Val mutation. Mov. Disord. 2007, 22, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Puschmann, A.; Englund, E.; Ross, O.A.; Vilarino-Guell, C.; Lincoln, S.J.; Kachergus, J.M.; Cobb, S.A.; Tornqvist, A.L.; Rehncrona, S.; Widner, H.; et al. First neuropathological description of a patient with Parkinson’s disease and LRRK2 p.N1437H mutation. Parkinsonism Relat. Disord. 2012, 18, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Marti-Masso, J.F.; Ruiz-Martinez, J.; Bolano, M.J.; Ruiz, I.; Gorostidi, A.; Moreno, F.; Ferrer, I.; Lopez de Munain, A. Neuropathology of Parkinson’s disease with the R1441G mutation in LRRK2. Mov. Disord. 2009, 24, 1998–2001. [Google Scholar] [CrossRef] [PubMed]
- Wszolek, Z.K.; Pfeiffer, B.; Fulgham, J.R.; Parisi, J.E.; Thompson, B.M.; Uitti, R.J.; Calne, D.B.; Pfeiffer, R.F. Western Nebraska family (family D) with autosomal dominant parkinsonism. Neurology 1995, 45, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.L.; Jain, S.; Lynch, J.M.; Pavese, N.; Abou-Sleiman, P.; Holton, J.L.; Healy, D.G.; Gilks, W.P.; Sweeney, M.G.; Ganguly, M.; et al. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: Clinical, pathological, olfactory and functional imaging and genetic data. Brain 2005, 128, 2786–2796. [Google Scholar] [CrossRef]
- Wszolek, Z.K.; Vieregge, P.; Uitti, R.J.; Gasser, T.; Yasuhara, O.; McGeer, P.; Berry, K.; Calne, D.B.; Vingerhoets, F.J.; Klein, C.; et al. German-Canadian family (family A) with parkinsonism, amyotrophy, and dementia—Longitudinal observations. Parkinsonism Relat. Disord. 1997, 3, 125–139. [Google Scholar] [CrossRef]
- Giasson, B.I.; Covy, J.P.; Bonini, N.M.; Hurtig, H.I.; Farrer, M.J.; Trojanowski, J.Q.; Van Deerlin, V.M. Biochemical and pathological characterization of Lrrk2. Ann. Neurol. 2006, 59, 315–322. [Google Scholar] [CrossRef]
- Gaig, C.; Marti, M.J.; Ezquerra, M.; Rey, M.J.; Cardozo, A.; Tolosa, E. G2019S LRRK2 mutation causing Parkinson’s disease without Lewy bodies. J. Neurol. Neurosurg. Psychiatry 2007, 78, 626–628. [Google Scholar] [CrossRef]
- Gaig, C.; Ezquerra, M.; Marti, M.J.; Valldeoriola, F.; Munoz, E.; Llado, A.; Rey, M.J.; Cardozo, A.; Molinuevo, J.L.; Tolosa, E. Screening for the LRRK2 G2019S and codon-1441 mutations in a pathological series of parkinsonian syndromes and frontotemporal lobar degeneration. J. Neurol. Sci. 2008, 270, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Kowa, H. Autosomal Dominant Familial Parkinson Disease: Older Onset of Age, and Good Response to Levodopa Therapy. Eur. Neurol. 1997, 38 (Suppl. 1), 39–43. [Google Scholar] [CrossRef] [PubMed]
- Funayama, M.; Hasegawa, K.; Kowa, H.; Saito, M.; Tsuji, S.; Obata, F. A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann. Neurol. 2002, 51, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Covy, J.P.; Yuan, W.; Waxman, E.A.; Hurtig, H.I.; Van Deerlin, V.M.; Giasson, B.I. Clinical and pathological characteristics of patients with leucine-rich repeat kinase-2 mutations. Mov. Disord. 2009, 24, 32–39. [Google Scholar] [CrossRef]
- Jellinger, K.A.; Seppi, K.; Wenning, G.K.; Poewe, W. Impact of coexistent Alzheimer pathology on the natural history of Parkinson’s disease. J. Neural. Transm. (Vienna) 2002, 109, 329–339. [Google Scholar] [CrossRef]
- Melrose, H.L.; Dächsel, J.C.; Behrouz, B.; Lincoln, S.J.; Yue, M.; Hinkle, K.M.; Kent, C.B.; Korvatska, E.; Taylor, J.P.; Witten, L.; et al. Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol. Dis. 2010, 40, 503–517. [Google Scholar] [CrossRef]
- Goldman, S.M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr Med. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Caudle, W.M.; Guillot, T.S.; Lazo, C.R.; Miller, G.W. Industrial toxicants and Parkinson’s disease. Neurotoxicology 2012, 33, 178–188. [Google Scholar] [CrossRef]
- Tanner, C.M. Advances in environmental epidemiology. Mov. Disord. 2010, 25 (Suppl. 1), S58–S62. [Google Scholar] [CrossRef]
- Gubert, C.; Kong, G.; Renoir, T.; Hannan, A.J. Exercise, diet and stress as modulators of gut microbiota: Implications for neurodegenerative diseases. Neurobiol. Dis. 2020, 134, 104621. [Google Scholar] [CrossRef] [PubMed]
- Chittoor-Vinod, V.G.; Villalobos-Cantor, S.; Roshak, H.; Shea, K.; Abalde-Atristain, L.; Martin, I. Dietary Amino Acids Impact LRRK2-Induced Neurodegeneration in Parkinson’s Disease Models. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 6234–6249. [Google Scholar] [CrossRef] [PubMed]
