
Spatially Resolved Expression of Transposable Elements in Disease and Somatic Tissue with SpatialTE

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Spatially Resolved TE Expression of p120 Spinal Cord Sections from SOD1G93A Mice
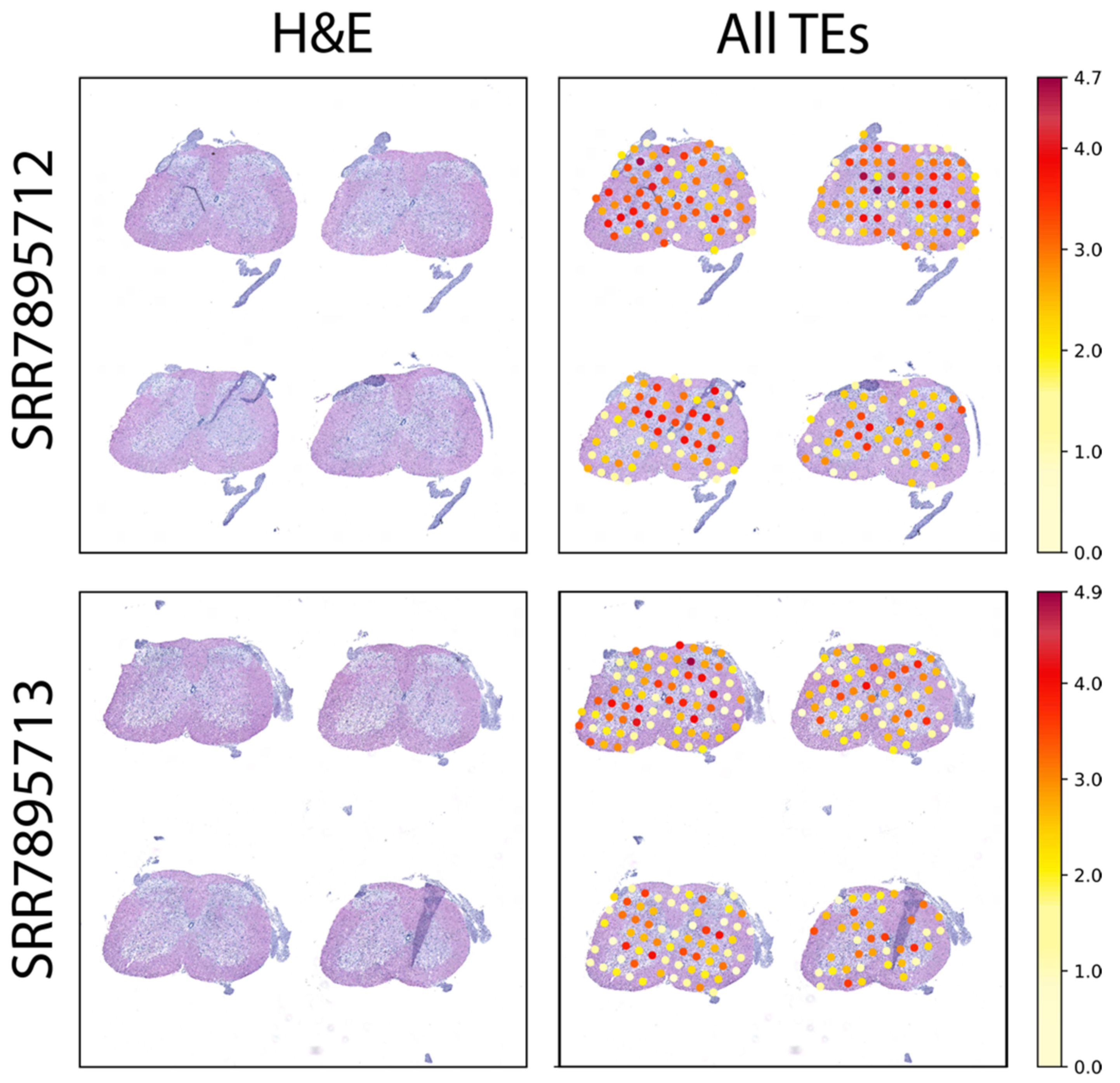
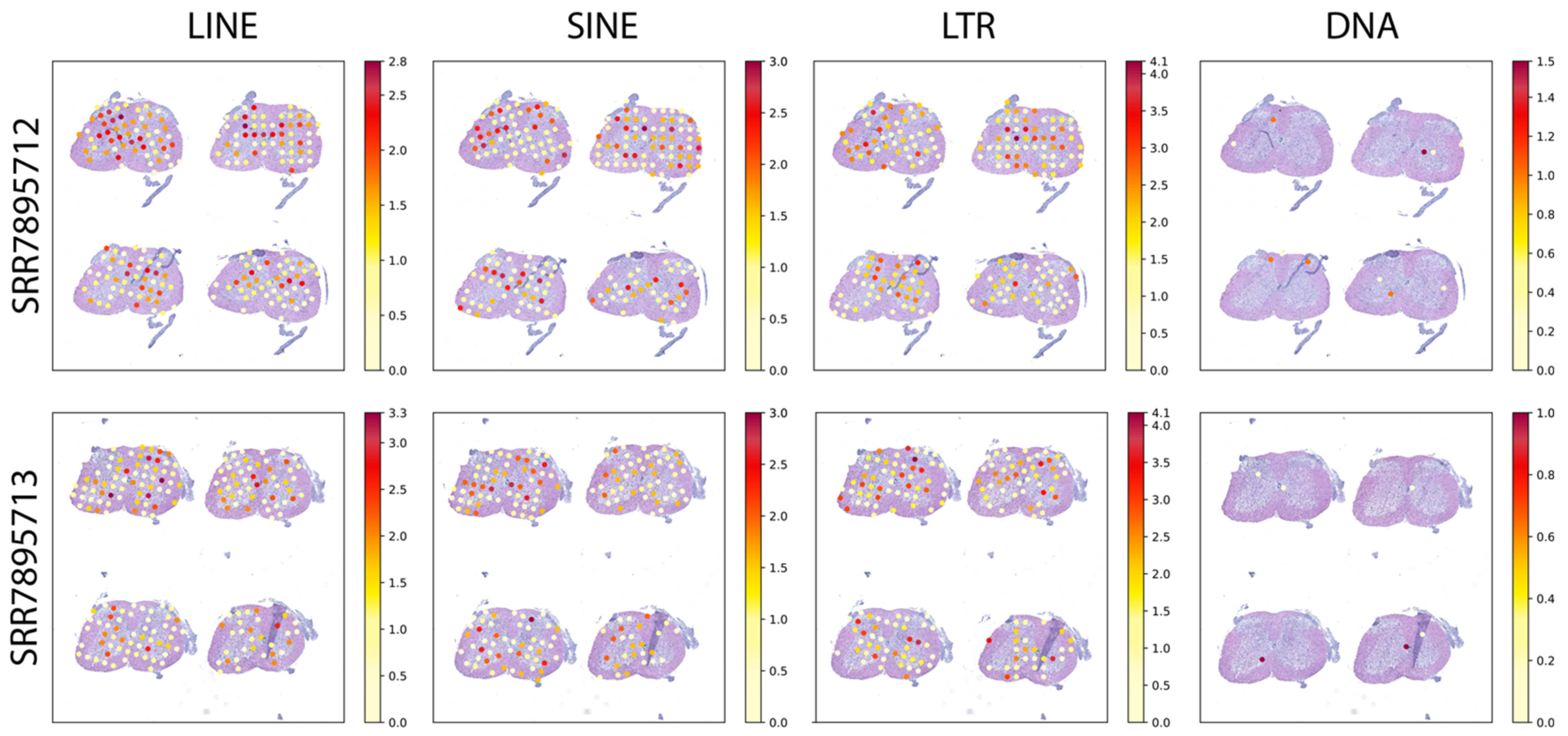
2.2. Spatially Resolved TE Expression in the Adult Mouse Brain
2.3. Spatially Resolved TE Expression in the Adult Mouse Kidney
3. Methods
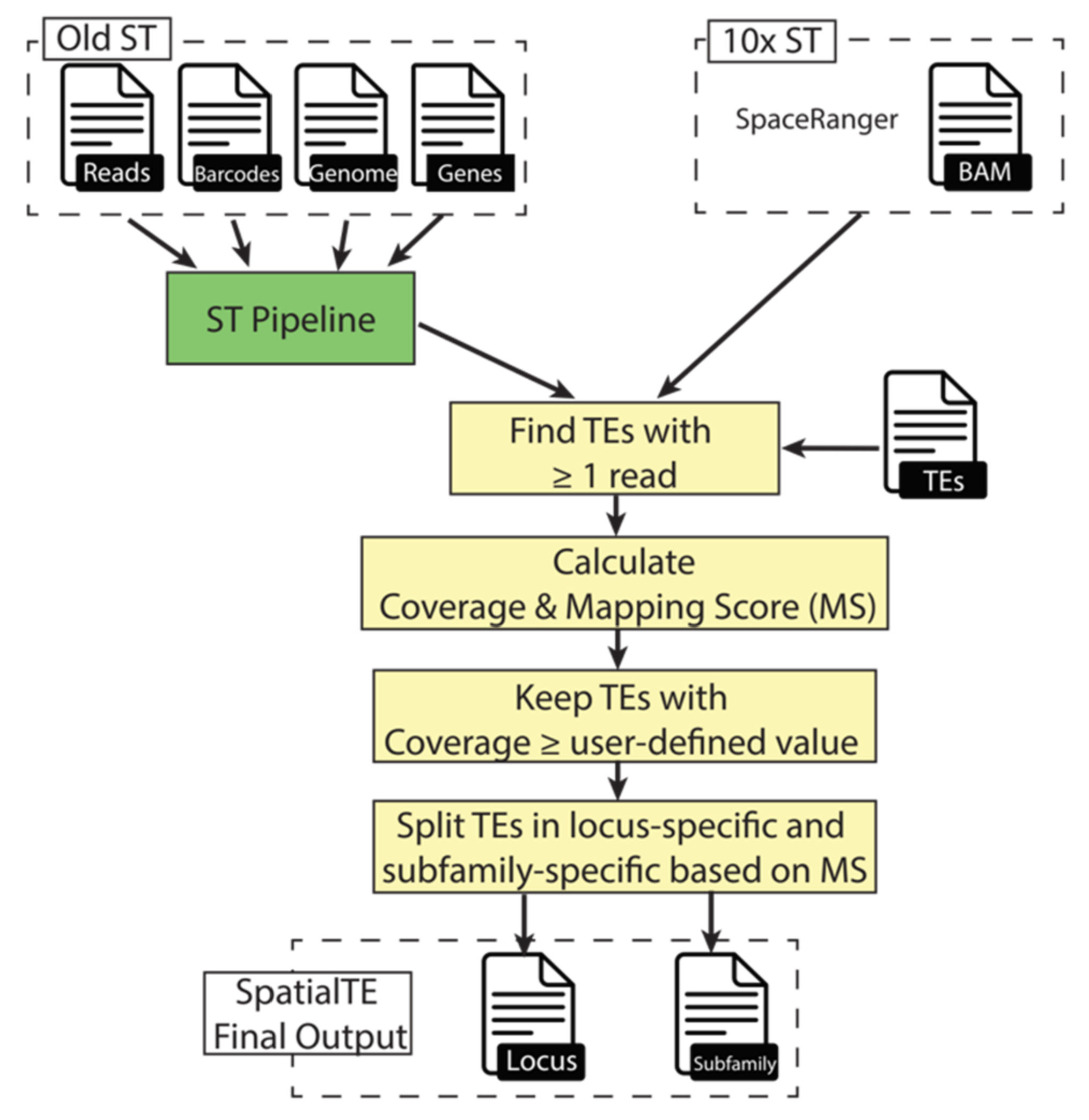
3.1. SpatialTE Implementation
3.2. Benchmarking and Validation
3.3. SpatialTE Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Slowikowski, K.; Zhang, F. Single-cell transcriptomics in cancer: Computational challenges and opportunities. Exp. Mol. Med. 2020, 52, 1452–1465. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, S.; Petrescu, J.; Phatnani, H. Spatially resolved transcriptomics and its applications in cancer. Curr. Opin. Genet. Dev. 2021, 66, 70–77. [Google Scholar] [CrossRef]
- Berglund, E.; Maaskola, J.; Schultz, N.; Friedrich, S.; Marklund, M.; Bergenstråhle, J.; Tarish, F.; Tanoglidi, A.; Vickovic, S.; Larsson, L.; et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat. Commun. 2018, 9, 2419. [Google Scholar] [CrossRef]
- Thrane, K.; Eriksson, H.; Maaskola, J.; Hansson, J.; Lundeberg, J. Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma. Cancer Res. 2018, 78, 5970–5979. [Google Scholar] [CrossRef] [Green Version]
