
APOE: The New Frontier in the Development of a Therapeutic Target towards Precision Medicine in Late-Onset Alzheimer’s




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
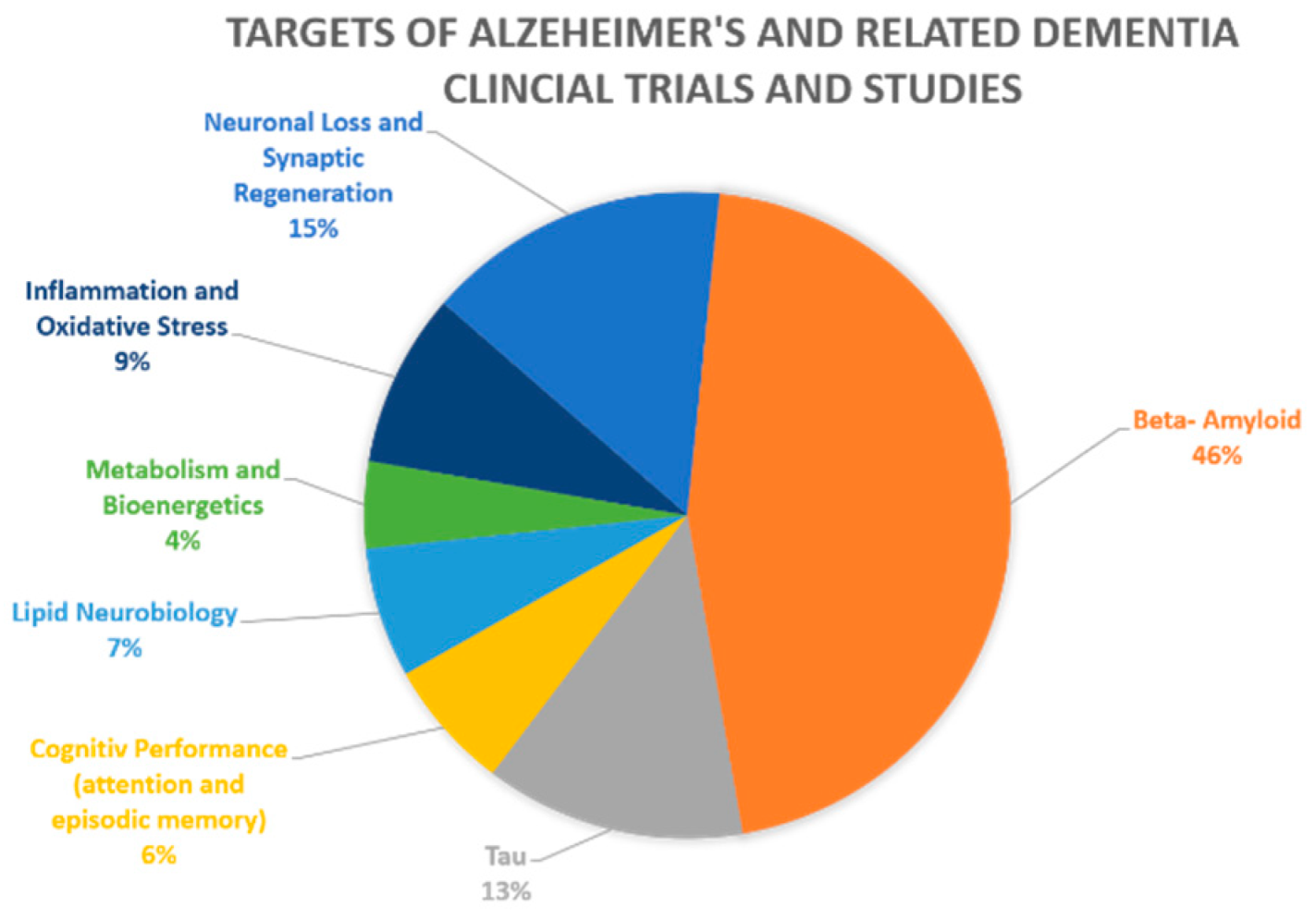
1. Introduction
2. The APOE Locus Is the Strongest Genetic Risk for LOAD
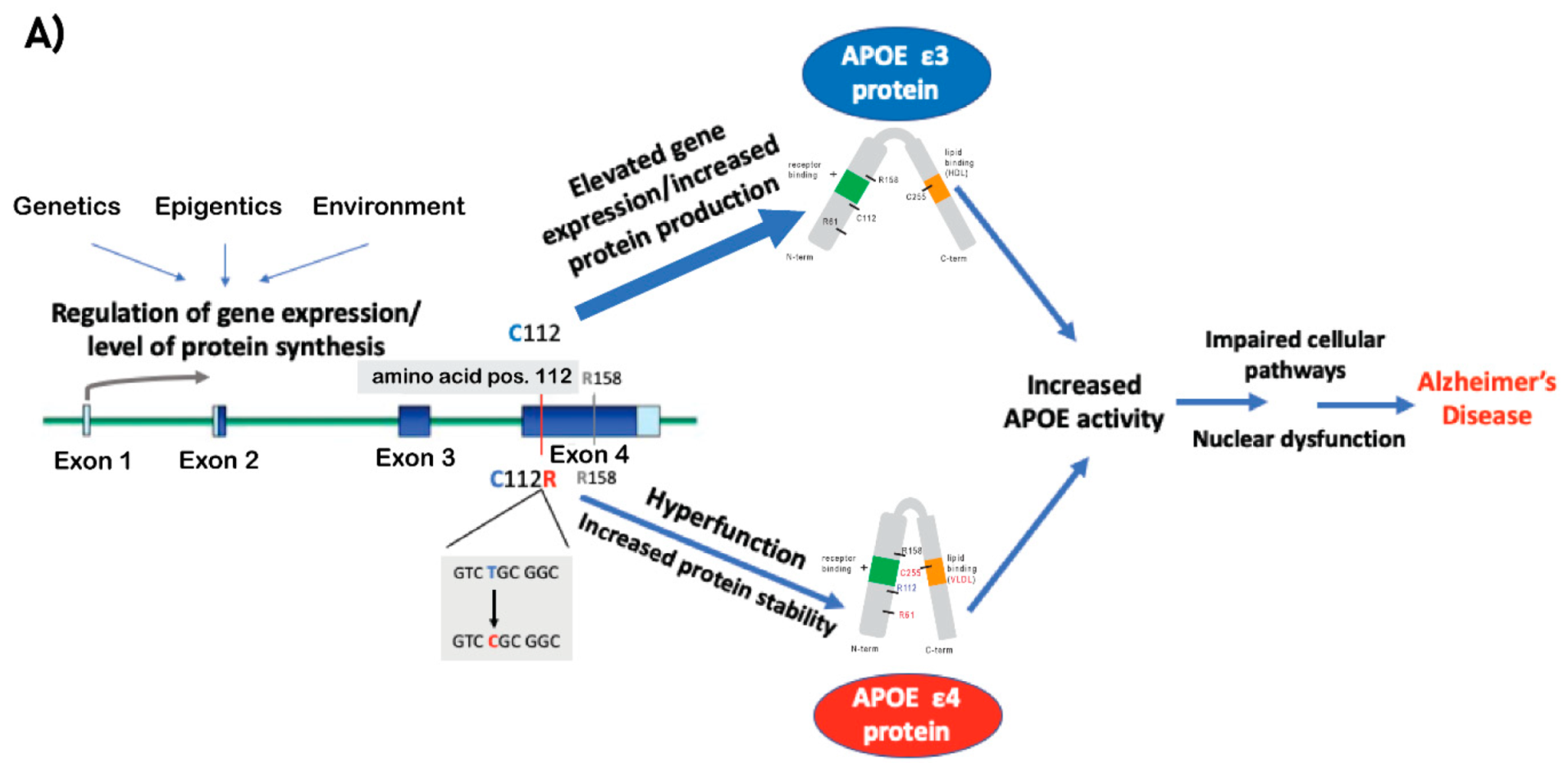
2.1. ApoE Protein: Function and Isoforms
2.2. APOE e4
2.3. Dysregulation of APOE Expression
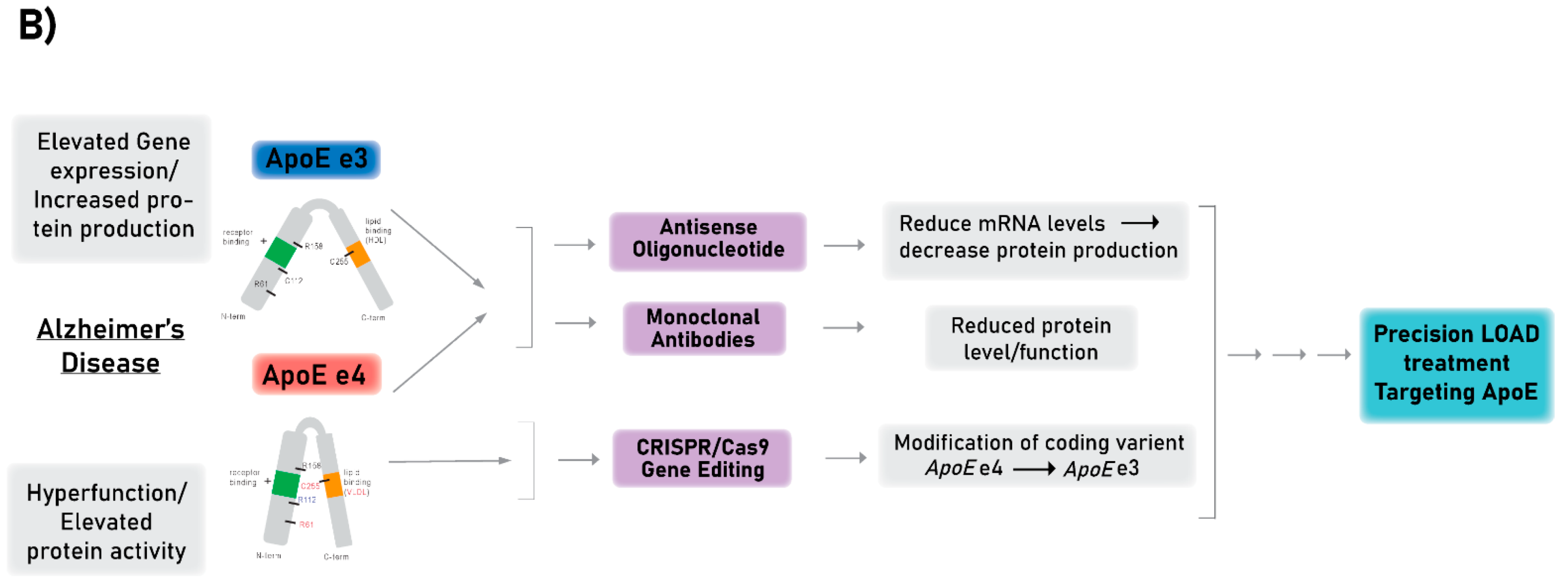
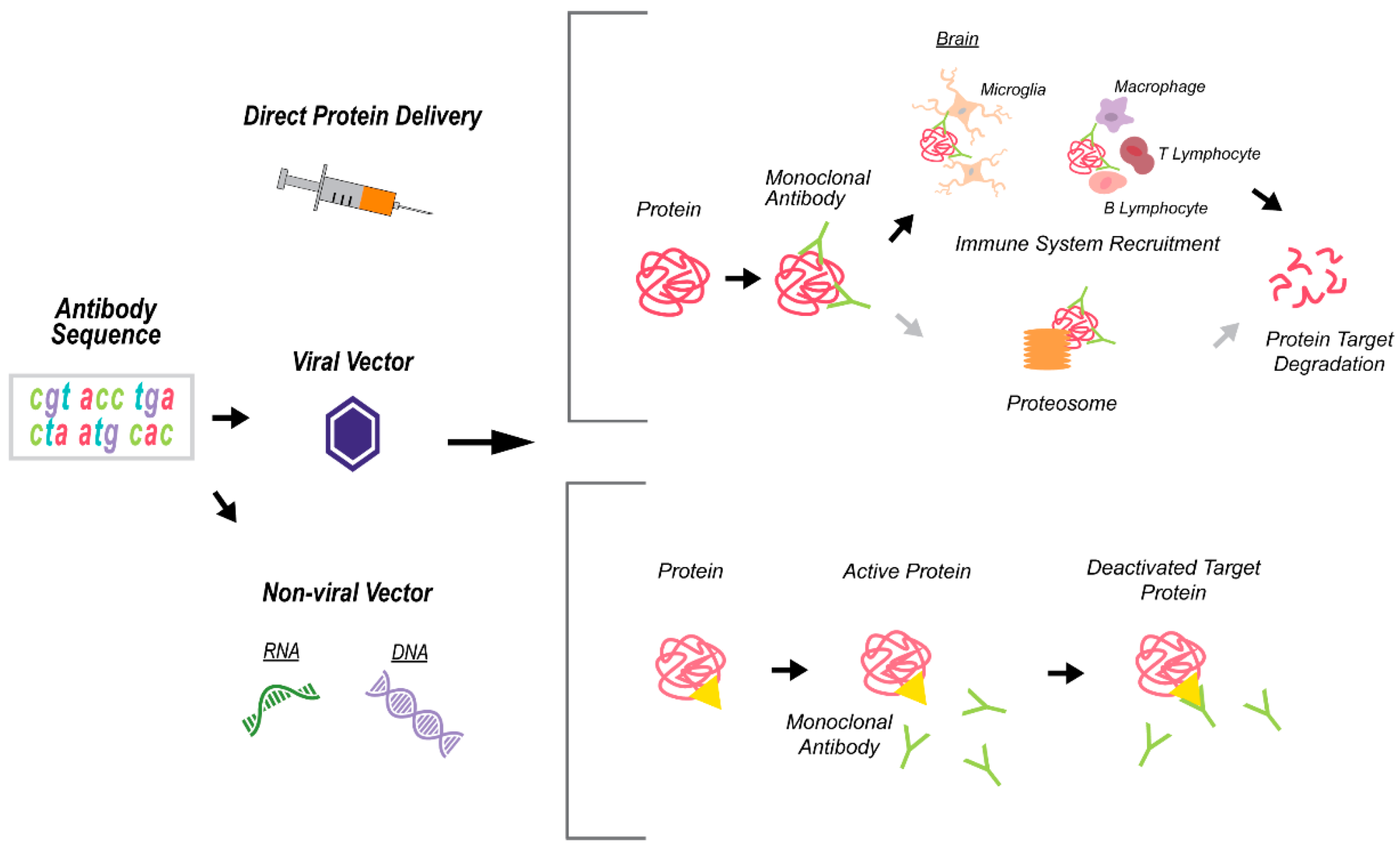
3. Technologies for Targeting APOE as a Proof-of-Concept for LOAD Therapies
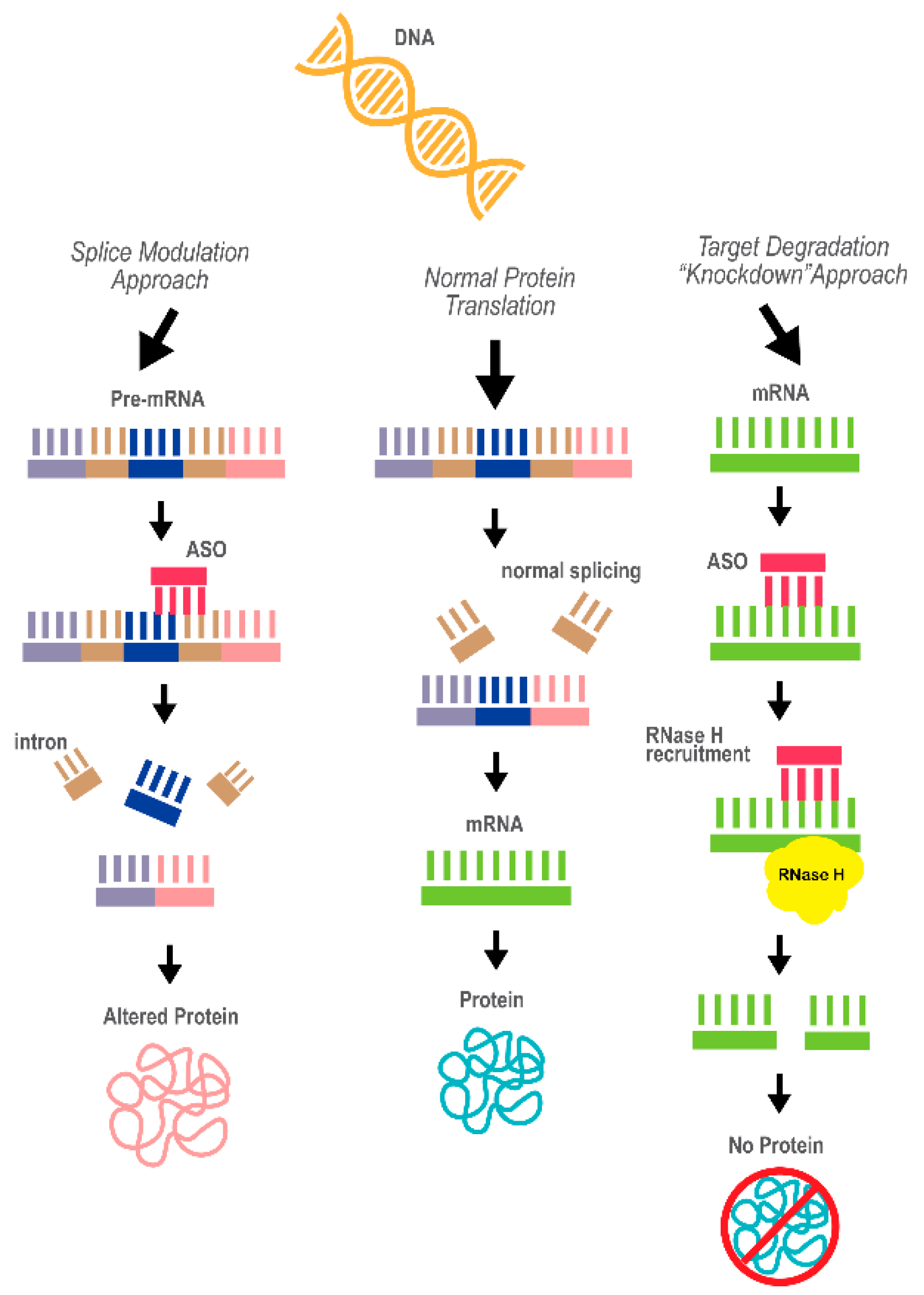
3.1. Antisense Oligonucleotide Therapy
3.1.1. ASO Technology and Application in Disease Therapy
