
DSResSol: A Sequence-Based Solubility Predictor Created with Dilated Squeeze Excitation Residual Networks


Abstract
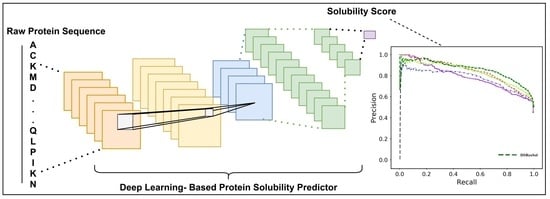
:
1. Introduction
2. Results
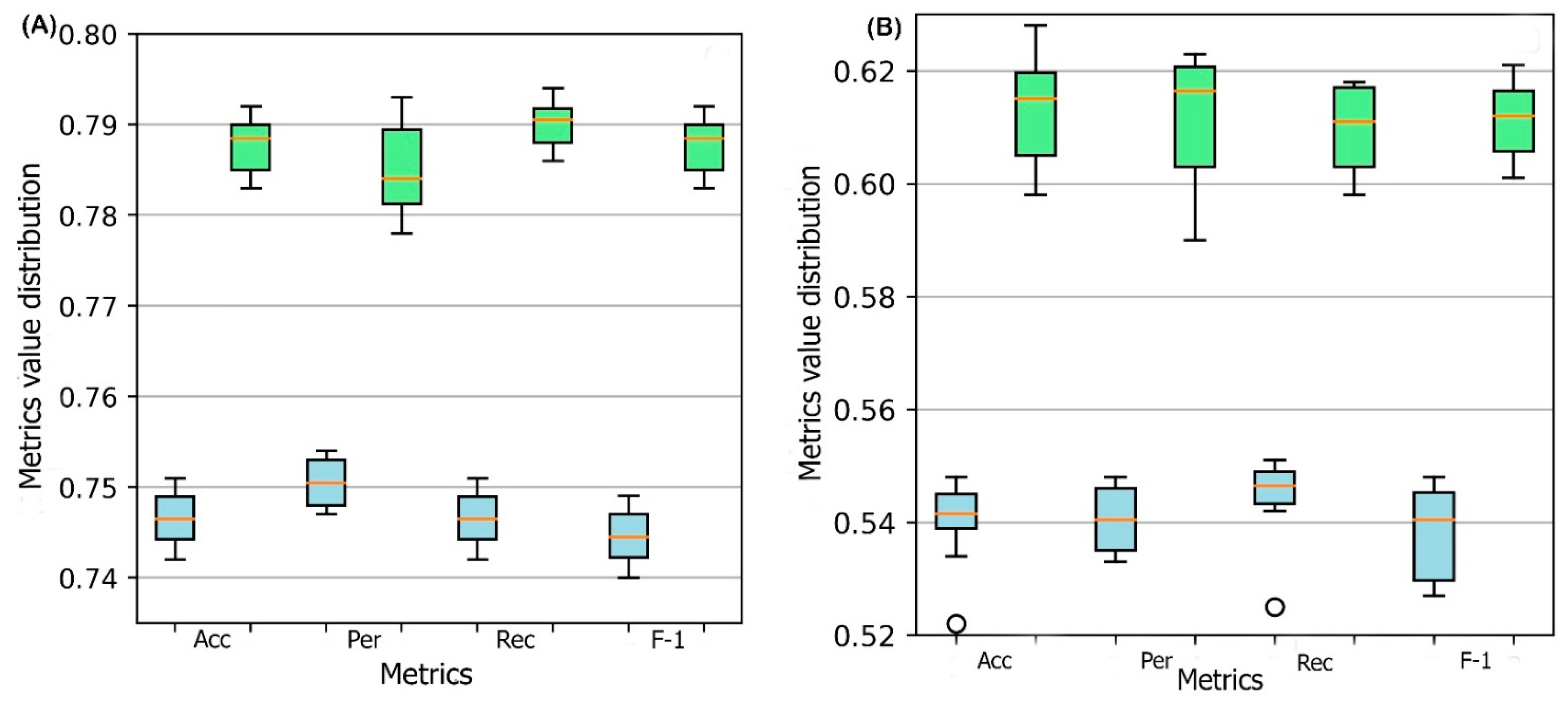
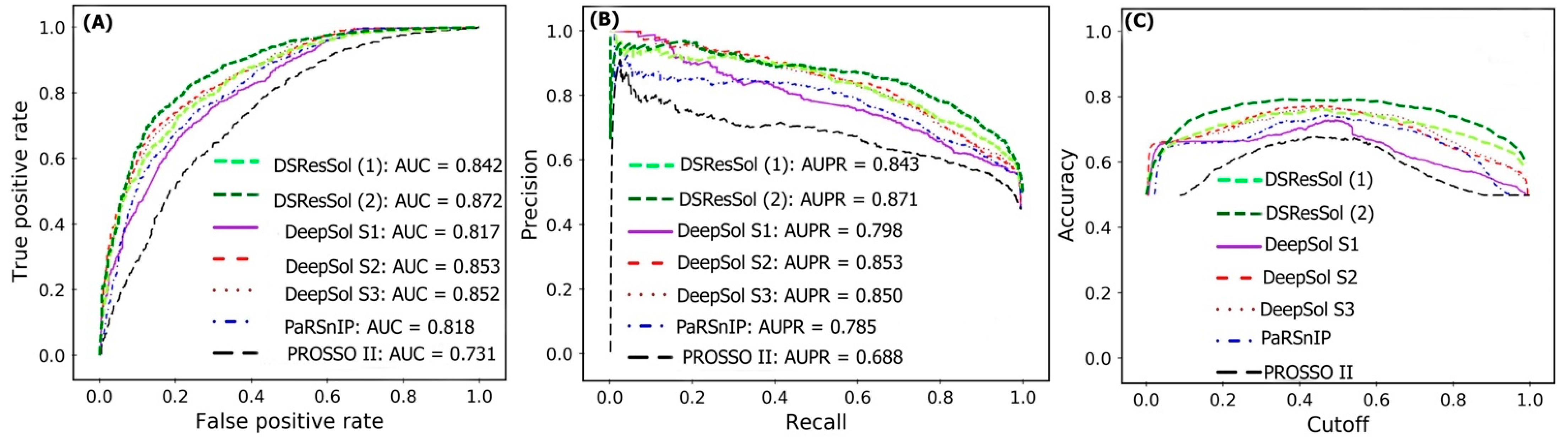
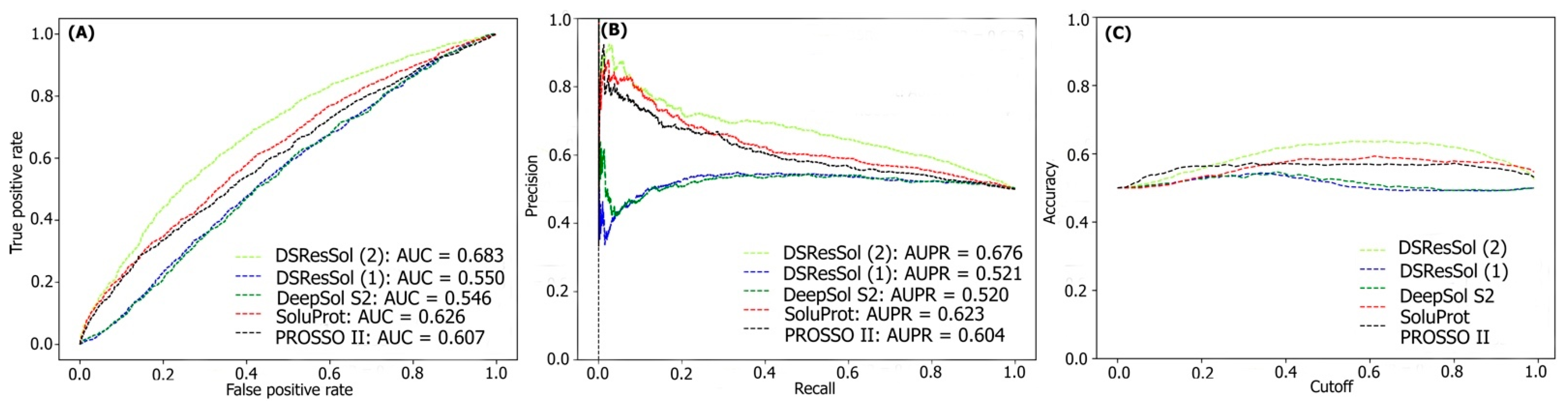
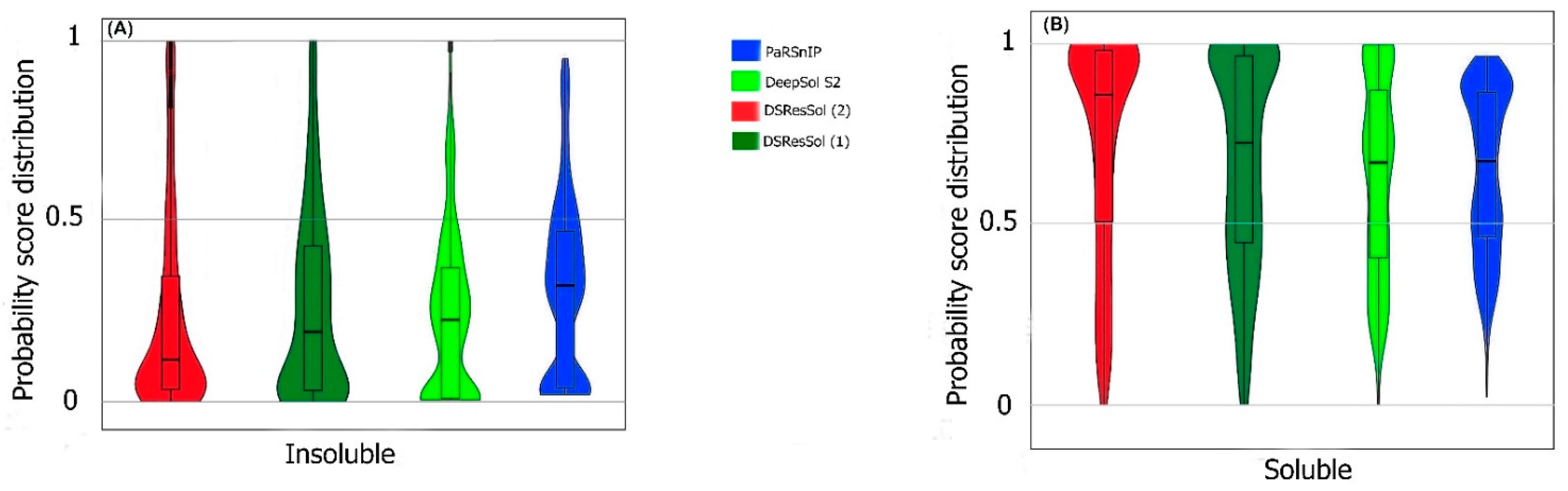
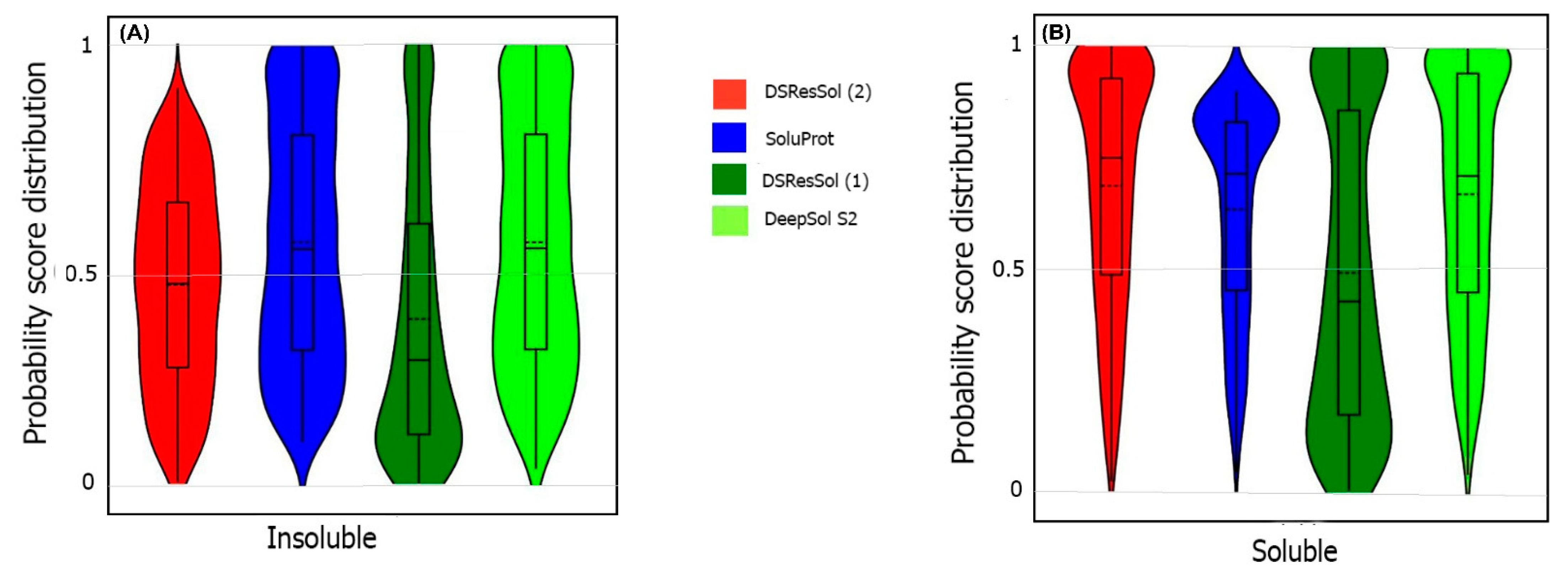
2.1. Model Performance
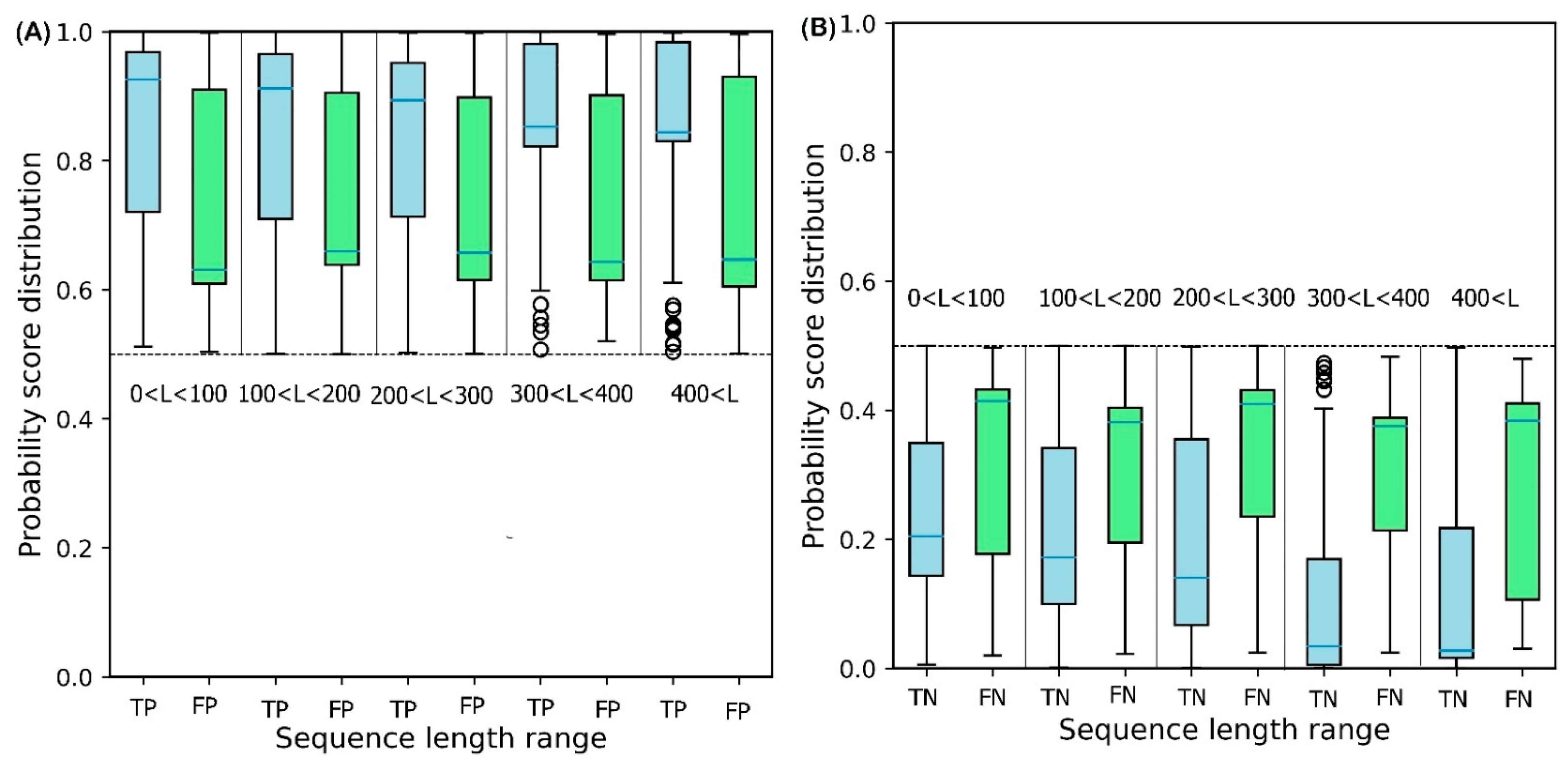
2.2. Effect of Sequence Length on Solubility Prediction
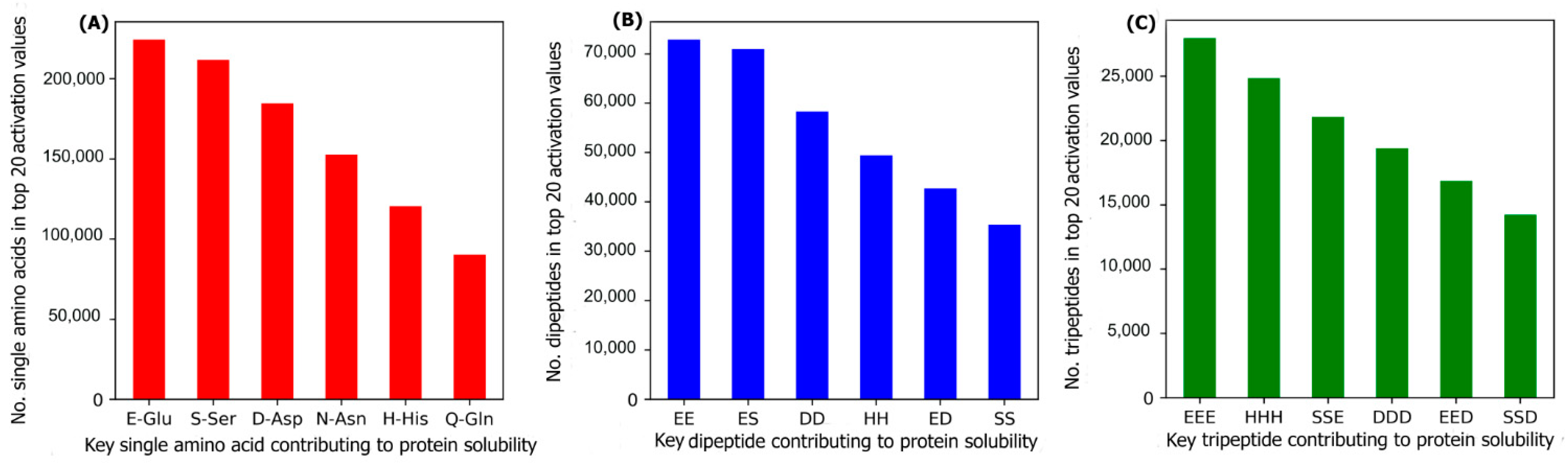
2.3. Key Amino Acids, Dipeptides, and Tripeptides for Protein Solubility
2.4. Effect of Additional Biological Features on DSResSol Performance
2.5. Effect of Sequence Identity Cutoff on DSResSol Performance
3. Discussion
4. Materials and Methods
4.1. Data Preparation and Feature Engineering
- The first test dataset was proposed by Chang et al. [25]. This dataset includes 2001 protein sequences and their corresponding solubility values;