- Yahalom, G.; Rigbi, A.; Israeli-Korn, S.; Krohn, L.; Rudakou, U.; Ruskey, J.A.; Benshimol, L.; Tsafnat, T.; Gan-Or, Z.; Hassin-Baer, S.; et al. Age at Onset of Parkinson’s Disease Among Ashkenazi Jewish Patients: Contribution of Environmental Factors, LRRK2 p.G2019S and GBA p.N370S Mutations. J. Parkinsons Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- San Luciano, M.; Tanner, C.M.; Meng, C.; Marras, C.; Goldman, S.M.; Lang, A.E.; Tolosa, E.; Schüle, B.; Langston, J.W.; Brice, A.; et al. Nonsteroidal Anti-Inflammatory Use and LRRK2 Parkinson’s Disease Penetrance. Mov. Disord. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, T.; Funakawa, K.; Komori, T.; Sakurai, M.; Yoshii, G.; Eguchi, T.; Fukuda, M.; Iwatsubo, T. Roles of lysosomotropic agents on LRRK2 activation and Rab10 phosphorylation. Neurobiol. Dis. 2020, 145, 105081. [Google Scholar] [CrossRef]
- Herbst, S.; Campbell, P.; Harvey, J.; Bernard, E.M.; Papayannopoulos, V.; Wood, N.W.; Morris, H.R.; Gutierrez, M.G. LRRK2 activation controls the repair of damaged endomembranes in macrophages. EMBO J. 2020, 39, e104494. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem. Cell 2016, 19, 709–724. [Google Scholar] [CrossRef]
- Cabezudo, D.; Baekelandt, V.; Lobbestael, E. Multiple-Hit Hypothesis in Parkinson’s Disease: LRRK2 and Inflammation. Front. Neurosci. 2020, 14, 376. [Google Scholar] [CrossRef]
- Patrick, K.L.; Bell, S.L.; Weindel, C.G.; Watson, R.O. Exploring the “Multiple-Hit Hypothesis” of Neurodegenerative Disease: Bacterial Infection Comes Up to Bat. Front. Cell Infect. Microbiol. 2019, 9, 138. [Google Scholar] [CrossRef]
- Perez-Rodriguez, D.; Kalyva, M.; Leija-Salazar, M.; Lashley, T.; Tarabichi, M.; Chelban, V.; Gentleman, S.; Schottlaender, L.; Franklin, H.; Vasmatzis, G.; et al. Investigation of somatic CNVs in brains of synucleinopathy cases using targeted SNCA analysis and single cell sequencing. Acta Neuropathol. Commun. 2019, 7, 219. [Google Scholar] [CrossRef]
- Albanese, F.; Novello, S.; Morari, M. Autophagy and LRRK2 in the Aging Brain. Front. Neurosci. 2019, 13, 1352. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P. Parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): Implications for treatment and the pathogenesis of Parkinson’s disease. Can. J. Neurol. Sci. 1984, 11, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Forno, L.S.; Tetrud, J.; Reeves, A.G.; Kaplan, J.A.; Karluk, D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999, 46, 598–605. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Xiong, Y.; Lee, Y.; Lawless, M.C.; Kim, D.; Nordquist, E.; Martin, I.; Ge, P.; Brahmachari, S.; Jhaldiyal, A.; et al. LRRK2 G2019S transgenic mice display increased susceptibility to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-mediated neurotoxicity. J. Chem. Neuroanat. 2016, 76, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Arbez, N.; He, X.; Huang, Y.; Ren, M.; Liang, Y.; Nucifora, F.C.; Wang, X.; Pei, Z.; Tessarolo, L.; Smith, W.W.; et al. G2019S-LRRK2 mutation enhances MPTP-linked Parkinsonism in mice. Hum. Mol. Genet. 2020, 29, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Andres-Mateos, E.; Mejias, R.; Sasaki, M.; Li, X.; Lin, B.M.; Biskup, S.; Zhang, L.; Banerjee, R.; Thomas, B.; Yang, L.; et al. Unexpected lack of hypersensitivity in LRRK2 knock-out mice to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine). J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 15846–15850. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, X.; Guo, Y.; Rong, H.; Liu, T. The long noncoding RNA HOTAIR promotes Parkinson’s disease by upregulating LRRK2 expression. Oncotarget 2017, 8, 24449–24456. [Google Scholar] [CrossRef]
- Shi, L.; Huang, C.; Luo, Q.; Xia, Y.; Liu, H.; Li, L.; Liu, W.; Ma, W.; Fang, J.; Tang, L.; et al. Pilot study: Molecular risk factors for diagnosing sporadic Parkinson’s disease based on gene expression in blood in MPTP-induced rhesus monkeys. Oncotarget 2017, 8, 105606–105614. [Google Scholar] [CrossRef]
- Rudyk, C.; Dwyer, Z.; Hayley, S.; Membership, C. Leucine-rich repeat kinase-2 (LRRK2) modulates paraquat-induced inflammatory sickness and stress phenotype. J. Neuroinflammation 2019, 16, 120. [Google Scholar] [CrossRef]
