
DNA Methylation Analysis Identifies Patterns in Progressive Glioma Grades to Predict Patient Survival


Abstract
1. Introduction
2. Results
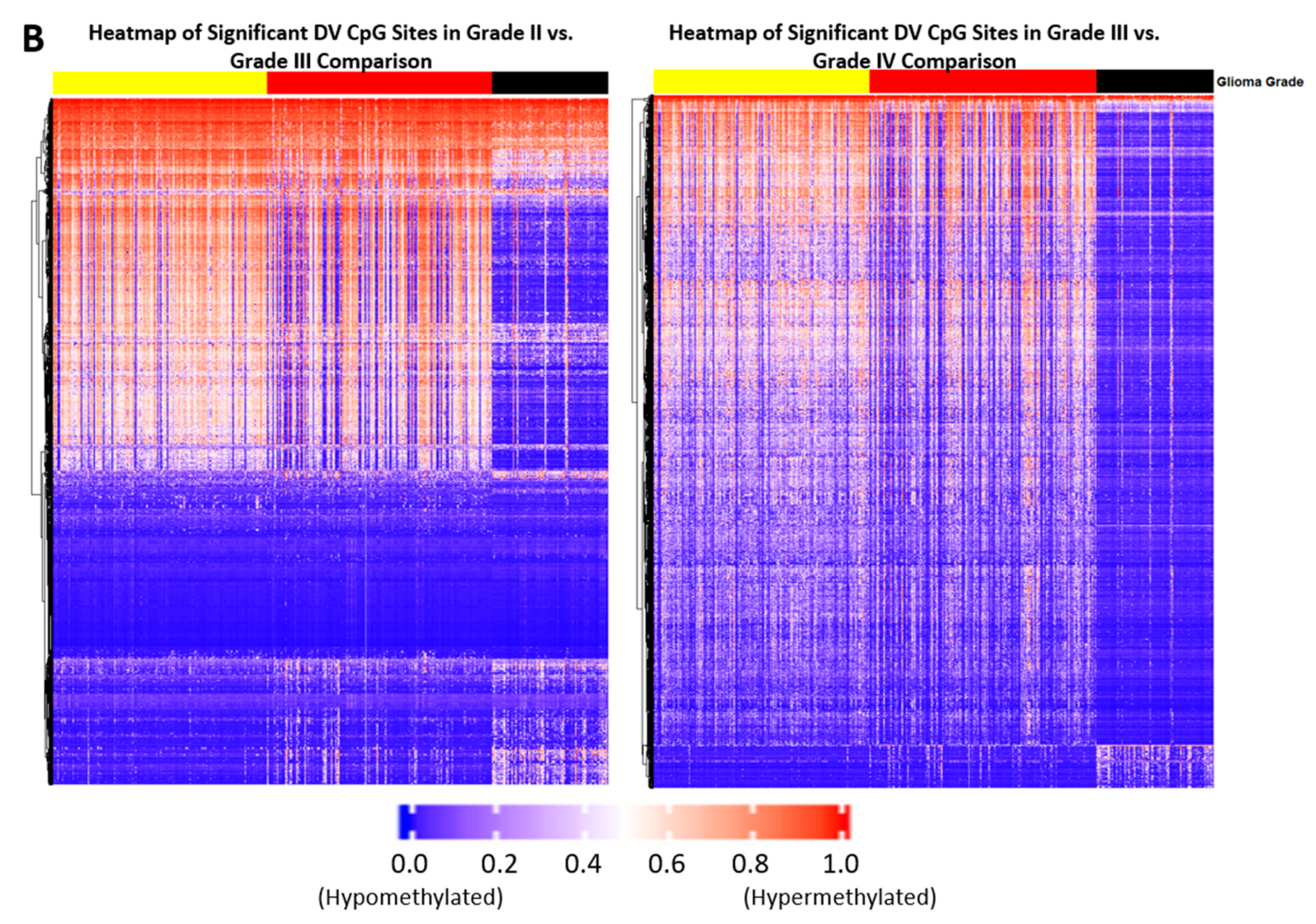
2.1. Differentially Variable Cytosine-phosphate-Guanine Sites Identified through Sample Comparison of Different Glioma Grade
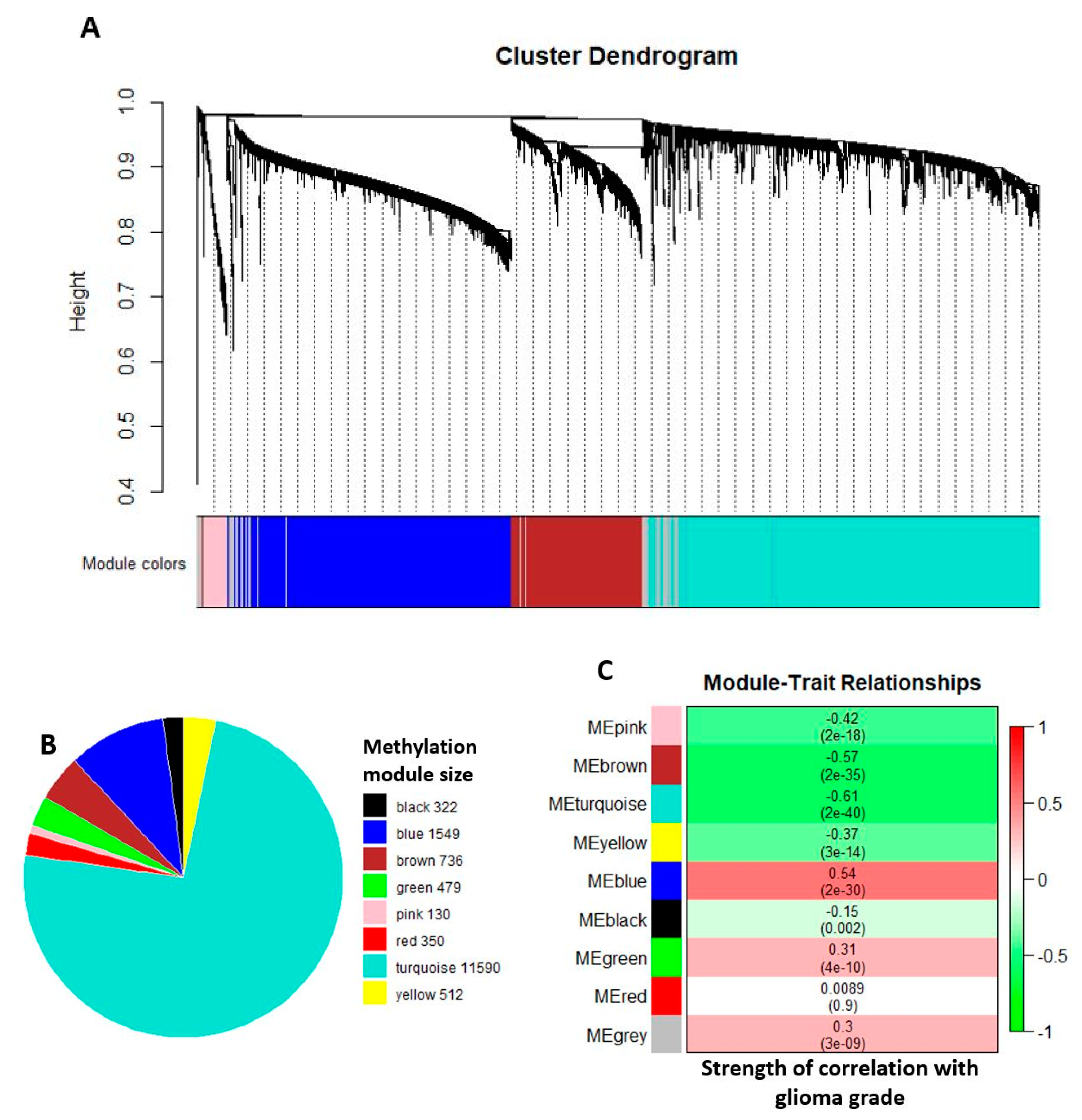
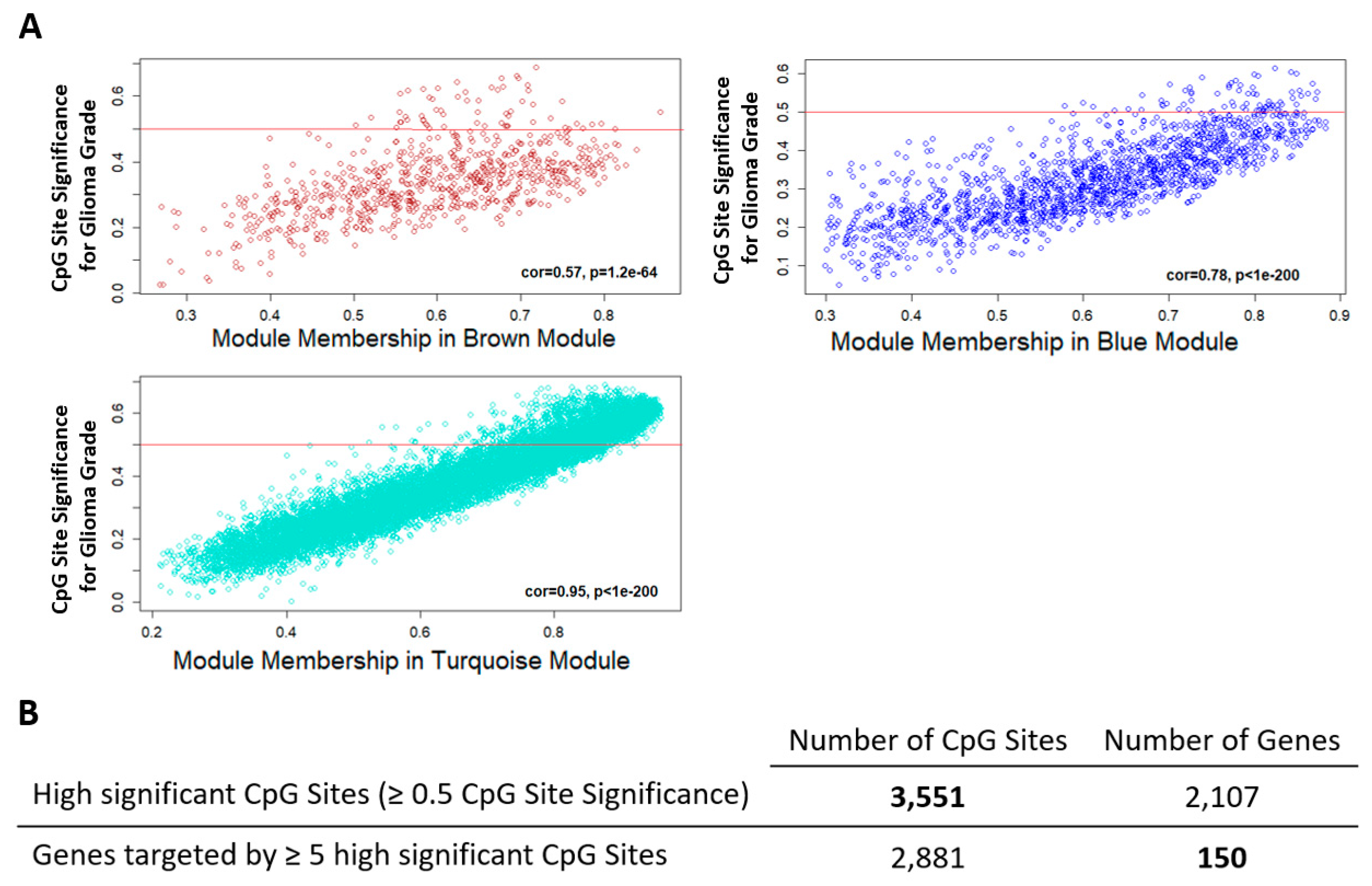
2.2. Comethylation Modules Displaying Correlation with Glioma Grade Progression Identified through Weighted Correlation Network Analysis
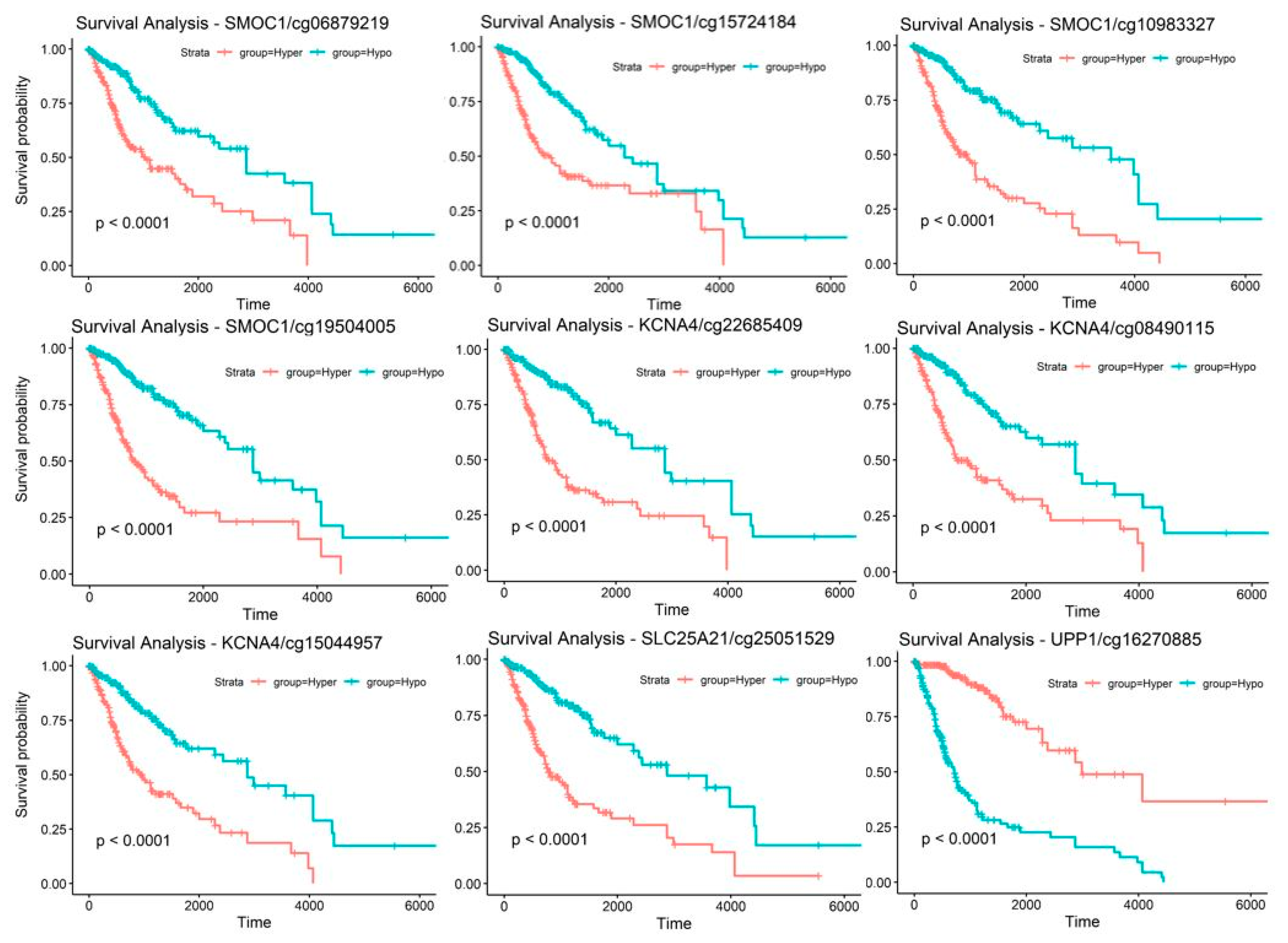
2.3. Candidate Genes and CpG Sites that Contribute to Tumor Progression
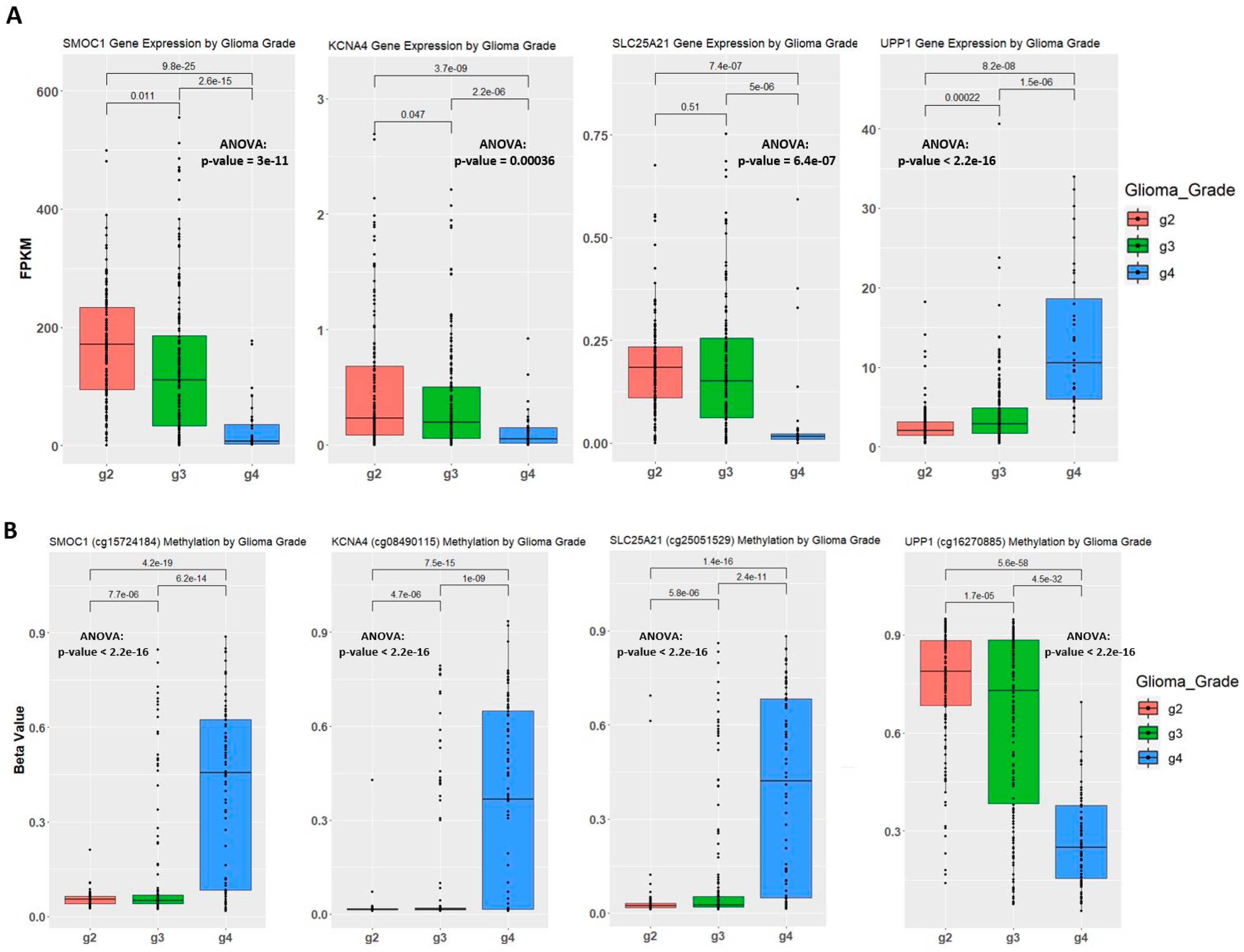
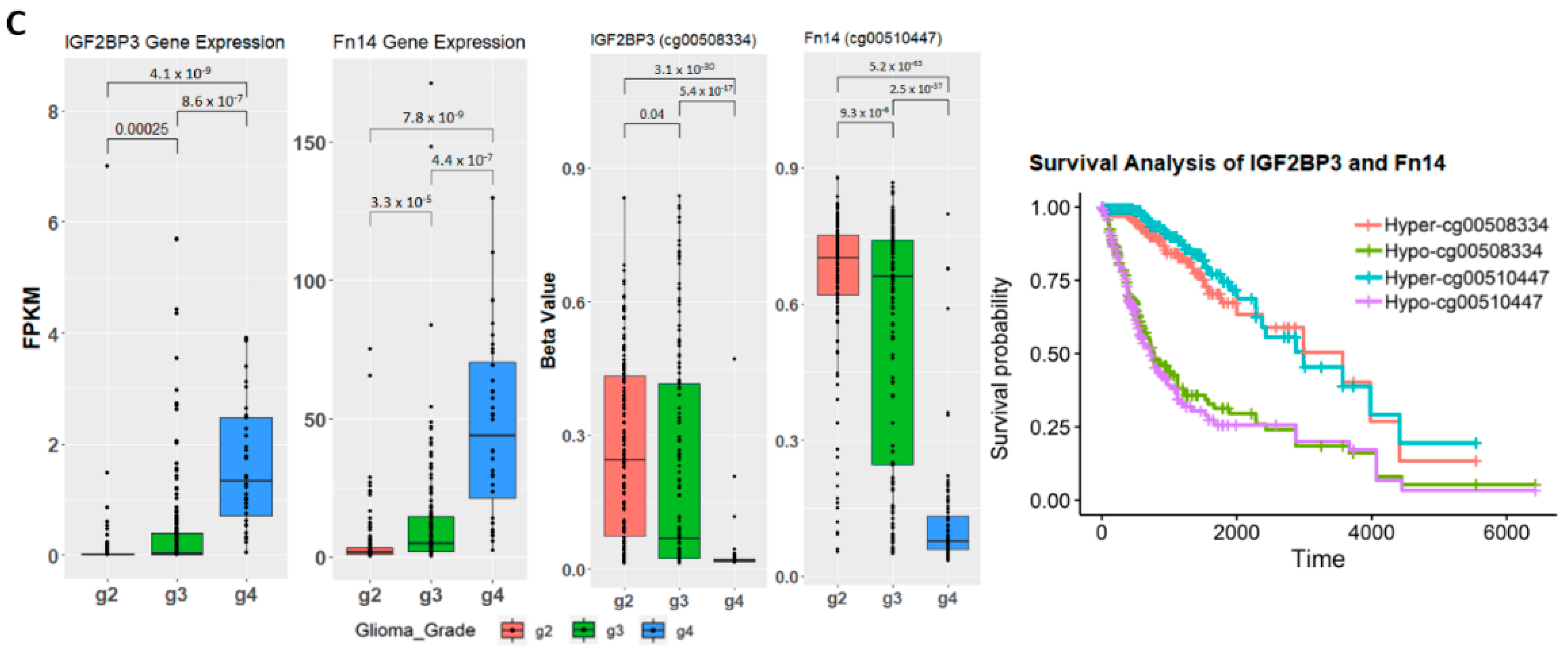
2.4. Establishing Correlation between Gene Expression, Methylation Level, and Glioma Grade
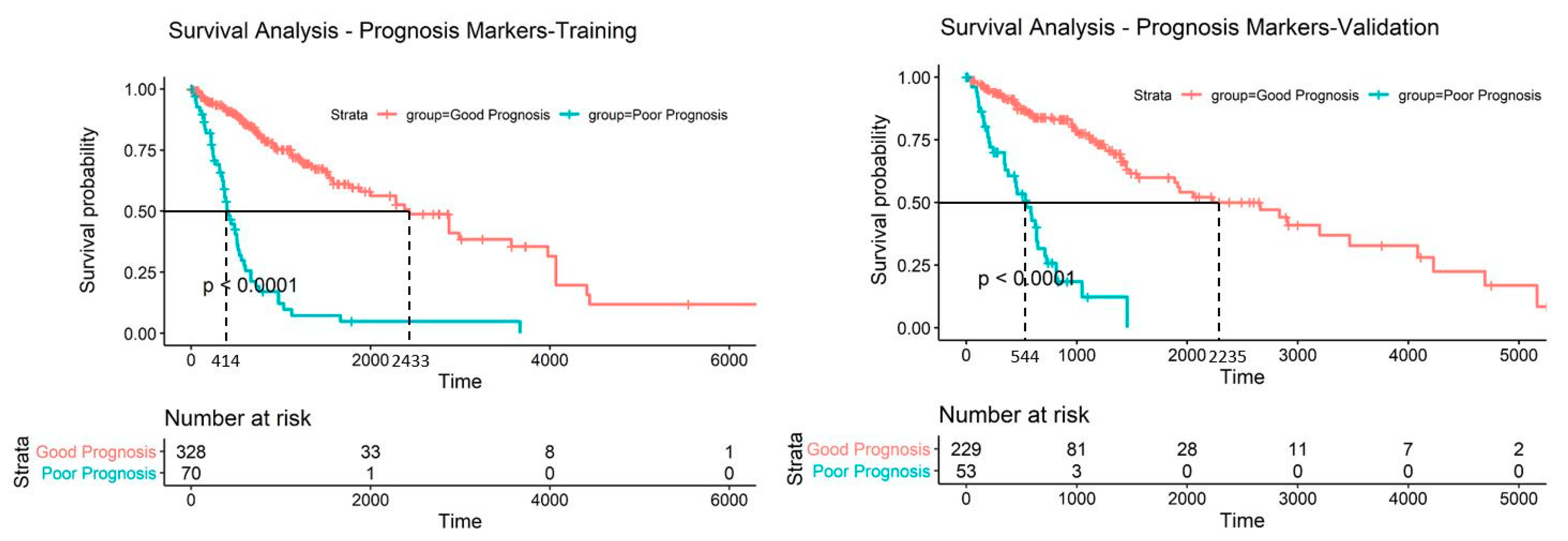
2.5. Discovery and Validation of Prognosis Markers Associated with Tumor Progression
2.6. Establishing a Prognostic Model from the Methylation Markers
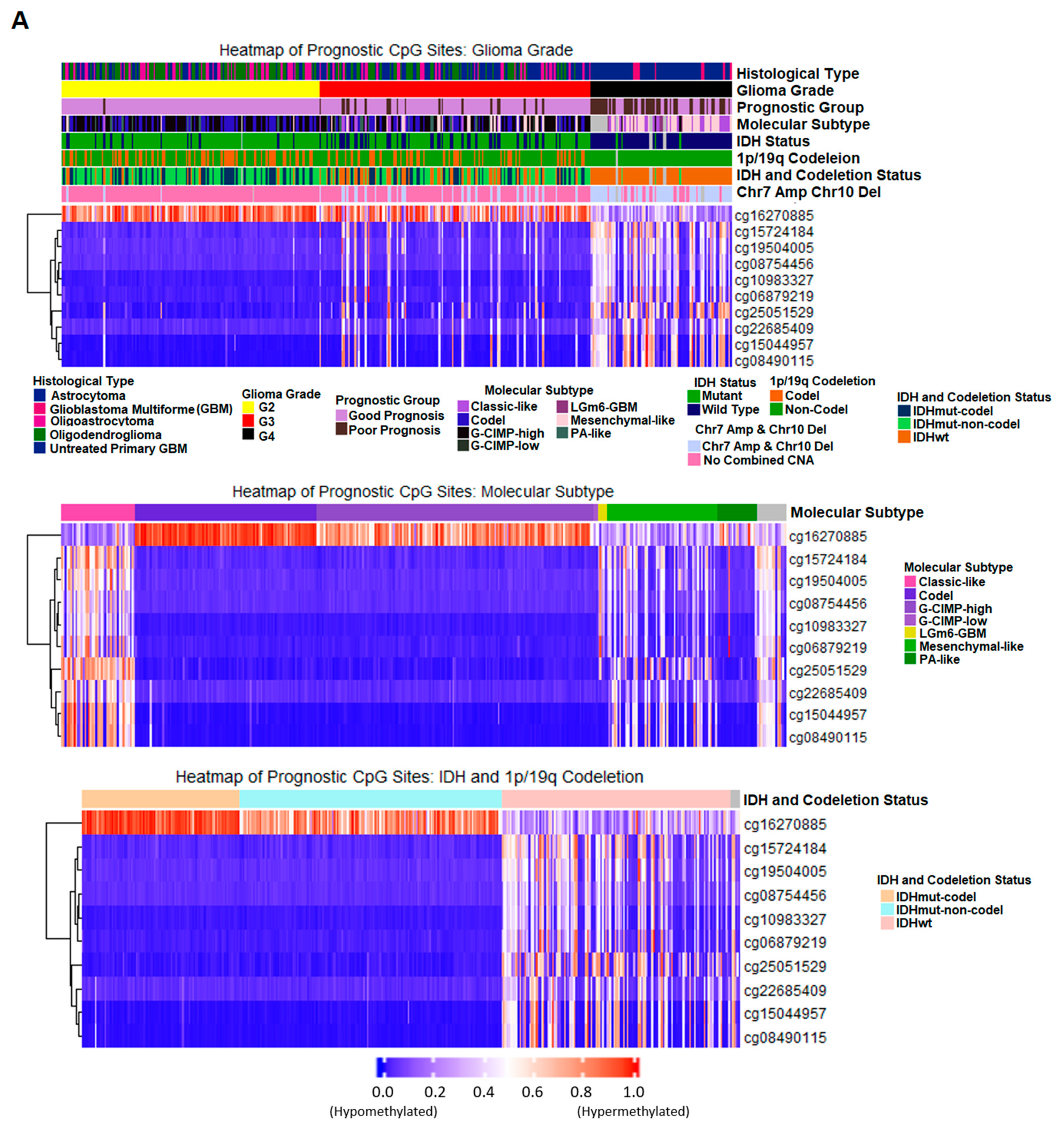
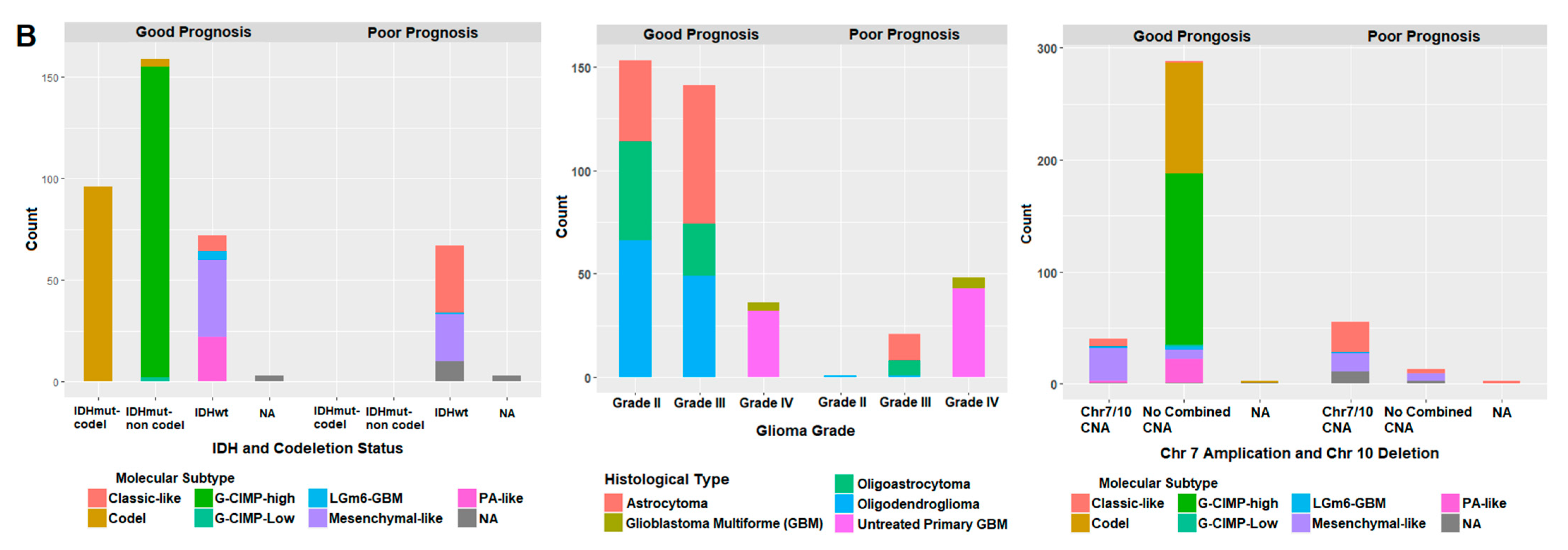
2.7. Correlation of Prognostic Markers to Clinical and Molecular Data
3. Discussion
4. Materials and Methods
4.1. Data Sources
4.2. Methylation Data Manipulation
4.3. Differential Variability Analysis
4.4. Weighted Correlation Network Analysis (WGCNA)
4.5. Gene Expression Data
4.6. Survival Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Choy, J.S.; Wei, S.; Lee, J.Y.; Tan, S.; Chu, S.; Lee, T.H. DNA methylation increases nucleosome compaction and rigidity. J. Am. Chem. Soc. 2010, 132, 1782–1783. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Chen, T. DNA Methylation Reprogramming during Mammalian Development. Genes 2019, 10, 257. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 1–8. [Google Scholar] [CrossRef]