- The second test dataset, first proposed by Hon et al. [15], has been constructed from a dataset generated by the North East Structural Consortium (NESG) [26] and includes 9644 proteins expressed in E. coli. The original dataset consists of integer values in the range from 0 to 5 for levels of expression and soluble fraction recovery. We maintained consistency between the procedure for constructing the training set and the test set. Finally, similar to the SoluProt test set [15], the solubility level of each protein within the NESG test set was transferred to a binary value. In the original NESG dataset, the solubility values for protein sequences are an integer value in the range from 0 to 4. To transfer these values to binary values, similar to the SoluProt method, we considered protein sequences having a solubility of value 0, 1, 2, as insoluble and those sequences have solubility values of 3 or 4 as soluble proteins.
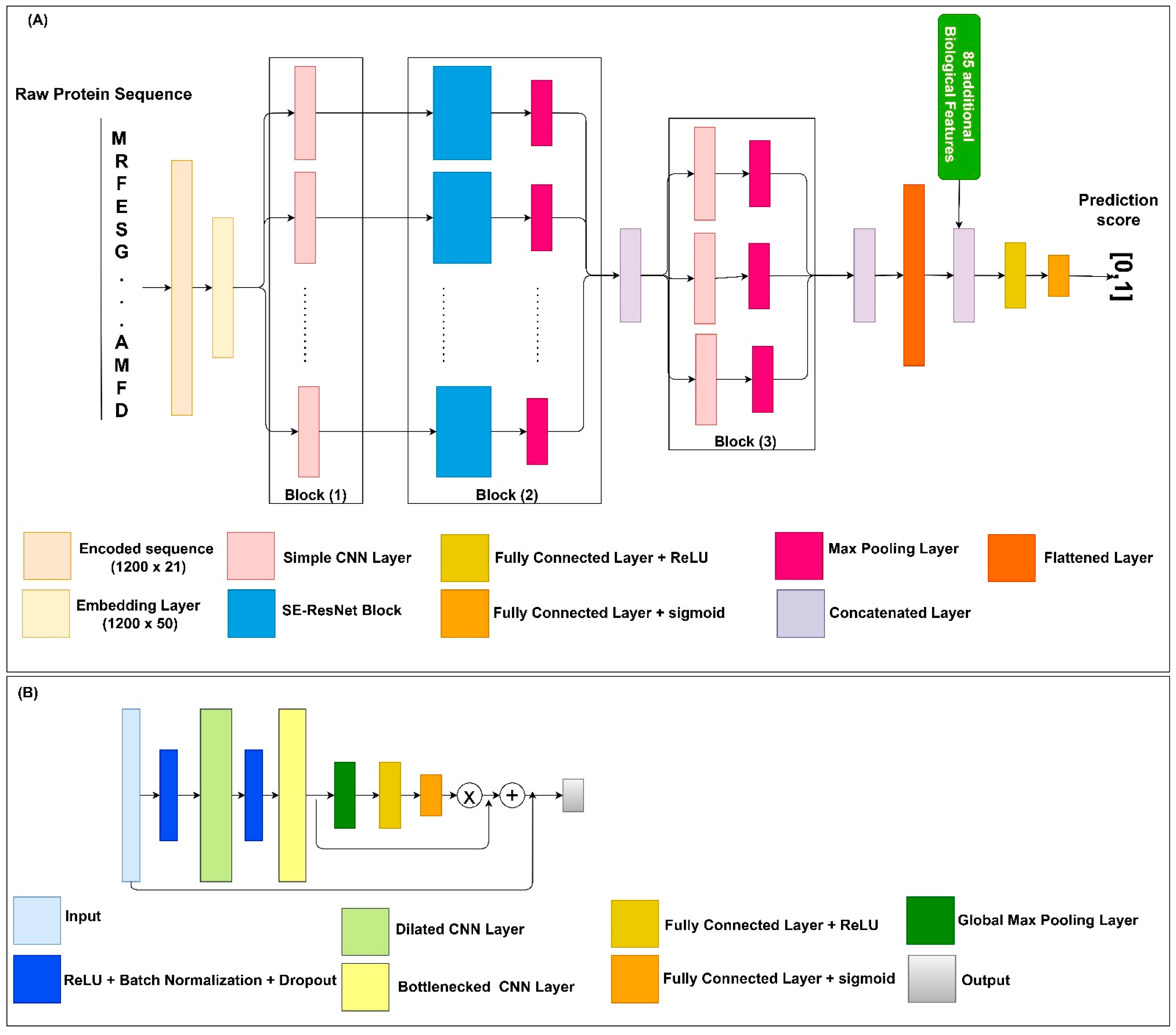
4.2. Model Architecture
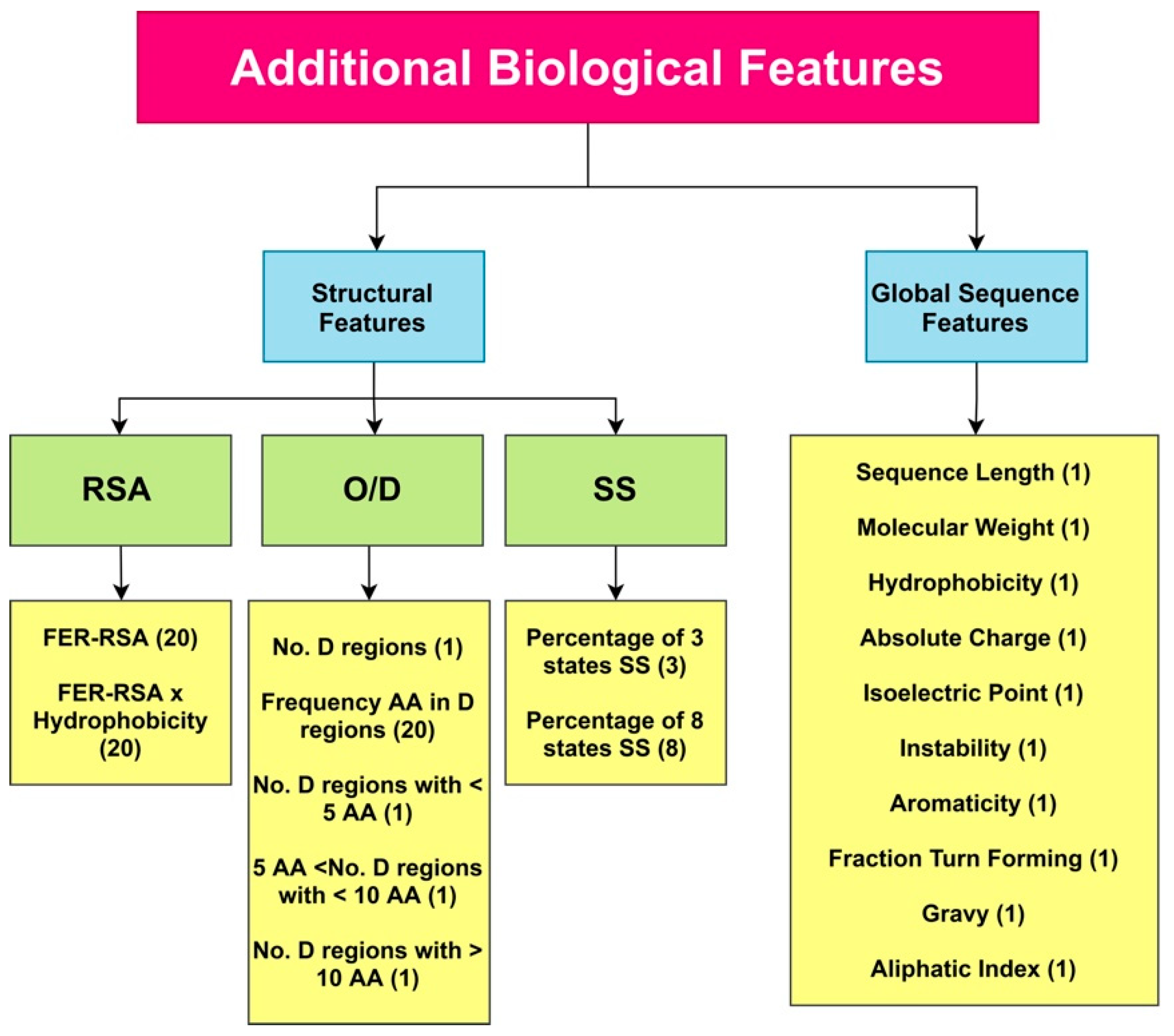
4.3. Additional Features
4.4. Training and Hyperparameter Tuning