- Maniatis, S.; Äijö, T.; Vickovic, S.; Braine, C.; Kang, K.; Mollbrink, A.; Fagegaltier, D.; Andrusivová, Ž.; Saarenpää, S.; Saiz-Castro, G.; et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science 2019, 364, 89–93. [Google Scholar] [CrossRef]
- Lanciano, S.; Cristofari, G. Measuring and interpreting transposable element expression. Nat. Rev. Genet. 2020, 21, 721–736. [Google Scholar] [CrossRef]
- Todd, C.D.; Deniz, Ö.; Taylor, D.; Branco, M.R. Functional evaluation of transposable elements as enhancers in mouse embryonic and trophoblast stem cells. Elife 2019, 8, e44344. [Google Scholar] [CrossRef]
- Jin, Y.; Tam, O.H.; Paniagua, E.; Hammell, M. TEtranscripts: A package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics 2015, 31, 3593–3599. [Google Scholar] [CrossRef]
- Valdebenito-Maturana, B.; Riadi, G. TEcandidates: Prediction of genomic origin of expressed transposable elements using RNA-seq data. Bioinformatics 2018, 34, 3915–3916. [Google Scholar] [CrossRef]
- Valdebenito-Maturana, B.; Torres, F.; Carrasco, M.; Tapia, J.C. Differential regulation of transposable elements (TEs) during the murine submandibular gland development. Mob. DNA 2021, 12, 23. [Google Scholar] [CrossRef]
- Valdebenito-Maturana, B.; Arancibia, E.; Riadi, G.; Tapia, J.C.; Carrasco, M. Locus-specific analysis of Transposable Elements during the progression of ALS in the SOD1G93A mouse model. PLoS ONE 2021, 16, e0258291. [Google Scholar]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Phatnani, H.P.; Guarnieri, P.; Friedman, B.A.; Carrasco, M.A.; Muratet, M.; Keeffe, S.O. Intricate interplay between astrocytes and motor neurons in ALS. Proc. Natl. Acad. Sci. USA 2013, 110, E756–E765. [Google Scholar] [CrossRef] [Green Version]
