
Targeting AVIL, a New Cytoskeleton Regulator in Glioblastoma


Abstract
:1. Introduction
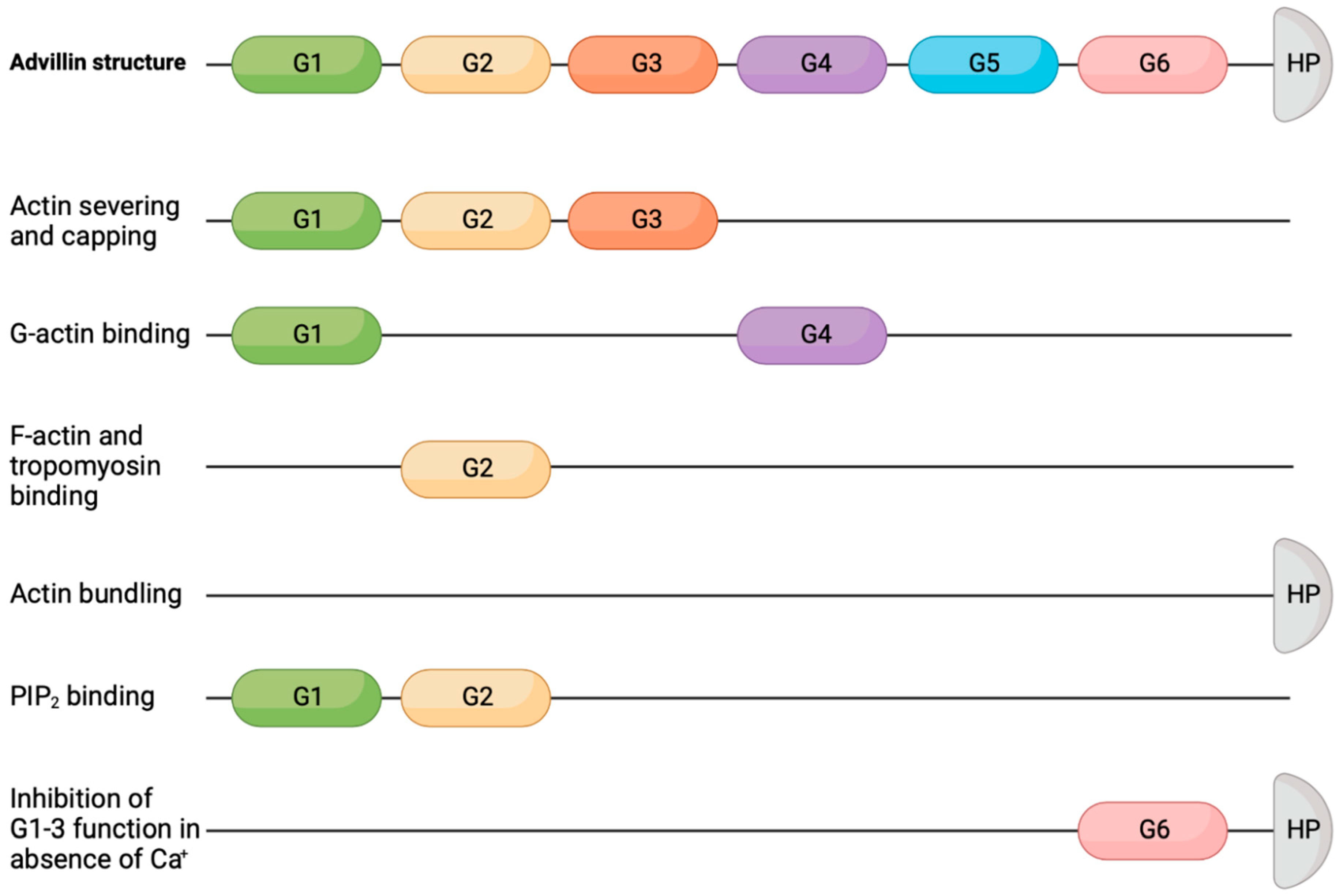
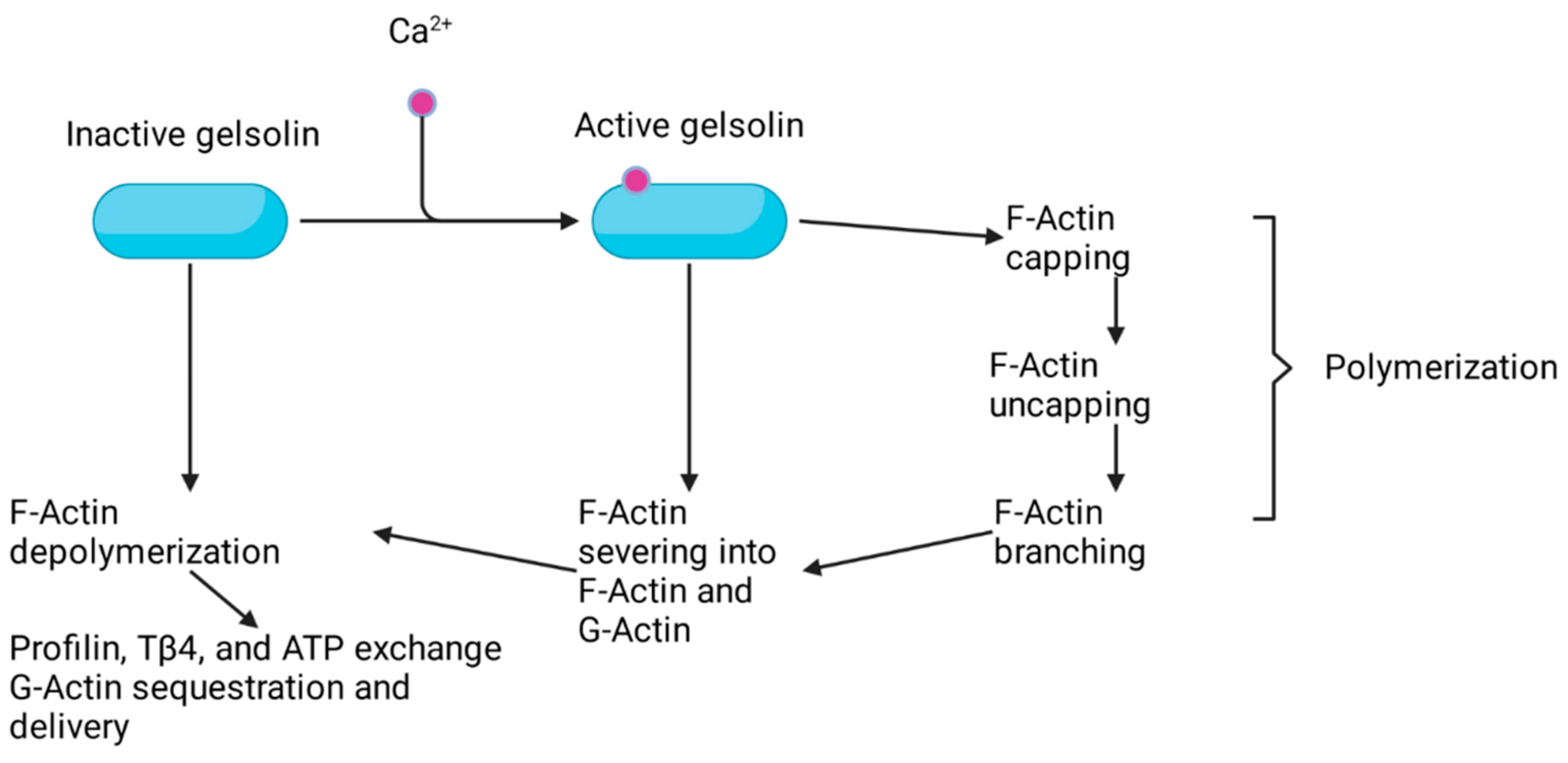
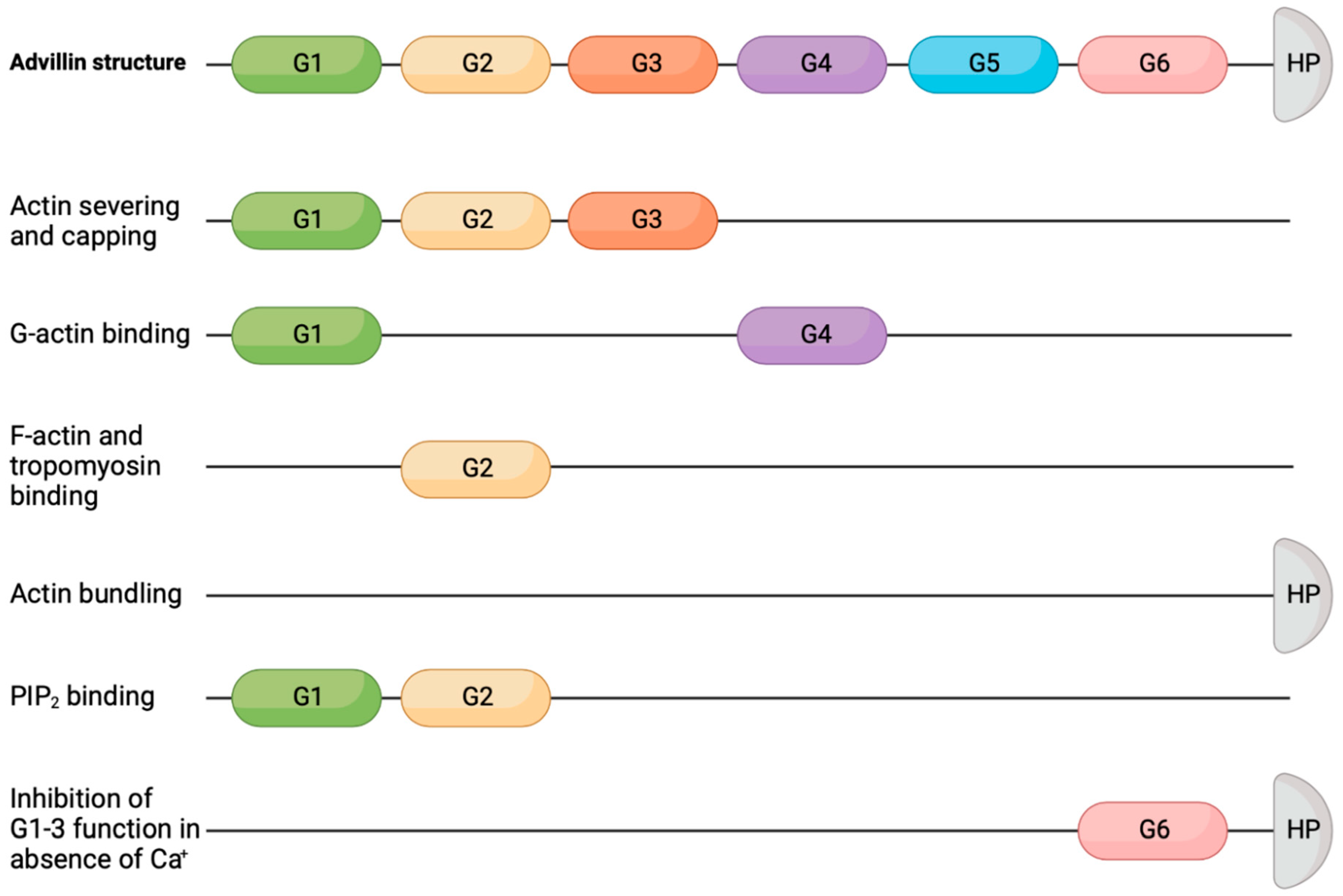
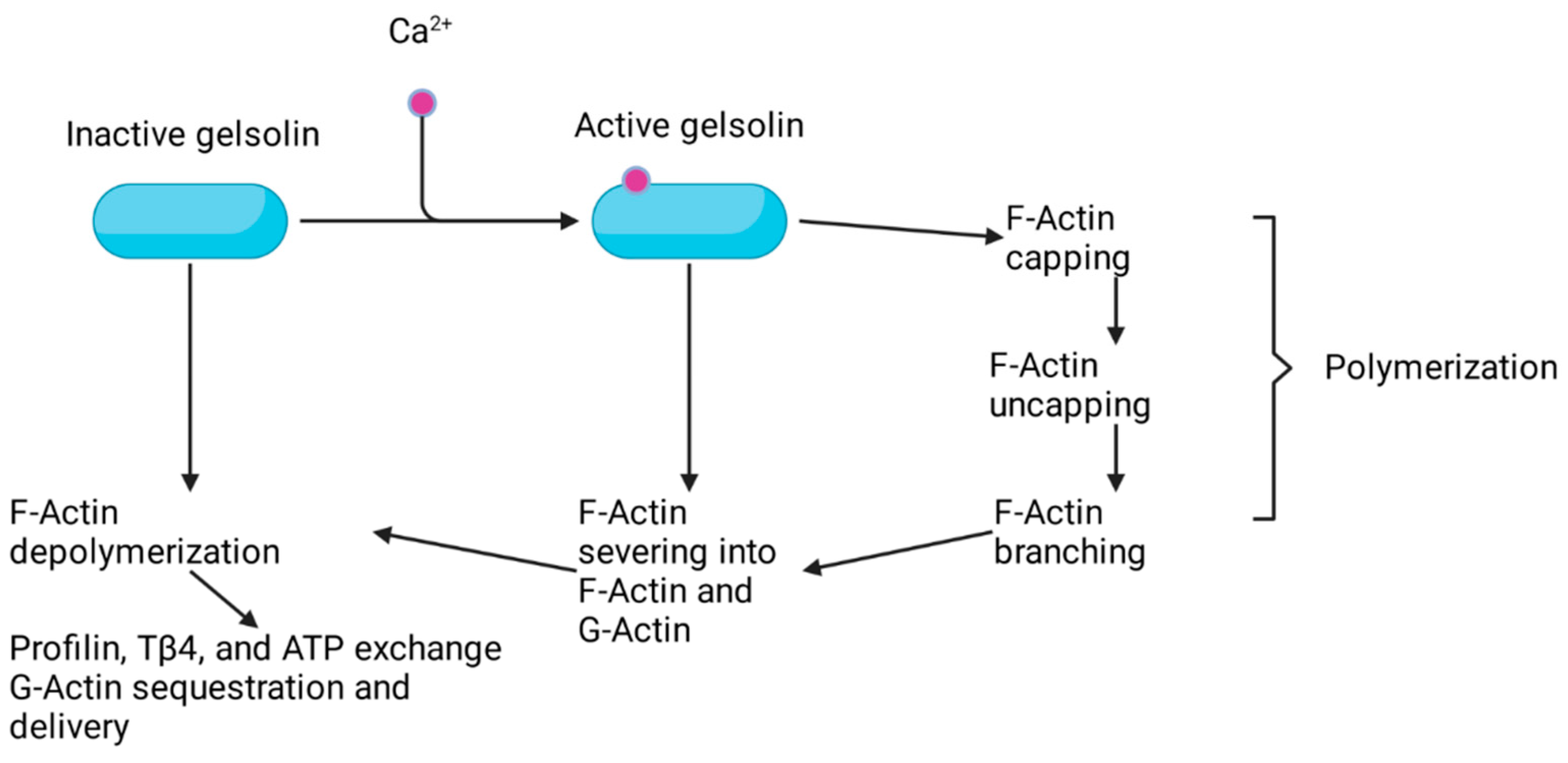
2. Advillin Overview
3. Advillin Family Members and Their Association with Cancer
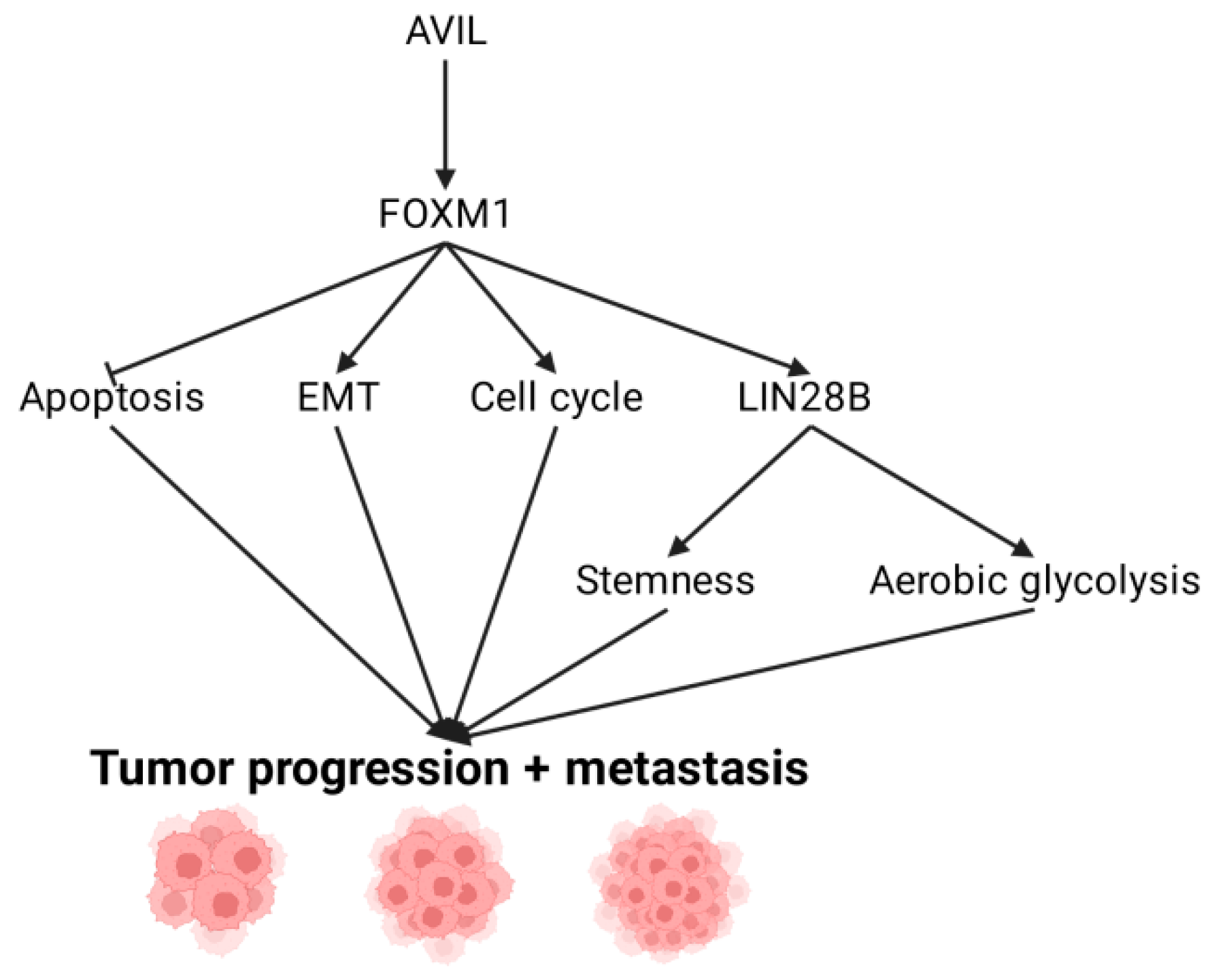
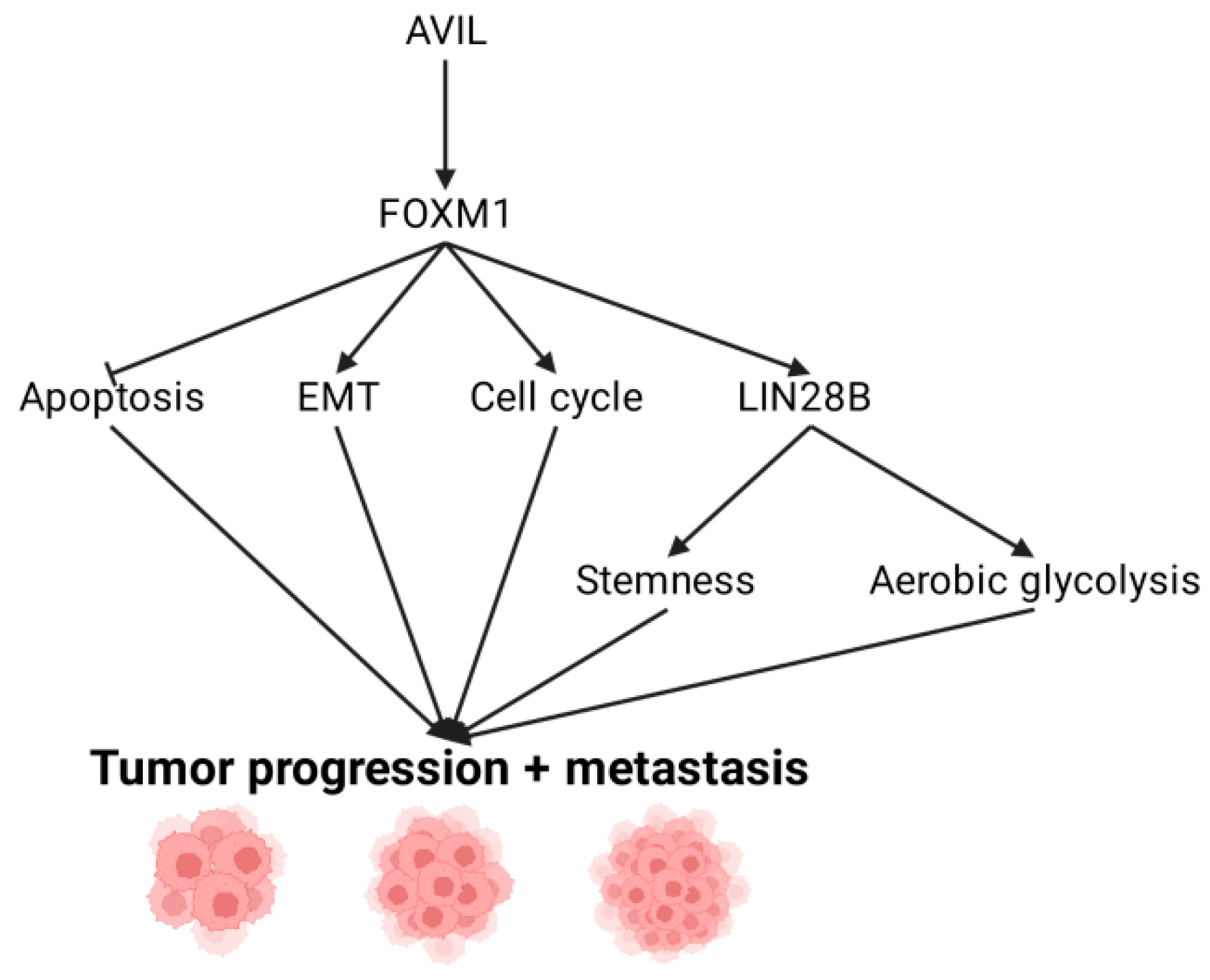
4. Downstream Targets of AVIL: FOXM1
5. FOXM1 Regulation of LIN28B and Its Role in GBM
6. Activating Invasion and Metastasis through EMT
7. Escaping Immune Surveillance
8. Discussion
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [Green Version]
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef]
- Batich, K.A.; Sampson, J.H. Standard of care and future pharmacological treatment options for malignant glioma: An urgent need for screening and identification of novel tumor-specific antigens. Expert Opin. Pharmacother. 2014, 15, 2047–2061. [Google Scholar] [CrossRef]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Huang, B.; Li, X.; Li, Y.; Zhang, J.; Zong, Z.; Zhang, H. Current Immunotherapies for Glioblastoma Multiforme. Front. Immunol. 2020, 11, 603911. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma-are we there yet? Neuro. Oncol. 2013, 15, 4–27. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.R.; O’Neill, B.P. Glioblastoma survival in the United States before and during the temozolomide era. J. Neurooncol. 2012, 107, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.-L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef] [Green Version]
- Kleihues, P.; Ohgaki, H. Primary and secondary glioblastomas: From concept to clinical diagnosis. Neuro. Oncol. 1999, 1, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Das, P.; Puri, T.; Jha, P.; Pathak, P.; Joshi, N.; Suri, V.; Sharma, M.C.; Sharma, B.S.; Mahapatra, A.K.; Suri, A.; et al. A clinicopathological and molecular analysis of glioblastoma multiforme with long-term survival. J. Clin. Neurosci. 