4.5. Evaluation Metrics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zayas, J.F. Solubility of Proteins. In Functionality of Proteins in Food; Zayas, J.F., Ed.; Springer: Berlin/Heidelberg, Germany, 1997; pp. 6–75. ISBN 978-3-642-59116-7. [Google Scholar]
- Jain, A.; Jain, A.; Gulbake, A.; Shilpi, S.; Hurkat, P.; Jain, S.K. Peptide and protein delivery using new drug delivery systems. Crit. Rev. Ther. Drug Carr. Syst. 2013, 30, 293–329. [Google Scholar] [CrossRef] [PubMed]
- Madani, M.; Tarakanova, A. Molecular Design of Soluble Zein Protein Sequences. Biophys. J. 2020, 118, 45a. [Google Scholar] [CrossRef]
- Idicula-Thomas, S.; Balaji, P.V. Understanding the relationship between the primary structure of proteins and its propensity to be soluble on overexpression in Escherichia coli. Protein Sci. Publ. Protein Soc. 2005, 14, 582–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef]
- Chan, W.-C.; Liang, P.-H.; Shih, Y.-P.; Yang, U.-C.; Lin, W.; Hsu, C.-N. Learning to predict expression efficacy of vectors in recombinant protein production. BMC Bioinform. 2010, 11, S21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiti, F.; Stefani, M.; Taddei, N.; Ramponi, G.; Dobson, C.M. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 2003, 424, 805–808. [Google Scholar] [CrossRef]
- Bhandari, B.K.; Gardner, P.P.; Lim, C.S. Solubility-Weighted Index: Fast and accurate prediction of protein solubility. Bioinformatics 2020, 36, 4691–4698. [Google Scholar] [CrossRef]
- Diaz, A.A.; Tomba, E.; Lennarson, R.; Richard, R.; Bagajewicz, M.J.; Harrison, R.G. Prediction of protein solubility in Escherichia coli using logistic regression. Biotechnol. Bioeng. 2010, 105, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Friedman, J.H. Greedy Function Approximation: A Gradient Boosting Machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Babich, G.A.; Camps, O.I. Weighted Parzen windows for pattern classification. IEEE Trans. Pattern Anal. Mach. Intell. 1996, 18, 567–570. [Google Scholar] [CrossRef]
- Smialowski, P.; Doose, G.; Torkler, P.; Kaufmann, S.; Frishman, D. PROSO II—A new method for protein solubility prediction. FEBS J. 2012, 279, 2192–2200. [Google Scholar] [CrossRef]
- Rawi, R.; Mall, R.; Kunji, K.; Shen, C.-H.; Kwong, P.D.; Chuang, G.-Y. PaRSnIP: Sequence-based protein solubility prediction using gradient boosting machine. Bioinforma. Oxf. Engl. 2018, 34, 1092–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hon, J.; Marusiak, M.; Martinek, T.; Kunka, A.; Zendulka, J.; Bednar, D.; Damborsky, J. SoluProt: Prediction of soluble protein expression in Escherichia coli. Bioinformatics 2021, 37, 23–28. [Google Scholar] [CrossRef]
- Khurana, S.; Rawi, R.; Kunji, K.; Chuang, G.-Y.; Bensmail, H.; Mall, R. DeepSol: A deep learning framework for sequence-based protein solubility prediction. Bioinformatics 2018, 34, 2605–2613. [Google Scholar] [CrossRef] [PubMed]
- Lecun, Y.; Bengio, Y. Convolutional networks for images, speech, and time-series. Handb. Brain Theory Neural Netw. 1995, 3361, 1995. Available online: https://nyuscholars.nyu.edu/en/publications/convolutional-networks-for-images-speech-and-time-series (accessed on 21 March 2021).