- Picardi, G.; Spalloni, A.; Generosi, A.; Paci, B.; Mercuri, N.B.; Luce, M.; Longone, P.; Cricenti, A. Tissue degeneration in ALS affected spinal cord evaluated by Raman spectroscopy. Sci. Rep. 2018, 8, 13110. [Google Scholar] [CrossRef]
- Savage, A.L.; Schumann, G.G.; Breen, G.; Bubb, V.J.; Al-Chalabi, A.; Quinn, J.P. Retrotransposons in the development and progression of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2019, 90, 284–293. [Google Scholar] [CrossRef]
- Liu, E.Y.; Russ, J.; Cali, C.P.; Phan, J.M.; Amlie-Wolf, A.; Lee, E.B. Loss of Nuclear TDP-43 Is Associated with Decondensation of LINE Retrotransposons. Cell Rep. 2019, 27, 1409–1421. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Lee, M.-H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; Von Geldern, G.; Johnson, K.; et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 2015, 7, 307ra153. [Google Scholar] [CrossRef]
- Gerdes, P.; Richardson, S.R.; Mager, D.L.; Faulkner, G.J. Transposable elements in the mammalian embryo: Pioneers surviving through stealth and service. Genome Biol. 2016, 17, 100. [Google Scholar] [CrossRef] [Green Version]
