
Benzimidazole Derivatives as New and Selective Inhibitors of Arginase from Leishmania mexicana with Biological Activity against Promastigotes and Amastigotes

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results
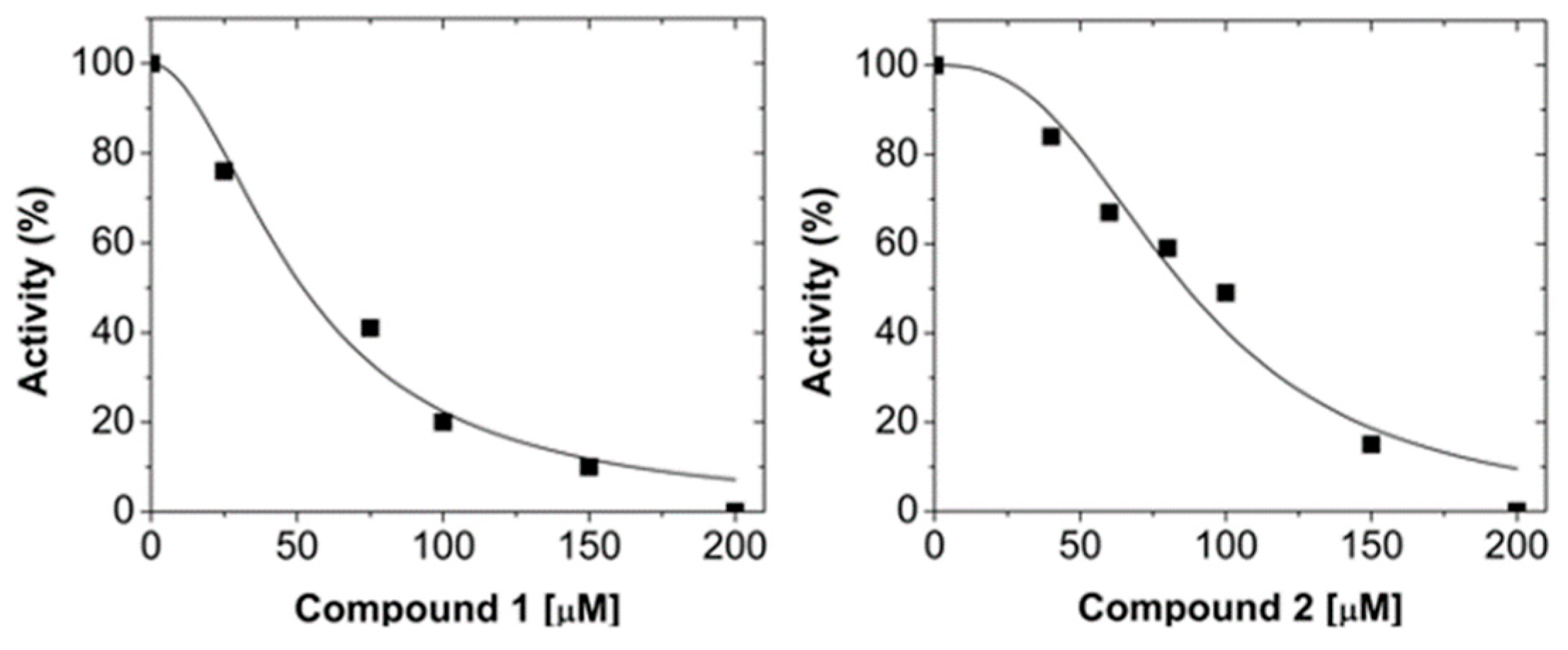
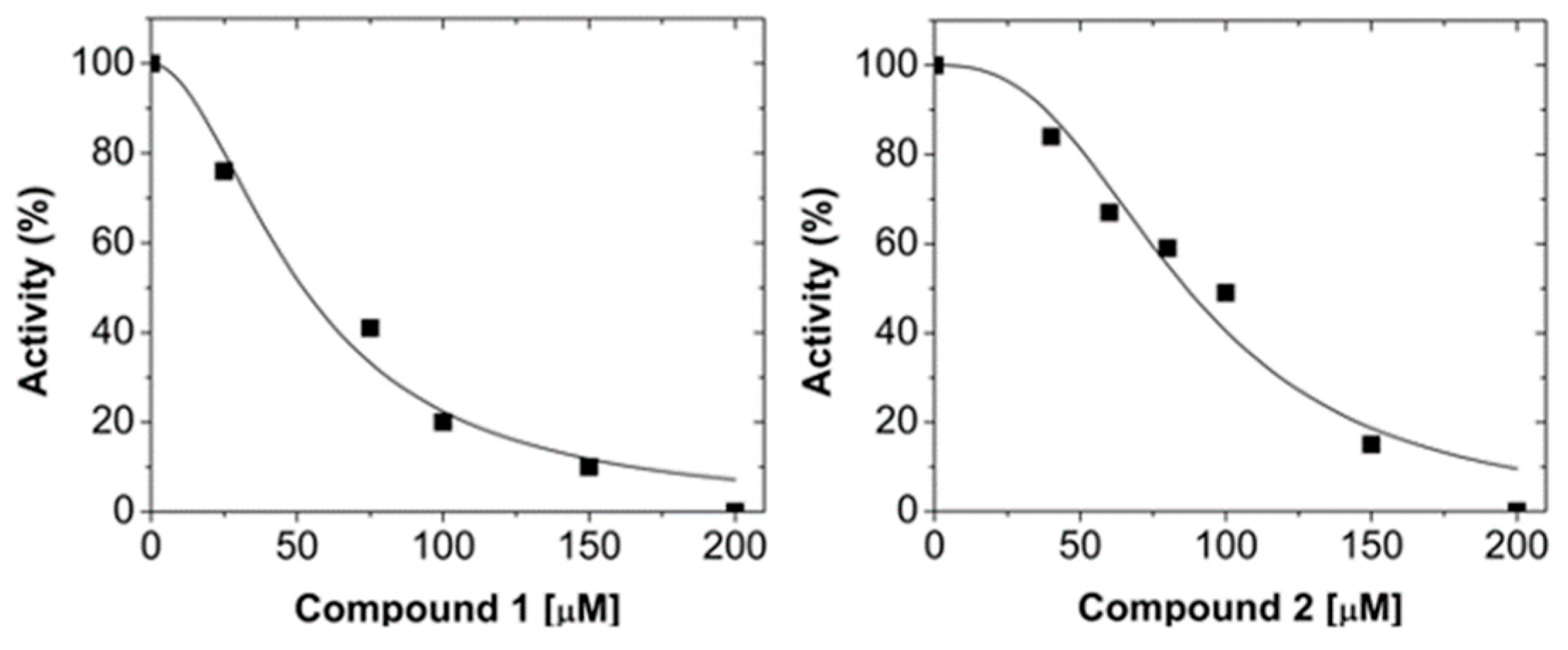
2.1. Virtual Screening and LmARG Inhibition
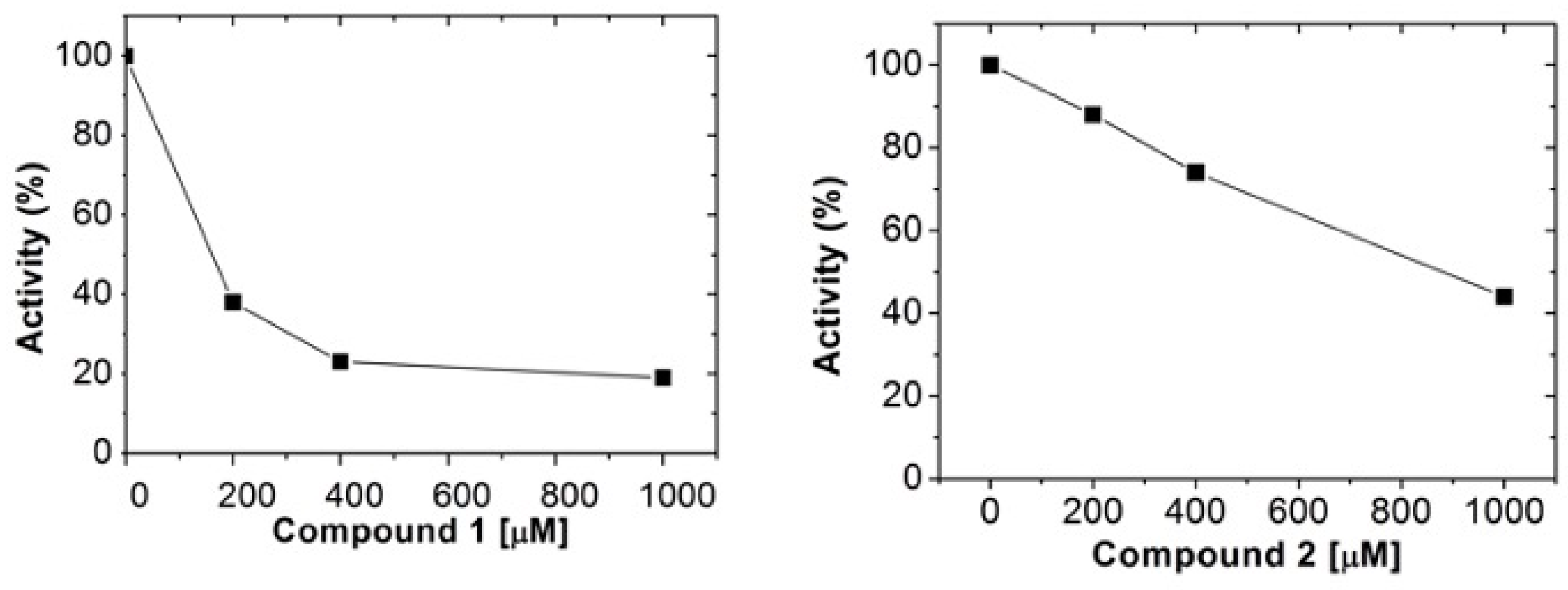
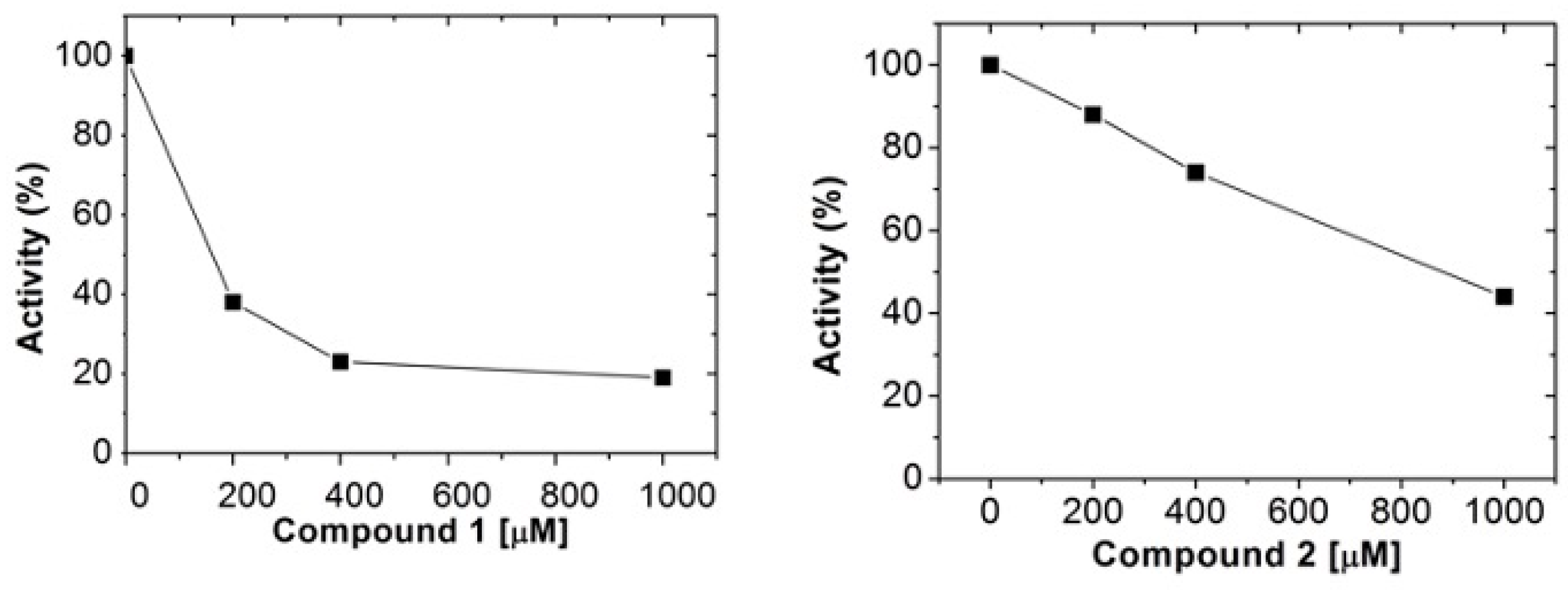