- Shaikh, K.T.; Yang, A.; Youshin, E.; Schmid, S. Transgenic LRRK2 (R1441G) rats-a model for Parkinson disease? PeerJ 2015, 3, e945. [Google Scholar] [CrossRef]
- Quintero-Espinosa, D.; Jimenez-Del-Rio, M.; Velez-Pardo, C. Knockdown transgenic Lrrk Drosophila resists paraquat-induced locomotor impairment and neurodegeneration: A therapeutic strategy for Parkinson’s disease. Brain Res. 2017, 1657, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Gehrke, S.; Wang, H.Q.; Takahashi, R.; Hasegawa, K.; Oota, E.; Lu, B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008, 27, 2432–2443. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Tapias, V.; Na, H.M.; Honick, A.S.; Drolet, R.E.; Greenamyre, J.T. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 2009, 34, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T.B.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-Yagi, A.; Greenamyre, J.T. Mechanism of toxicity in rotenone models of Parkinson’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [CrossRef]
- Liu, H.F.; Ho, P.W.; Leung, G.C.; Lam, C.S.; Pang, S.Y.; Li, L.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Combined LRRK2 mutation, aging and chronic low dose oral rotenone as a model of Parkinson’s disease. Sci. Rep. 2017, 7, 40887. [Google Scholar] [CrossRef] [PubMed]
- Venderova, K.; Kabbach, G.; Abdel-Messih, E.; Zhang, Y.; Parks, R.J.; Imai, Y.; Gehrke, S.; Ngsee, J.; Lavoie, M.J.; Slack, R.S.; et al. Leucine-Rich Repeat Kinase 2 interacts with Parkin, DJ-1 and PINK-1 in a Drosophila melanogaster model of Parkinson’s disease. Hum. Mol. Genet. 2009, 18, 4390–4404. [Google Scholar] [CrossRef]
- Ng, C.H.; Mok, S.Z.; Koh, C.; Ouyang, X.; Fivaz, M.L.; Tan, E.K.; Dawson, V.L.; Dawson, T.M.; Yu, F.; Lim, K.L. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 11257–11262. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.R.; Castro, S.; Drolet, R.; Hatcher, N.G.; Yao, L.; Smith, S.M.; Keeney, M.T.; Di Maio, R.; Kofler, J.; et al. LRRK2 inhibition prevents endolysosomal deficits seen in human Parkinson’s disease. Neurobiol. Dis. 2020, 134, 104626. [Google Scholar] [CrossRef]
- Mendivil-Perez, M.; Velez-Pardo, C.; Jimenez-Del-Rio, M. Neuroprotective Effect of the LRRK2 Kinase Inhibitor PF-06447475 in Human Nerve-Like Differentiated Cells Exposed to Oxidative Stress Stimuli: Implications for Parkinson’s Disease. Neurochem Res. 2016, 41, 2675–2692. [Google Scholar] [CrossRef]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schondorf, D.C.; Wagner, L.; Glatza, M.; Hoing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem. Cell 2013, 12, 354–367. [Google Scholar] [CrossRef]
- Cannon, J.R.; Greenamyre, J.T. Gene-environment interactions in Parkinson’s disease: Specific evidence in humans and mammalian models. Neurobiol. Dis. 2013, 57, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Manning-Bog, A.B.; Langston, J.W. Model fusion, the next phase in developing animal models for Parkinson’s disease. Neurotox Res. 2007, 11, 219–240. [Google Scholar] [CrossRef] [PubMed]
- Varçin, M.; Bentea, E.; Michotte, Y.; Sarre, S. Oxidative Stress in Genetic Mouse Models of Parkinson’s Disease. Oxidative Med. Cell. Longev. 2012, 2012, 624925. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.H.; Matsuoka, Y.; Sziráki, I.; Hashim, A.; Lafrancois, J.; Sershen, H.; Duff, K.E. Increased dopaminergic neuron sensitivity to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in transgenic mice expressing mutant A53T alpha-synuclein. Neurochem. Res. 2008, 33, 902–911. [Google Scholar] [CrossRef]
- Lee, S.; Oh, S.T.; Jeong, H.J.; Pak, S.C.; Park, H.J.; Kim, J.; Cho, H.S.; Jeon, S. MPTP-induced vulnerability of dopamine neurons in A53T alpha-synuclein overexpressed mice with the potential involvement of DJ-1 downregulation. Korean J. Physiol. Pharmacol. 2017, 21, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Hayakawa, H.; Nihira, T.; Ren, Y.R.; Nakata, Y.; Nagai, M.; Hattori, N.; Miyake, K.; Takada, M.; Shimada, T.; et al. Parkin-mediated protection of dopaminergic neurons in a chronic MPTP-minipump mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 2011, 70, 686–697. [Google Scholar] [CrossRef]
- Bieri, G.; Brahic, M.; Bousset, L.; Couthouis, J.; Kramer, N.J.; Ma, R.; Nakayama, L.; Monbureau, M.; Defensor, E.; Schüle, B.; et al. LRRK2 modifies alpha-syn pathology and spread in mouse models and human neurons. Acta Neuropathol. 2019, 137, 961–980. [Google Scholar] [CrossRef]