- Yu, F.; Koltun, V. Multi-Scale Context Aggrgation by Dilated Convolutions. arXiv 2016, arXiv:1511.07122. Available online: http://arxiv.org/abs/1511.07122 (accessed on 21 March 2021).
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep Residual Learning for Image Recognition. In Proceedings of the 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Hu, J.; Shen, L.; Sun, G. Squeeze-and-Excitation Networks. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Salt Lake City, UT, USA, 18–23 June 2018; pp. 7132–7141. Available online: https://openaccess.thecvf.com/content_cvpr_2018/html/Hu_Squeeze-and-Excitation_Networks_CVPR_2018_paper.html (accessed on 12 December 2020).
- Yang, Z.; Hu, Z.; Salakhutdinov, R.; Berg-Kirkpatrick, T. Improved Variational Autoencoders for Text Modeling using Dilated Convolutions. In Proceedings of the International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017; pp. 3881–3890. Available online: http://proceedings.mlr.press/v70/yang17d.html (accessed on 17 March 2021).
- Bileschi, M.L.; Belanger, D.; Bryant, D.; Sanderson, T.; Carter, B.; Sculley, D.; DePristo, M.A.; Colwell, L.J. Using Deep Learning to Annotate the Protein Universe. bioRxiv 2019, 20, 626507. [Google Scholar] [CrossRef] [Green Version]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Shi, W.; Tian, J.; Qi, Z.; Li, B.; Hao, H.; Xu, B. Attention-Based Bidirectional Long Short-Term Memory Networks for Relation Classification. In Proceedings of the 54th Annual Meeting of the Association for Computational Linguistics (Volume 2: Short Papers), Berlin/Heidelberg, Germany, 7–12 August 2016; pp. 207–212. [Google Scholar]
- Chang, C.C.H.; Song, J.; Tey, B.T.; Ramanan, R.N. Bioinformatics approaches for improved recombinant protein production in Escherichia coli: Protein solubility prediction. Brief. Bioinform. 2014, 15, 953–962. [Google Scholar] [CrossRef] [Green Version]
- Price, W.N.; Handelman, S.K.; Everett, J.K.; Tong, S.N.; Bracic, A.; Luff, J.D.; Naumov, V.; Acton, T.; Manor, P.; Xiao, R.; et al. Large-scale experimental studies show unexpected amino acid effects on protein expression and solubility in vivo in E. coli. Microb. Inform. Exp. 2011, 1, 6. [Google Scholar] [CrossRef] [Green Version]
- Kramer, R.M.; Shende, V.R.; Motl, N.; Pace, C.N.; Scholtz, J.M. Toward a Molecular Understanding of Protein Solubility: Increased Negative Surface Charge Correlates with Increased Solubility. Biophys. J. 2012, 102, 1907–1915. [Google Scholar] [CrossRef] [Green Version]
- Trevino, S.R.; Scholtz, J.M.; Pace, C.N. Amino acid contribution to protein solubility: Asp, Glu, and Ser contribute more favorably than the other hydrophilic amino acids in RNase Sa. J. Mol. Biol. 2007, 366, 449–460. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.M.; Khan, M.A.; Kuroda, Y. Analysis of amino acid contributions to protein solubility using short peptide tags fused to a simplified BPTI variant. Biochim. Biophys. Acta 2012, 1824, 1144–1150. [Google Scholar] [CrossRef]
- Kuntz, I.D. Hydration of macromolecules. III. Hydration of polypeptides. J. Am. Chem. Soc. 1971, 93, 514–516. [Google Scholar] [CrossRef]
- Chan, P.; Curtis, R.A.; Warwicker, J. Soluble expression of proteins correlates with a lack of positively-charged surface. Sci. Rep. 2013, 3, 3333. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.K.M.; Ki, M.R.; Son, R.G.; Pack, S.P. The NT11, a novel fusion tag for enhancing protein expression in Escherichia coli. Appl. Microbiol. Biotechnol. 2019, 103, 2205–2216. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, R.; Zhu, D.; Wang, W.; Yi, L.; Ma, L. Non-peptide guided auto-secretion of recombinant proteins by super-folder green fluorescent protein in Escherichia coli. Sci. Rep. 2017, 7, 6990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.; Hong, P.; Yang, P.; Zhou, C.; Xiao, D.; Zhong, T. Correlation Between the Water Solubility and Secondary Structure of Tilapia-Soybean Protein Co-Precipitates. Molecules 2019, 24, 4337. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Adhikari, B.; Cheng, J. DeepSF: Deep convolutional neural network for mapping protein sequences to folds. Bioinformatics 2018, 34, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lin, Y.; Elofsson, A.; Yao, Y. Protein Contact Map Prediction Based on ResNet and DenseNet. BioMed Res. Int. 2020, 2020, e7584968. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.D.; Gabanyi, M.J.; Tao, W.; Shah, R.; Kouranov, A.; Schwede, T.; Arnold, K.; Kiefer, F.; Bordoli, L.; et al. The protein structure initiative structural genomics knowledgebase. Nucleic Acids Res. 2009, 37, D365–D368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Harris, D.; Harris, S. Digital Design and Computer Architecture; Morgan Kaufmann: Burlington, MA, USA, 2010; ISBN 978-0-08-054706-0. [Google Scholar]
- Conneau, A.; Schwenk, H.; Barrault, L.; Lecun, Y. Very Deep Convolutional Networks for Natural Language Processing. KI—Künstl. Intell. 2016, 26, 1. [Google Scholar]
- Chang, S.; Zhang, Y.; Han, W.; Yu, M.; Guo, X.; Tan, W.; Cui, X.; Witbrock, M.; Hasegawa-Johnson, M.; Huang, T.S. Dilated Recurrent Neural Networks. arXiv 2017, arXiv:1710.02224. Available online: http://arxiv.org/abs/1710.02224 (accessed on 25 March 2021).