3.1.2. Current Progress in ASO Therapy Targeting APOE
3.2. Monoclonal Antibody Therapy
3.2.1. Monoclonal Antibody (mAb) Approach and Application in Disease Therapy
3.2.2. Monoclonal Antibodies Targeting ApoE
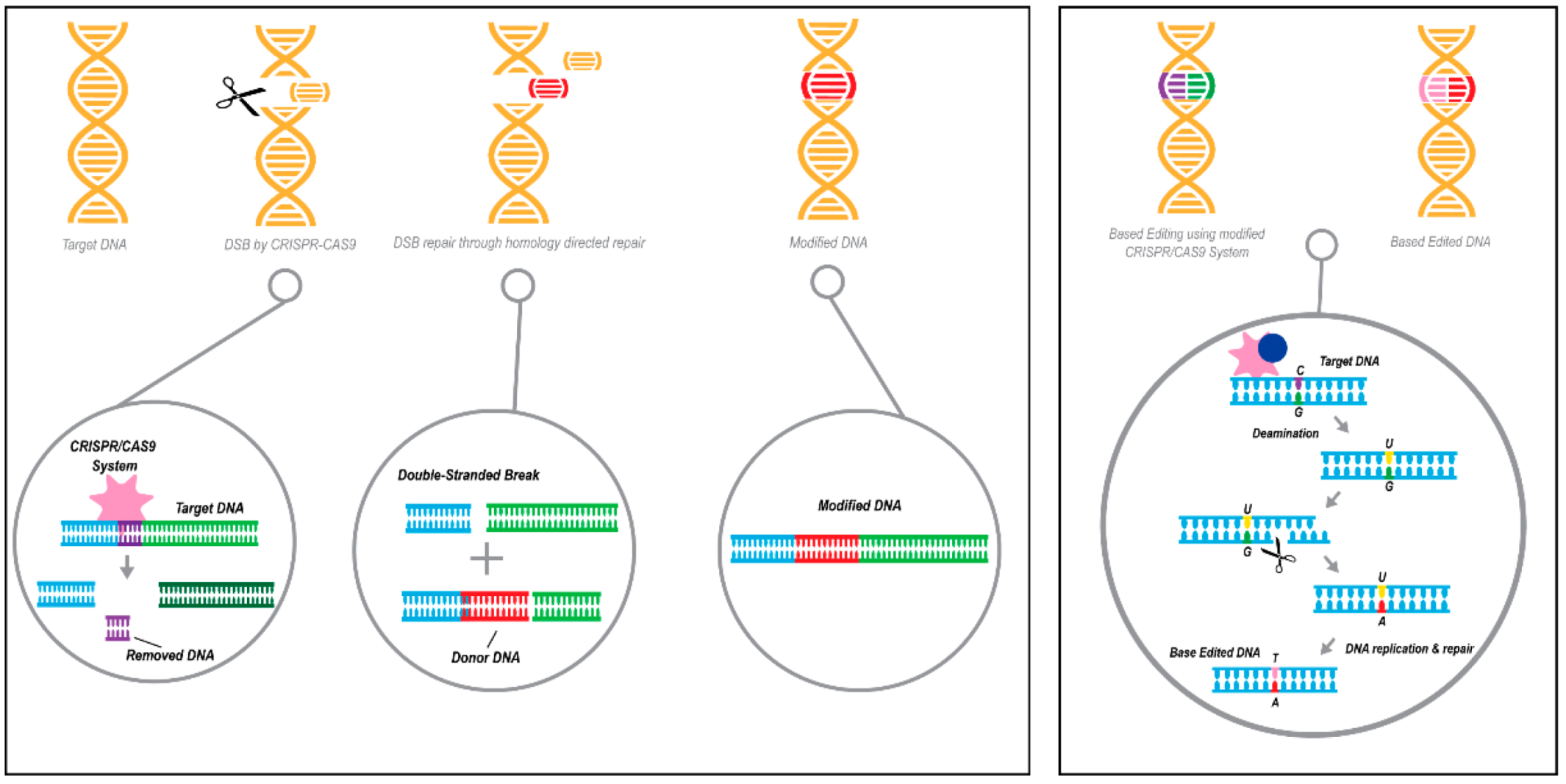
3.3. Gene Editing
3.3.1. Applications of Gene Editing in Disease Therapy
3.3.2. Using CRISPR/Cas Technologies to Target APOE
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| LOAD | Late-onset Alzheimer’s Disease |
| APOE | Apolipoprotein E |
| ASO | Antisense Oligonucleotides |
| mAB | Monoclonal Antibodies |
References
- Costs of Alzheimer’s to Medicare and Medicaid; Alzheimer’s Association: Alzheimer’s Imipact Movement: Factsheet March 2020; Alzheimer’s Association: Chicago, IL, USA, 2020.