- Purlyte, E.; Dhekne, H.S.; Sarhan, A.R.; Gomez, R.; Lis, P.; Wightman, M.; Martinez, T.N.; Tonelli, F.; Pfeffer, S.R.; Alessi, D.R. Rab29 activation of the Parkinson’s disease-associated LRRK2 kinase. EMBO J. 2018, 37, 1–18. [Google Scholar] [CrossRef]
- Botta-Orfila, T.; Ezquerra, M.; Pastor, P.; Fernandez-Santiago, R.; Pont-Sunyer, C.; Compta, Y.; Lorenzo-Betancor, O.; Samaranch, L.; Marti, M.J.; Valldeoriola, F.; et al. Age at onset in LRRK2-associated PD is modified by SNCA variants. J. Mol. Neurosci. 2012, 48, 245–247. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Bar-Shira, A.; Mirelman, A.; Gurevich, T.; Giladi, N.; Orr-Urtreger, A. The age at motor symptoms onset in LRRK2-associated Parkinson’s disease is affected by a variation in the MAPT locus: A possible interaction. J. Mol. Neurosci. 2012, 46, 541–544. [Google Scholar] [CrossRef]
- Trinh, J.; Gustavsson, E.K.; Guella, I.; Vilarino-Guell, C.; Evans, D.; Encarnacion, M.; Sherman, H.; Hentati, F.; Farrer, M.J. The role of SNCA and MAPT in Parkinson disease and LRRK2 parkinsonism in the Tunisian Arab-Berber population. Eur. J. Neurol. 2014, 21, e91–e92. [Google Scholar] [CrossRef] [PubMed]
- Trinh, J.; Gustavsson, E.K.; Vilarino-Guell, C.; Bortnick, S.; Latourelle, J.; McKenzie, M.B.; Tu, C.S.; Nosova, E.; Khinda, J.; Milnerwood, A.; et al. DNM3 and genetic modifiers of age of onset in LRRK2 Gly2019Ser parkinsonism: A genome-wide linkage and association study. Lancet Neurol. 2016, 15, 1248–1256. [Google Scholar] [CrossRef]
- Fernandez-Santiago, R.; Garrido, A.; Infante, J.; Gonzalez-Aramburu, I.; Sierra, M.; Fernandez, M.; Valldeoriola, F.; Munoz, E.; Compta, Y.; Marti, M.J.; et al. Alpha-synuclein (SNCA) but not dynamin 3 (DNM3) influences age at onset of leucine-rich repeat kinase 2 (LRRK2) Parkinson’s disease in Spain. Mov. Disord. 2018, 33, 637–641. [Google Scholar] [CrossRef]
- Brown, E.E.; Blauwendraat, C.; Trinh, J.; Rizig, M.; Nalls, M.A.; Leveille, E.; Ruskey, J.A.; Jonvik, H.; Tan, M.M.X.; Bandres-Ciga, S.; et al. Analysis of DNM3 and VAMP4 as genetic modifiers of LRRK2 Parkinson’s disease. Neurobiol. Aging 2021, 97, 148.e117–148.e124. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, Y.; Wang, C.; Wang, T.; Zheng, Z.; Chan, P. Brain-derived neurotrophic factor (BDNF) genetic polymorphism greatly increases risk of leucine-rich repeat kinase 2 (LRRK2) for Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Dan, X.; Wang, C.; Ma, J.; Feng, X.; Wang, T.; Zheng, Z.; Chan, P. MAPT IVS1+124 C>G modifies risk of LRRK2 G2385R for Parkinson’s disease in Chinese individuals. Neurobiol. Aging 2014, 35, 1780.e7–1780.e10. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Cai, Y.; Zheng, Z.; Tang, B.S.; Xu, Y.; Wang, T.; Ma, J.; Chen, S.D.; Langston, J.W.; Tanner, C.M.; et al. Penetrance of LRRK2 G2385R and R1628P is modified by common PD-associated genetic variants. Parkinsonism Relat. Disord. 2012, 18, 958–963. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H.; Li, Y.S.; Shi, M.M.; Yang, J.; Liu, Y.T.; Mao, C.Y.; Fan, Y.; Hu, X.C.; Shi, C.H.; Xu, Y.M. SNCA but not DNM3 and GAK modifies age at onset of LRRK2-related Parkinson’s disease in Chinese population. J. Neurol. 2019, 266, 1796–1800. [Google Scholar] [CrossRef] [PubMed]
- Soto-Ortolaza, A.I.; Heckman, M.G.; Labbe, C.; Serie, D.J.; Puschmann, A.; Rayaprolu, S.; Strongosky, A.; Boczarska-Jedynak, M.; Opala, G.; Krygowska-Wajs, A.; et al. GWAS risk factors in Parkinson’s disease: LRRK2 coding variation and genetic interaction with PARK16. Am. J. Neurodegener Dis. 2013, 2, 287–299. [Google Scholar]
- Wang, L.; Heckman, M.G.; Aasly, J.O.; Annesi, G.; Bozi, M.; Chung, S.J.; Clarke, C.; Crosiers, D.; Eckstein, G.; Garraux, G.; et al. Evaluation of the interaction between LRRK2 and PARK16 loci in determining risk of Parkinson’s disease: Analysis of a large multicenter study. Neurobiol. Aging 2017, 49, 217.e211–217.e214. [Google Scholar] [CrossRef]
- Golub, Y.; Berg, D.; Calne, D.B.; Pfeiffer, R.F.; Uitti, R.J.; Stoessl, A.J.; Wszolek, Z.K.; Farrer, M.J.; Mueller, J.C.; Gasser, T.; et al. Genetic factors influencing age at onset in LRRK2-linked Parkinson disease. Parkinsonism Relat. Disord. 2009, 15, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Nalls, M.A.; Hallgrimsdottir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team; Kerchner, G.A.; Ayalon, G.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Expanding Parkinson’s disease genetics: Novel risk loci, genomic context, causal insights and heritable risk. bioRxiv 2019, 388165. [Google Scholar] [CrossRef]
