
The Effect of a Sustained High-Fat Diet on the Metabolism of White and Brown Adipose Tissue and Its Impact on Insulin Resistance: A Selected Time Point Cross-Sectional Study

 ,
,
Abstract
:1. Introduction
2. Results
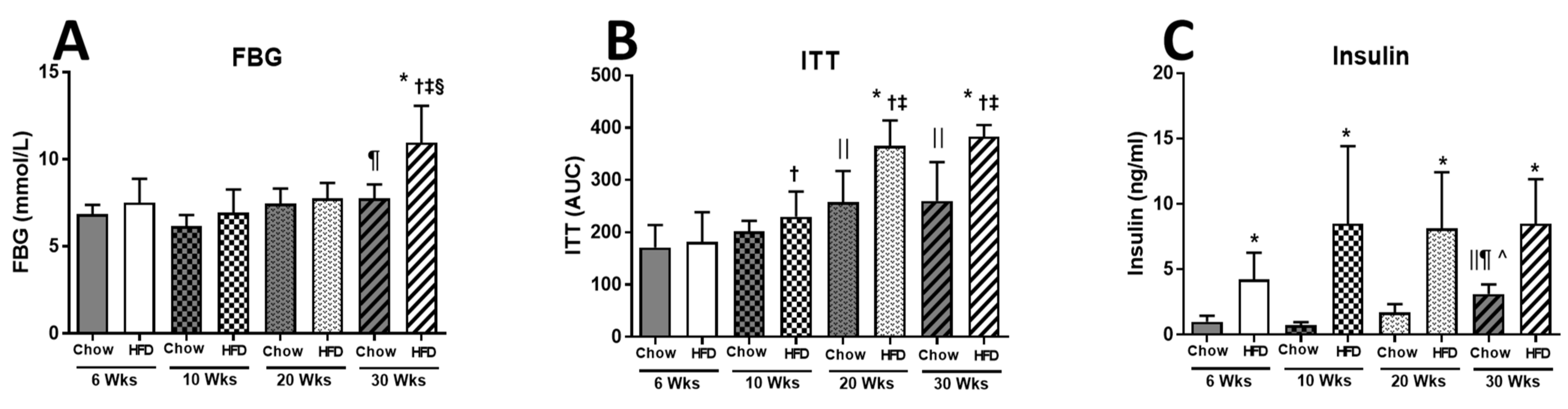
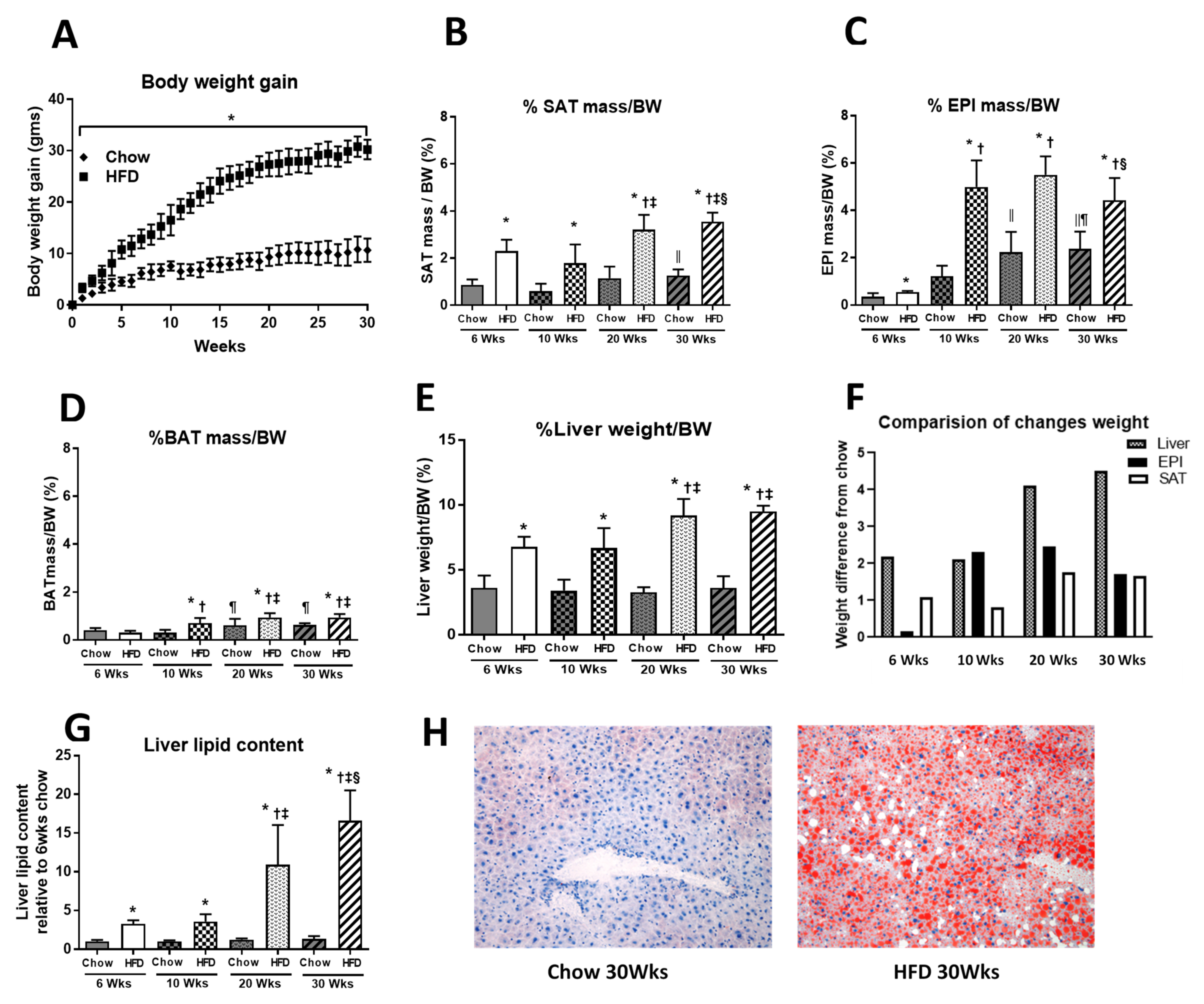
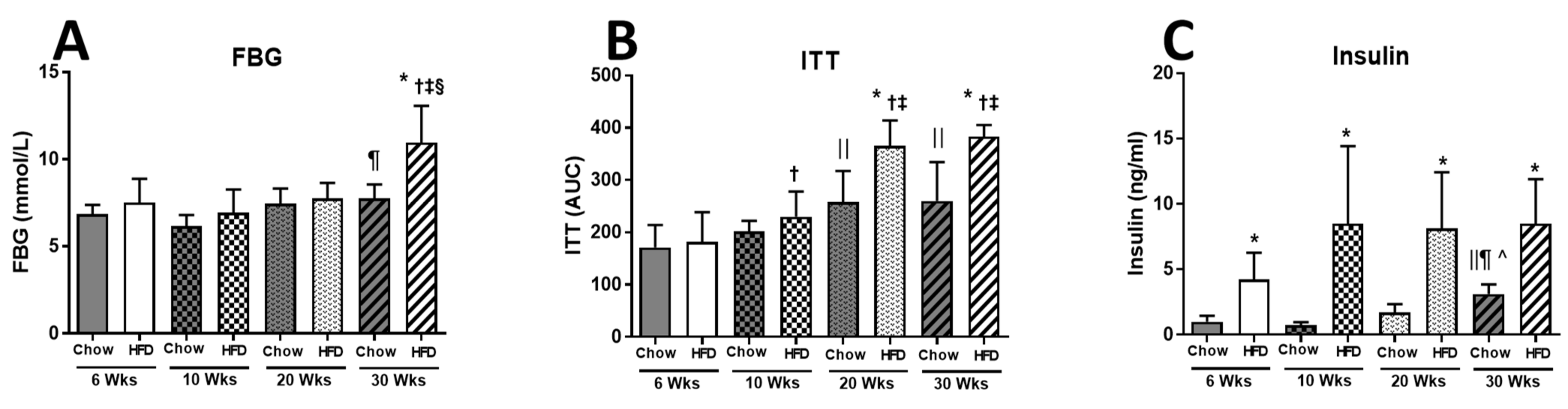
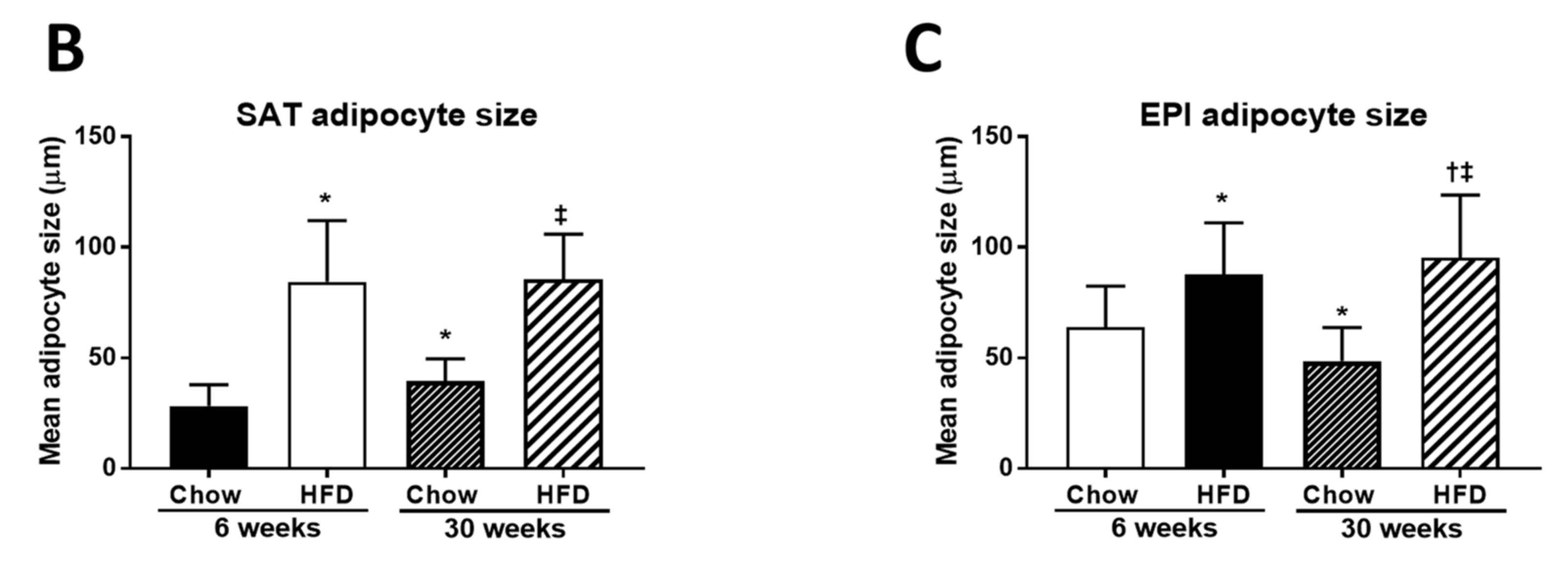
2.1. Anthropometric Measurements
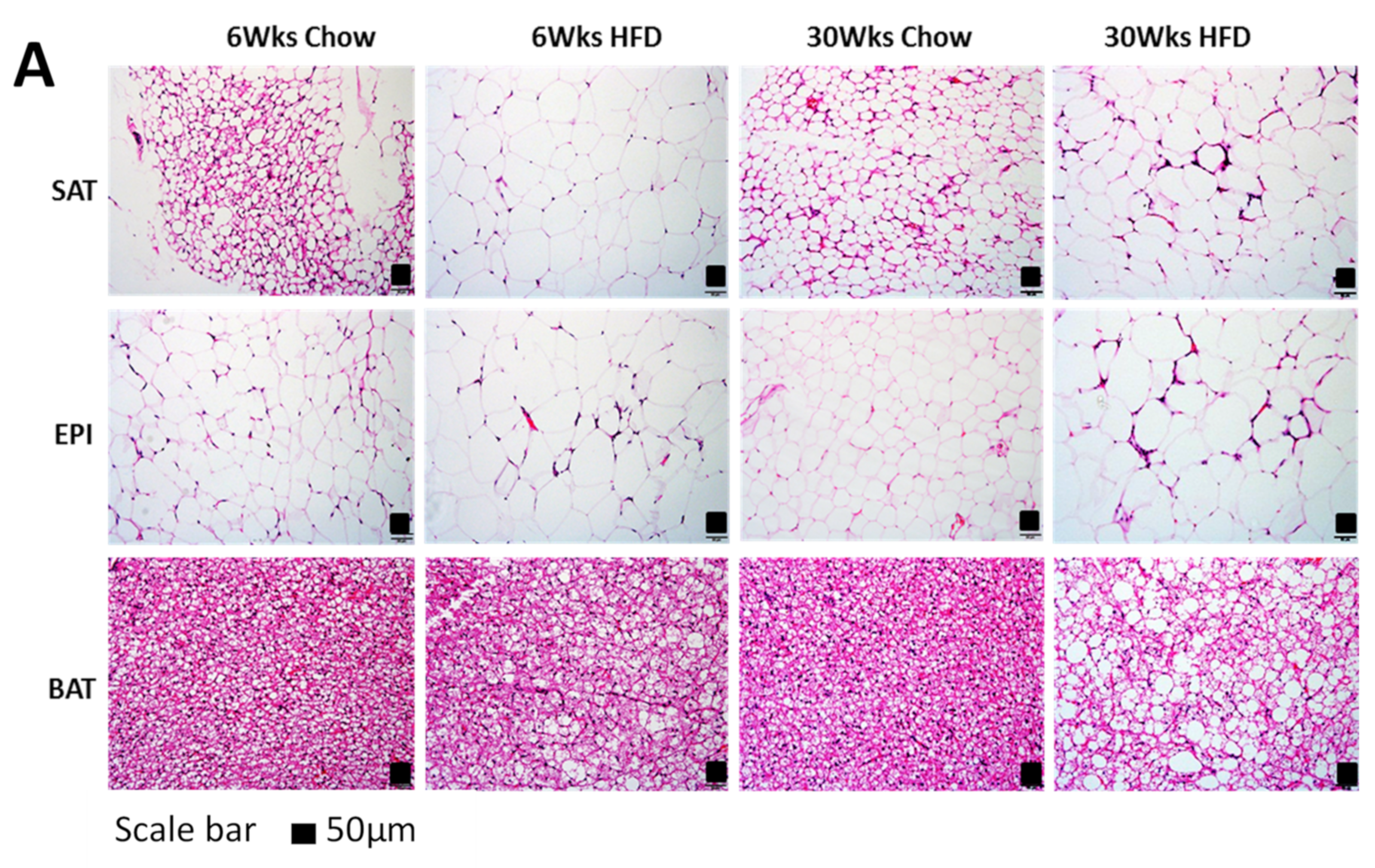
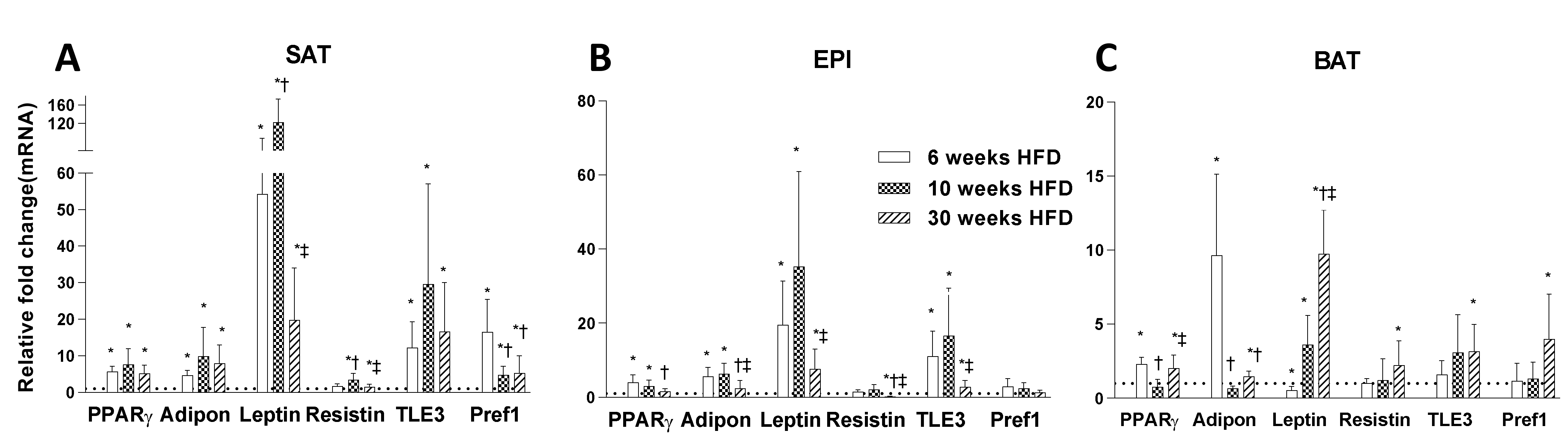
2.2. Adipogenesis Markers
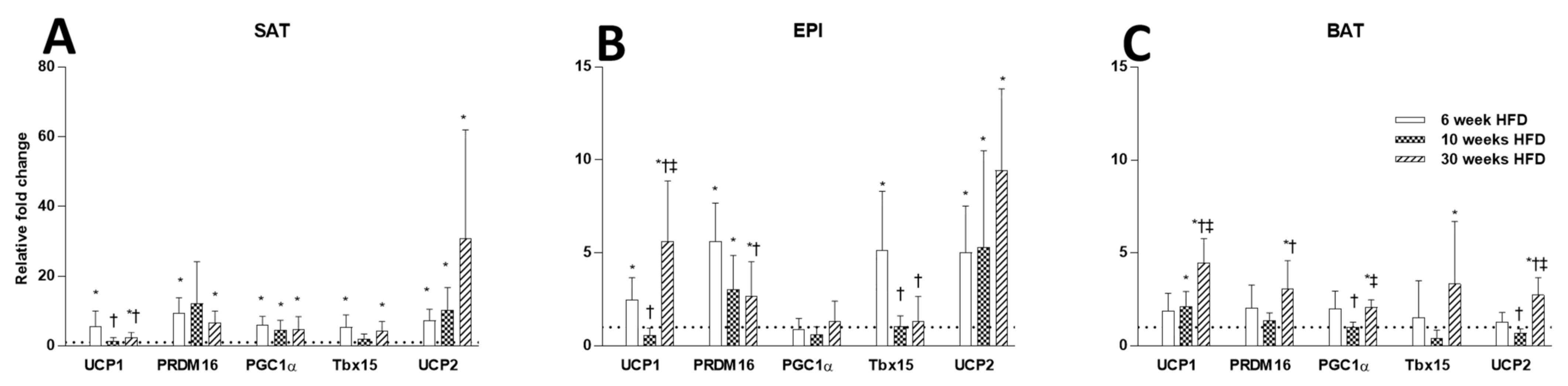
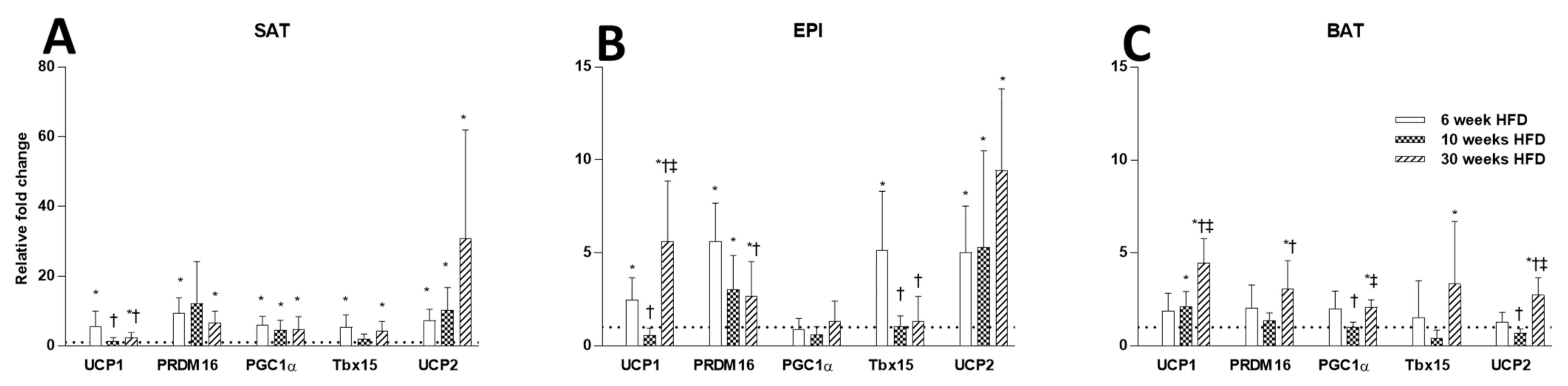
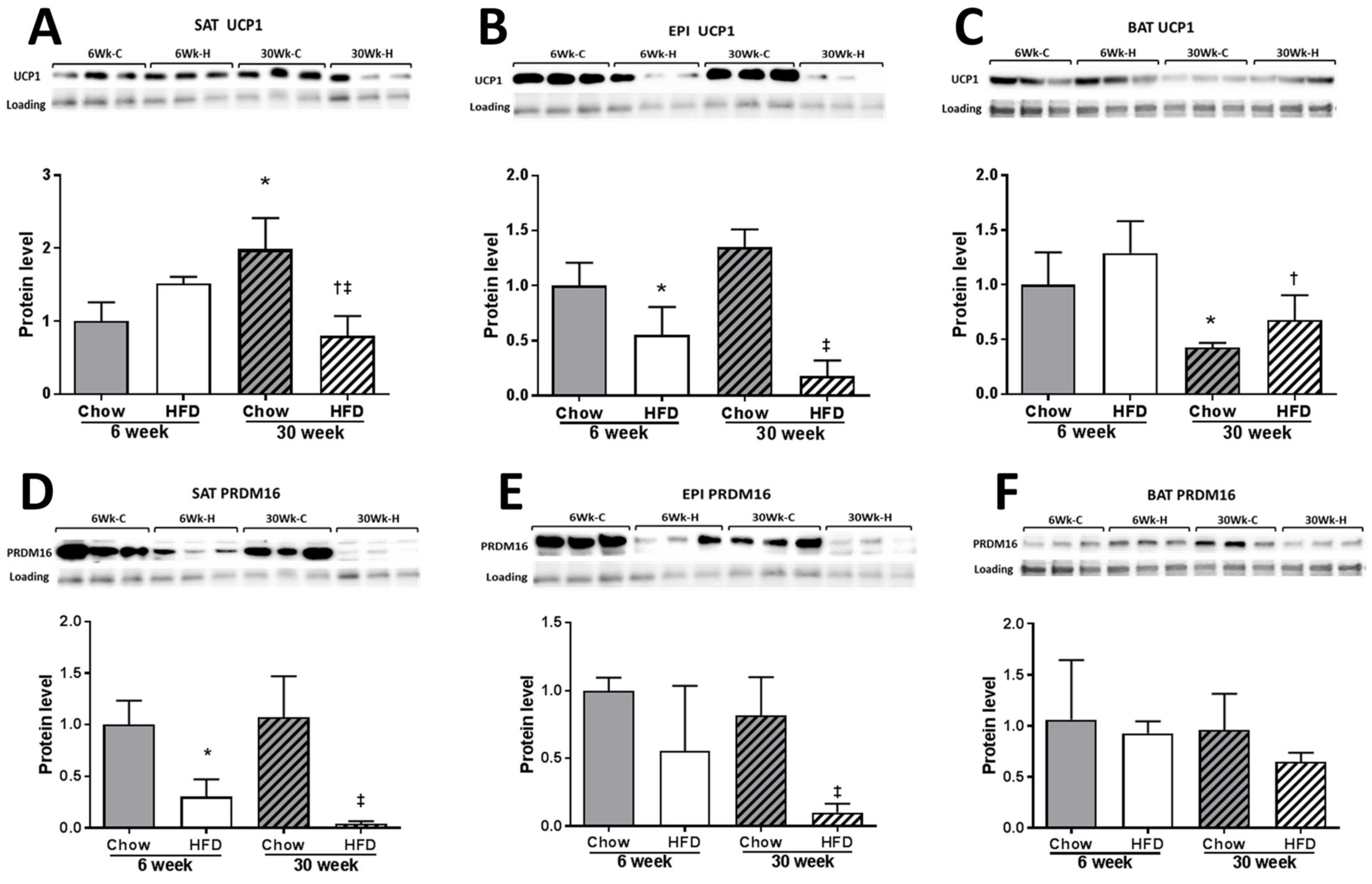
2.3. Thermogenesis Markers
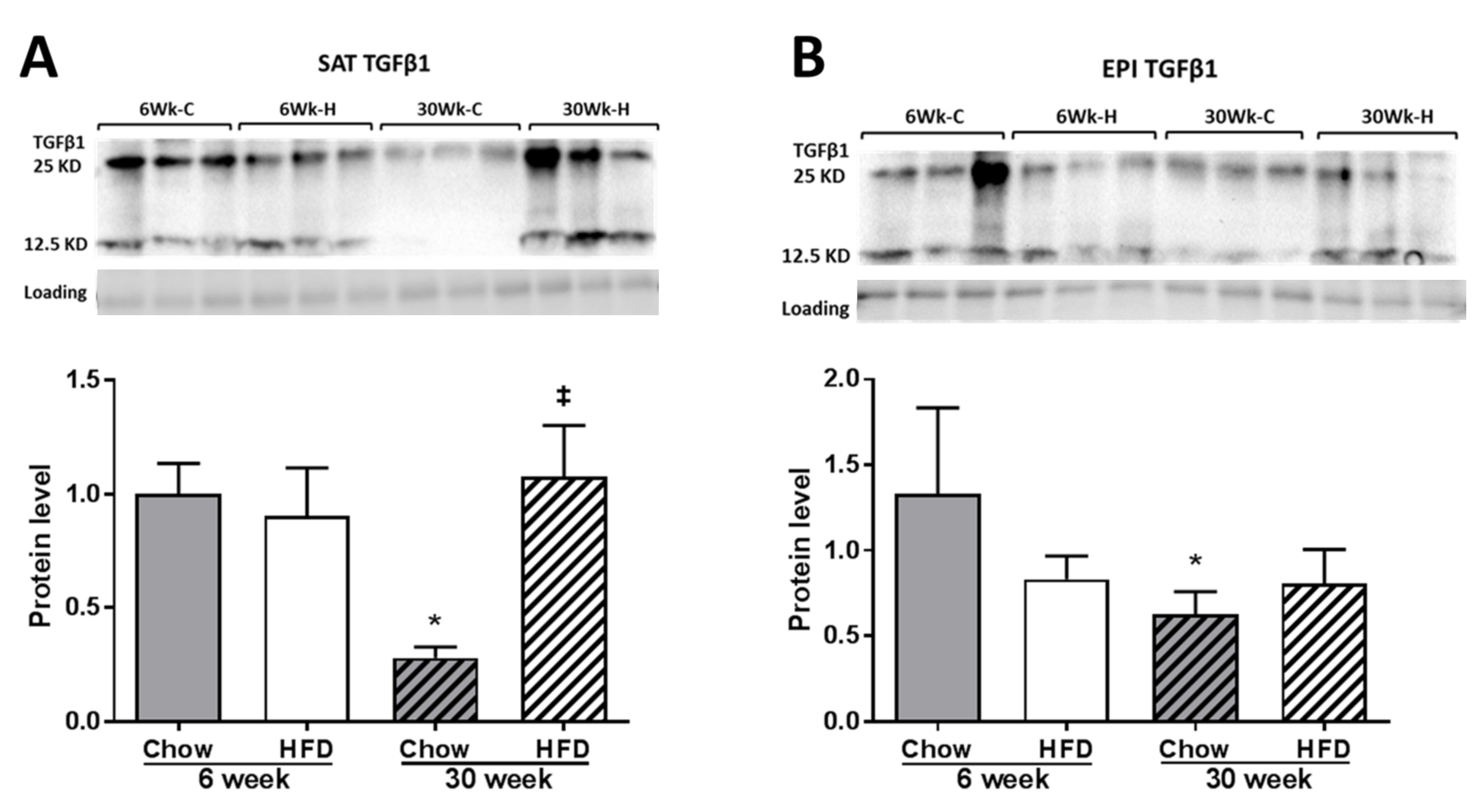
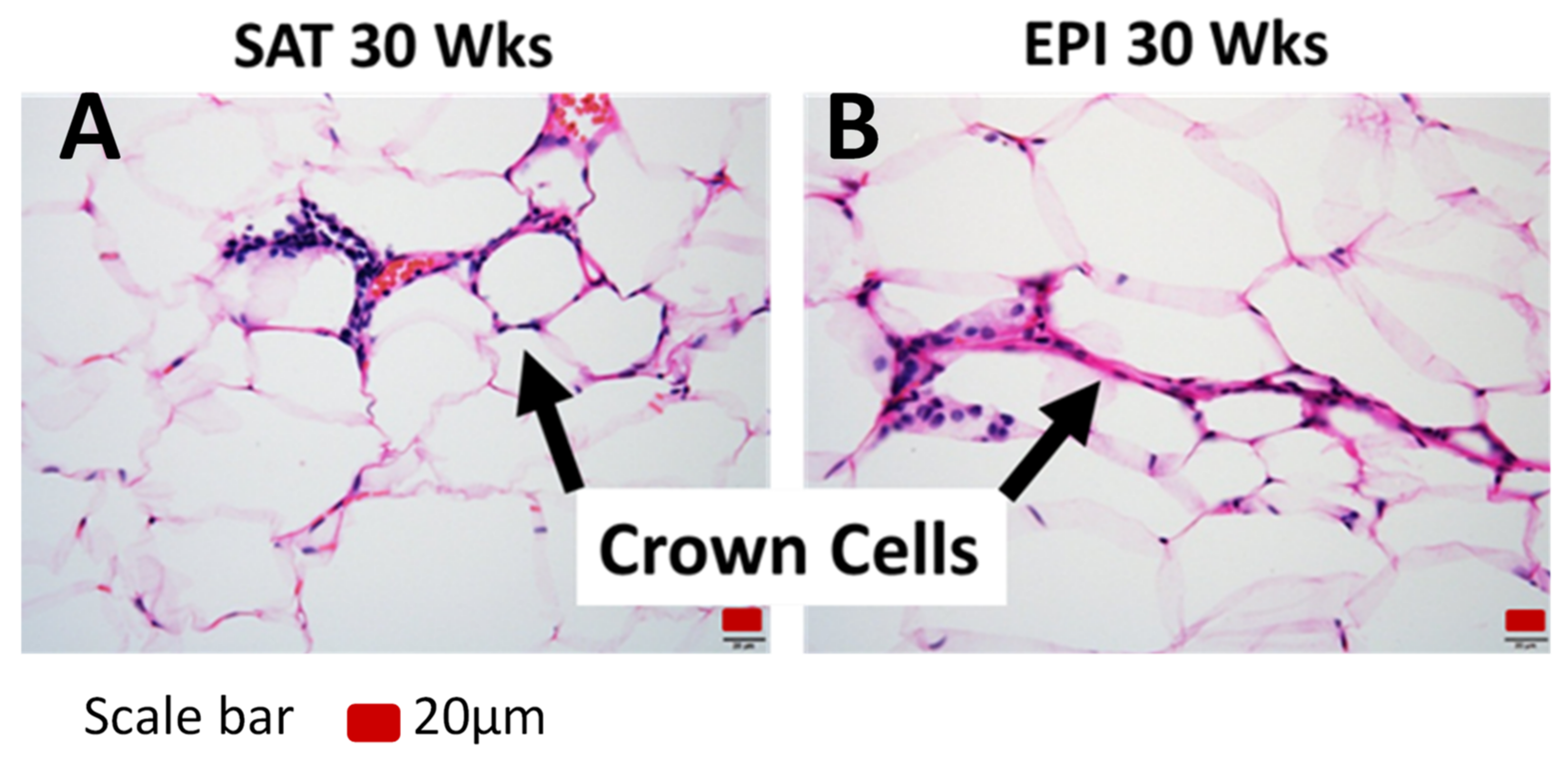
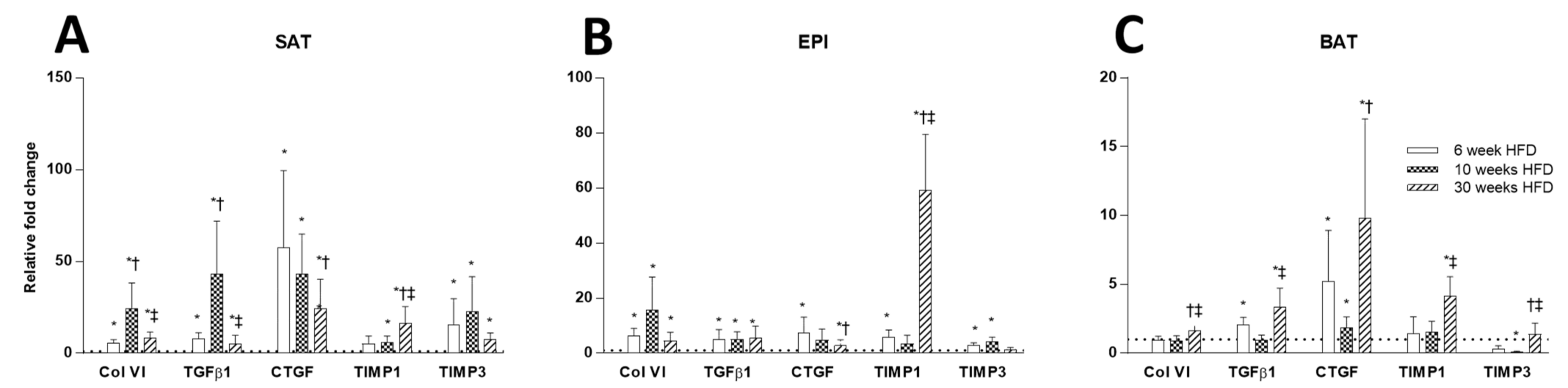
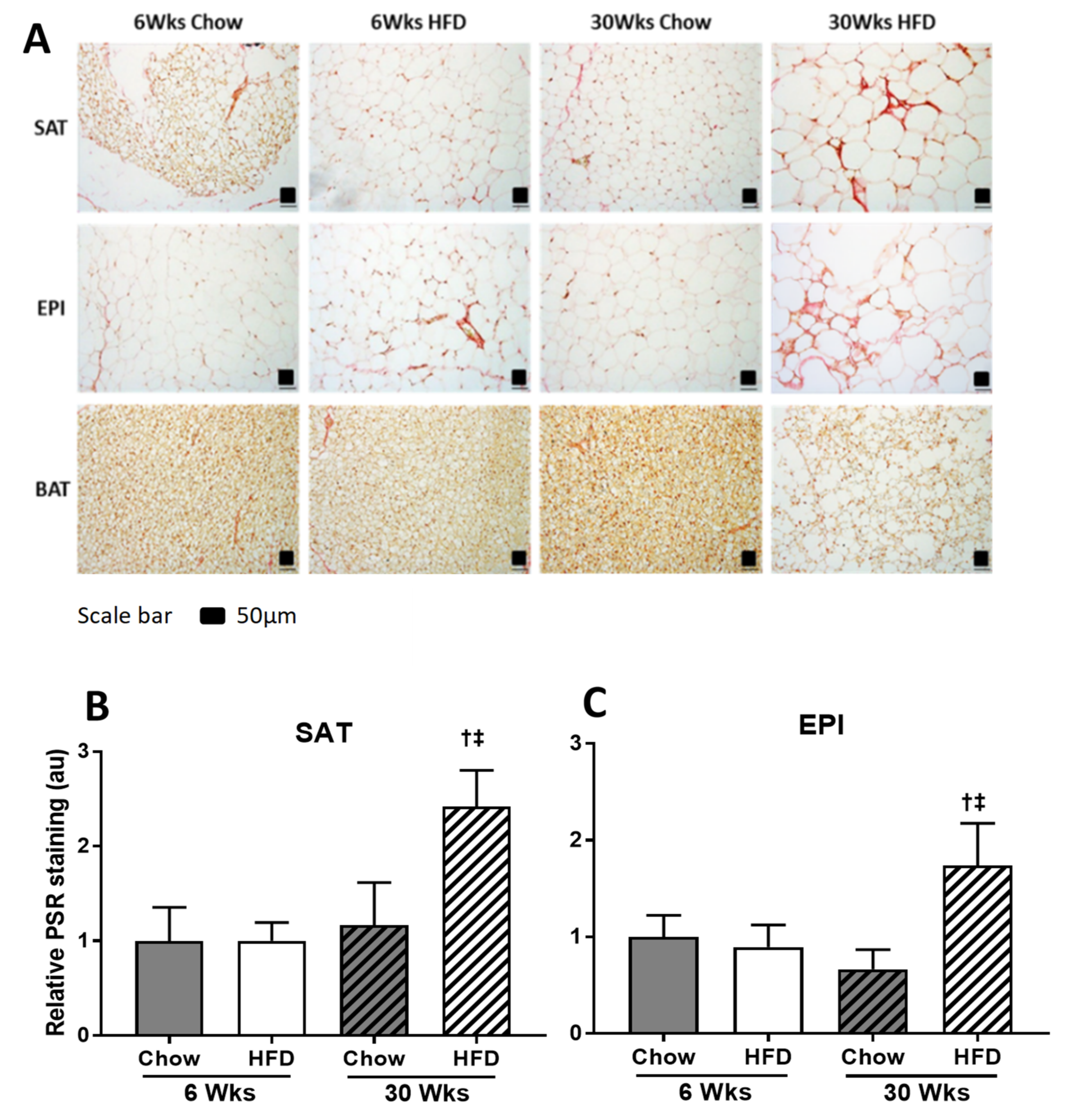
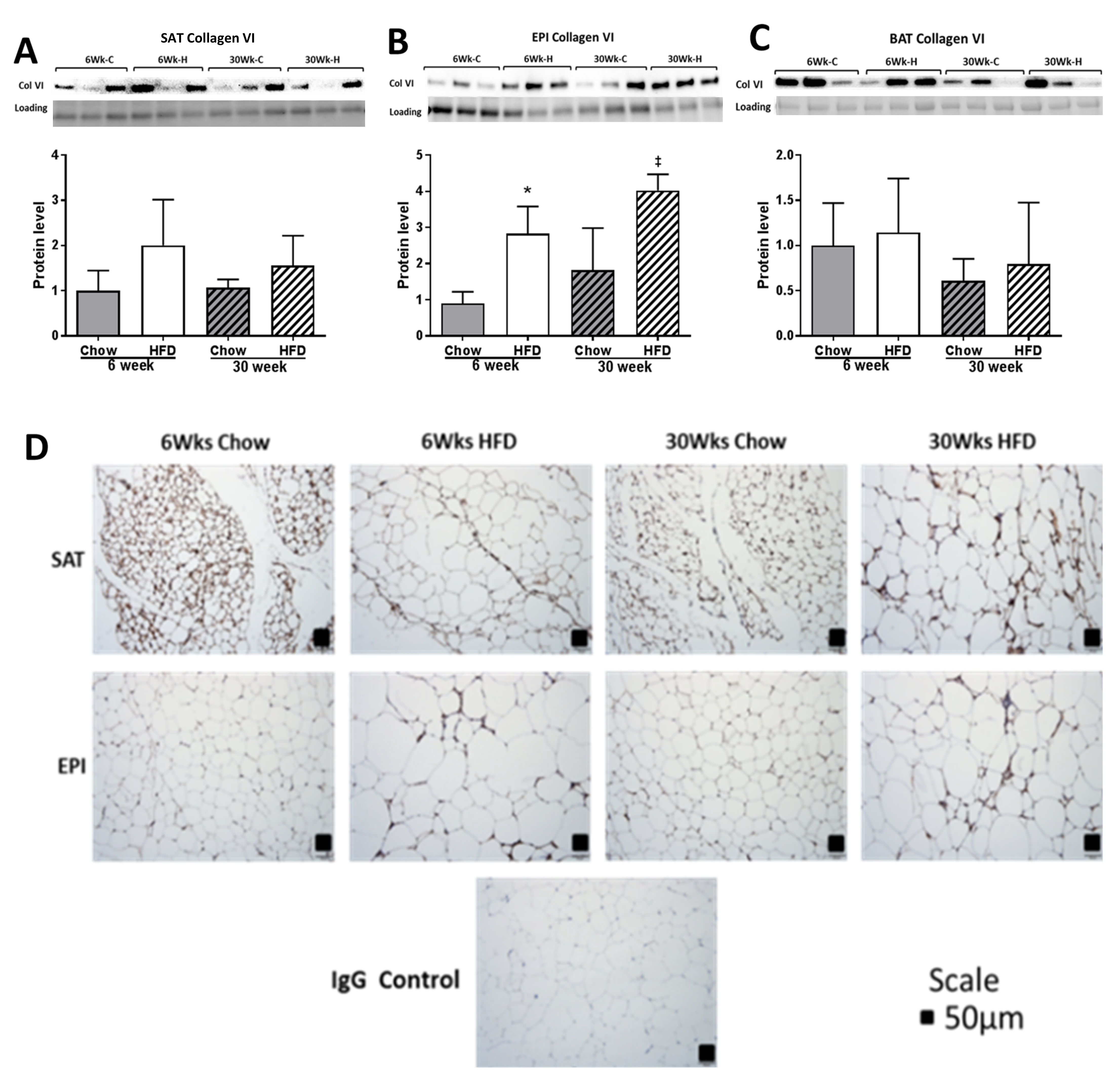
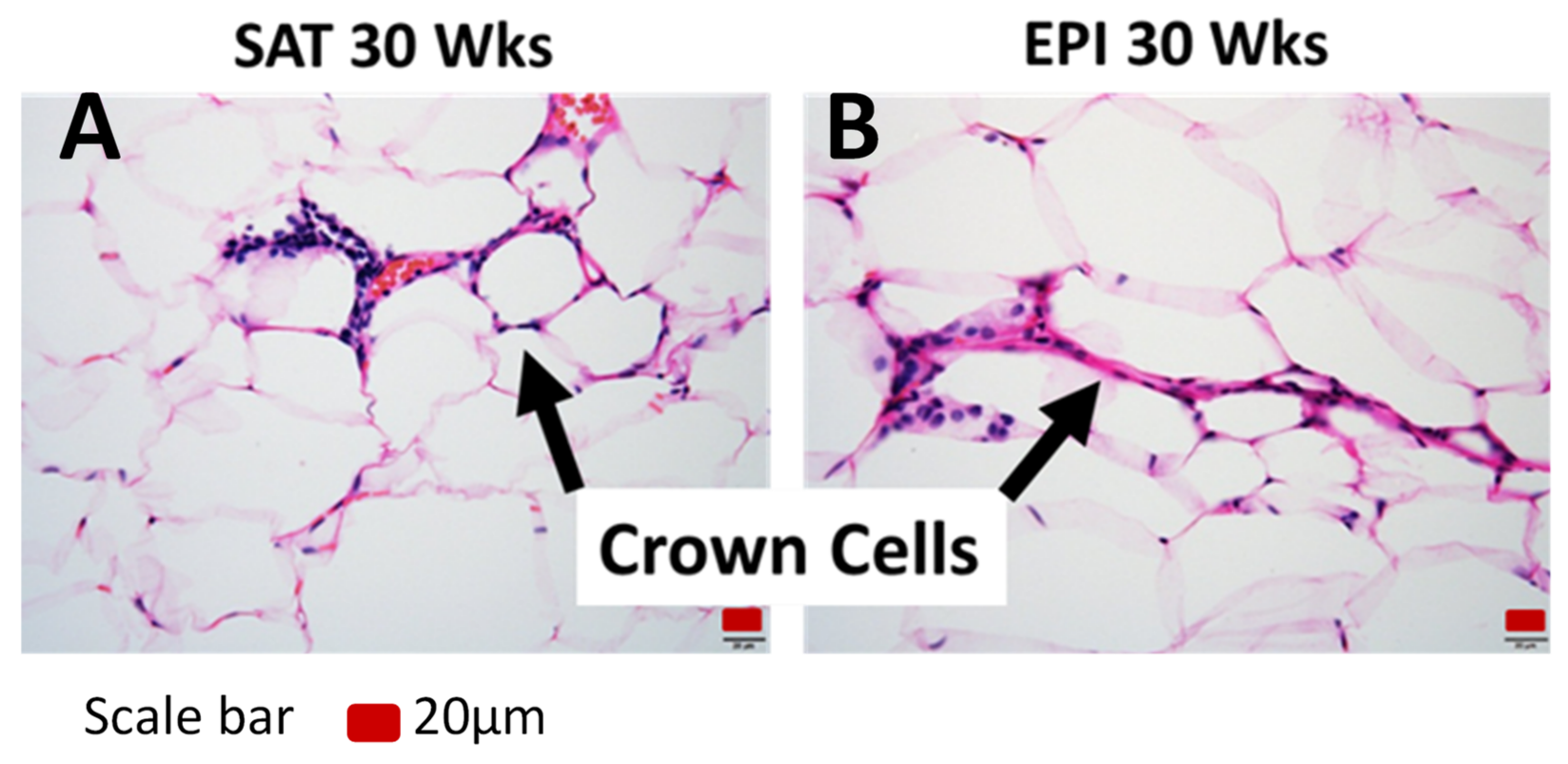
2.4. ECM Remodeling Markers
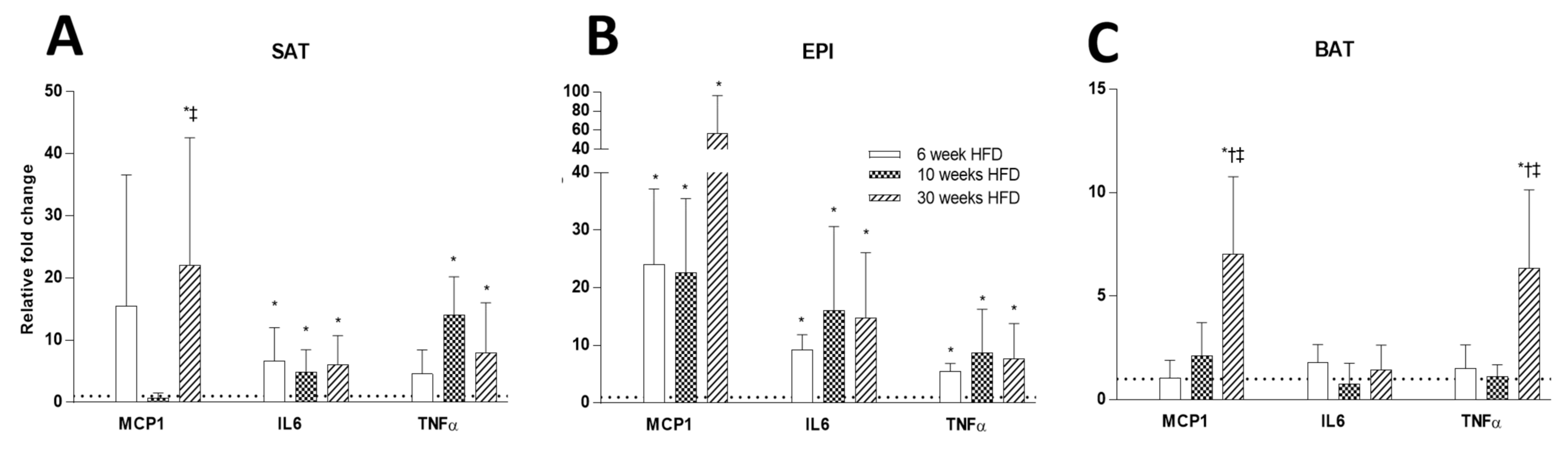
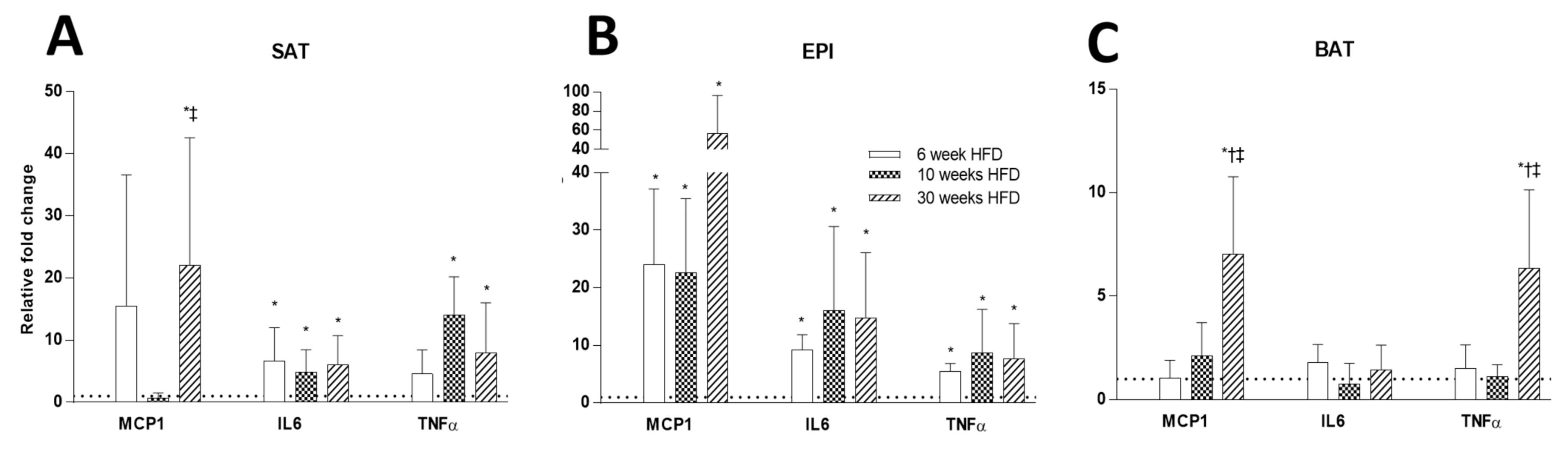
2.5. Inflammatory Markers
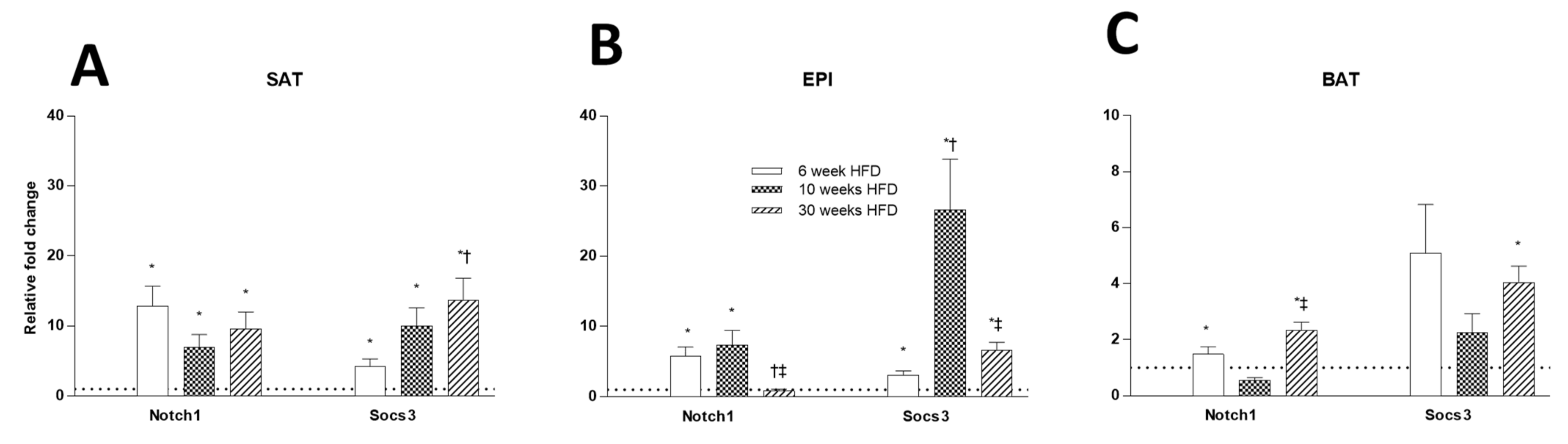
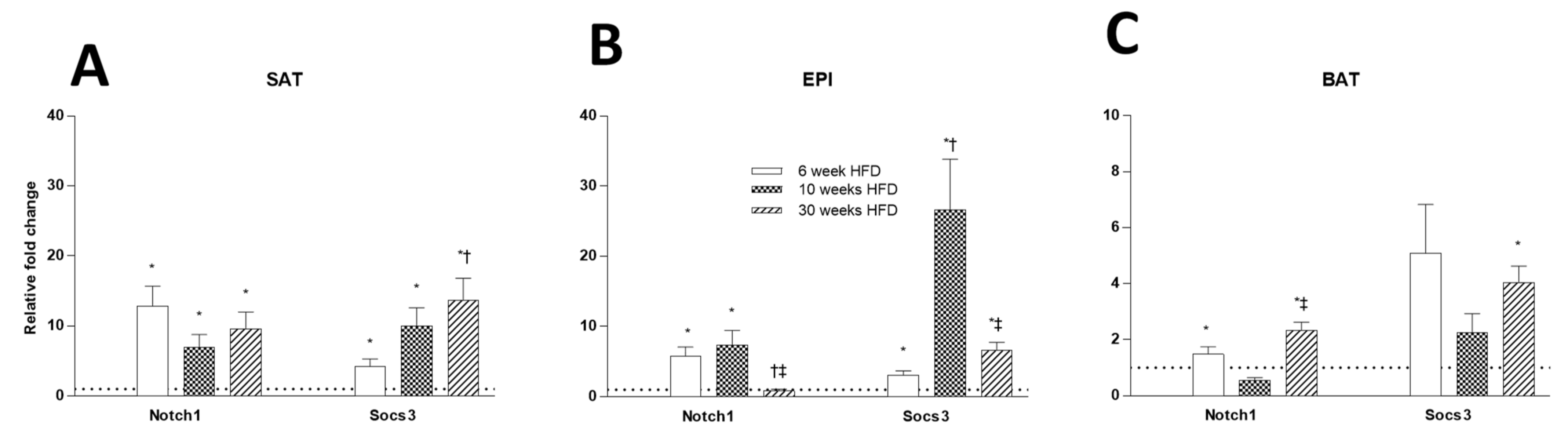
2.6. Tissue Insulin Resistance Markers
3. Discussion
4. Materials and Methods
4.1. Animals and Diet
4.2. Measurement of Gene Expression
4.3. Protein Quantification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward | Reverse | Accession Number |
|---|---|---|---|
| Pparg | 5′-CTGTCGGTTTCAGAAGTGCCT-3′ | 5′-CCCAAACCTGATGGCATTGTGAGACA-3′ | NM_011671.4 |
| Tle3 | 5′-TTGTCACAGGAGCATCAGCAG-3′ | 5′-CAGATTGGGGAGTCCACGTA-3′ | NM_001083927.1 |
| Pref1 | 5′-CCTGGCTGTGTCAATGGAGT-3′ | 5′-CTTGTGCTGGCAGTCCTTTC-3′ | NM_001190705.1 |
| Adiponectin | 5′-CGACACCAAAAGGGCTCAGG-3′ | 5′-ACGTCATCTTCGGCATGACT-3′ | NM_009605.4 |
| Leptin | 5′-GCTGCAAGGTGCAAGAAGAAG-3′ | 5′-TAGGACCAAAGCCACAGGAAC-3′ | NM_008493.3 |