- Ehrlich, M.; Gama-Sosa, M.A.; Huang, L.H.; Midgett, R.M.; Kuo, K.C.; McCune, R.A.; Gehrke, C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982, 10, 2709–2721. [Google Scholar] [CrossRef]
- Van Tongelen, A.; Loriot, A.; de Smet, C. Oncogenic roles of DNA hypomethylation through the activation of cancer-germline genes. Cancer Lett. 2017, 396, 130–137. [Google Scholar] [CrossRef]
- Pfeifer, G.P. Defining Driver DNA Methylation Changes in Human Cancer. Int. J. Mol. Sci. 2018, 19, 1166. [Google Scholar] [CrossRef]
- Klutstein, M.; Nejman, D.; Greenfield, R.; Cedar, H. DNA Methylation in Cancer and Aging. Cancer Res. 2016, 76, 3446–3450. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA methylation in cancer: Too much, but also too little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Gibney, E.; Nolan, C. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.W.; Huang, K.; Yang, C.; Kang, C.S. Non-coding RNAs as regulators in epigenetics (Review). Oncol. Rep. 2016, 37, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Nachtergaele, S.; Moshitch-Moshkovitz, S.; Peer, E.; Kol, N.; Ben-Haim, M.S.; Dai, Q.; Di Segni, A.; Salmon-Divon, M.; Clark, W.C.; et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature 2016, 530, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Dwivedi, T.A. Simplified Overview of World Health Organization Classification Update of Central Nervous System Tumors 2016. J. Neurosci. Rural. Pract. 2017, 8, 629–641. [Google Scholar] [CrossRef]