- Germer, E.L.; Imhoff, S.; Vilariño-Güell, C.; Kasten, M.; Seibler, P.; Brüggemann, N.; Klein, C.; Trinh, J. The Role of Rare Coding Variants in Parkinson’s Disease GWAS Loci. Front. Neurol. 2019, 10, 1284. [Google Scholar] [CrossRef]
- International Parkinson’s Disease Genomics Consortium; Nalls, M.A.; Plagnol, V.; Hernandez, D.G.; Sharma, M.; Sheerin, U.M.; Saad, M.; Simon-Sanchez, J.; Schulte, C.; Lesage, S.; et al. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet 2011, 377, 641–649. [Google Scholar] [CrossRef]
- Lill, C.M.; Roehr, J.T.; McQueen, M.B.; Kavvoura, F.K.; Bagade, S.; Schjeide, B.M.; Schjeide, L.M.; Meissner, E.; Zauft, U.; Allen, N.C.; et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: The PDGene database. PLoS Genet. 2012, 8, e1002548. [Google Scholar] [CrossRef]
- Lai, D.; Alipanahi, B.; Fontanillas, P.; Schwantes-An, T.-H.; Aasly, J.; Alcalay, R.N.; Beecham, G.W.; Berg, D.; Bressman, S.; Brice, A.; et al. Genome-wide association studies of LRRK2 modifiers of Parkinson’s disease. medRxiv 2020. [Google Scholar] [CrossRef]
- Iwaki, H.; Blauwendraat, C.; Makarious, M.B.; Bandres-Ciga, S.; Leonard, H.L.; Gibbs, J.R.; Hernandez, D.G.; Scholz, S.W.; Faghri, F.; International Parkinson’s Disease Genomics Consortium; et al. Penetrance of Parkinson’s Disease in LRRK2 p.G2019S Carriers Is Modified by a Polygenic Risk Score. Mov. Disord. 2020. [Google Scholar] [CrossRef]
- Yahalom, G.; Greenbaum, L.; Israeli-Korn, S.; Fay-Karmon, T.; Livneh, V.; Ruskey, J.A.; Roncière, L.; Alam, A.; Gan-Or, Z.; Hassin-Baer, S. Carriers of both GBA and LRRK2 mutations, compared to carriers of either, in Parkinson’s disease: Risk estimates and genotype-phenotype correlations. Parkinsonism Relat. Disord. 2019, 62, 179–184. [Google Scholar] [CrossRef]
- Omer, N.; Giladi, N.; Gurevich, T.; Bar-Shira, A.; Gana-Weisz, M.; Goldstein, O.; Kestenbaum, M.; Cedarbaum, J.M.; Orr-Urtreger, A.; Mirelman, A.; et al. A Possible Modifying Effect of the G2019S Mutation in the LRRK2 Gene on GBA Parkinson’s Disease. Mov. Disord. 2020, 35, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Ross, O.A.; Soto-Ortolaza, A.I.; Heckman, M.G.; Aasly, J.O.; Abahuni, N.; Annesi, G.; Bacon, J.A.; Bardien, S.; Bozi, M.; Brice, A.; et al. Association of LRRK2 exonic variants with susceptibility to Parkinson’s disease: A case-control study. Lancet Neurol. 2011, 10, 898–908. [Google Scholar] [CrossRef]
- Tan, E.K.; Peng, R.; Teo, Y.Y.; Tan, L.C.; Angeles, D.; Ho, P.; Chen, M.L.; Lin, C.H.; Mao, X.Y.; Chang, X.L.; et al. Multiple LRRK2 variants modulate risk of Parkinson disease: A Chinese multicenter study. Hum. Mutat 2010, 31, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.R.; Chang, K.H.; Chang, W.T.; Hsiao, Y.C.; Hsu, H.C.; Jiang, P.R.; Chen, Y.C.; Chao, C.Y.; Chang, Y.C.; Lee, B.H.; et al. Genetic variants ofLRRK2 in Taiwanese Parkinson’s disease. PLoS ONE 2013, 8, e82001. [Google Scholar] [CrossRef]
- Gopalai, A.A.; Lim, J.L.; Li, H.H.; Zhao, Y.; Lim, T.T.; Eow, G.B.; Puvanarajah, S.; Viswanathan, S.; Norlinah, M.I.; Abdul Aziz, Z.; et al. LRRK2 N551K and R1398H variants are protective in Malays and Chinese in Malaysia: A case-control association study for Parkinson’s disease. Mol. Genet. Genomic Med. 2019, 7, e604. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, S.; Liu, Y.; Hong, H.; Wang, H.; Zheng, Y.; Zhou, H.; Chen, J.; Xian, W.; He, Y.; et al. LRRK2 R1398H polymorphism is associated with decreased risk of Parkinson’s disease in a Han Chinese population. Parkinsonism Relat. Disord. 2011, 17, 291–292. [Google Scholar] [CrossRef]
- Heckman, M.G.; Schottlaender, L.; Soto-Ortolaza, A.I.; Diehl, N.N.; Rayaprolu, S.; Ogaki, K.; Fujioka, S.; Murray, M.E.; Cheshire, W.P.; Uitti, R.J.; et al. LRRK2 exonic variants and risk of multiple system atrophy. Neurology 2014, 83, 2256–2261. [Google Scholar] [CrossRef]
- Ng, A.S.L.; Ng, E.Y.L.; Tan, Y.J.; Prakash, K.M.; Au, W.L.; Tan, L.C.S.; Tan, E.K. Case-control analysis of LRRK2 protective variants in Essential Tremor. Sci. Rep. 2018, 8, 5346. [Google Scholar] [CrossRef]