- Jachowicz, J.W.; Torres-Padilla, M.E. LINEs in mice: Features, families, and potential roles in early development. Chromosoma 2016, 125, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.R.; Morell, S.; Faulkner, G.J. L1 Retrotransposons and Somatic Mosaicism in the Brain. Annu. Rev. Genet. 2014, 9, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, G.J.; Billon, V. L1 retrotransposition in the soma: A field jumping ahead. Mob. DNA 2018, 9, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misiak, B.; Ricceri, L.; Sasiadek, M.M. Transposable elements and their epigenetic regulation in mental disorders: Current evidence in the field. Front. Genet. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Song, R.; Yosypiv, I.V. Development of the kidney medulla. Organogenesis 2012, 8, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Navarro, J.F.; Sjöstrand, J.; Salmén, F.; Lundeberg, J.; Ståhl, P.L. ST Pipeline: An automated pipeline for spatial mapping of unique transcripts. Bioinformatics 2017, 33, 2591–2593. [Google Scholar] [CrossRef]
- Navarro Gonzalez, J.; Zweig, A.S.; Speir, M.L.; Schmelter, D.; Rosenbloom, K.R.; Raney, B.J.; Powell, C.C.; Nassar, L.R.; Maulding, N.D.; Lee, C.M.; et al. The UCSC Genome Browser database: 2021 update. Nucleic Acids Res. 2021, 49, D1046–D1057. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Frazee, A.C.; Jaffe, A.E.; Langmead, B.; Leek, J.T. Polyester: Simulating RNA-seq datasets with differential transcript expression. Bioinformatics 2015, 31, 2778–2784. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 15 October 2021).
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organ | Section | LINE | SINE | LTR | DNA |
|---|---|---|---|---|---|