- Xu, B.; Wang, N.; Chen, T.; Li, M. Empirical Evaluation of Rectified Activations in Convolutional Network. arXiv 2015, arXiv:1505.00853. [Google Scholar]
- Han, D.; Yun, S.; Heo, B.; Yoo, Y. ReXNet: Diminishing Representational Bottleneck on Convolutional Neural Network. arXiv 2020, arXiv:2007.00992. Available online: http://arxiv.org/abs/2007.00992 (accessed on 25 March 2021).
- Hu, J.; Shen, L.; Albanie, S.; Sun, G.; Wu, E. Squeeze-and-Excitation Networks. IEEE Trans. Pattern Anal. Mach. Intell. 2020, 42, 2011–2023. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.G.; Navab, N.; Wachinger, C. Recalibrating Fully Convolutional Networks With Spatial and Channel “Squeeze and Excitation” Blocks. IEEE Trans. Med. Imaging 2019, 38, 540–549. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, I.; Martin, A.J.M.; Di Domenico, T.; Tosatto, S.C.E. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Müller, A.T.; Gabernet, G.; Hiss, J.A.; Schneider, G. modlAMP: Python for antimicrobial peptides. Bioinformatics 2017, 33, 2753–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, D.; Franco-Pedroso, J.; Lozano-Diez, A.; Gonzalez-Rodriguez, J. Deconstructing Cross-Entropy for Probabilistic Binary Classifiers. Entropy 2018, 20, 208. [Google Scholar] [CrossRef] [Green Version]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2014, arXiv:1412.6980. Available online: http://arxiv.org/abs/1412.6980 (accessed on 5 April 2021).
- Li, W.; Xing, X.; Liu, F.; Zhang, Y. Application of Improved Grid Search Algorithm on SVM for Classification of Tumor Gene. Int. J. Multimed. Ubiquitous Eng. 2014, 9, 181–188. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | ACC | MMC | Selectivity (Soluble) | Selectivity (Insoluble) | Sensitivity (Soluble) | Sensitivity (Insoluble) | Gain (Soluble) | Gain (Insoluble) |
|---|---|---|---|---|---|---|---|---|
| DSResSol (2) | 0.796 | 0.589 | 0.817 | 0.782 | 0.769 | 0.823 | 1.634 | 1.564 |
| DSResSol (1) | 0.751 | 0.508 | 0.786 | 0.722 | 0.691 | 0.813 | 1.572 | 1.445 |
| SoluProt | 0.682 | 0.382 | 0.701 | 0.670 | 0.722 | 0.643 | 1.403 | 1.342 |
| DeepSol S2 | 0.762 | 0.546 | 0.821 | 0.721 | 0.681 | 0.843 | 1.642 | 1.442 |
| DeepSol S3 | 0.760 | 0.543 | 0.801 | 0.725 | 0.707 | 0.822 | 1.602 | 1.451 |
| PaRSnIP | 0.720 | 0.472 | 0.761 | 0.723 | 0.698 | 0.743 | 1.522 | 1.446 |
| DeepSol S1 | 0.720 | 0.471 | 0.752 | 0.706 | 0.691 | 0.749 | 1.504 | 1.412 |
| PROSSO II | 0.638 | 0.345 | 0.671 | 0.682 | 0.693 | 0.662 | 1.342 | 1.365 |
| SCM | 0.600 | 0.214 | 0.650 | 0.572 | 0.422 | 0.773 | 1.301 | 1.145 |
| PROSO | 0.581 | 0.161 | 0.582 | 0.575 | 0.541 | 0.622 | 1.164 | 1.151 |
| CCSOL | 0.543 | 0.083 | 0.543 | 0.539 | 0.514 | 0.572 | 1.087 | 1.081 |
| RPSP | 0.520 | 0.032 | 0.522 | 0.517 | 0.447 | 0.588 | 1.044 | 1.035 |
| Method | ACC | MCC | Selectivity (Soluble) | Selectivity (Insoluble) | Sensitivity (Soluble) | Sensitivity (Insoluble) | Gain (Soluble) | Gain (Insoluble) |
|---|---|---|---|---|---|---|---|---|
| DSResSol (2) | 0.629 | 0.273 | 0.606 | 0.663 | 0.73 | 0.53 | 1.212 | 1.326 |
| DSResSol (1) | 0.557 | 0.169 | 0.558 | 0.555 | 0.54 | 0.58 | 1.117 | 1.11 |
| SoluProt | 0.578 | 0.189 | 0.575 | 0.581 | 0.6 | 0.56 | 1.15 | 1.162 |
| PROSSO II | 0.565 | 0.143 | 0.578 | 0.555 | 0.48 | 0.66 | 1.157 | 1.11 |
| SWI | 0.558 | 0.142 | 0.545 | 0.58 | 0.7 | 0.42 | 1.09 | 1.16 |
| CamSol | 0.535 | 0.115 | 0.548 | 0.527 | 0.39 | 0.68 | 1.097 | 1.054 |
| ESPRESSO | 0.493 | 0.093 | 0.493 | 0.492 | 0.55 | 0.44 | 0.987 | 0.984 |
| rWH | 0.519 | 0.133 | 0.532 | 0.513 | 0.31 | 0.73 | 1.065 | 1.026 |
| DeepSol S2 | 0.546 | 0.132 | 0.894 | 0.56 | 0.22 | 0.88 | 1.788 | 1.12 |
| SOLpro | 0.5 | 0.089 | 0.5 | 0.5 | 0.48 | 0.52 | 1 | 1 |
| Sequence Length Range | M(TP)—M(FP) | M(FN)—M(TN) |
|---|---|---|
| (0, 100) | 0.29 | 0.23 |
| (100, 200) | 0.24 | 0.22 |
| (200, 300) | 0.24 | 0.26 |
| (300, 400) | 0.22 | 0.31 |
| (400, ∞) | 0.22 | 0.34 |
| Model | ACC DSResSol (1) after Adding the Additional Biological Features | ACC Improvement |
|---|---|---|
| DSResSol (1) + Solvent accessibility related features | 0.787 | 3.7% |