| Resistin | 5′-TTCCTGATGTCGGGGAAGTGA-3′ | 5′-GACCGGAGGACATCAGACATC-3′ | NM_001204959.1 |
| Pgca1 | 5′-CTGCGGGATGATGGAGACAG-3′ | 5′-TCGTTCGACCTGCGTAAAGT-3′ | NM_008904.2 |
| Prdm16 | 5′-TGACCATACCCGGAGGCATA-3′ | 5′-CTGACGAGGGTCCTGTGATG-3′ | NM_001177995.1 |
| Tbx15 | 5′-TGGCAGAAACAGAACTGGACT-3′ | 5′-CCTTGCTGCTTTTGCATGGT-3′ | NM_009323.2 |
| Ucp1 | 5′-CATGGGATCAAACCCCGCTA-3′ | 5′-ATTAGGGGTCGTCCCTTTCC-3′ | NM_009463.3 |
| Ucp2 | 5′-GGCCTCTGGAAAGGGACTTCT-3′ | 5′-TTGGCTTTCAGGAGAGTATCTTT-3′ | NM_011671.4 |
| Mcp1 | 5′-CACTCACCTGCTGCTACTCA-3′ | 5′-GCTTGGTGACAAAAACTACAGC-3′ | NM_011333.3 |
| Il6 | 5′-TCCTCTCTGCAAGAGACTTCC-3′ | 5′-TTGTGAAGTAGGGAAGGCCG-3′ | NM_031168.1 |
| Tnfa | 5′-GACCCTCACACTCACAAACCA-3′ | 5′-ACAAGGTACAACCCATCGGC-3′ | NM_001278601.1 |
| Collagen VI | 5′-GAACTTCCCTGCCAAACAGA-3′ | 5′-CACCTTGTGGAAGTTCTGCTC-3′ | NM_146007.2 |
| Tgfb1 | 5′-ACCGCAACAACGCCATCTAT-3′ | 5′-TGCTTCCCGAATGTCTGACG-3′ | NM_011577.1 |
| Ccn2/ctgf | 5′-GAGTGTGCACTGCCAAAGATG-3′ | 5′-TCCAGGCAAGTGCATTGG T-3′ | NM_010217.2 |
| Timp1 | 5′-CACAAGTCCCAGAACCGC-3′ | 5′-GGATTCCGTGGCAGGC-3′ | NM_001294280.2 |
| Timp3 | 5′-CTTCTGCAACTCCGACATCGTGAT-3′ | 5′-CAGCAGGTACTGGTACTTGTTGAC-3′ | NM_011595.2 |
| Notch1 | 5′-ACAGTGCAACCCCCTGTATG-3′ | 5′-TCTAGGCCATCCCACTCACA-3′ | NM_008714.3 |
| Socs3 | 5′-TAGACTTCACGGCTGCCAAC-3′ | 5′-CGGGGAGCTAGTCCCGAA-3′ | NM_007707.3 |
| Nono | 5′-TGCTCCTGTGCCACCTGGTACTC-3′ | 5′-CCGGAGCTGGACGGTTGAATGC-3′ | NM_146007.2 |
4.4. Analysis of Tissue Structural Changes by Histochemistry
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bellisari, A. Evolutionary origins of obesity. Obes. Rev. 2008, 9, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Pellegrinelli, V.; Carobbio, S.; Vidal-Puig, A. Adipose tissue plasticity: How fat depots respond differently to pathophysiological cues. Diabetologia 2016, 59, 1075–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, P.J. Culture and the evolution of obesity. Hum. Nat. 1991, 2, 31–57. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.; Yang, W.; Chen, C.S.; Reynolds, K.; He, J. Global burden of obesity in 2005 and projections to 2030. Int. J. Obes. 2008, 32, 1431–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, A.; McGraw, T.E.; Kahn, B.B. Insulin action in adipocytes, adipose remodeling, and systemic effects. Cell Metab. 2021, 33, 748–757. [Google Scholar] [CrossRef] [PubMed]
- He, M.Q.; Wang, J.Y.; Wang, Y.; Sui, J.; Zhang, M.; Ding, X.; Zhao, Y.; Chen, Z.Y.; Ren, X.X.; Shi, B.Y. High-fat diet-induced adipose tissue expansion occurs prior to insulin resistance in C57BL/6J mice. Chronic Dis. Transl. Med. 2020, 6, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, Q.; Bai, Z.; Zhou, W.; Semenza, G.L.; Ge, R.L. Chronic cold exposure results in subcutaneous adipose tissue browning and altered global metabolism in Qinghai-Tibetan plateau pika (Ochotona curzoniae). Biochem. Biophys. Res. Commun. 2018, 500, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.K.; Hallahan, N.L.; Brown, S.H.; Liu, M.; Mitchell, T.W.; Cooney, G.J.; Turner, N. Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia 2013, 56, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, N.J.; Stock, M.J. A role for brown adipose tissue in diet-induced thermogenesis. Nature 1979, 281, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.S.; Kim, Y.J.; Kwon, E.Y.; Ryoo, J.Y.; Kim, S.R.; Jung, U.J. High-fat diet decreases energy expenditure and expression of genes controlling lipid metabolism, mitochondrial function and skeletal system development in the adipose tissue, along with increased expression of extracellular matrix remodelling- and