- Ng, A.S.L.; Ng, E.Y.L.; Tan, Y.J.; Kandiah, N.; Zhou, J.; Hameed, S.; Ting, S.K.S.; Tan, E.K. Case-control analysis of leucine-rich repeat kinase 2 protective variants in Alzheimer’s disease. Neurobiol. Aging 2018, 64, e157–e159. [Google Scholar] [CrossRef]
- Ma, D.; Tio, M.; Ng, S.H.; Li, Z.; Lim, C.Y.; Zhao, Y.; Tan, E.K. Derivation of human induced pluripotent stem cell (iPSC) line with LRRK2 gene R1398H variant in Parkinson’s disease. Stem. Cell Res. 2017, 18, 48–50. [Google Scholar] [CrossRef]
- Ma, D.; Ng, E.Y.; Zeng, L.; Lim, C.Y.; Zhao, Y.; Tan, E.K. Development of a human induced pluripotent stem cell (iPSC) line from a Parkinson’s disease patient carrying the N551K variant in LRRK2 gene. Stem. Cell Res. 2017, 18, 51–53. [Google Scholar] [CrossRef]
- Madzar, D.; Schulte, C.; Gasser, T. Screening for LRRK2 R1441 mutations in a cohort of PSP patients from Germany. Eur. J. Neurol. 2009, 16, 1230–1232. [Google Scholar] [CrossRef] [PubMed]
- Ross, O.A.; Whittle, A.J.; Cobb, S.A.; Hulihan, M.M.; Lincoln, S.J.; Toft, M.; Farrer, M.J.; Dickson, D.W. Lrrk2 R1441 substitution and progressive supranuclear palsy. Neuropathol. Appl. Neurobiol. 2006, 32, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Chen-Plotkin, A.S. LRRK2 and survival in progressive supranuclear palsy. Lancet Neurol. 2020. [Google Scholar] [CrossRef]
- Jabbari, E.; Koga, S.; Valentino, R.R.; Reynolds, R.H.; Ferrari, R.; Tan, M.M.X.; Rowe, J.B.; Dalgard, C.L.; Scholz, S.W.; Dickson, D.W.; et al. Genetic determinants of survival in progressive supranuclear palsy: A genome-wide association study. Lancet Neurol. 2020. [Google Scholar] [CrossRef]
- Heckman, M.G.; Soto-Ortolaza, A.I.; Contreras, M.Y.S.; Murray, M.E.; Pedraza, O.; Diehl, N.N.; Walton, R.; Labbe, C.; Lorenzo-Betancor, O.; Uitti, R.J.; et al. LRRK2 variation and dementia with Lewy bodies. Parkinsonism Relat. Disord. 2016, 31, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Chen, Y.; Cao, B.; Zhao, B.; Wei, Q.; Guo, X.; Yang, Y.; Yuan, L.; Shang, H. An association analysis of the R1628P and G2385R polymorphisms of the LRRK2 gene in multiple system atrophy in a Chinese population. Parkinsonism Relat. Disord. 2015, 21, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Ozelius, L.J.; Foroud, T.; May, S.; Senthil, G.; Sandroni, P.; Low, P.A.; Reich, S.; Colcher, A.; Stern, M.B.; Ondo, W.G.; et al. G2019S mutation in the leucine-rich repeat kinase 2 gene is not associated with multiple system atrophy. Mov. Disord. 2007, 22, 546–549. [Google Scholar] [CrossRef]
- Cho, J.W.; Kim, S.Y.; Park, S.S.; Jeon, B.S. The G2019S LRRK2 Mutation is Rare in Korean Patients with Parkinson’s Disease and Multiple System Atrophy. J. Clin. Neurol. 2009, 5, 29–32. [Google Scholar] [CrossRef]
- Fatahian, R.; Bagheri, S.R.; Sadeghi, M. A meta-analysis of leucine-rich repeat kinase 2 (LRRK2) polymorphisms in Alzheimer’s disease. Folia Neuropathol. 2019, 57, 1–5. [Google Scholar] [CrossRef]
- Deng, H.; Le, W.; Davidson, A.L.; Xie, W.; Jankovic, J. The LRRK2 I2012T, G2019S and I2020T mutations are not common in patients with essential tremor. Neurosci. Lett. 2006, 407, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.K.; Lee, J.; Lim, H.Q.; Yuen, Y.; Zhao, Y. Essential tremor and the common LRRK2 G2385R variant. Parkinsonism Relat. Disord. 2008, 14, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Vitale, C.; Ciotti, P.; Gulli, R.; Bellone, E.; Scaglione, C.; Abbruzzese, G.; Martinelli, P.; Barone, P.; Mandich, P. Common mutations in the LRRK2 exon 41 are not responsible for essential tremor in Italian patients. Parkinsonism Relat. Disord. 2009, 15, 162–163. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.N.; Kisselev, S.; Park, N.; Ross, B.; Verbitsky, M.; Rios, E.; Alcalay, R.N.; Lee, J.H.; Louis, E.D. Mutations in the Parkinson’s disease genes, Leucine Rich Repeat Kinase 2 (LRRK2) and Glucocerebrosidase (GBA), are not associated with essential tremor. Parkinsonism Relat. Disord. 2010, 16, 132–135. [Google Scholar] [CrossRef][Green Version]
- Chen, H.; Yuan, L.; Song, Z.; Deng, X.; Yang, Z.; Gong, L.; Zi, X.; Deng, H. Genetic Analysis of LRRK1 and LRRK2 Variants in Essential Tremor Patients. Genet. Test. Mol. Biomarkers 2018, 22, 398–402. [Google Scholar] [CrossRef]
- Chao, Y.X.; Ng, E.Y.; Tan, L.; Prakash, K.M.; Au, W.L.; Zhao, Y.; Tan, E.K. Lrrk2 R1628P variant is a risk factor for essential tremor. Sci. Rep. 2015, 5, 9029. [Google Scholar] [CrossRef]