| Brain | Coronal | 0.170 | −0.189 | 0.200 | −0.245 |
| Sagittal Anterior 1 | 0.320 | 0.266 | 0.678 | 0.483 | |
| Sagittal Anterior 2 | 0.364 | 0.473 | 0.262 | 0.622 | |
| Sagital Posterior 1 | 0.269 | 0.264 | 0.506 | 0.739 | |
| Sagital Posterior 2 | 0.318 | 0.527 | 0.403 | 0.751 | |
| Kidney | Coronal | 0.729 | 0.311 | 0.519 | 0.253 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdebenito-Maturana, B.; Guatimosim, C.; Carrasco, M.A.; Tapia, J.C. Spatially Resolved Expression of Transposable Elements in Disease and Somatic Tissue with SpatialTE. Int. J. Mol. Sci. 2021, 22, 13623. https://doi.org/10.3390/ijms222413623
Valdebenito-Maturana B, Guatimosim C, Carrasco MA, Tapia JC. Spatially Resolved Expression of Transposable Elements in Disease and Somatic Tissue with SpatialTE. International Journal of Molecular Sciences. 2021; 22(24):13623. https://doi.org/10.3390/ijms222413623
Chicago/Turabian StyleValdebenito-Maturana, Braulio, Cristina Guatimosim, Mónica Alejandra Carrasco, and Juan Carlos Tapia. 2021. "Spatially Resolved Expression of Transposable Elements in Disease and Somatic Tissue with SpatialTE" International Journal of Molecular Sciences 22, no. 24: 13623. https://doi.org/10.3390/ijms222413623
APA StyleValdebenito-Maturana, B., Guatimosim, C., Carrasco, M. A., & Tapia, J. C. (2021). Spatially Resolved Expression of Transposable Elements in Disease and Somatic Tissue with SpatialTE. International Journal of Molecular Sciences, 22(24), 13623. https://doi.org/10.3390/ijms222413623