inflammation-related genes. Br. J. Nutr. 2015, 113, 867–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keophiphath, M.; Achard, V.; Henegar, C.; Rouault, C.; Clement, K.; Lacasa, D. Macrophage-secreted factors promote a profibrotic phenotype in human preadipocytes. Mol. Endocrinol. 2009, 23, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Wernstedt Asterholm, I.; Tao, C.; Morley, T.S.; Wang, Q.A.; Delgado-Lopez, F.; Wang, Z.V.; Scherer, P.E. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014, 20, 103–118. [Google Scholar] [CrossRef] [Green Version]
- Despre’s, J.P.; Lemieux, I.; Bergeron, J.; Pibarot, P.; Mathieu, P.; Larose, E.; Rodés-Cabau, J.; Bertrand, O.F.; Poirier, P.R. Abdominal obesity and the metabolic syndrome: Contribution to global cardiometabolic risk. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1039–1049. [Google Scholar] [CrossRef]
- Tchkonia, T.; Thomou, T.; Zhu, Y.; Karagiannides, I.; Pothoulakis, C.; Jensen, M.D.; Kirkland, J.L. Mechanisms and Metabolic Implications of Regional Differences among Fat Depots. Cell Metab. 2013, 17, 644–656. [Google Scholar] [CrossRef] [Green Version]
- Spencer, M.; Yao-Borengasser, A.; Unal, R.; Rasouli, N.; Gurley, C.M.; Zhu, B.; Peterson, C.A.; Kern, P.A. Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E1016–E1027. [Google Scholar] [CrossRef]
- Jee, S.H.; Sull, J.W.; Park, J.; Lee, S.Y.; Ohrr, H.; Guallar, E.; Samet, J.M. Body-mass index and mortality in Korean men and women. N. Engl. J. Med. 2006, 355, 779–787. [Google Scholar] [CrossRef] [Green Version]
- Wildman, R.P.; Muntner, P.; Reynolds, K.; McGinn, A.P.; Rajpathak, S.; Wylie-Rosett, J.; Sowers, M.R. The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: Prevalence and correlates of 2 phenotypes among the US population (NHANES 1999–2004). Arch. Intern. Med. 2008, 168, 1617–1624. [Google Scholar] [CrossRef] [Green Version]