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Olivares, D.; Deshpande, V.K.; Shi, Y.; Lahiri, D.K.; Greig, N.H.; Rogers, J.T.; Huang, X. N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer’s disease, vascular dementia and Parkinson’s disease. Curr. Alzheimer Res. 2012, 9, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Mawuenyega, K.G.; Kasten, T.; Sigurdson, W.; Bateman, R.J. Amyloid-beta isoform metabolism quantitation by stable isotope-labeled kinetics. Anal. Biochem. 2013, 440, 56–62. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reitz, C. Toward precision medicine in Alzheimer’s disease. Ann. Transl. Med. 2016, 4, 107. [Google Scholar] [CrossRef]
- Chiba-Falek, O.; Lutz, M.W. Towards precision medicine in Alzheimer’s disease: Deciphering genetic data to establish informative biomarkers. Expert Rev. Precis Med. Drug Dev. 2017, 2, 47–55. [Google Scholar] [CrossRef]
- NIA-Funded Active Alzheimer’s and Related Dementias Clinical Trials and Studies. National Institute on Aging, 15 July 2020; Volume NIA-Funded Active Alzheimer’s and Related Dementias Clinical Trials and Studies. Available online: https://www.nia.nih.gov/research/ongoing-AD-trials#:~:text=The%20National%20Institute%20on%20Aging,AD%2FADRD%20that%20are%20addressed (accessed on 26 October 2020).