2.2. Human Arginase 1 Inhibition
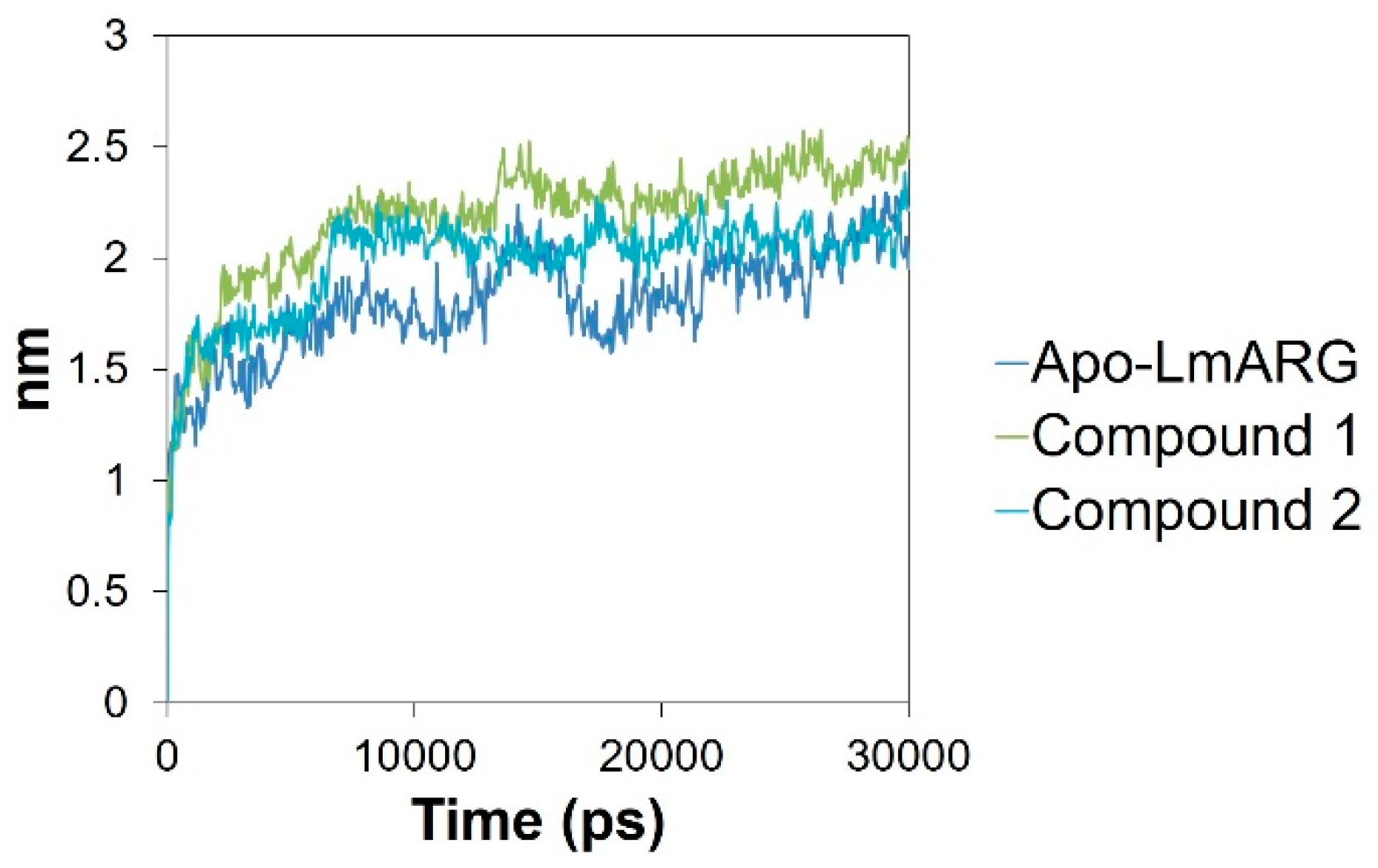
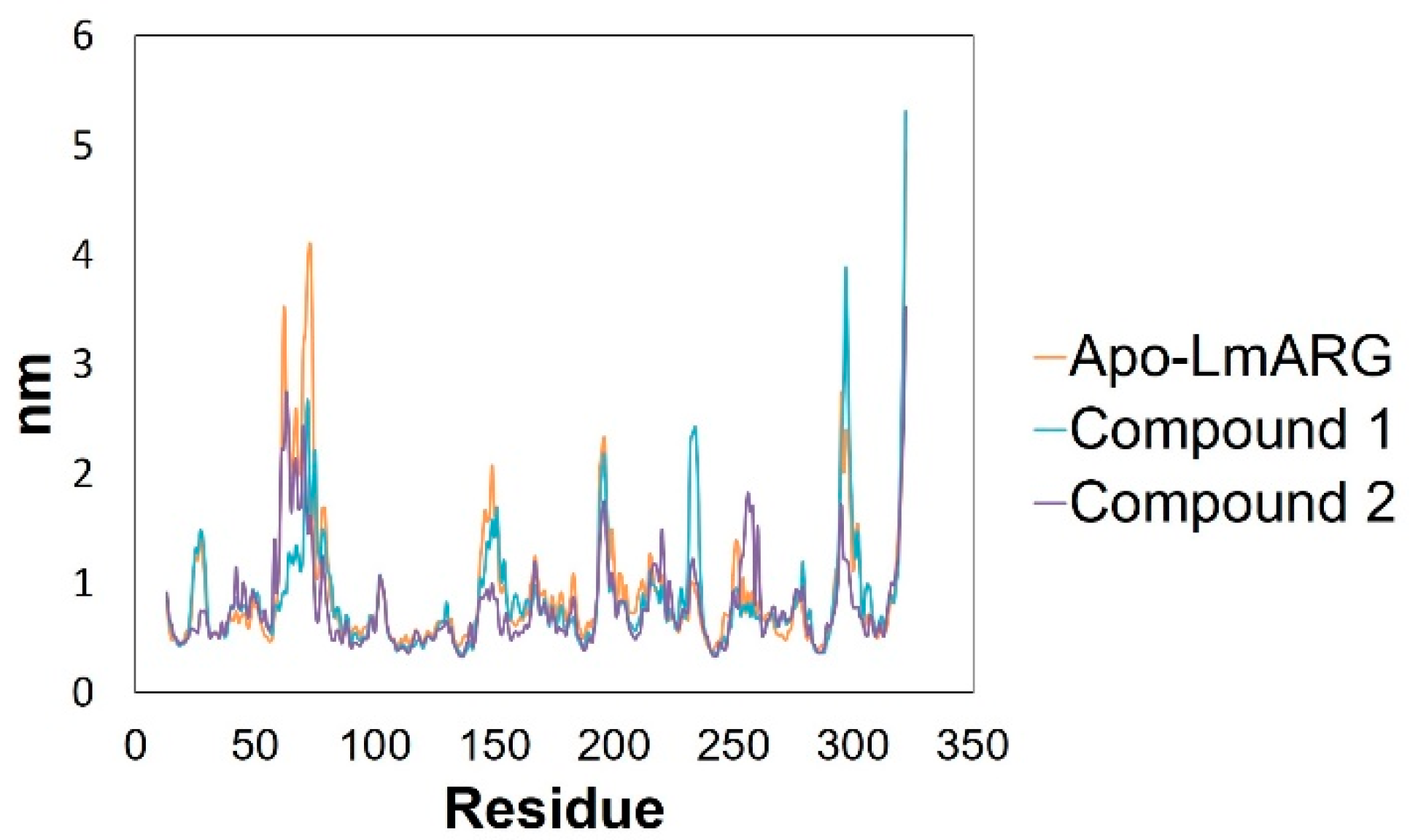
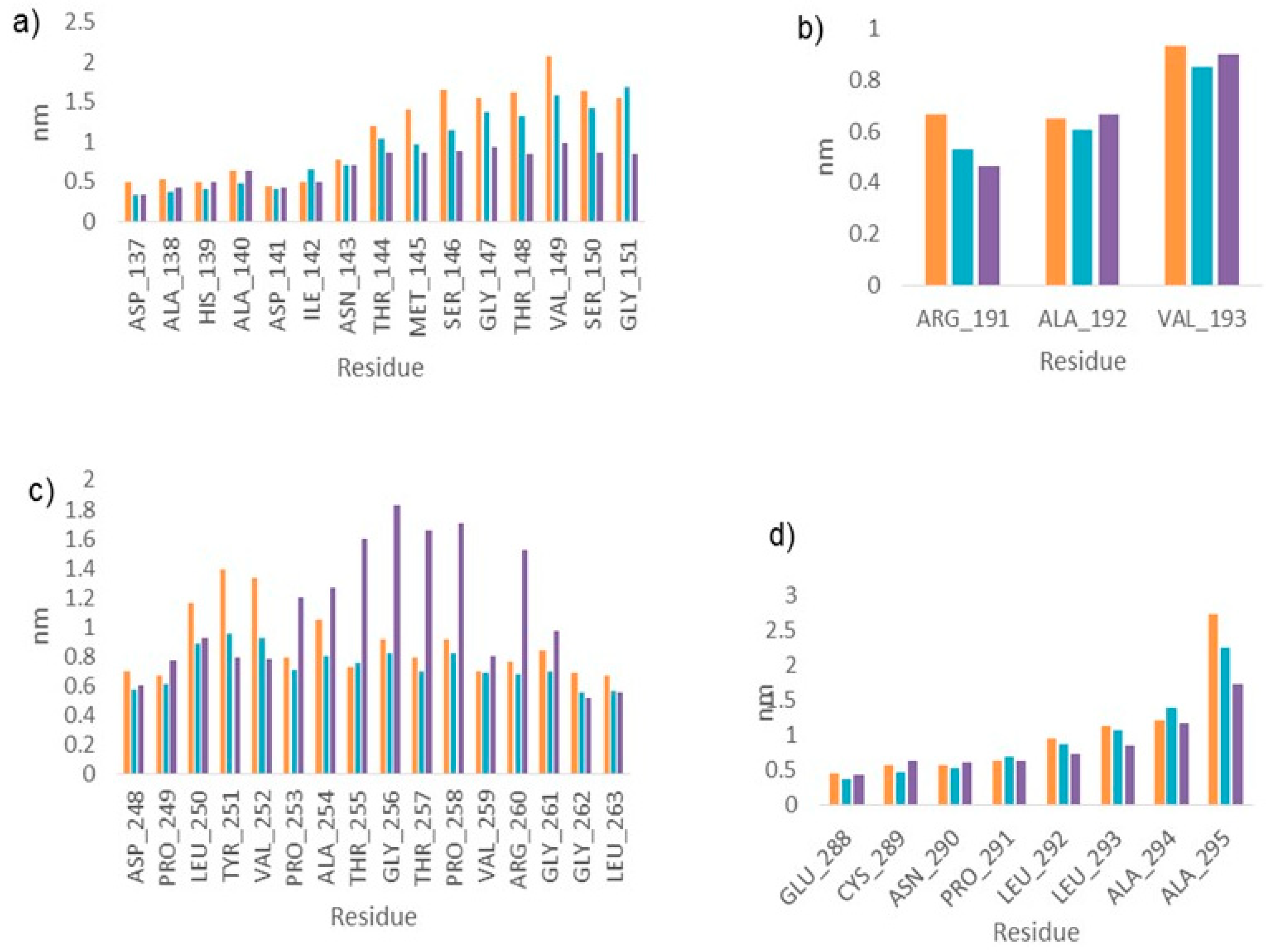
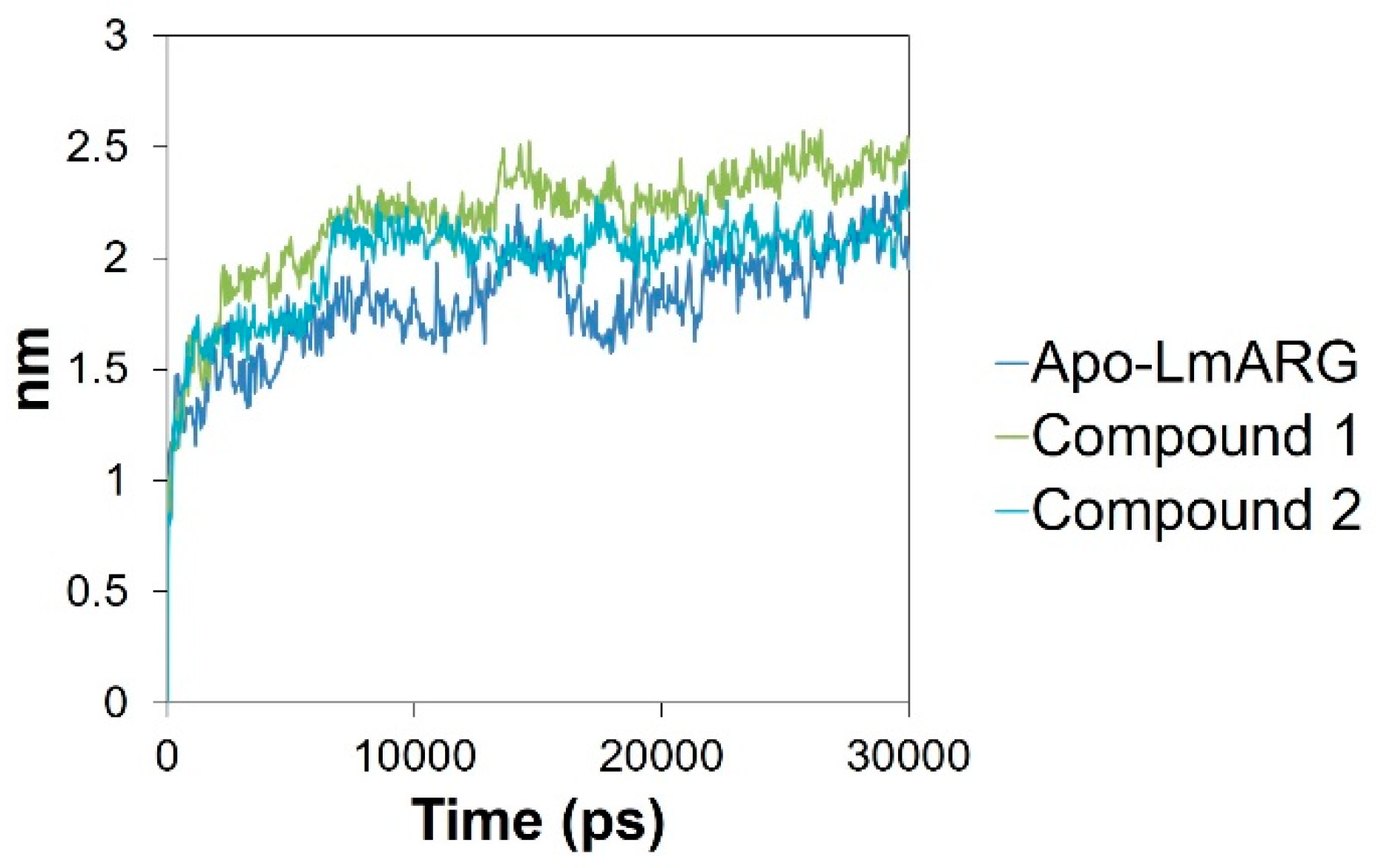
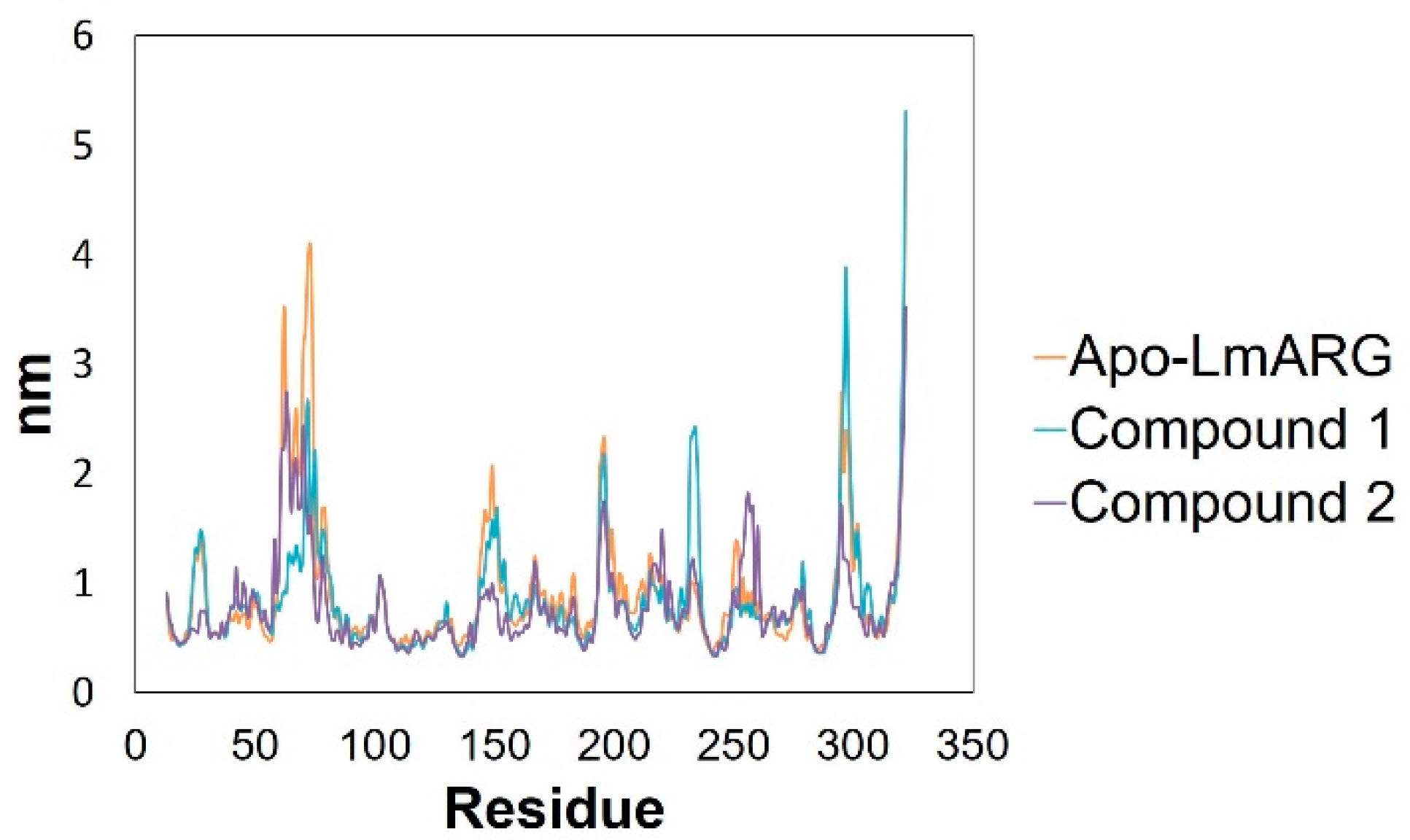
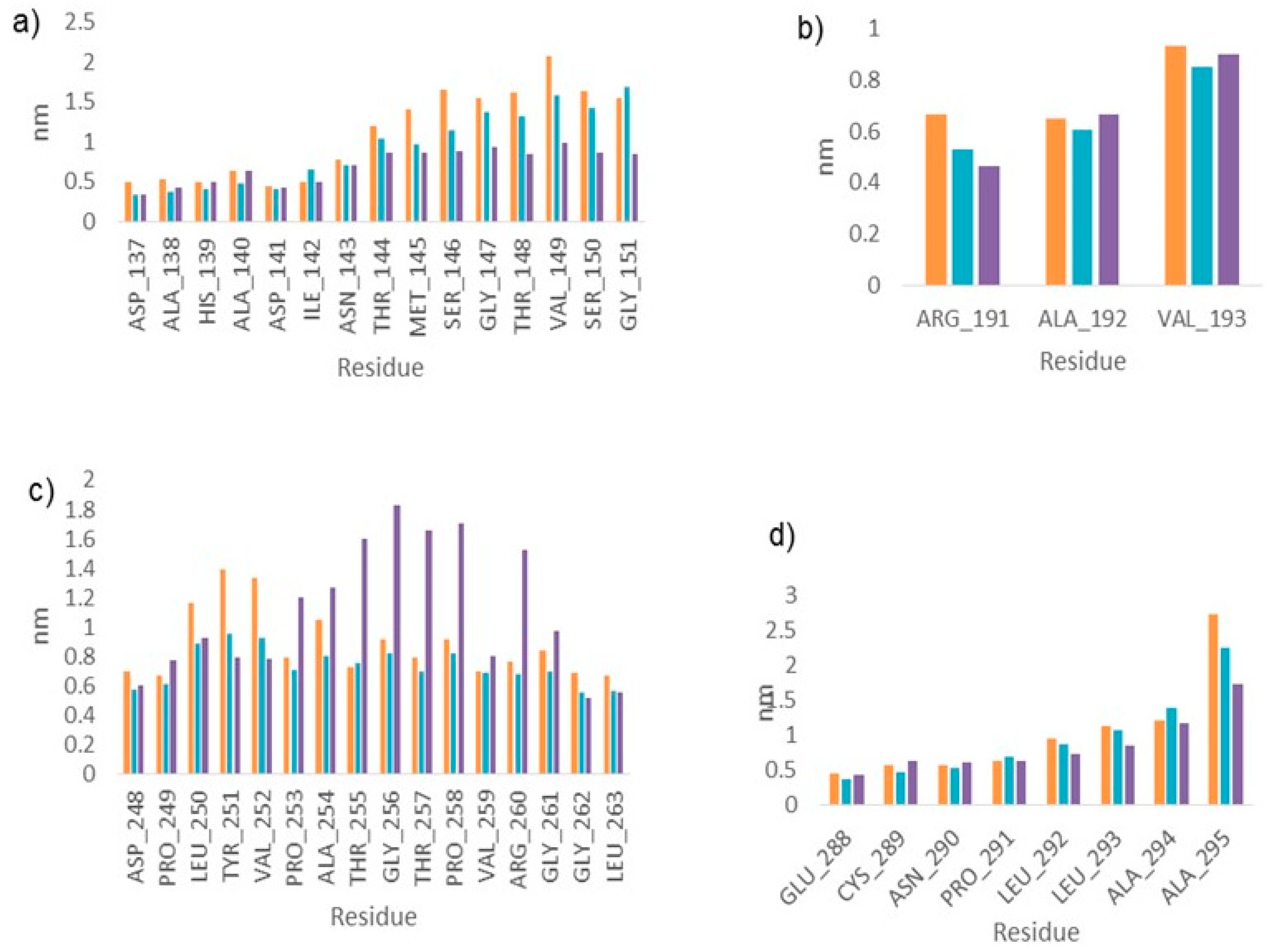
2.3. Molecular Dynamics
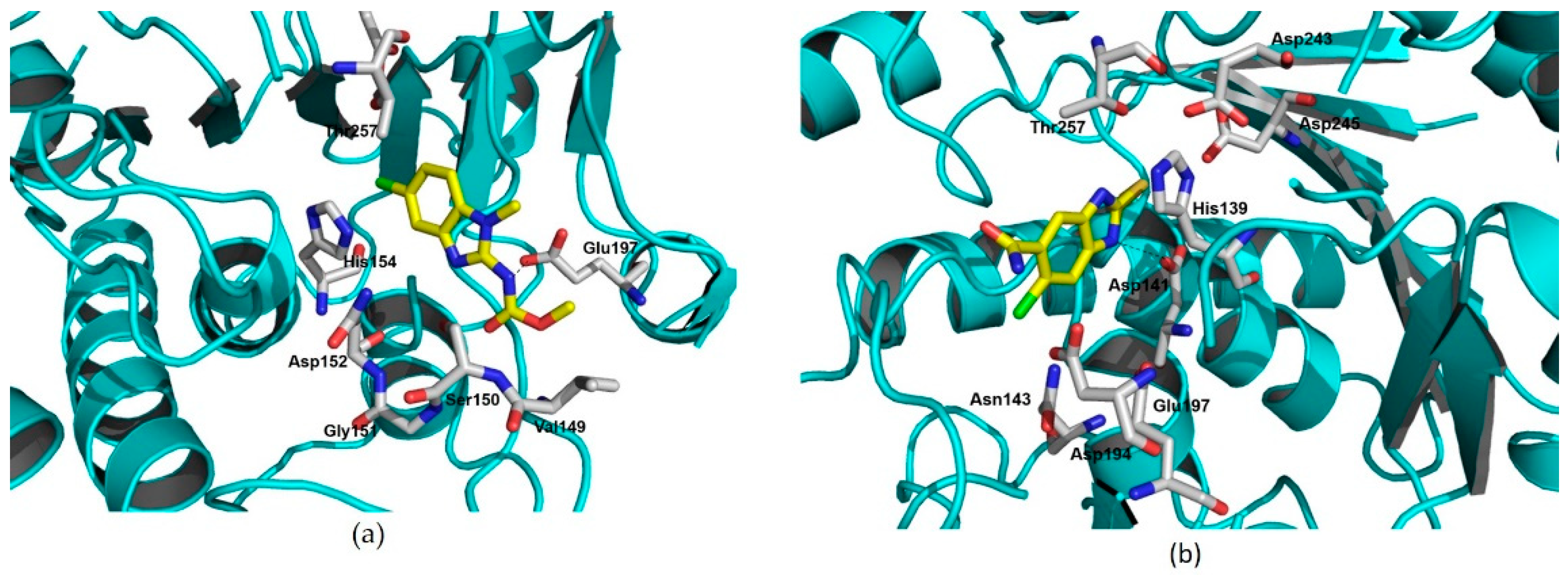
2.4. LmARG-Inhibitor Interactions
2.5. Biological Activity Evaluation and Cytotoxicity