2011, 18, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Krex, D.; Klink, B.; Hartmann, C.; von Deimling, A.; Pietsch, T.; Simon, M.; Sabel, M.; Steinbach, J.P.; Heese, O.; Reifenberger, G.; et al. German Glioma Network Long-term survival with glioblastoma multiforme. Brain 2007, 130, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brat, D.J.; Van Meir, E.G. Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab. Investig. 2004, 84, 397–405. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef]
- Zhang, P.; Xia, Q.; Liu, L.; Li, S.; Dong, L. Current Opinion on Molecular Characterization for GBM Classification in Guiding Clinical Diagnosis, Prognosis, and Therapy. Front. Mol. Biosci. 2020, 7, 241. [Google Scholar] [CrossRef]
- Komori, T. Grading of adult diffuse gliomas according to the 2021 WHO Classification of Tumors of the Central Nervous System. Lab. Investig. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro. Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Le Rhun, E. How did lomustine become standard of care in recurrent glioblastoma? Cancer Treat. Rev. 2020, 87, 102029. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Ren, X.; Ai, D.; Li, T.; Xia, L.; Sun, L. Effectiveness of Lomustine Combined With Bevacizumab in Glioblastoma: A Meta-Analysis. Front. Neurol. 2021, 11, 1893. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Sane, R.; Oberoi, R.; Ohlfest, J.R.; Elmquist, W. Delivery of Molecularly Targeted Therapy to Malignant Glioma, a Disease of the Whole Brain. Expert Rev. Mol. Med. 2011, 13, e17. [Google Scholar] [CrossRef] [Green Version]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebelt, B.D.; Shingu, T.; Zhou, X.; Ren, J.; Shin, S.A.; Hu, J. Glioma Stem Cells: Signaling, Microenvironment, and Therapy. Stem Cells Int. 2016, 2016, 7849890. [Google Scholar] [CrossRef] [Green Version]