- Brudek, T. Inflammatory Bowel Diseases and Parkinson’s Disease. J. Parkinsons Dis. 2019, 9, S331–S344. [Google Scholar] [CrossRef]
- Zhu, F.; Li, C.; Gong, J.; Zhu, W.; Gu, L.; Li, N. The risk of Parkinson’s disease in inflammatory bowel disease: A systematic review and meta-analysis. Dig. Liver Dis. 2019, 51, 38–42. [Google Scholar] [CrossRef]
- Fuji, R.N.; Flagella, M.; Baca, M.; MA, S.B.; Brodbeck, J.; Chan, B.K.; Fiske, B.K.; Honigberg, L.; Jubb, A.M.; Katavolos, P.; et al. Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci. Transl. Med. 2015, 7, 273ra15. [Google Scholar] [CrossRef]
- Baptista, M.A.S.; Merchant, K.; Barrett, T.; Bhargava, S.; Bryce, D.K.; Ellis, J.M.; Estrada, A.A.; Fell, M.J.; Fiske, B.K.; Fuji, R.N.; et al. LRRK2 inhibitors induce reversible changes in nonhuman primate lungs without measurable pulmonary deficits. Sci. Transl. Med. 2020, 12, eaav0820. [Google Scholar] [CrossRef]
- Ahadi, S.; Zhou, W.; Schussler-Fiorenza Rose, S.M.; Sailani, M.R.; Contrepois, K.; Avina, M.; Ashland, M.; Brunet, A.; Snyder, M. Personal aging markers and ageotypes revealed by deep longitudinal profiling. Nat. Med. 2020, 26, 83–90. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| SNP (rs ID) | cDNA NM_198578.4 | Protein XP_5268686.1 | Odds Ratio (95% Confidence Interval) | Allele Frequency | Pathogenicity Score | Domain | Enzymatic Impact/Function |
|---|---|---|---|---|---|---|---|
| rs34594498 | c.1256C > T | p.A419V | 2.45 (1.43, 4.2) East Asian | 0.0004854 | Not reported | ARM | Does not alter kinase activity [70] |
| rs7308720 | c.1653C > A, C > G | p.N551K | Not reported | 0.000003985 | Not reported | No domain | Does not alter kinase activity [70,71]; CD protection [71] |
| rs17466213 | c.4111A > G | p.I1371V | Not reported | 0.0008429 | Probable | ROC | Increased GTP binding [72] |
| rs7133914 | c.4193G > A | p.R1398H | 0.81 (0.75, 0.89) Mixed | 0.00001772 | Not reported | ROC | Increased GTP binding and GTPase activity [73]; CD protection [71] |
| rs74163686 | c.4309A > C | p.N1437H | Low frequency, not reported | Not found | Definite | ROC | Disrupts GTP hydrolysis, increased kinase activity [42,74] |
| rs33939927 | c.4321C > T/A/G | p.R1441C/G/H | 12.75 (3.11, 52.27) | 0.00001195 | Definite | ROC | Disrupts GTP hydrolysis; increased kinase activity [42,75,76] |
| rs33949390 | c.4883G > T | p.R1628P | 2.13 (Asian) | 0.0001491 | Not reported | COR | Increased kinase activity [77]; protective for T1R [78] |
| rs35801418 | c.5096A > G | p.Y1699C | Low frequency, not reported | Not found | Definite | COR | Disrupts GTP hydrolysis; increased kinase activity [42,79] |
| rs34637584 | c.6055G > A | p.G2019S | 13.16 (10.16, 17.04) Mixed ethnicities | 0.0004884 | Definite | Kinase | Increased kinase activity [28,42,47,75] |
| rs35870237 | c.6059T > C | p.I2020T | Low frequency, not reported | Not found | Definite | Kinase | Increased kinase activity in cells [42,50,72] |
| rs33995883 | c.6241A > G | p.N2081D | Not reported | 0.01685 | Not reported | Kinase | CD risk and increased kinase activity [71] |
| rs34778348 | c.7153G > A | p.G2385R | 2.27 (2.03, 2.53) East Asian | 0.001680 | Possible | WD40 | Increased kinase activity in cells [30,54] |
| rs3761863 | c.7190T > C | p.M2397T | Not reported | 0.6191 | Not reported | WD40 | CD risk enhances IFN-γ response [80]; T1R proinflammatory [81]; destabilizes protein [82] |
| LRRK2 Mutation | Clinical Presentation | # of Cases | SND | LBD | MSA | PSP | CBD | TDP-43 | Low AD | Interim AD | High AD | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c.4111A > G (p.I1371V) | PD | 1 | - | 1 | - | - | - | - | - | - | - | [55,113] |
| MSA | 1 | - | - | 1 | - | - | - | - | - | - | [58] | |
| c.4309C > A (p.N1437H) | PD | 1 | - | 1 | - | - | - | - | 1 | - | - | [55,114] |
| c.4321C > T/G (p.R1441C/G) | PD | 6 | 3 | 2 | - | 1 | - | - | - | - | - | [115] |
| PD | 4 | 1 | 2 | - | 1 | - | - | - | - | - | [1,116] (Family D, R1441C) | |
| PD | 6 | 3 | 2 | - | 1 | - | - | - | - | - | [1,55,115] | |
| c.4322 G > A (p.R1441H) | PD | 3 | 3 | - | - | - | - | - | - | - | - | [57] |
| c.5096A > G (p.Y1699C) | PD | 3 | 2 | 1 | - | - | - | - | - | - | 1 | [55,117] (Lincolnshire, III.13), [1,118] (Fam A, III.27, III.29) |
| c.6055G > A (p.G2019S) | 2 PD, 3 PDD | 5 | 1 | 2 | - | 2 | - | - | 1 | 3 | 1 | [24] |
| 7 PD, 2 PDD | 9 | 4 | 5 | - | - | - | - | - | - | 3 | [56,119] (3 cases) | |