2.6. In Silico Physichemical and ADME-Tox Predictions
3. Discussion
4. Materials and Methods
4.1. Virtual Screening
4.2. Cloning of L. mexicana Arginase Gene
4.3. Cloning of Human Arginase 1 Gene
4.4. Expression and Purification of Arginases
4.5. Enzyme Activity
4.6. Inhibition Assays
4.7. Molecular Dynamics
4.8. In Silico ADME-Tox Prediction
4.9. Biological Activity against Leishmania mexicana Promastigotes
4.10. Biological Activity against Leishmania mexicana Amastigotes
4.11. Cytotoxicity Assays
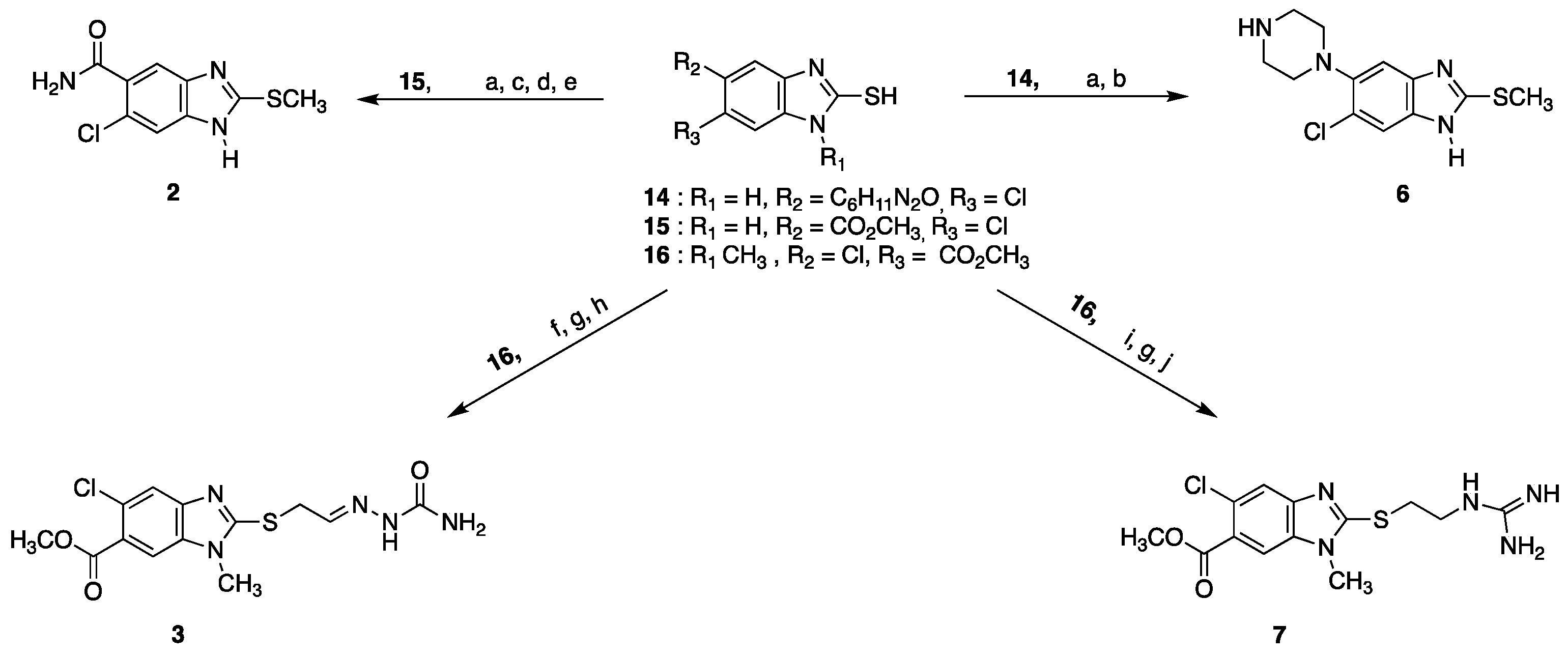
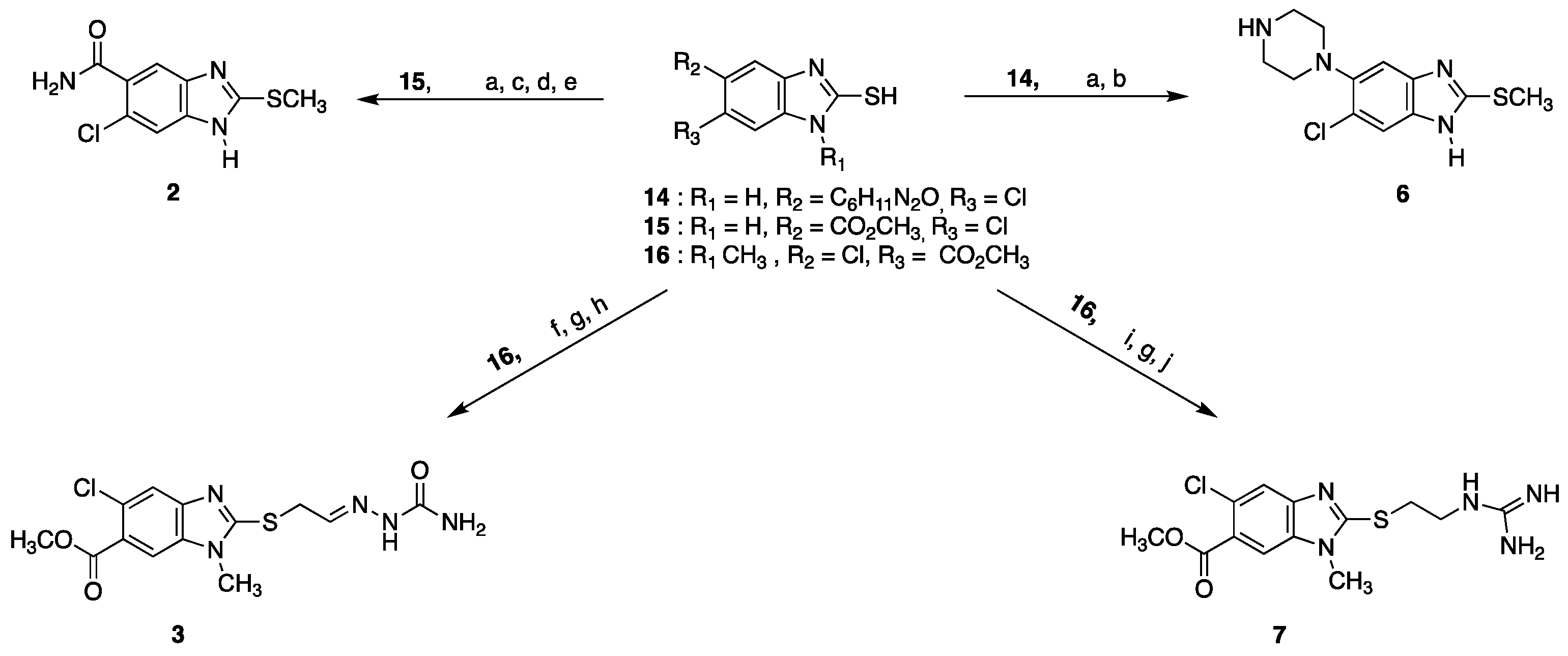
4.12. Synthesis of Benzimidazole Derivatives
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reithinger, R.; Dujardin, J.C. Molecular Diagnosis of Leishmaniasis: Current Status and Future Applications. J. Clin. Microbiol. 2007, 45, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Schriefer, A.; Wilson, M.E.; Carvalho, E.M. Recent Developments Leading toward a Paradigm Switch in the Diagnostic and Therapeutic Approach to Human Leishmaniasis. Curr. Opin. Infect. Dis. 2008, 21, 483–488. [Google Scholar] [CrossRef]
- World Health Organization Leishmaniasis. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 9 February 2021).