- Mair, M.J.; Geurts, M.; van den Bent, M.J.; Berghoff, A.S. A basic review on systemic treatment options in WHO grade II-III gliomas. Cancer Treat. Rev. 2021, 92, 102124. [Google Scholar] [CrossRef]
- Bi, W.L.; Beroukhim, R. Beating the odds: Extreme long-term survival with glioblastoma. Neuro-Oncology 2014, 16, 1159–1160. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. The Cancer Genome Atlas Research Network. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, Z.; Cai, X. Identification of epigenetic modulators in human breast cancer by integrated analysis of DNA methylation and RNA-Seq data. Epigenetics 2018, 13, 473–489. [Google Scholar] [CrossRef]
- Vrba, L.; Futscher, B.W. DNA methylation changes in biomarker loci occur early in cancer progression. F1000Research 2020, 8, 2106. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Saito, K.; Aihara, K.; Nagae, G.; Yamamoto, S.; Tatsuno, K.; Ueda, H.; Fukuda, S.; Umeda, T.; Tanaka, S.; et al. DNA demethylation is associated with malignant progression of lower-grade gliomas. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Zhao, C.; Zhao, Z.; Hu, L.; Ye, F.; Wang, H.; Fang, Z.; Wu, Y.; Chen, X. Specific glioblastoma multiforme prognostic-subtype distinctions based on DNA methylation patterns. Cancer Gene Ther. 2020, 27, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, C.; Zhao, Z.; Wang, H.; Fang, Z. Specific Glioma Prognostic Subtype Distinctions Based on DNA Methylation Patterns. Front. Genet. 2019, 10, 786. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhu, H.; Yang, M.; Zhu, C. DNA methylation profiling analysis identifies a DNA methylation signature for predicting prognosis and recurrence of lung adenocarcinoma. Oncol. Lett. 2019, 18, 5831–5842. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Tan, D.; Pang, F.M.; Li, D.; Zhang, L.; Wu, J.; Liu, Z.Q.; Li, X.; Yan, H. Overexpression of Fn14 in gliomas: Tumor progression and poor prognosis. Futur. Oncol. 2018, 14, 1273–1284. [Google Scholar] [CrossRef]
- Cadieux, B.; Ching, T.T.; VandenBerg, S.R.; Costello, J.F. Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res. 2006, 66, 8469–8476. [Google Scholar] [CrossRef]
- Vannahme, C.; Smyth, N.; Miosge, N.; Gösling, S.; Frie, C.; Paulsson, M.; Maurer, P.; Hartmann, U. Characterization of SMOC-1, a novel modular calcium-binding protein in basement membranes. J. Biol. Chem. 2002, 277, 37977–37986. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, X. SMOC1 silencing suppresses the angiotensin II-induced myocardial fibrosis of mouse myocardial fibroblasts via affecting the BMP2/Smad pathway. Oncol. Lett. 2018, 16, 2903–2910. [Google Scholar] [CrossRef]
- Thomas, J.T.; Eric Dollins, D.; Andrykovich, K.R.; Chu, T.; Stultz, B.G.; Hursh, D.A.; Moos, M. SMOC can act as both an antagonist and an expander of BMP signaling. eLife 2017, 6, e17935. [Google Scholar] [CrossRef] [PubMed]
- Awwad, K.; Hu, J.; Shi, L.; Mangels, N.; Abdel Malik, R.; Zippel, N.; Fisslthaler, B.; Eble, J.A.; Pfeilschifter, J.; Popp, R.; et al. Role of secreted modular calcium-binding protein 1 (SMOC1) in transforming growth factor β signalling and angiogenesis. Cardiovasc. Res. 2015, 106, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Boon, K.; Edwards, J.B.; Eberhart, C.G.; Riggins, G.J. Identification of astrocytoma associated