- Huynh, T.V.; Liao, F.; Francis, C.M.; Robinson, G.O.; Serrano, J.R.; Jiang, H.; Roh, J.; Finn, M.B.; Sullivan, P.M.; Esparza, T.J.; et al. Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of beta-amyloidosis. Neuron 2017, 96, 1013–1023.e1014. [Google Scholar] [CrossRef]
- Brody, D.L.; Holtzman, D.M. Active and passive immunotherapy for neurodegenerative disorders. Annu. Rev. Neurosci. 2008, 31, 175–193. [Google Scholar] [CrossRef]
- Kim, J.; Eltorai, A.E.; Jiang, H.; Liao, F.; Verghese, P.B.; Kim, J.; Stewart, F.R.; Basak, J.M.; Holtzman, D.M. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Abeta amyloidosis. J. Exp. Med. 2012, 209, 2149–2156. [Google Scholar] [CrossRef]
- Shen, L.; Jia, J. An Overview of Genome-Wide Association Studies in Alzheimer’s Disease. Neurosci. Bull. 2016, 32, 183–190. [Google Scholar] [CrossRef]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72 Pt. A, 3–12. [Google Scholar] [CrossRef]
- Frieden, C.; Garai, K. Concerning the structure of apoE. Protein Sci. 2013, 22, 1820–1825. [Google Scholar] [CrossRef] [PubMed]
- Frieden, C.; Garai, K. Structural differences between apoE3 and apoE4 may be useful in developing therapeutic agents for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2012, 109, 8913–8918. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, K.; Zhao, H. Haplotype-association analysis. Adv. Genet. 2008, 60, 335–405. [Google Scholar] [CrossRef] [PubMed]
- Schmechel, D.E.; Saunders, A.M.; Strittmatter, W.J.; Crain, B.J.; Hulette, C.M.; Joo, S.H.; Pericak-Vance, M.A.; Goldgaber, D.; Roses, A.D. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 9649–9653. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J.; et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat. Commun. 2020, 11, 667. [Google Scholar] [CrossRef]
- Gottschalk, W.K.; Mihovilovic, M.; Roses, A.D.; Chiba-Falek, O. The Role of Upregulated APOE in Alzheimer’s Disease Etiology. J. Alzheimers Dis. Parkinsonism 2016, 6. [Google Scholar] [CrossRef]
- Huang, Y.A.; Zhou, B.; Wernig, M.; Sudhof, T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Abeta Secretion. Cell 2017, 168, 427–441 e421. [Google Scholar] [CrossRef]
- Sen, A.; Nelson, T.J.; Alkon, D.L. ApoE4 and Abeta Oligomers Reduce BDNF Expression via HDAC Nuclear Translocation. J. Neurosci. 2015, 35, 7538–7551. [Google Scholar] [CrossRef] [PubMed]
- Theendakara, V.; Patent, A.; Peters Libeu, C.A.; Philpot, B.; Flores, S.; Descamps, O.; Poksay, K.S.; Zhang, Q.; Cailing, G.; Hart, M.; et al. Neuroprotective Sirtuin ratio reversed by ApoE4. Proc. Natl. Acad. Sci. USA 2013, 110, 18303–18308. [Google Scholar] [CrossRef] [PubMed]
- Theendakara, V.; Peters-Libeu, C.A.; Spilman, P.; Poksay, K.S.; Bredesen, D.E.; Rao, R.V. Direct Transcriptional Effects of Apolipoprotein E. J. Neurosci. 2016, 36, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef]
- Tambini, M.D.; Pera, M.; Kanter, E.; Yang, H.; Guardia-Laguarta, C.; Holtzman, D.; Sulzer, D.; Area-Gomez, E.; Schon, E.A. ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 2016, 17, 27–36. [Google Scholar] [CrossRef]
- Hatters, D.M.; Zhong, N.; Rutenber, E.; Weisgraber, K.H. Amino-terminal domain stability mediates apolipoprotein E aggregation into neurotoxic fibrils. J. Mol. Biol. 2006, 361, 932–944. [Google Scholar] [CrossRef][Green Version]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Heinzen, E.L.; Need, A.C.; Hayden, K.M.; Chiba-Falek, O.; Roses, A.D.; Strittmatter, W.J.; Burke, J.R.; Hulette, C.M.; Welsh-Bohmer, K.A.; Goldstein, D.B. Genome-wide scan of copy number variation in late-onset Alzheimer’s disease. J. Alzheimers Dis. 2010, 19, 69–77. [Google Scholar] [CrossRef]
- Kamboh, M.I.; Barmada, M.M.; Demirci, F.Y.; Minster, R.L.; Carrasquillo, M.M.; Pankratz, V.S.; Younkin, S.G.; Saykin, A.J.; Sweet, R.A.; Alzheimer’s Disease Neuroimaging Initiative; et al. Genome-wide association analysis of age-at-onset in Alzheimer’s disease. Mol. Psychiatry 2012, 17, 1340–1346. [Google Scholar] [CrossRef]
- Kamboh, M.I.; Demirci, F.Y.; Wang, X.; Minster, R.L.; Carrasquillo, M.M.; Pankratz, V.S.; Younkin, S.G.; Saykin, A.J.; Jun, G.; Alzheimer’s Disease Neuroimaging Initiative; et al. Genome-wide association study of Alzheimer’s disease. Transl. Psychiatry 2012, 2, e117. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Fitzpatrick, A.L.; Ikram, M.A.; DeStefano, A.L.; Gudnason, V.; Boada, M.; Bis, J.C.; Smith, A.V.; Carassquillo, M.M.; Lambert, J.C.; et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010, 303, 1832–1840. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Coon, K.D.; Myers, A.J.; Craig, D.W.; Webster, J.A.; Pearson, J.V.; Lince, D.H.; Zismann, V.L.; Beach, T.G.; Leung, D.; Bryden, L.; et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J. Clin. Psychiatry 2007, 68, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Lutz, F.; Yu, C.E. Functional analysis of APOE locus genetic variation implicates regional enhancers in the regulation of both TOMM40 and APOE. J. Hum. Genet. 2012, 57, 18–25. [Google Scholar] [CrossRef]