- Baglioni, S.; Cantini, G.; Poli, G.; Francalanci, M.; Squecco, R.; Di Franco, A.; Borgogni, E.; Frontera, S.; Nesi, G.; Liotta, F.; et al. Functional differences in visceral and subcutaneous fat pads originate from differences in the adipose stem cell. PLoS ONE 2012, 7, e36569. [Google Scholar] [CrossRef]
- Wueest, S.; Yang, X.; Liu, J.; Schoenle, E.J.; Konrad, D. Inverse regulation of basal lipolysis in perigonadal and mesenteric fat depots in mice. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E153–E160. [Google Scholar] [CrossRef]
- Porter, S.A.; Massaro, J.M.; Hoffmann, U.; Vasan, R.S.; O’Donnel, C.J.; Fox, C.S. Abdominal subcutaneous adipose tissue: A protective fat depot? Diabetes Care 2009, 32, 1068–1075. [Google Scholar] [CrossRef] [Green Version]
- Tchoukalova, Y.D.; Votruba, S.B.; Tchkonia, T.; Giorgadze, N.; Kirkland, J.L.; Jensen, M.D. Regional differences in cellular mechanisms of adipose tissue gain with overfeeding. Proc. Natl. Acad. Sci. USA 2010, 107, 18226–18231. [Google Scholar] [CrossRef] [Green Version]
- Laurencikiene, J.; Skurk, T.; Kulyte, A.; Heden, P.; Astrom, G.; Sjolin, E.; Ryden, M.; Hauner, H.; Arner, P. Regulation of lipolysis in small and large fat cells of the same subject. J. Clin. Endocrinol. Metab. 2011, 96, E2045–E2049. [Google Scholar] [CrossRef] [Green Version]
- Hudak, C.S.; Sul, H.S. Pref-1, a gatekeeper of adipogenesis. Front. Endocrinol. 2013, 4, 79. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, I.; Aprahamian, T.; Kikuchi, R.; Shimizu, A.; Papanicolaou, K.N.; MacLauchlan, S.; Maruyama, S.; Walsh, K. Vascular rarefaction mediates whitening of brown fat in obesity. J. Clin. Investig. 2014, 124, 2099–2112. [Google Scholar] [CrossRef] [Green Version]