- Livshits, Z.; Rao, R.B.; Smith, S.W. An approach to chemotherapy-associated toxicity. Emerg. Med. Clin. North Am. 2014, 32, 167–203. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Sawyers, C.L.; Kantarjian, H.; Resta, D.J.; Reese, S.F.; Ford, J.M.; Capdeville, R.; Talpaz, M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N. Engl. J. Med. 2001, 344, 1038–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet. Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Greco, A.; Safi, D.; Swami, U.; Ginader, T.; Milhem, M.; Zakharia, Y. Efficacy and Adverse Events in Metastatic Melanoma Patients Treated with Combination BRAF Plus MEK Inhibitors Versus BRAF Inhibitors: A Systematic Review. Cancers (Basel) 2019, 11, 1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet. Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Perez, E.A. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Padilla, I.; Oza, A.M. Epothilones in the treatment of ovarian cancer. Futur. Oncol. 2011, 7, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Zagouri, F.; Sergentanis, T.N.; Chrysikos, D.; Dimopoulos, M.A.; Bamias, A. Epothilones in epithelial ovarian, fallopian tube, or primary peritoneal cancer: A systematic review. Onco. Targets. Ther. 2015, 8, 2187–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Janczyk, P.Ł.; Zhang, Y.; Liu, A.; Shi, X.; Singh, S.; Facemire, L.; Kubow, K.; Li, Z.; Jia, Y.; et al. A cytoskeleton regulator AVIL drives tumorigenesis in glioblastoma. Nat. Commun. 2020, 11, 3457. [Google Scholar] [CrossRef]
- Marks, P.W.; Arai, M.; Bandura, J.L.; Kwiatkowski, D.J. Advillin (p92): A new member of the gelsolin/villin family of actin regulatory proteins. J. Cell Sci. 1998, 111. [Google Scholar] [CrossRef]
- Silacci, P.; Mazzolai, L.; Gauci, C.; Stergiopulos, N.; Yin, H.L.; Hayoz, D. Gelsolin superfamily proteins: Key regulators of cellular functions. Cell. Mol. Life Sci. 2004, 61, 2614–2623. [Google Scholar] [CrossRef] [Green Version]
- Nag, S.; Larsson, M.; Robinson, R.C.; Burtnick, L.D. Gelsolin: The tail of a molecular gymnast. Cytoskeleton 2013, 70, 360–384. [Google Scholar] [CrossRef]