- Croft, S.L.; Olliaro, P. Leishmaniasis Chemotherapy-Challenges and Opportunities. Clin. Microbiol. Infect. 2011, 17, 1478–1483. [Google Scholar] [CrossRef] [Green Version]
- Mohapatra, S. Drug Resistance in Leishmaniasis: Newer Developments. Trop. Parasitol. 2014, 4, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Bray, P.G.; Barrett, M.P.; Ward, S.A.; de Koning, H.P. Pentamidine Uptake and Resistance in Pathogenic Protozoa: Past, Present and Future. Trends Parasitol. 2003, 19, 232–239. [Google Scholar] [CrossRef]
- Ghorbani, M.; Farhoudi, R. Leishmaniasis in Humans: Drug or Vaccine Therapy? Drug Des. Dev. Ther. 2018, 12, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Rijal, S.; Ostyn, B.; Uranw, S.; Rai, K.; Bhattarai, N.R.; Dorlo, T.P.C.; Beijnen, J.H.; Vanaerschot, M.; Decuypere, S.; Dhakal, S.S.; et al. Increasing Failure of Miltefosine in the Treatment of Kala-Azar in Nepal and the Potential Role of Parasite Drug Resistance, Reinfection, or Noncompliance. Clin. Infect. Dis. 2013, 56, 1530–1538. [Google Scholar] [CrossRef] [Green Version]
- Lodi, G.; Sannino, M.; Caterino, P.; Cannarozzo, G.; Bennardo, L.; Nisticò, S.P. Fractional CO2 Laser-Assisted Topical Rifamycin Drug Delivery in the Treatment of Pediatric Cutaneous Leishmaniasis. Pediatric Dermatol. 2021, 38, 1–4. [Google Scholar] [CrossRef]
- Ponte-sucre, A.; Gamarro, F.; Dujardin, J.; Barrett, M.P.; Garcı, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug Resistance and Treatment Failure in Leishmaniasis: A 21st Century Challenge. PLoS Negl. Trop. Dis. 2017, 11, 1–24. [Google Scholar] [CrossRef]
- Heby, O.; Persson, L.; Rentala, M. Targeting the Polyamine Biosynthetic Enzymes: A Promising Approach to Therapy of African Sleeping Sickness, Chagas’ Disease, and Leishmaniasis. Amino Acids 2007, 33, 359–366. [Google Scholar] [CrossRef]
- Ilari, A.; Fiorillo, A.; Genovese, I.; Colotti, G. Polyamine-Trypanothione Pathway:An Update. Future Med. Chem. 2017, 9, 61–77. [Google Scholar] [CrossRef]
- Reguera, R.M.; Balaña-Fouce, R.; Showalter, M.; Hickerson, S.; Beverley, S.M. Leishmania Major Lacking Arginase (ARG) Are Auxotrophic for Polyamines but Retain Infectivity to Susceptible BALB/c Mice. Mol. Biochem. Parasitol. 2009, 165, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Colotti, G.; Ilari, A. Polyamine Metabolism in Leishmania: From Arginine to Trypanothione. Amino Acids 2011, 40, 269–285. [Google Scholar] [CrossRef] [PubMed]
- D’Antonio, E.L.; Ullman, B.; Roberts, S.C.; Dixit, U.G.; Wilson, M.E.; Hai, Y.; Christianson, D.W. Crystal Structure of Arginase from Leishmania Mexicana and Implications for the Inhibition of Polyamine Biosynthesis in Parasitic Infections. Arch. Biochem. Biophys. 2013, 535, 163–176. [Google Scholar] [CrossRef] [Green Version]
- Roberts, S.C.; Tancer, M.J.; Polinsky, M.R.; Michael Gibson, K.; Heby, O.; Ullman, B. Arginase Plays a Pivotal Role in Polyamine Precursor Metabolism in Leishmania: Characterization of Gene Deletion Mutants. J. Biol. Chem. 2004, 279, 23668–23678. [Google Scholar] [CrossRef] [Green Version]
- da Silva, E.R.; Boechat, N.; Pinheiro, L.C.S.; Bastos, M.M.; Costa, C.C.P.; Bartholomeu, J.C.; da Costa, T.H. Novel Selective Inhibitor of Leishmania (Leishmania) Amazonensis Arginase. Chem. Biol. Drug Des. 2015, 86, 969–978. [Google Scholar] [CrossRef]
- da Silva, E.R.; Maquiaveli, C.d.C.; Magalhães, P.P. The Leishmanicidal Flavonols Quercetin and Quercitrin Target Leishmania (Leishmania) Amazonensis Arginase. Exp. Parasitol. 2012, 130, 183–188. [Google Scholar] [CrossRef]
- dos Reis, M.B.G.; Manjolin, L.C.; Maquiaveli, C.D.C.; Santos-Filho, O.A.; da Silva, E.R. Inhibition of Leishmania (Leishmania) Amazonensis and Rat Arginases by Green Tea EGCG, (+)-Catechin and (−)-Epicatechin: A Comparative Structural Analysis of Enzyme-Inhibitor Interactions. PLoS ONE 2013, 8, e78387. [Google Scholar]
- da Silva, E.R.; Come, J.A.A.D.S.S.; Brogi, S.; Calderone, V.; Chemi, G.; Campiani, G.; Oliveira, T.M.F.D.S.; Pham, T.N.; Pudlo, M.; Girard, C.; et al. Cinnamides Target Leishmania Amazonensis Arginase Selectively. Molecules 2020, 25, 5271. [Google Scholar] [CrossRef]
- Garcia, A.R.; Oliveira, D.M.P.; Claudia, F.; Amaral, A.; Jesus, J.B.; Rennó Sodero, A.C.; Souza, A.M.T.; Supuran, C.T.; Vermelho, A.B.; Rodrigues, I.A.; et al. Leishmania Infantum Arginase: Biochemical Characterization and Inhibition by Naturally Occurring Phenolic Substances. J. Enzym. Inhib. Med. Chem. 2019, 34, 1100–1109. [Google Scholar] [CrossRef] [Green Version]
- Garcia, A.R.; Oliveira, D.M.P.; Jesus, J.B.; Souza, A.M.T.; Sodero, A.C.R.; Vermelho, A.B.; Leal, I.C.R.; Souza, R.O.M.A.; Miranda, L.S.M.; Pinheiro, A.S.; et al. Identification of Chalcone Derivatives as Inhibitors of Leishmania Infantum Arginase and Promising Antileishmanial Agents. Front. Chem. 2021, 8, 624678. [Google Scholar] [CrossRef]
- Riley, E.; Roberts, S.C.; Ullman, B. Inhibition Profile of Leishmania Mexicana Arginase Reveals Differences with Human Arginase I. Int. J. Parasitol. 2011, 41, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Hai, Y.; Christianson, D.W. Crystal Structures of Leishmania Mexicana Arginase Complexed with α,α-Disubstituted Boronic Amino-Acid Inhibitors. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 300–306. [Google Scholar] [CrossRef]
- Tellez-Valencia, A.; Avila-Ríos, S.A.; Perez-Montfort, R.; Rodríguez-Romero, A.; Tuena de Gomez-Puyou, M.; Lopez-Calahorra, F.; Gomez-Puyou, A. Highly Specific Inactivation of Triosephosphate Isomerase from Trypanosoma Cruzi. Biochem. Biophys. Res. Commun. 2002, 295, 958–963. [Google Scholar] [CrossRef]