- Lescai, F.; Chiamenti, A.M.; Codemo, A.; Pirazzini, C.; D’Agostino, G.; Ruaro, C.; Ghidoni, R.; Benussi, L.; Galimberti, D.; Esposito, F.; et al. An APOE haplotype associated with decreased epsilon4 expression increases the risk of late onset Alzheimer’s disease. J. Alzheimers Dis. 2011, 24, 235–245. [Google Scholar] [CrossRef]
- Xin, X.Y.; Ding, J.Q.; Chen, S.D. Apolipoprotein E promoter polymorphisms and risk of Alzheimer’s disease: Evidence from meta-analysis. J. Alzheimers Dis. 2010, 19, 1283–1294. [Google Scholar] [CrossRef]
- Laws, S.M.; Hone, E.; Gandy, S.; Martins, R.N. Expanding the association between the APOE gene and the risk of Alzheimer’s disease: Possible roles for APOE promoter polymorphisms and alterations in APOE transcription. J. Neurochem. 2003, 84, 1215–1236. [Google Scholar] [CrossRef]
- Nicodemus, K.K.; Stenger, J.E.; Schmechel, D.E.; Welsh-Bohmer, K.A.; Saunders, A.M.; Roses, A.D.; Gilbert, J.R.; Vance, J.M.; Haines, J.L.; Pericak-Vance, M.A.; et al. Comprehensive association analysis of APOE regulatory region polymorphisms in Alzheimer disease. Neurogenetics 2004, 5, 201–208. [Google Scholar] [CrossRef]
- Lambert, J.C.; Araria-Goumidi, L.; Myllykangas, L.; Ellis, C.; Wang, J.C.; Bullido, M.J.; Harris, J.M.; Artiga, M.J.; Hernandez, D.; Kwon, J.M.; et al. Contribution of APOE promoter polymorphisms to Alzheimer’s disease risk. Neurology 2002, 59, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Tycko, B.; Lee, J.H.; Ciappa, A.; Saxena, A.; Li, C.M.; Feng, L.; Arriaga, A.; Stern, Y.; Lantigua, R.; Shachter, N.; et al. APOE and APOC1 promoter polymorphisms and the risk of Alzheimer disease in African American and Caribbean Hispanic individuals. Arch. Neurol. 2004, 61, 1434–1439. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.E.; Seltman, H.; Peskind, E.R.; Galloway, N.; Zhou, P.X.; Rosenthal, E.; Wijsman, E.M.; Tsuang, D.W.; Devlin, B.; Schellenberg, G.D. Comprehensive analysis of APOE and selected proximate markers for late-onset Alzheimer’s disease: Patterns of linkage disequilibrium and disease/marker association. Genomics 2007, 89, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Linnertz, C.; Anderson, L.; Gottschalk, W.; Crenshaw, D.; Lutz, M.W.; Allen, J.; Saith, S.; Mihovilovic, M.; Burke, J.R.; Welsh-Bohmer, K.A.; et al. The cis-regulatory effect of an Alzheimer’s disease-associated poly-T locus on expression of TOMM40 and apolipoprotein E genes. Alzheimers Dement. 2014, 10, 541–551. [Google Scholar] [CrossRef]
- Zarow, C.; Victoroff, J. Increased apolipoprotein E mRNA in the hippocampus in Alzheimer disease and in rats after entorhinal cortex lesioning. Exp. Neurol. 1998, 149, 79–86. [Google Scholar] [CrossRef]
- Matsui, T.; Ingelsson, M.; Fukumoto, H.; Ramasamy, K.; Kowa, H.; Frosch, M.P.; Irizarry, M.C.; Hyman, B.T. Expression of APP pathway mRNAs and proteins in Alzheimer’s disease. Brain Res. 2007, 1161, 116–123. [Google Scholar] [CrossRef]
- Akram, A.; Schmeidler, J.; Katsel, P.; Hof, P.R.; Haroutunian, V. Association of ApoE and LRP mRNA levels with dementia and AD neuropathology. Neurobiol. Aging 2012, 33, 628 e1. [Google Scholar] [CrossRef]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Hashemiaghdam, A.; Mroczek, M. Microglia heterogeneity and neurodegeneration: The emerging paradigm of the role of immunity in Alzheimer’s disease. J. Neuroimmunol. 2020, 341, 577185. [Google Scholar] [CrossRef]
- Zheng, J.Y.; Sun, J.; Ji, C.M.; Shen, L.; Chen, Z.J.; Xie, P.; Sun, Y.Z.; Yu, R.T. Selective deletion of apolipoprotein E in astrocytes ameliorates the spatial learning and memory deficits in Alzheimer’s disease (APP/PS1) mice by inhibiting TGF-beta/Smad2/STAT3 signaling. Neurobiol. Aging 2017, 54, 112–132. [Google Scholar] [CrossRef] [PubMed]
- Bien-Ly, N.; Gillespie, A.K.; Walker, D.; Yoon, S.Y.; Huang, Y. Reducing human apolipoprotein E levels attenuates age-dependent Abeta accumulation in mutant human amyloid precursor protein transgenic mice. J. Neurosci. 2012, 32, 4803–4811. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jiang, H.; Park, S.; Eltorai, A.E.; Stewart, F.R.; Yoon, H.; Basak, J.M.; Finn, M.B.; Holtzman, D.M. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J. Neurosci. 2011, 31, 18007–18012. [Google Scholar] [CrossRef] [PubMed]
- Foraker, J.; Millard, S.P.; Leong, L.; Thomson, Z.; Chen, S.; Keene, C.D.; Bekris, L.M.; Yu, C.E. The APOE Gene is Differentially Methylated in Alzheimer’s Disease. J. Alzheimers Dis. 2015, 48, 745–755. [Google Scholar] [CrossRef]
- Shao, Y.; Shaw, M.; Todd, K.; Khrestian, M.; D’Aleo, G.; Barnard, P.J.; Zahratka, J.; Pillai, J.; Yu, C.E.; Keene, C.D.; et al. DNA methylation of TOMM40-APOE-APOC2 in Alzheimer’s disease. J. Hum. Genet. 2018, 63, 459–471. [Google Scholar] [CrossRef]
- Tulloch, J.; Leong, L.; Thomson, Z.; Chen, S.; Lee, E.G.; Keene, C.D.; Millard, S.P.; Yu, C.E. Glia-specific APOE epigenetic changes in the Alzheimer’s disease brain. Brain Res. 2018, 1698, 179–186. [Google Scholar] [CrossRef]