- Izdebska, M.; Zielińska, W.; Grzanka, D.; Gagat, M. The Role of Actin Dynamics and Actin-Binding Proteins Expression in Epithelial-to-Mesenchymal Transition and Its Association with Cancer Progression and Evaluation of Possible Therapeutic Targets. Biomed Res. Int. 2018, 2018, 4578373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, J.; Ashraf, S.; Tan, W.; Van Der Ven, A.T.; Gee, H.Y.; Braun, D.A.; Fehér, K.; George, S.P.; Esmaeilniakooshkghazi, A.; Choi, W.; et al. Advillin acts upstream of phospholipase C ϵ1 in steroid-resistant nephrotic syndrome. J. Clin. Invest. 2017, 127, 4257–4269. [Google Scholar] [CrossRef]
- George, S.P.; Esmaeilniakooshkghazi, A.; Roy, S.; Khurana, S. F-actin-bundling sites are conserved in proteins with villin-type headpiece domains. Mol. Biol. Cell 2020, 31, 1857–1866. [Google Scholar] [CrossRef]
- Vermeulen, W.; Vanhaesebrouck, P.; Van Troys, M.; Verschueren, M.; Fant, F.; Goethals, M.; Ampe, C.; Martins, J.C.; Borremans, F.A.M. Solution structures of the C-terminal headpiece subdomains of human villin and advillin, evaluation of headpiece F-actin-binding requirements. Protein Sci. 2004, 13, 1276–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matarrese, P.; Vona, R.; Ascione, B.; Paggi, M.G.; Mileo, A.M. Physical Interaction between HPV16E7 and the Actin-Binding Protein Gelsolin Regulates Epithelial-Mesenchymal Transition via HIPPO-YAP Axis. Cancers (Basel) 2021, 13, 353. [Google Scholar] [CrossRef]
- Oelrich, F.; Junker, H.; Stope, M.; Erb, H.H.; Walther, R.; Venz, S.; Zimmermann, U. Gelsolin governs the neuroendocrine transdifferentiation of prostate cancer cells and suppresses the apoptotic machinery. Anticancer Res. 2021, 41, 3717–3729. [Google Scholar] [CrossRef]
- Miura, N.; Takemori, N.; Kikugawa, T.; Tanji, N.; Higashiyama, S.; Yokoyama, M. Adseverin: A novel cisplatin-resistant marker in the human bladder cancer cell line HT1376 identified by quantitative proteomic analysis. Mol. Oncol. 2012, 6, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Li, J.; Zhu, D.; Niu, Z.; Pan, X.; Xu, P.; Ji, M.; Wei, Y.; Xu, J. Aberrant Scinderin Expression Correlates With Liver Metastasis and Poor Prognosis in Colorectal Cancer. Front. Pharmacol. 2019, 10, 1183. [Google Scholar] [CrossRef]