| DSResSol (1) + Secondary structure related features | 0.762 | 1.1% |
| DSResSol (1) + order/disorder related features | 0.757 | 0.6% |
| DSResSol (1) + global sequence features | 0.756 | 0.5% |
| Model | ACC DSResSol (1) after Adding the Additional Biological Features | ACC Improvement |
|---|---|---|
| DSResSol (1) + Solvent accessibility related features | 0.618 | 6.1% |
| DSResSol (1) + Secondary structure related features | 0.582 | 2.5% |
| DSResSol (1) + order/disorder related features | 0.564 | 0.7% |
| DSResSol (1) + global sequence features | 0.561 | 0.4% |
| Model | ACC | MMC | Sensitivity (Soluble) | Sensitivity (Insoluble) |
|---|---|---|---|---|
| DSResSol (1) Cutoff 25% | 0.751 | 0.508 | 0.691 | 0.813 |
| DSResSol (1) Cutoff 15% | 0.744 | 0.491 | 0.686 | 0.805 |
| DSResSol (1) Cutoff 20% | 0.743 | 0.488 | 0.701 | 0.795 |
| DSResSol (1) Cutoff 30% | 0.744 | 0.492 | 0.688 | 0.801 |
| Model | ACC | MCC | Sensitivity (Soluble) | Sensitivity (Insoluble) |
|---|---|---|---|---|
| DSResSol (1) Cutoff 25% | 0.557 | 0.166 | 0.545 | 0.568 |
| DSResSol (1) Cutoff 15% | 0.553 | 0.157 | 0.542 | 0.567 |
| DSResSol (1) Cutoff 20% | 0.555 | 0.164 | 0.547 | 0.563 |
| DSResSol (1) Cutoff 30% | 0.552 | 0.159 | 0.541 | 0.567 |
| Construction Step | Training Set | Soluble | Insoluble | Test Set 1 | Soluble | Insoluble | Test Set 2 | Soluble | Insoluble |
|---|---|---|---|---|---|---|---|---|---|
| Input | 129,593 | - | - | 2001 | 1000 | 1001 | 9703 | - | - |
| Pre-processing and solubility assignment | 109,648 | - | - | 2001 | 1000 | 1001 | - | - | - |
| Redundancy removal | 87,969 | 40,905 | 14,064 | 2001 | 1000 | 1001 | 9423 | 5718 | 3705 |
| Removal of short sequences and sequences with unknown residues | 82,902 | 50,004 | 32,898 | 2001 | 1000 | 1001 | 9420 | 5715 | 3705 |
| Removal of transmembrane proteins | 76,274 | 45,603 | 30,671 | 2001 | 1000 | 1001 | 8769 | 5421 | 3348 |
| Removal of insoluble sequences with available PDB structure | 72,756 | 42,530 | 30,226 | 2001 | 1000 | 1001 | 8754 | 5421 | 3333 |
| Clustering to 25% identity | 49,369 | 26,422 | 22,947 | 2001 | 1000 | 1001 | 3945 | 2078 | 1867 |
| Overlap removal with test sets 15% identity | 46,028 | 24,920 | 21,108 | 2001 | 1000 | 1001 | 3945 | 2078 | 1867 |
| Class and length balancing | 40,317 | 19,718 | 20,599 | 2001 | 1000 | 1001 | 3729 | 1864 | 1865 |
| Layers | Number of Tested Units or Filters | Optimal Value | Filter Size | Parameters | Tested Values | Optimal Value |
|---|---|---|---|---|---|---|
| Embedding layer | (50, 100, 150) | 50 | - | Epochs | 50 | 50 |
| Initial CNNs | (32, 64, 128, 256) | 32 | {1, 2, …, 9} | Learning rate | (0.005, 0.008, 0.01, 0.02) | 0.008 |
| Dilated CNN | (32, 64, 128, 256) | 32 | 3 | Batch size | (32, 64, 128, 256) | 64 |
| Bottlenecked CNN | (32, 64, 128, 256) | 32 | 1 | Decay rate | ) | |
| Final CNNs | (32, 64, 128, 256) | 32 | {11, 13, 15} | Early stopping value | (3, 4, 5, 6) | 5 |
| FC | (64, 128, 256, 512) | 128 | - | - | - | - |
| MaxPooling | (2, 3, 5, 7) | 3 | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madani, M.; Lin, K.; Tarakanova, A. DSResSol: A Sequence-Based Solubility Predictor Created with Dilated Squeeze Excitation Residual Networks. Int. J. Mol. Sci. 2021, 22, 13555. https://doi.org/10.3390/ijms222413555
Madani M, Lin K, Tarakanova A. DSResSol: A Sequence-Based Solubility Predictor Created with Dilated Squeeze Excitation Residual Networks. International Journal of Molecular Sciences. 2021; 22(24):13555. https://doi.org/10.3390/ijms222413555
Chicago/Turabian StyleMadani, Mohammad, Kaixiang Lin, and Anna Tarakanova. 2021. "DSResSol: A Sequence-Based Solubility Predictor Created with Dilated Squeeze Excitation Residual Networks" International Journal of Molecular Sciences 22, no. 24: 13555. https://doi.org/10.3390/ijms222413555
APA StyleMadani, M., Lin, K., & Tarakanova, A. (2021). DSResSol: A Sequence-Based Solubility Predictor Created with Dilated Squeeze Excitation Residual Networks. International Journal of Molecular Sciences, 22(24), 13555. https://doi.org/10.3390/ijms222413555