- Vázquez-Raygoza, A.; Cano-González, L.; Velázquez-Martínez, I.; Trejo-Soto, P.J.; Castillo, R.; Hernández-Campos, A.; Hernández-Luis, F.; Oria-Hernández, J.; Castillo-Villanueva, A.; Avitia-Domínguez, C.; et al. Species-Specific Inactivation of Triosephosphate Isomerase from Trypanosoma Brucei: Kinetic and Molecular Dynamics Studies. Molecules 2017, 22, 2055. [Google Scholar] [CrossRef] [Green Version]
- Mortier, J.; Prévost, J.R.C.; Sydow, D.; Teuchert, S.; Omieczynski, C.; Bermudez, M.; Frédérick, R.; Wolber, G. Arginase Structure and Inhibition: Catalytic Site Plasticity Reveals New Modulation Possibilities. Sci. Rep. 2017, 7, 13616. [Google Scholar] [CrossRef] [Green Version]
- Matadamas-Martínez, F.; Hernández-Campos, A.; Téllez-Valencia, A.; Vázquez-Raygoza, A.; Comparán-Alarcón, S.; Yépez-Mulia, L.; Castillo, R. Leishmania Mexicana Trypanothione Reductase Inhibitors: Computational and Biological Studies. Molecules 2019, 24, 3216. [Google Scholar] [CrossRef] [Green Version]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Mariappan, G.; Hazarika, R.; Alam, F.; Karki, R.; Patangia, U.; Nath, S. Synthesis and Biological Evaluation of 2-Substituted Benzimidazole Derivatives. Arab. J. Chem. 2015, 8, 715–719. [Google Scholar] [CrossRef] [Green Version]
- Chintakunta, R.; Meka, G. Synthesis, in Silico Studies and Antibacterial Activity of Some Novel 2-Substituted Benzimidazole Derivatives. Future J. Pharm. Sci. 2020, 6, 10921–10945. [Google Scholar] [CrossRef]
- Aroso, R.T.; Guedes, R.C.; Pereira, M.M. Synthesis of Computationally Designed 2,5(6)-Benzimidazole Derivatives via Pd-Catalyzed Reactions for Potential E. Coli DNA Gyrase B Inhibition. Molecules 2021, 26, 1326. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Chhabra, S.; Shrivastava, S.K.; Mishra, P. Benzimidazole: A Promising Pharmacophore. Med. Chem. Res. 2013, 22, 5077–5104. [Google Scholar] [CrossRef]
- Nieto-Meneses, R.; Castillo, R.; Hernández-Campos, A.; Maldonado-Rangel, A.; Matius-Ruiz, J.B.; Trejo-Soto, P.J.; Nogueda-Torres, B.; Dea-Ayuela, M.A.; Bolás-Fernández, F.; Méndez-Cuesta, C.; et al. In Vitro Activity of New N-Benzyl-1H-Benzimidazol-2-Amine Derivatives against Cutaneous, Mucocutaneous and Visceral Leishmania Species. Exp. Parasitol. 2018, 184, 82–89. [Google Scholar] [CrossRef]
- de Almeida, L.; Alves, K.F.; Maciel-Rezende, C.M.; Jesus, L.D.O.P.; Pires, F.R.; Junior, C.V.; Izidoro, M.A.; Júdice, W.A.D.S.; dos Santos, M.H.; Marques, M.J. Benzophenone Derivatives as Cysteine Protease Inhibitors and Biological Activity against Leishmania (L.) Amazonensis Amastigotes. Biomed. Pharmacother. 2015, 75, 93–99. [Google Scholar] [CrossRef]
- Martín-Montes, Á.; Aguilera-Venegas, B.; Ma Morales-Martín, R.; Martín-Escolano, R.; Zamora-Ledesma, S.; Marín, C.; Arán, V.J.; Sánchez-Moreno, M. In Vitro Assessment of 3-Alkoxy-5-Nitroindazole-Derived Ethylamines and Related Compounds as Potential Antileishmanial Drugs. Bioorganic Chem. 2019, 92, 103274. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies Using Molecular Dynamics Free Energy Perturbation and the Opls Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Alcántara, G.; Torres-Larios, A.; Enríquez-Flores, S.; García-Torres, I.; Castillo-Villanueva, A.; Méndez, S.T.; de la Mora-de la Mora, I.; Gómez-Manzo, S.; Torres-Arroyo, A.; López-Velázquez, G.; et al. Structural and Functional Perturbation of Giardia Lamblia Triosephosphate Isomerase by Modification of a Non-Catalytic, Non-Conserved Region. PLoS ONE 2013, 8, e69031. [Google Scholar] [CrossRef]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Corraliza, I.M.; Campo, M.L.; Soler, G.; Modolell, M. Determination of Arginase Activity in Macrophages: A Micromethod. J. Immunol. Methods 1994, 174, 231–235. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the SC ’06: 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: Free ADME-Tox Filtering Compu-Tations for Chemical Biology and Early Stages Drug Discovery. Bioinformatics 2017, 33, 3658–3660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Modeling 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Drwal, M.N.; Banerjee, P.; Dunkel, M.; Wettig, M.R.; Preissner, R. ProTox: A Web Server for the in Silico Prediction of Rodent Oral Toxicity. Nucleic Acids Res. 2014, 42, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Pum, J. A Practical Guide to Validation and Verification of Analytical Methods in the Clinical Laboratory. Adv. Clin. Chem. 2019, 90, 215–281. [Google Scholar]
- Soria-Arteche, O.; Castillo, R.; Hernández-Campos, A.; Hurtado-De La Peña, M.; Navarrete-Vázquez, G.; Luis Medina-Franco, J.; Gómez-Flores, K. Studies on the Selective S-Oxidation of Albendazole, Fenbendazole, Triclabendazole, and Other Benzimidazole Sulfides. J. Mex. Chem. Soc. 2005, 49, 353–358. [Google Scholar]
- Soria-Arteche, O.; Hernández-Campos, A.; Yépez-Mulia, L.; Trejo-Soto, P.J.; Hernández-Luis, F.; Gres-Molina, J.; Maldonado, L.A.; Castillo, R. Synthesis and Antiprotozoal Activity of Nitazoxanide-N-Methylbenzimidazole Hybrids. Bioorganic Med. Chem. Lett. 2013, 23, 6838–6841. [Google Scholar] [CrossRef] [PubMed]
- Valdez, J.; Cedillo, R.; Hernandez-Campos, A.; Yepez, L.; Hernandez-Luis, F.; Navarrete-Vazquez, G.; Tapia, A.; Cortes, R.; Hernandez, M.; Castillo, R. Synthesis and Antiparasitic Activity of 1H-Benzimidazole Derivatives. Bioorganic Med. Chem. Lett. 2002, 12, 2221–2224. [Google Scholar] [CrossRef]