- Maklad, A.; Sharma, A.; Azimi, I. Calcium Signaling in Brain Cancers: Roles and Therapeutic Targeting. Cancers (Basel) 2019, 11, 145. [Google Scholar] [CrossRef] [Green Version]
- Strudwick, X.L.; Cowin, A.J. Multifunctional Roles of the Actin-Binding Protein Flightless I in Inflammation, Cancer and Wound Healing. Front. Cell Dev. Biol. 2020, 8, 1394. [Google Scholar] [CrossRef] [PubMed]
- Gdynia, G.; Grund, K.; Eckert, A.; Böck, B.C.; Funke, B.; Macher-Goeppinger, S.; Sieber, S.; Herold-Mende, C.; Wiestler, B.; Wiestler, O.D.; et al. Basal caspase activity promotes migration and invasiveness in glioblastoma cells. Mol. Cancer Res. 2007, 5, 1232–1240. [Google Scholar] [CrossRef] [Green Version]
- Qiao, X.; Zhou, Y.; Xie, W.; Wang, Y.; Zhang, Y.; Tian, T.; Dou, J.; Yang, X.; Shen, S.; Hu, J.; et al. Scinderin is a novel transcriptional target of BRMS1 involved in regulation of hepatocellular carcinoma cell apoptosis. Am. J. Cancer Res. 2018, 8, 1008–1018. [Google Scholar] [PubMed]
- Asare-Werehene, M.; Communal, L.; Carmona, E.; Han, Y.; Song, Y.S.; Burger, D.; Mes-Masson, A.M.; Tsang, B.K. Plasma gelsolin inhibits CD8þ t-cell function and regulates glutathione production to confer chemoresistance in ovarian cancer. Cancer Res. 2020, 80, 3959–3971. [Google Scholar] [CrossRef] [PubMed]
- Asare-Werehene, M.; Nakka, K.; Reunov, A.; Chiu, C.T.; Lee, W.T.; Abedini, M.R.; Wang, P.W.; Shieh, D.-B.; Dilworth, F.J.; Carmona, E.; et al. The exosome-mediated autocrine and paracrine actions of plasma gelsolin in ovarian cancer chemoresistance. Oncogene 2020, 39, 1600–1616. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.T.; Wang, P.W.; Asare-Werehene, M.; Tsang, B.K.; Shieh, D. Bin Circulating plasma gelsolin: A predictor of favorable clinical outcomes in head and neck cancer and sensitive biomarker for early disease diagnosis combined with soluble fas ligand. Cancers (Basel) 2020, 12, 1569. [Google Scholar] [CrossRef]
