
Sparsely Connected Autoencoders: A Multi-Purpose Tool for Single Cell omics Analysis

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
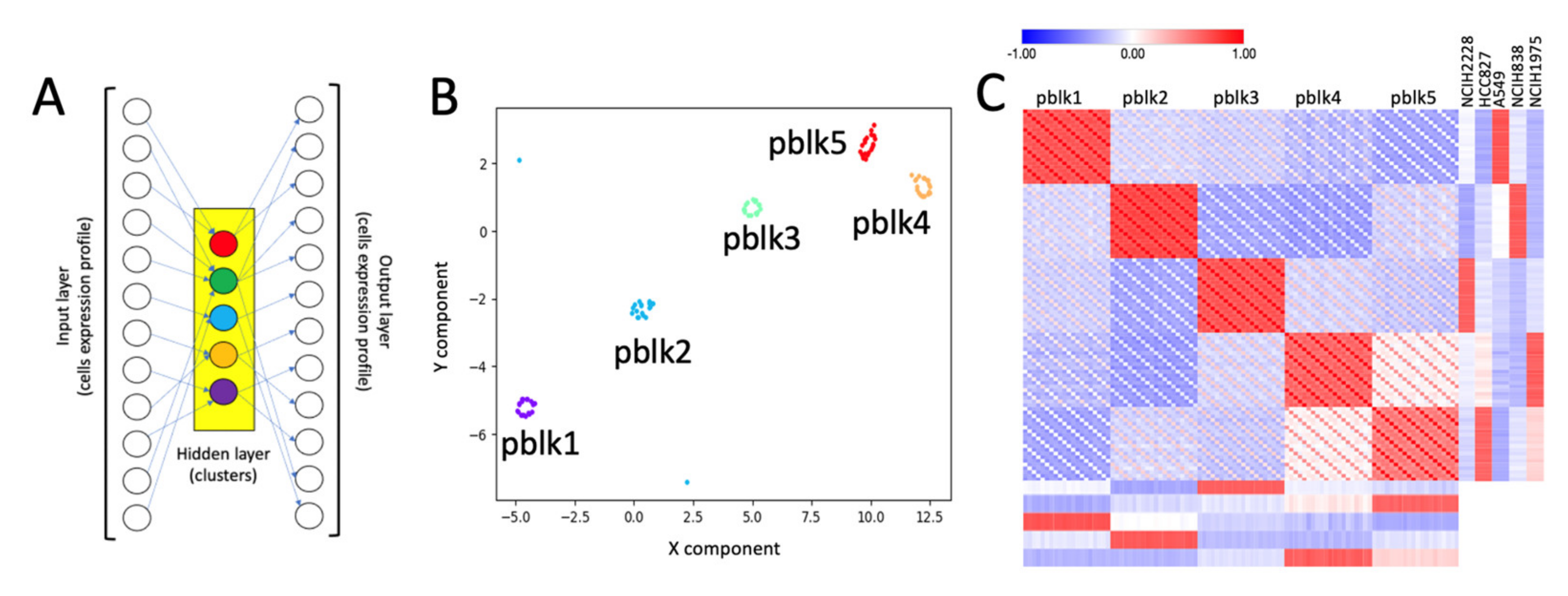
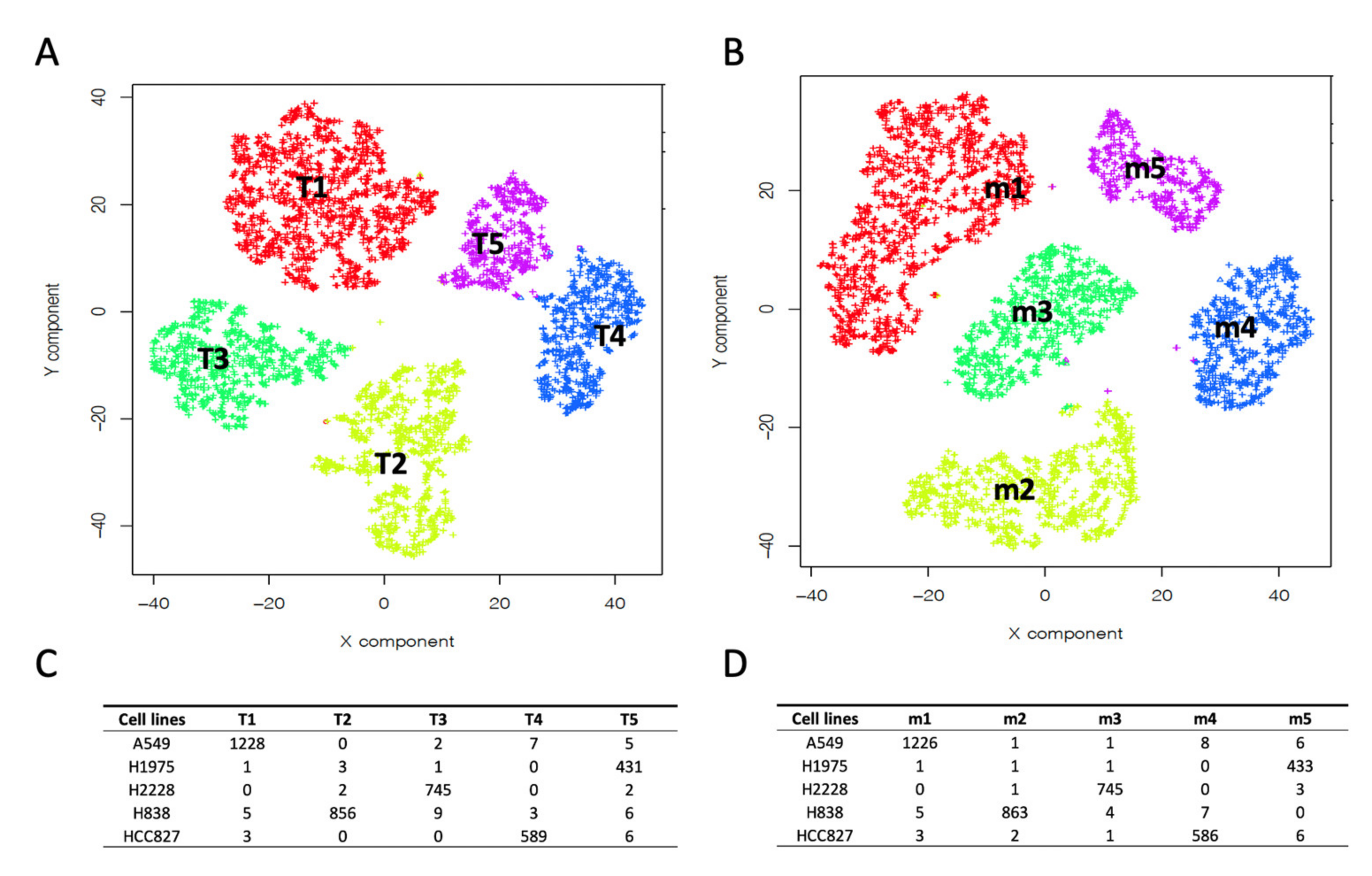