- Fromme, T.; Klingenspor, M. Uncoupling protein 1 expression and high-fat diets. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1–R8. [Google Scholar] [CrossRef]
- De Sousa Abreu, R.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009, 5, 1512–1526. [Google Scholar] [CrossRef] [Green Version]
- Fleury, C.; Neverova, M.; Collins, S.; Raimbault, S.; Champigny, O.; Levi-Meyrueis, C.; Bouillaud, F.; Seldin, M.F.; Surwit, R.S.; Ricquier, D.; et al. Uncoupling protein-2: A novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 1997, 15, 269–272. [Google Scholar] [CrossRef]
- Brand, M.D.; Esteves, T.C. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2005, 2, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Malemud, C.J. Matrix metalloproteinases (MMPs) in health and disease: An overview. Front. Biosci. 2006, 11, 1696–1701. [Google Scholar] [CrossRef]
- Lijnen, H.R.; Demeulemeester, D.; Van Hoef, B.; Collen, D.; Maquoi, E. Deficiency of tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) impairs nutritionally induced obesity in mice. Thromb. Haemost. 2003, 89, 249–255. [Google Scholar] [PubMed]
- Menghini, R.; Menini, S.; Amoruso, R.; Fiorentino, L.; Casagrande, V.; Marzano, V.; Tornei, F.; Bertucci, P.; Iacobini, C.; Serino, M.; et al. Tissue inhibitor of metalloproteinase 3 deficiency causes hepatic steatosis and adipose tissue inflammation in mice. Gastroenterology 2009, 136, 663–672.e4. [Google Scholar] [CrossRef] [PubMed]
- Sam, S.; Haffner, S.; Davidson, M.H.; D’Agostino, R.B.; Feinstein, S.; Kondos, G.; Perez, A.; Mazzone, T. Relation of abdominal fat depots to systemic markers of inflammation in type 2 diabetes. Diabetes Care 2009, 32, 932–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, H.; Karasawa, T.; Usui, F.; Kawashima, A.; Endo, Y.; Kobayashi, M.; Sadatomo, A.; Nakamura, J.; Iwasaki, Y.; Yada, T.; et al. Caspase-1 deficiency promotes high-fat diet-induced adipose tissue inflammation and the development of obesity. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E881–E890. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Ishibashi, J.; Trefely, S.; Shao, M.; Cowan, A.J.; Sakers, A.; Lim, H.W.; O’Connor, S.; Doan, M.T.; Cohen, P.; et al. A PRDM16-Driven Metabolic Signal from Adipocytes Regulates Precursor Cell Fate. Cell Metab. 2019, 30, 174–189.e5. [Google Scholar] [CrossRef]
- Minh, H.V.; Tien, H.A.; Sinh, C.T.; Thang, D.C.; Chen, C.H.; Tay, J.C.; Siddique, S.; Wang, T.D.; Sogunuru, G.P.; Chia, Y.C.; et al. Assessment of preferred methods to measure insulin resistance in Asian patients with hypertension. J. Clin. Hypertens. 2021, 23, 529–537. [Google Scholar] [CrossRef]
- Chau, Y.Y.; Bandiera, R.; Serrels, A.; Martinez-Estrada, O.M.; Qing, W.; Lee, M.; Slight, J.; Thornburn, A.; Berry, R.; McHaffie, S.; et al. Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nat. Cell Biol. 2014, 16, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Lo, L.; McLennan, S.V.; Williams, P.F.; Bonner, J.; Chowdhury, S.; McCaughan, G.W.; Gorrell, M.D.; Yue, D.K.; Twigg, S.M. Diabetes is a progression factor for hepatic fibrosis in a high fat fed mouse obesity model of non-alcoholic steatohepatitis. J. Hepatol. 2011, 55, 435–444. [Google Scholar] [CrossRef]
- Martinez-Huenchullan, S.F.; Maharjan, B.R.; Williams, P.F.; Tam, C.S.; McLennan, S.V.; Twigg, S.M. Differential metabolic effects of constant moderate versus high intensity interval training in high-fat fed mice: Possible role of muscle adiponectin. Physiol. Rep. 2018, 6, e13599. [Google Scholar] [CrossRef] [Green Version]