genes including cell surface markers. BMC Cancer 2004, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Brellier, F.; Ruggiero, S.; Zwolanek, D.; Martina, E.; Hess, D.; Brown-Luedi, M.; Hartmann, U.; Koch, M.; Merlo, A.; Lino, M.; et al. SMOC1 is a tenascin-C interacting protein over-expressed in brain tumors. Matrix Biol. J. Int. Soc. Matrix Biol. 2011, 30, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Niwa, N.; Nerbonne, J.M. Molecular determinants of cardiac transient outward potassium current (I(to)) expression and regulation. J. Mol. Cell. Cardiol. 2010, 48, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chen, L.; Li, J.; Yu, B.; Su, L.; Chen, X.; Yu, Y.; Yan, M.; Liu, B.; Zhu, Z. Hypermethylated DNA as potential biomarkers for gastric cancer diagnosis. Clin. Biochem. 2011, 44, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Coma, M.; Vicente, R.; Busquets, S.; Carbó, N.; Tamkun, M.M.; López-Soriano, F.J.; Argilés, J.M.; Felipe, A. Impaired voltage-gated K+ channel expression in brain during experimental cancer cachexia. FEBS Lett. 2003, 536, 45–50. [Google Scholar] [CrossRef]
- Comes, N.; Bielanska, J.; Vallejo-Gracia, A.; Serrano-Albarrás, A.; Marruecos, L.; Gómez, D.; Soler, C.; Condom, E.; Ramón, Y.; Cajal, S.; et al. The voltage-dependent K(+) channels Kv1.3 and Kv1.5 in human cancer. Front. Physiol. 2013, 4, 283. [Google Scholar] [CrossRef]
- Preussat, K.; Beetz, C.; Schrey, M.; Kraft, R.; Wölfl, S.; Kalff, R.; Patt, S. Expression of voltage-gated potassium channels Kv1.3 and Kv1.5 in human gliomas. Neurosci. Lett. 2003, 346, 33–36. [Google Scholar] [CrossRef]