- Mancera-Paez, O.; Estrada-Orozco, K.; Mahecha, M.F.; Cruz, F.; Bonilla-Vargas, K.; Sandoval, N.; Guerrero, E.; Salcedo-Tacuma, D.; Melgarejo, J.D.; Vega, E.; et al. Differential Methylation in APOE (Chr19; Exon Four; from 44,909,188 to 44,909,373/hg38) and Increased Apolipoprotein E Plasma Levels in Subjects with Mild Cognitive Impairment. Int J. Mol. Sci. 2019, 20, 1394. [Google Scholar] [CrossRef]
- Babenko, V.N.; Afonnikov, D.A.; Ignatieva, E.V.; Klimov, A.V.; Gusev, F.E.; Rogaev, E.I. Haplotype analysis of APOE intragenic SNPs. BMC Neurosci. 2018, 19, 16. [Google Scholar] [CrossRef]
- Li, Z.; Shue, F.; Zhao, N.; Shinohara, M.; Bu, G. APOE2: Protective mechanism and therapeutic implications for Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 63. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Rinaldi, C.; Wood, M.J.A. Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Scoles, D.R.; Minikel, E.V.; Pulst, S.M. Antisense oligonucleotides: A primer. Neurol. Genet. 2019, 5, e323. [Google Scholar] [CrossRef] [PubMed]
- Schoch, K.M.; Miller, T.M. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron 2017, 94, 1056–1070. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, O.S.; Li, S.; Nash, K.; Gorbatyuk, M.; Lewin, A.S.; Sullivan, L.F.; Mandel, R.J.; Chen, W.; Meyers, C.; Manfredsson, F.P.; et al. In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1450–1457. [Google Scholar] [CrossRef]
- Khodr, C.E.; Sapru, M.K.; Pedapati, J.; Han, Y.; West, N.C.; Kells, A.P.; Bankiewicz, K.S.; Bohn, M.C. An alpha-synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson’s disease, but displays toxicity in dopamine neurons. Brain Res. 2011, 1395, 94–107. [Google Scholar] [CrossRef]
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 2020, 94, e2270–e2282. [Google Scholar] [CrossRef]
- Mercuri, E.; Darras, B.T.; Chiriboga, C.A.; Day, J.W.; Campbell, C.; Connolly, A.M.; Iannaccone, S.T.; Kirschner, J.; Kuntz, N.L.; Saito, K.; et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2018, 378, 625–635. [Google Scholar] [CrossRef]
- Wild, E.J.; Tabrizi, S.J. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017, 16, 837–847. [Google Scholar] [CrossRef]
- van Roon-Mom, W.M.C.; Roos, R.A.C.; de Bot, S.T. Dose-Dependent Lowering of Mutant Huntingtin Using Antisense Oligonucleotides in Huntington Disease Patients. Nucleic Acid Ther. 2018, 28, 59–62. [Google Scholar] [CrossRef]
- McCampbell, A.; Cole, T.; Wegener, A.J.; Tomassy, G.S.; Setnicka, A.; Farley, B.J.; Schoch, K.M.; Hoye, M.L.; Shabsovich, M.; Sun, L.; et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J. Clin. Investig. 2018, 128, 3558–3567. [Google Scholar] [CrossRef]
- Miller, T.M.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: A phase 1, randomised, first-in-man study. Lancet Neurol. 2013, 12, 435–442. [Google Scholar] [CrossRef]
- Grainger, D.W. Controlled-release and local delivery of therapeutic antibodies. Expert Opin. Biol. Ther. 2004, 4, 1029–1044. [Google Scholar] [CrossRef] [PubMed]
- Bakker, J.M.; Bleeker, W.K.; Parren, P.W. Therapeutic antibody gene transfer: An active approach to passive immunity. Mol. Ther. 2004, 10, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Hollevoet, K.; Declerck, P.J. State of play and clinical prospects of antibody gene transfer. J. Transl. Med. 2017, 15, 131. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.; Borchelt, D.R.; Chakrabarty, P. Therapeutic approaches targeting Apolipoprotein E function in Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 8. [Google Scholar] [CrossRef]
- Guijarro-Munoz, I.; Compte, M.; Alvarez-Vallina, L.; Sanz, L. Antibody gene therapy: Getting closer to clinical application? Curr. Gene Ther. 2013, 13, 282–290. [Google Scholar] [CrossRef]
- Yu, Y.J.; Watts, R.J. Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics 2013, 10, 459–472. [Google Scholar] [CrossRef]
- Samaranayake, H.; Wirth, T.; Schenkwein, D.; Raty, J.K.; Yla-Herttuala, S. Challenges in monoclonal antibody-based therapies. Ann. Med. 2009, 41, 322–331. [Google Scholar] [CrossRef]
- Zheng, D. Antibody gene therapy: An attractive approach for the treatment of cancers and other chronic diseases. Cell Res. 2007, 17, 303–306. [Google Scholar] [CrossRef]
- Kotsovilis, S.; Andreakos, E. Therapeutic human monoclonal antibodies in inflammatory diseases. Methods Mol. Biol. 2014, 1060, 37–59. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Goodman, I.; Safirstein, B.; Marmon, T.K.; Schenk, D.B.; Koller, M.; Zago, W.; Ness, D.K.; Griffith, S.G.; Grundman, M.; et al. Safety and Tolerability of Multiple Ascending Doses of PRX002/RG7935, an Anti-alpha-Synuclein Monoclonal Antibody, in Patients with Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2018, 75, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- A Study to Evaluate the Efficacy of Prasinezumab (RO7046015/PRX002) in Participants with Early Parkinson’s Disease. Available online: https://ClinicalTrials.gov/show/NCT03100149 (accessed on 26 January 2021).
- Evaluating the Efficacy, Safety, Pharmacokinetics, and Pharmacodynamics of BIIB054 in Participants With Parkinson’s Disease. Available online: https://ClinicalTrials.gov/show/NCT03318523 (accessed on 26 January 2021).