- Prescher, N.; Hänsch, S.; Knobbe-Thomsen, C.B.; Stühler, K.; Poschmann, G. The migration behavior of human glioblastoma cells is influenced by the redox-sensitive human macrophage capping protein CAPG. Free Radic. Biol. Med. 2021, 167, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Lang, Z.; Chen, Y.; Zhu, H.; Sun, Y.; Zhang, H.; Huang, J.; Zou, Z. Prognostic and clinicopathological significance of CapG in various cancers: Evidence from a meta-analysis. Pathol. Res. Pract. 2019, 215, 152683. [Google Scholar] [CrossRef] [PubMed]
- Yun, D.-P.; Wang, Y.-Q.; Meng, D.-L.; Ji, Y.-Y.; Chen, J.-X.; Chen, H.-Y.; Lu, D.-R. Actin-capping protein CapG is associated with prognosis, proliferation and metastasis in human glioma. Oncol. Rep. 2018, 39, 1011–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Chi, Y.; Qin, Y.; Wang, Z.; Xiu, B.; Su, Y.; Guo, R.; Guo, L.; Sun, H.; Zeng, C.; et al. CAPG enhances breast cancer metastasis by competing with PRMT5 to modulate STC-1 transcription. Theranostics 2018, 8, 2549–2564. [Google Scholar] [CrossRef]
- Koya, R.C.; Fujita, H.; Shimizu, S.; Ohtsu, M.; Takimoto, M.; Tsujimoto, Y.; Kuzumaki, N. Gelsolin Inhibits Apoptosis by Blocking Mitochondrial Membrane Potential Loss and Cytochrome c Release. J. Biol. Chem. 2000, 275, 15343–15349. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, S.; Siu, T.L.; Huang, S. Glioblastoma Multiforme Formation and EMT: Role of FoxM1 Transcription Factor. Curr. Pharm. Des. 2015, 21, 1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myatt, S.S.; Lam, E.W.-F. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Dai, B.; Kang, S.-H.; Ban, K.; Huang, F.-J.; Lang, F.F.; Aldape, K.D.; Xie, T.; Pelloski, C.E.; Xie, K.; et al. FoxM1B Is Overexpressed in Human Glioblastomas and Critically Regulates the Tumorigenicity of Glioma Cells. Cancer Res. 2006, 66, 3593–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, R.; Zhou, J.; Chen, C.; Xu, T.; Yan, Y.; Ma, Y.; Zheng, Z.; Shen, Y.; Lu, Y.; Fu, D.; et al. LIN28 is involved in glioma carcinogenesis and predicts outcomes of glioblastoma multiforme patients. PLoS ONE 2014, 9, e86446. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, S.R.; Powers, J.T.; Einhorn, W.; Hoshida, Y.; Ng, T.L.; Toffanin, S.; O’Sullivan, M.; Lu, J.; Phillips, L.A.; Lockhart, V.L.; et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat. Genet. 2009, 41, 843–848. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Hütt-Cabezas, M.; Orr, B.A.; Weingart, M.; Taylor, I.; Rajan, A.K.D.; Odia, Y.; Kahlert, U.; Maciaczyk, J.; Nikkhah, G.; et al. LIN28A facilitates the transformation of human neural stem cells and promotes glioblastoma tumorigenesis through a pro-invasive genetic program. Oncotarget 2013, 4, 1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Li, W.; Yan, Y.; Yao, X.; Gu, W.; Zhang, H. LINC01094 triggers radio-resistance in clear cell renal cell carcinoma via miR-577/CHEK2/FOXM1 axis. Cancer Cell Int. 2020, 20, 274. [Google Scholar] [CrossRef]
- Luo, C.; Lin, K.; Hu, C.; Zhu, X.; Zhu, J.; Zhu, Z. LINC01094 promotes pancreatic cancer progression by sponging miR-577 to regulate LIN28B expression and the PI3K/AKT pathway. Mol. Ther.-Nucleic Acids 2021, 26, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [Green Version]
- Hall, A. The cytoskeleton and cancer. Cancer Metastasis Rev. 2009, 28, 5–14. [Google Scholar] [CrossRef]
- Klymkowsky, M.W.; Savagner, P. Epithelial-mesenchymal transition: A cancer researcher’s conceptual friend and foe. Am. J. Pathol. 2009, 174, 1588–1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, M.J.; Hayakawa, Y.; Takeda, K.; Yagita, H. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat. Rev. Cancer 2002, 2, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Al Absi, A.; Wurzer, H.; Guerin, C.; Hoffmann, C.; Moreau, F.; Mao, X.; Brown-Clay, J.; Petrolli, R.; Casellas, C.P.; Dieterle, M.; et al. Actin Cytoskeleton Remodeling Drives Breast Cancer Cell Escape from Natural Killer-Mediated Cytotoxicity. Cancer Res. 2018, 78, 5631–5643. [Google Scholar] [CrossRef] [Green Version]