- Fiermonte, G.; Dolce, V.; Palmieri, L.; Ventura, M.; Runswick, M.J.; Palmieri, F.; Walker, J.E. Identification of the human mitochondrial oxodicarboxylate carrier. Bacterial expression, reconstitution, functional characterization, tissue distribution, and chromosomal location. J. Biol. Chem. 2001, 276, 8225–8230. [Google Scholar] [CrossRef]
- Rochette, L.; Meloux, A.; Zeller, M.; Malka, G.; Cottin, Y.; Vergely, C. Mitochondrial SLC25 Carriers: Novel Targets for Cancer Therapy. Molecules 2020, 25, 2417. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Dituri, F.; Convertini, P.; Santarsiero, A.; Palmieri, F.; Todisco, S.; Mancarella, S.; Giannelli, G.; Iacobazzi, V. Epigenetic upregulation and functional role of the mitochondrial aspartate/glutamate carrier isoform 1 in hepatocellular carcinoma. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Uchida, T. Cloning and expression of human uridine phosphorylase. Biochem. Biophys. Res. Commun. 1995, 216, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Roosild, T.P.; Castronovo, S.; Fabbiani, M.; Pizzorno, G. Implications of the structure of human uridine phosphorylase 1 on the development of novel inhibitors for improving the therapeutic window of fluoropyrimidine chemotherapy. BMC Struct. Biol. 2009, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Bhandari, A.; Zhang, X.; Wang, O. Uridine phosphorylase 1 associates to biological and clinical significance in thyroid carcinoma cell lines. J. Cell. Mol. Med. 2019, 23, 7438–7448. [Google Scholar] [CrossRef]
- Wang, J.; Xu, S.; Lv, W.; Shi, F.; Mei, S.; Shan, A.; Xu, J.; Yang, Y. Uridine phosphorylase 1 is a novel immune-related target and predicts worse survival in brain glioma. Cancer Med. 2020, 9, 5940–5947. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2015, 44, e71. [Google Scholar] [CrossRef]