- Maharjan, B.R.; Martinez-Huenchullan, S.F.; McLennan, S.V.; Twigg, S.M.; Williams, P.F. Exercise induces favorable metabolic changes in white adipose tissue preventing high-fat diet obesity. Physiol Rep. 2021, 9, e14929. [Google Scholar] [CrossRef]
- Maharjan, B.R.; McLennan, S.V.; Twigg, S.M.; Williams, P.F. The effect of TGFbeta1 on thermogenic markers is dependent on the degree of adipocyte differentiation. Biosci. Rep. 2020, 40, BSR20194262. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maharjan, B.R.; McLennan, S.V.; Yee, C.; Twigg, S.M.; Williams, P.F. The Effect of a Sustained High-Fat Diet on the Metabolism of White and Brown Adipose Tissue and Its Impact on Insulin Resistance: A Selected Time Point Cross-Sectional Study. Int. J. Mol. Sci. 2021, 22, 13639. https://doi.org/10.3390/ijms222413639
Maharjan BR, McLennan SV, Yee C, Twigg SM, Williams PF. The Effect of a Sustained High-Fat Diet on the Metabolism of White and Brown Adipose Tissue and Its Impact on Insulin Resistance: A Selected Time Point Cross-Sectional Study. International Journal of Molecular Sciences. 2021; 22(24):13639. https://doi.org/10.3390/ijms222413639
Chicago/Turabian StyleMaharjan, Babu Raja, Susan V. McLennan, Christine Yee, Stephen M. Twigg, and Paul F. Williams. 2021. "The Effect of a Sustained High-Fat Diet on the Metabolism of White and Brown Adipose Tissue and Its Impact on Insulin Resistance: A Selected Time Point Cross-Sectional Study" International Journal of Molecular Sciences 22, no. 24: 13639. https://doi.org/10.3390/ijms222413639
APA StyleMaharjan, B. R., McLennan, S. V., Yee, C., Twigg, S. M., & Williams, P. F. (2021). The Effect of a Sustained High-Fat Diet on the Metabolism of White and Brown Adipose Tissue and Its Impact on Insulin Resistance: A Selected Time Point Cross-Sectional Study. International Journal of Molecular Sciences, 22(24), 13639. https://doi.org/10.3390/ijms222413639