| MSA | 1 | - | - | 1 | - | - | - | - | - | - | [105] | |
| PSP | - | - | - | - | 2 | - | - | - | - | - | [108] | |
| PD | 1 | 1 | - | - | - | - | - | 1 | - | - | [103] | |
| PSP | 1 | - | - | - | 1 | - | - | 1 | - | - | [112] | |
| PD | 18 | 5 | 9 | - | 1 | - | - | - | - | 3 | [55,120,121], 3 cases excluded from [119] | |
| PD | 1 | - | - | - | - | - | 1 | - | - | 1 | [107], patient also carries MAPT c.370C > G, (p.Q124E) variant | |
| c.6059T > C (p.I2020T) | PD/MSA/PSP | 9 | 5 | 1 | 1 | 4 | - | - | - | - | - | Sagamihara [55,104,109,122,123]; (4 cases with tau pathology) |
| c.3494T > C, p.L1165P | PDD, Pat. E | 1 | - | 1 | - | - | - | - | - | - | 1 | [56,124] |
| c.2378 G > T, p.R793M | PD, Pat. D | 1 | - | 1 | - | - | - | - | - | 1 | - | [56,124] |
| c.5120G > A, p.R1707K | CBD | 1 | - | - | - | - | 1 | - | - | - | 1 | [112] |
| c.4883G > T, p.R1628P | PSP/CBD | 3 | - | - | - | 2 | 1 | - | - | - | - | [112] |
| c.4237G > A, p.A1413T | PSP | 1 | - | - | - | 1 | - | - | - | - | - | [112] |
| Total cases | 74 | 24 | 28 | 3 | 16 | 2 | 1 | 4 | 4 | 9 |
| Association with PD Risk Alleles | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LRRK2 Variants | Sample Size | Population | SNCA | DNM3 | GAK | BDNF | MAPT | BST1 | Rab29/Rab7L1 | VAMP4 | PD-PRS | Ref. |
| rs1491942 (intronic) | 1381 PD, 1328 ctrl | North America, Irish, Polish | rs356219, no | N/A | rs6599388 no | N/A | rs2942168 no | rs11724635 no | rs708723 no | N/A | N/A | [179] |
| rs1491942, rs7133914 (R1398H) | not reported | Caucasian, Asian (GEOPD) | N/A | N/A | N/A | N/A | N/A | N/A | No | N/A | N/A | [180] |
| p.R1441C, p.Y1699C, p.G2019S, p.I2020T | 44 carriers, 19 families | European, North American | No | N/A | N/A | N/A | rs2435207, AAO | N/A | N/A | N/A | N/A | [181] |
| p.G2385R, p.R1628P | 231 PD G2385R, 65 PD R1628P | Chinese | No | N/A | N/A | N/A | No | No | No | N/A | N/A | [177] |
| p.G2385R | 64 PD | Chinese | N/A | N/A | N/A | AAO | N/A | N/A | N/A | N/A | N/A | [175] |
| p.G2385R | 53 PD | Chinese | N/A | N/A | N/A | N/A | IVS1 + 124C > G increased risk (major allele) | N/A | N/A | N/A | N/A | [176] |
| p.G2385R, p.R1628P | 82 PD G2385R, 46 PD R1628P | Han Chinese | rs356219, risk and AAO (OR: 1.5) | rs2421947 No | rs1564282 No | N/A | N/A | N/A | N/A | N/A | N/A | [178] |
| p.G2019S | 84 PD | European | rs356219, AAO | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | [169] |
| p.G2019S | 84 PD | Ashkenazi | N/A | N/A | N/A | N/A | rs11079727, AAO (older in minor allele) | N/A | N/A | N/A | N/A | [170] |
| p.G2019S | 101 PD | Arab-Berber | No | N/A | N/A | N/A | No | N/A | N/A | N/A | N/A | [171] |
| p.G2019S | 41 families: 150 PD, 103 unaffected, 232 unrelated | Arab-Berber | N/A | rs2421947, AAO earlier with GG allele | N/A | N/A | N/A | N/A | N/A | N/A | N/A | [172] |
| p.G2019S | 210 PD, 119 unaffected | European (Spain) | rs356219, AAO | No | N/A | N/A | N/A | N/A | N/A | N/A | N/A | [173] |
| p.G2019S, rs10878226 (2 kb upstream) | 724 PD p.G2019S, 4882 PD rs10878226 | IPDGC and other | N/A | No AAO | N/A | N/A | N/A | N/A | N/A | No | N/A | [174] |
| p.G2019S | 841 (439 PD, 394 unaffected) | European, North America | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | P (OR: 1.34) | [189] |
| # of Significant Studies (# Total Studies) | 3 (6) | 1 (4) | 0 (2) | 1 (1) | 3 (6) | 0 (2) | 0 (3) | 0 (1) | 1 (1) | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chittoor-Vinod, V.G.; Nichols, R.J.; Schüle, B. Genetic and Environmental Factors Influence the Pleomorphy of LRRK2 Parkinsonism. Int. J. Mol. Sci. 2021, 22, 1045. https://doi.org/10.3390/ijms22031045
Chittoor-Vinod VG, Nichols RJ, Schüle B. Genetic and Environmental Factors Influence the Pleomorphy of LRRK2 Parkinsonism. International Journal of Molecular Sciences. 2021; 22(3):1045. https://doi.org/10.3390/ijms22031045
Chicago/Turabian StyleChittoor-Vinod, Vinita G., R. Jeremy Nichols, and Birgitt Schüle. 2021. "Genetic and Environmental Factors Influence the Pleomorphy of LRRK2 Parkinsonism" International Journal of Molecular Sciences 22, no. 3: 1045. https://doi.org/10.3390/ijms22031045
APA StyleChittoor-Vinod, V. G., Nichols, R. J., & Schüle, B. (2021). Genetic and Environmental Factors Influence the Pleomorphy of LRRK2 Parkinsonism. International Journal of Molecular Sciences, 22(3), 1045. https://doi.org/10.3390/ijms22031045