- Silva, T.C.; Colaprico, A.; Olsen, C.; D’Angelo, F.; Bontempi, G.; Ceccarelli, M.; Noushmehr, H. TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Research 2016, 5, 1542. [Google Scholar] [CrossRef]
- Mounir, M.; Lucchetta, M.; Silva, T.C.; Olsen, C.; Bontempi, G.; Chen, X.; Noushmehr, H.; Colaprico, A.; Papaleo, E. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput. Biol. 2019, 15, e1006701. [Google Scholar] [CrossRef]
- Daca-Roszak, P.; Pfeifer, A.; Żebracka-Gala, J.; Rusinek, D.; Szybińska, A.; Jarząb, B.; Witt, M.; Ziętkiewicz, E. Impact of SNPs on methylation readouts by Illumina Infinium HumanMethylation450 BeadChip Array: Implications for comparative population studies. BMC Genom. 2015, 16, 1003. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Phipson, B.; Maksimovic, J.; Oshlack, A. missMethyl: An R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics (Oxford, England) 2016, 32, 286–288. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic, J.; Phipson, B.; Oshlack, A. A cross-package Bioconductor workflow for analysing methylation array data. F1000Research 2016, 5, 1281. [Google Scholar] [CrossRef]
- Rainer, J.; Gatto, L.; Weichenberger, C.X. ensembldb: An R package to create and use Ensembl-based annotation resources. Bioinformatics (Oxford, England) 2019, 35, 3151–3153. [Google Scholar] [CrossRef]
- Therneau, T. A Package for Survival Analysis in R. R package Version 3.2-7. 2020. Available online: https://CRAN.R-project.org/package=survival (accessed on 19 January 2021).