- Brys, M.; Fanning, L.; Hung, S.; Ellenbogen, A.; Penner, N.; Yang, M.; Welch, M.; Koenig, E.; David, E.; Fox, T.; et al. Randomized phase I clinical trial of anti-alpha-synuclein antibody BIIB054. Mov. Disord. 2019, 34, 1154–1163. [Google Scholar] [CrossRef]
- Siemers, E.R.; Friedrich, S.; Dean, R.A.; Gonzales, C.R.; Farlow, M.R.; Paul, S.M.; Demattos, R.B. Safety and changes in plasma and cerebrospinal fluid amyloid beta after a single administration of an amyloid beta monoclonal antibody in subjects with Alzheimer disease. Clin. Neuropharmacol. 2010, 33, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Progress of Mild Alzheimer’s Disease in Participants on Solanezumab Versus Placebo. Available online: https://ClinicalTrials.gov/show/NCT01900665 (accessed on 26 January 2021).
- A Study to Evaluate the Efficacy and Safety of MABT5102A in Patients with Mild to Moderate Alzheimer’s Disease (ABBY). Available online: https://ClinicalTrials.gov/show/NCT01343966 (accessed on 26 January 2021).
- A Study Evaluating the Efficacy and Safety of Crenezumab Versus Placebo in Participants with Prodromal to Mild Alzheimer’s Disease (AD). Available online: https://ClinicalTrials.gov/show/NCT02670083 (accessed on 26 January 2021).
- Update on FDA Advisory Committee’s Meeting on Aducanumab in Alzheimer’s Disease; Biogen: Cambridge, MA, USA, 2020.
- Arndt, J.W.; Qian, F.; Smith, B.A.; Quan, C.; Kilambi, K.P.; Bush, M.W.; Walz, T.; Pepinsky, R.B.; Bussiere, T.; Hamann, S.; et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-beta. Sci. Rep. 2018, 8, 6412. [Google Scholar] [CrossRef]
- Biogen Plans Regulatory Filing for Aducanumab in Alzheimer’s Disease Based on New Analysis of Larger Dataset from Phase 3 Studies; Biogen: Cambridge, MA, USA, 2019.
- Bryne, J. Biogen ‘Continues to Work’ with FDA on Alzheimer’s Drug Candidate Despite Regulatory Setback. 2020. Available online: https://www.biopharma-reporter.com/Article/2020/2011/2010/Biogen-continues-to-work-with-FDA-on-Alzheimer-s-drug-candidate-despite-regulatory-setback (accessed on 26 January 2021).
- Liao, F.; Hori, Y.; Hudry, E.; Bauer, A.Q.; Jiang, H.; Mahan, T.E.; Lefton, K.B.; Zhang, T.J.; Dearborn, J.T.; Kim, J.; et al. Anti-ApoE antibody given after plaque onset decreases Abeta accumulation and improves brain function in a mouse model of Abeta amyloidosis. J. Neurosci. 2014, 34, 7281–7292. [Google Scholar] [CrossRef]
- Luz, I.; Liraz, O.; Michaelson, D.M. An Anti-apoE4 Specific Monoclonal Antibody Counteracts the Pathological Effects of apoE4 In Vivo. Curr. Alzheimer Res. 2016, 13, 918–929. [Google Scholar] [CrossRef]
- Liao, F.; Li, A.; Xiong, M.; Bien-Ly, N.; Jiang, H.; Zhang, Y.; Finn, M.B.; Hoyle, R.; Keyser, J.; Lefton, K.B.; et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Investig. 2018, 128, 2144–2155. [Google Scholar] [CrossRef]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal. Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef]
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature 2020, 578, 229–236. [Google Scholar] [CrossRef]
- Ruan, G.X.; Barry, E.; Yu, D.; Lukason, M.; Cheng, S.H.; Scaria, A. CRISPR/Cas9-Mediated Genome Editing as a Therapeutic Approach for Leber Congenital Amaurosis 10. Mol. Ther. 2017, 25, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Gyorgy, B.; Loov, C.; Zaborowski, M.P.; Takeda, S.; Kleinstiver, B.P.; Commins, C.; Kastanenka, K.; Mu, D.; Volak, A.; Giedraitis, V.; et al. CRISPR/Cas9 Mediated Disruption of the Swedish APP Allele as a Therapeutic Approach for Early-Onset Alzheimer’s Disease. Mol. Ther. Nucleic Acids 2018, 11, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1294. [Google Scholar] [CrossRef]
- Ravindran, S. Got mutation? ‘Base editors’ fix genomes one nucleotide at a time. Nature 2019, 575, 553–555. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Schene, I.F.; Joore, I.P.; Oka, R.; Mokry, M.; van Vugt, A.H.M.; van Boxtel, R.; van der Doef, H.P.J.; van der Laan, L.J.W.; Verstegen, M.M.A.; van Hasselt, P.M.; et al. Prime editing for functional repair in patient-derived disease models. Nat. Commun. 2020, 11, 5352. [Google Scholar] [CrossRef]
- Guerreiro, R.; Bilgic, B.; Guven, G.; Bras, J.; Rohrer, J.; Lohmann, E.; Hanagasi, H.; Gurvit, H.; Emre, M. Novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia-like family. Neurobiol. Aging 2013, 34, 2890.e1. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, A.; Kantor, B.; Chiba-Falek, O. APOE: The New Frontier in the Development of a Therapeutic Target towards Precision Medicine in Late-Onset Alzheimer’s. Int. J. Mol. Sci. 2021, 22, 1244. https://doi.org/10.3390/ijms22031244
Yang A, Kantor B, Chiba-Falek O. APOE: The New Frontier in the Development of a Therapeutic Target towards Precision Medicine in Late-Onset Alzheimer’s. International Journal of Molecular Sciences. 2021; 22(3):1244. https://doi.org/10.3390/ijms22031244
Chicago/Turabian StyleYang, Anna, Boris Kantor, and Ornit Chiba-Falek. 2021. "APOE: The New Frontier in the Development of a Therapeutic Target towards Precision Medicine in Late-Onset Alzheimer’s" International Journal of Molecular Sciences 22, no. 3: 1244. https://doi.org/10.3390/ijms22031244
APA StyleYang, A., Kantor, B., & Chiba-Falek, O. (2021). APOE: The New Frontier in the Development of a Therapeutic Target towards Precision Medicine in Late-Onset Alzheimer’s. International Journal of Molecular Sciences, 22(3), 1244. https://doi.org/10.3390/ijms22031244