- Iser, I.C.; Pereira, M.B.; Lenz, G.; Wink, M.R. The Epithelial-to-Mesenchymal Transition-Like Process in Glioblastoma: An Updated Systematic Review and In Silico Investigation. Med. Res. Rev. 2017, 37, 271–313. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Advillin (AVIL) as a Lead Target in GBM |
|---|
| Expression: AVIL is overexpressed in 100% GBM cell lines and clinical samples, but hardly expressed in astrocyte and normal brain tissues. |
| Loss-of-function systems: Silencing AVIL caused reduced proliferation and migration of GBM cell culture and xenograft, but had no effect on astrocytes. |
| Gain-of-function systems: Overexpressing AVIL promoted GBM and astrocyte cell proliferation and migration, and transformed astrocytes. |
| GBM stem cell/initiating cells: The major therapy resistant cells. GSC/GIC cells express even higher levels of AVIL. Silencing AVIL triggered reduced neurosphere formation and stemness. |
| Clinical correlation: High AVIL expression is correlated with worse patient survival. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cornelison, R.; Marrah, L.; Horter, D.; Lynch, S.; Li, H. Targeting AVIL, a New Cytoskeleton Regulator in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 13635. https://doi.org/10.3390/ijms222413635
Cornelison R, Marrah L, Horter D, Lynch S, Li H. Targeting AVIL, a New Cytoskeleton Regulator in Glioblastoma. International Journal of Molecular Sciences. 2021; 22(24):13635. https://doi.org/10.3390/ijms222413635
Chicago/Turabian StyleCornelison, Robert, Laine Marrah, Drew Horter, Sarah Lynch, and Hui Li. 2021. "Targeting AVIL, a New Cytoskeleton Regulator in Glioblastoma" International Journal of Molecular Sciences 22, no. 24: 13635. https://doi.org/10.3390/ijms222413635
APA StyleCornelison, R., Marrah, L., Horter, D., Lynch, S., & Li, H. (2021). Targeting AVIL, a New Cytoskeleton Regulator in Glioblastoma. International Journal of Molecular Sciences, 22(24), 13635. https://doi.org/10.3390/ijms222413635