- Alix-Panabières, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef]
- Chen, M.; Zhao, H. Next-generation sequencing in liquid biopsy: Cancer screening and early detection. Hum. Genom. 2019, 13, 34. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Training (400 Samples) | Validation (282 Samples) | |||
|---|---|---|---|---|
| Number of Samples | Percent of Samples | Number of Samples | Percent of Samples | |
| Age | 46.19 yo | N/A | 46.69 yo | N/A |
| Days to death | 888.7 days | N/A | 916.6 days | N/A |
| Days to last follow up | 834.9 days | N/A | 866.4 days | N/A |
| Grade II | 154 | 38.5% | 107 | 37.9% |
| Grade III | 162 | 40.5% | 109 | 38.7% |
| Grade IV | 84 | 21.0% | 66 | 23.4% |
| Astrocytoma | 119 | 29.7% | 78 | 27.7% |
| Oligoastrocytoma | 80 | 20.0% | 55 | 19.5% |
| Oligodendroglioma | 117 | 29.3% | 83 | 29.4% |
| Glioblastoma multiforme (GBM) | 84 | 21.0% | 66 | 23.4% |
| Alive | 259 | 64.8% | 182 | 64.5% |
| Dead | 141 | 35.2% | 100 | 35.5% |
| Probe ID | CpG Position Relative to Targeting Gene | Locus | Gene | CpG Position Relative to CpG Island | Methylation Status for Poor Prognosis * |
|---|---|---|---|---|---|
| cg16270885 | 5’UTR ** | chr7:48096722 | UPP1 | S_Shore † | Hypomethylation |
| cg08490115 | TSS200 ‡ | chr11:29995251 | KCNA4 | Island | Hypermethylation |
| cg15044957 | TSS200 | chr11:29995248 | KCNA4 | Island | Hypermethylation |
| cg22685409 | TSS1500 †† | chr11:29995268 | KCNA4 | Island | Hypermethylation |
| cg06879219 | TSS200 | chr14:69415835 | SMOC1 | Island | Hypermethylation |
| cg08754456 | TSS200 | chr14:69415812 | SMOC1 | Island | Hypermethylation |
| cg10983327 | TSS200 | chr14:69415825 | SMOC1 | Island | Hypermethylation |
| cg15724184 | 1st Exon | chr14:69416230 | SMOC1 | Island | Hypermethylation |
| cg19504005 | 1st Exon | chr14:69416170 | SMOC1 | Island | Hypermethylation |
| cg25051529 | Body | chr14:36711213 | SLC25A21 | Island | Hypermethylation |
| Training Set (400) | Validation Set (282) | |||
|---|---|---|---|---|
| Good Prognosis (330) | Poor Prognosis (70) | Good Prognosis (229) | Poor Prognosis (53) | |
| Clinical | ||||
| Grade | ||||
| Grade II | 153 | 1 | 104 | 3 |
| Grade III | 141 | 21 | 95 | 14 |
| Grade IV | 36 | 48 | 30 | 36 |
| Histology | ||||
| Astrocytoma | 106 | 13 | 69 | 9 |
| Oligodendroglioma | 115 | 2 | 78 | 5 |
| Oligoastrocytoma | 73 | 7 | 52 | 3 |
| Glioblastoma | 36 | 48 | 30 | 36 |
| Molecular | ||||
| Isocitrate Dehydrogenase (IDH) | ||||
| Mutant | 256 | 0 | 185 | 0 |
| Wild Type | 72 | 67 | 39 | 52 |
| NA | 2 | 3 | 5 | 1 |
| 1p/19q codeletion | ||||
| Codeletion | 96 | 0 | 76 | 0 |
| No codeletion | 233 | 70 | 153 | 51 |
| NA | 1 | 0 | 0 | 2 |
| IDH and 1p/19q codeletion status | ||||
| IDH mutant and codeletion | 96 | 0 | 76 | 0 |
| IDH mutant and no-codeletion | 159 | 0 | 109 | 0 |
| IDH wild type and no-codeletion | 72 | 67 | 39 | 50 |
| NA | 3 | 3 | 5 | 3 |
| Chr7 Amplification/Chr 10 Deletion | ||||
| Chr7 Amp/Chr 10 Del | 40 | 55 | 19 | 40 |
| No combined copy number alteration | 288 | 13 | 206 | 9 |
| NA | 2 | 0 | 4 | 4 |
| Molecular Subtype (Ceccarelli et al., 2016) | ||||
| Classic-like * | 8 | 33 | 6 | 31 |
| Codel | 100 | 0 | 76 | 0 |
| G-CIMP **-high | 153 | 0 | 97 | 0 |
| G-CIMP **-low | 2 | 0 | 10 | 0 |
| LGm6-GBM | 4 | 1 | 7 | 1 |
| Mesenchymal-like | 38 | 23 | 19 | 20 |
| Pilocytic Astrocytomas (PA) like | 22 | 0 | 7 | 0 |
| NA | 3 | 3 | 7 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weng, J.; Salazar, N. DNA Methylation Analysis Identifies Patterns in Progressive Glioma Grades to Predict Patient Survival. Int. J. Mol. Sci. 2021, 22, 1020. https://doi.org/10.3390/ijms22031020
Weng J, Salazar N. DNA Methylation Analysis Identifies Patterns in Progressive Glioma Grades to Predict Patient Survival. International Journal of Molecular Sciences. 2021; 22(3):1020. https://doi.org/10.3390/ijms22031020
Chicago/Turabian StyleWeng, Jingyin, and Nicole Salazar. 2021. "DNA Methylation Analysis Identifies Patterns in Progressive Glioma Grades to Predict Patient Survival" International Journal of Molecular Sciences 22, no. 3: 1020. https://doi.org/10.3390/ijms22031020
APA StyleWeng, J., & Salazar, N. (2021). DNA Methylation Analysis Identifies Patterns in Progressive Glioma Grades to Predict Patient Survival. International Journal of Molecular Sciences, 22(3), 1020. https://doi.org/10.3390/ijms22031020