- Trejo-Soto, P.; Flores-Gahona, A.; Castillo, R.; Hernández-Campos, A. Simplifying Nucleophilic Aromatic Substitutions: Microwave-Assisted Solvent Free Synthesis of 5-Alkylamino-2-Nitroanilines. Curr. Microw. Chem. 2016, 3, 258–265. [Google Scholar] [CrossRef]
- Flores-Carrillo, P.; Velázquez-López, J.M.; Aguayo-Ortiz, R.; Hernández-Campos, A.; Trejo-Soto, P.J.; Yépez-Mulia, L.; Castillo, R. Synthesis, Antiprotozoal Activity, and Chemoinformatic Analysis of 2-(Methylthio)-1H-Benzimidazole-5-Carboxamide Derivatives: Identification of New Selective Giardicidal and Trichomonicidal Compounds. Eur. J. Med. Chem. 2017, 137, 211–220. [Google Scholar] [CrossRef]
- Velázquez-López, J.M.; Hernández-Campos, A.; Yépez-Mulia, L.; Téllez-Valencia, A.; Flores-Carrillo, P.; Nieto-Meneses, R.; Castillo, R. Synthesis and Trypanocidal Activity of Novel Benzimidazole Derivatives. Bioorganic Med. Chem. Lett. 2016, 26, 4377–4381. [Google Scholar] [CrossRef]
- Mikhaleva, A.I.; Ivanov, A.V.; Vasil’tsov, A.M.; Ushakov, I.A.; Shi Ma, J.; Yang, G. Directed Synthesis of Semicarbazones, Thiosemicarbazones, and Guanylhydrazones of 1-Vinylpyrrole-2-Carbaldehydes. Chem. Heterocycl. Compd. 2008, 44, 1384–1390. [Google Scholar] [CrossRef]
- Bernatowicz, M.S.; Wu, Y.; Matsueda, G.R. 1H-Pyrazole-1-Carboxamidine Hydrochloride an Attractive Reagent for Guanylation of Amines and Its Application to Peptide Synthesis. J. Org. Chem. 1992, 57, 2497–2502. [Google Scholar] [CrossRef]
- Valdez-Padilla, D.; Rodríguez-Morales, S.; Hernández-Campos, A.; Hernández-Luis, F.; Yépez-Mulia, L.; Tapia-Contreras, A.; Castillo, R. Synthesis and Antiprotozoal Activity of Novel 1-Methylbenzimidazole Derivatives. Bioorganic Med. Chem. 2009, 17, 1724–1730. [Google Scholar] [CrossRef] [PubMed]
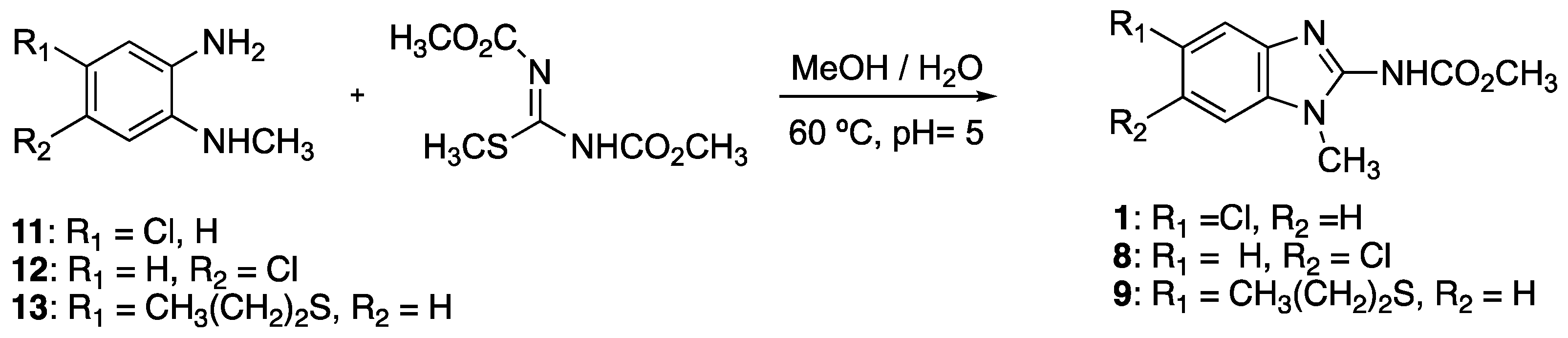

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
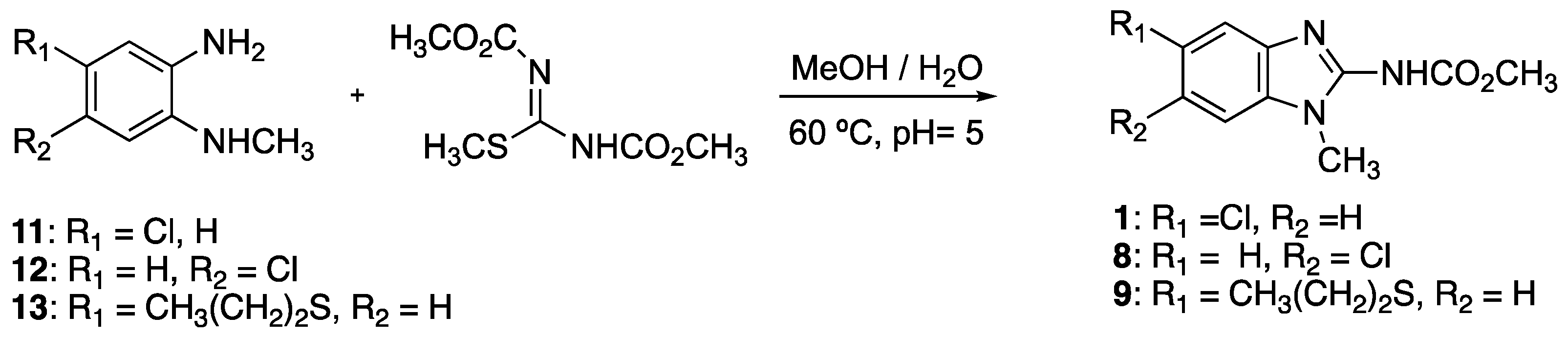{kind=link}
{kind=link}
| Compound | Chemical Structure | Docking Score (kcal/mol) | % Inhibition LmARG [200 μM] |
|---|---|---|---|
| 1 | 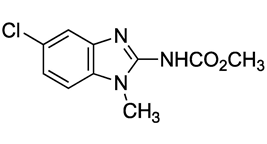 | −5.46 | 100% |
| 2 |  | −5.04 | 100% |
| 3 | 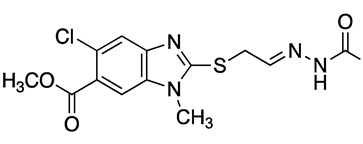 | −6.39 | 60% |
| 4 | 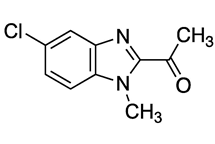 | −6.05 | 56% |
| 5 | 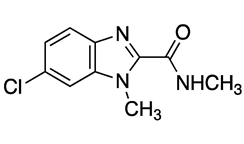 | −5.96 | 56% |
| 6 |  | −8.38 | 46% |
| 7 | 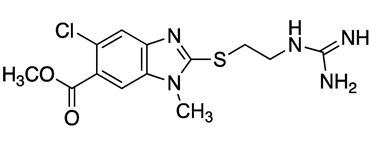 | −6.05 | 40% |
| 8 | 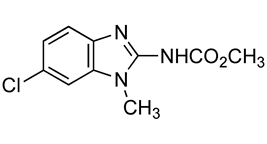 | −6.00 | 40% |
| 9 | 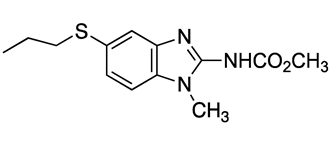 | −5.04 | 36% |
| 10 | 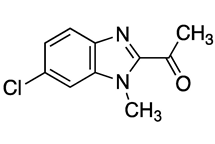 | −6.65 | 33% |
| Compound | IC50 (µM) | CC50 (µM) | SI 1 | ||
|---|---|---|---|---|---|
| Promastigotes | Amastigotes | Promastigotes | Amastigotes | ||
| 1 | 64 | 32 | >417 2 | 6.5 | 13 |
| 2 | 132 | 101 | >414 2 | 3.1 | 4.1 |
| Glucantime | 101 | 35 | >273 3 | 2.7 | 7.8 |
| Amphotericin B | 0.89 | 1.3 | 7.4 3 | 8.3 | 6.2 |
| Miltefosine | 8.1 | 8.6 | 141 3 | 17.4 | 2 |
| Descriptor | Compound 1 | Compound 2 |
|---|---|---|
| MW 1 | 239.66 | 241.7 |
| logP 1 | 2.35 | 2.1 |
| logD 1 | 2.63 | 2.03 |
| logSw 1 | −2.93 | −2.82 |
| tPSA 1 | 56.15 | 97.07 |
| RB 1 | 2 | 2 |
| HBD 1 | 1 | 3 |
| HBA 1 | 5 | 4 |
| Lipinski’s violations 1 | none | none |
| Veber Rules 1 | Good | Good |
| Egan Rules 1 | Good | Good |
| BBB+ 2 | 0.9584 | 0.9117 |
| HIA+ 2 | 0.9965 | 0.9819 |
| Caco2 2 | 0.6531 | 0.5468 |
| Lysosome 2 | 0.6217 | 0.6217 |
| Predicted LD50 3 | 5050 mg/kg | 705 mg/kg |
| Predicted toxicity class 3 | 6 | 4 |
| Toxicity targets 3 | no binding | no binding |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Betancourt-Conde, I.; Avitia-Domínguez, C.; Hernández-Campos, A.; Castillo, R.; Yépez-Mulia, L.; Oria-Hernández, J.; Méndez, S.T.; Sierra-Campos, E.; Valdez-Solana, M.; Martínez-Caballero, S.; et al. Benzimidazole Derivatives as New and Selective Inhibitors of Arginase from Leishmania mexicana with Biological Activity against Promastigotes and Amastigotes. Int. J. Mol. Sci. 2021, 22, 13613. https://doi.org/10.3390/ijms222413613
Betancourt-Conde I, Avitia-Domínguez C, Hernández-Campos A, Castillo R, Yépez-Mulia L, Oria-Hernández J, Méndez ST, Sierra-Campos E, Valdez-Solana M, Martínez-Caballero S, et al. Benzimidazole Derivatives as New and Selective Inhibitors of Arginase from Leishmania mexicana with Biological Activity against Promastigotes and Amastigotes. International Journal of Molecular Sciences. 2021; 22(24):13613. https://doi.org/10.3390/ijms222413613
Chicago/Turabian StyleBetancourt-Conde, Irene, Claudia Avitia-Domínguez, Alicia Hernández-Campos, Rafael Castillo, Lilián Yépez-Mulia, Jesús Oria-Hernández, Sara T. Méndez, Erick Sierra-Campos, Mónica Valdez-Solana, Siseth Martínez-Caballero, and et al. 2021. "Benzimidazole Derivatives as New and Selective Inhibitors of Arginase from Leishmania mexicana with Biological Activity against Promastigotes and Amastigotes" International Journal of Molecular Sciences 22, no. 24: 13613. https://doi.org/10.3390/ijms222413613
APA StyleBetancourt-Conde, I., Avitia-Domínguez, C., Hernández-Campos, A., Castillo, R., Yépez-Mulia, L., Oria-Hernández, J., Méndez, S. T., Sierra-Campos, E., Valdez-Solana, M., Martínez-Caballero, S., Hermoso, J. A., Romo-Mancillas, A., & Téllez-Valencia, A. (2021). Benzimidazole Derivatives as New and Selective Inhibitors of Arginase from Leishmania mexicana with Biological Activity against Promastigotes and Amastigotes. International Journal of Molecular Sciences, 22(24), 13613. https://doi.org/10.3390/ijms222413613