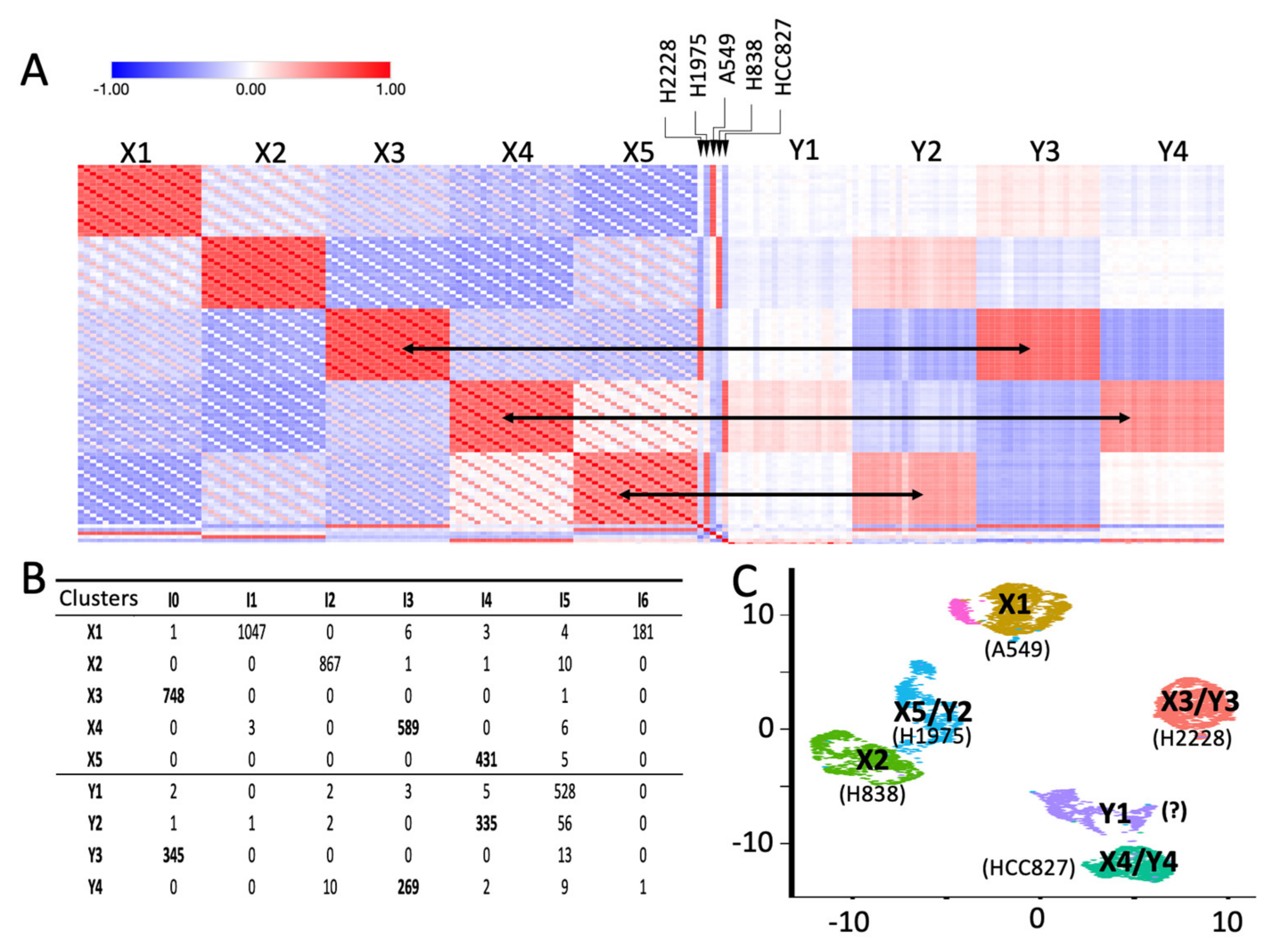
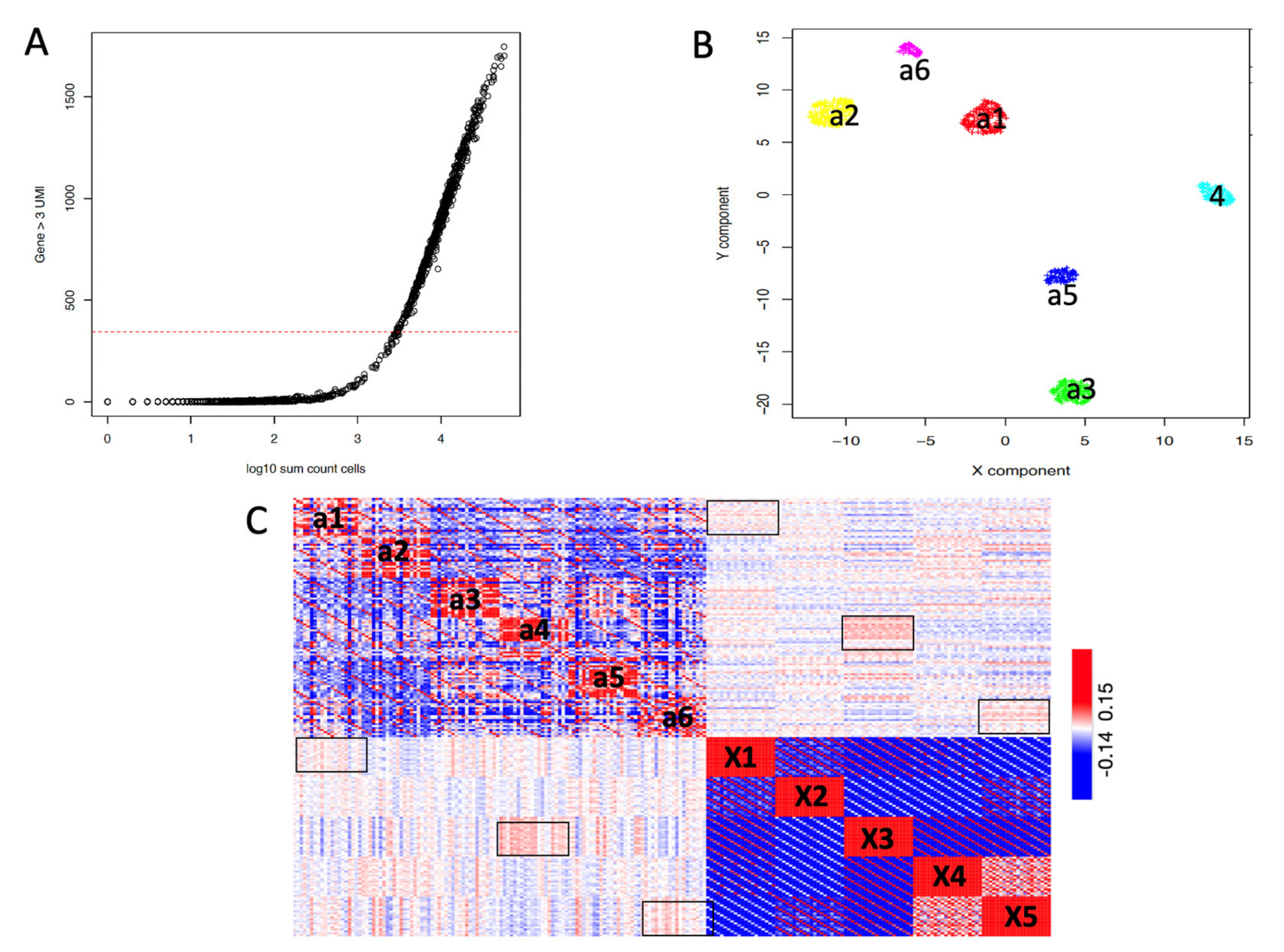
2.1. Building Clusters’ Specific Pseudo-Bulk Using SCA
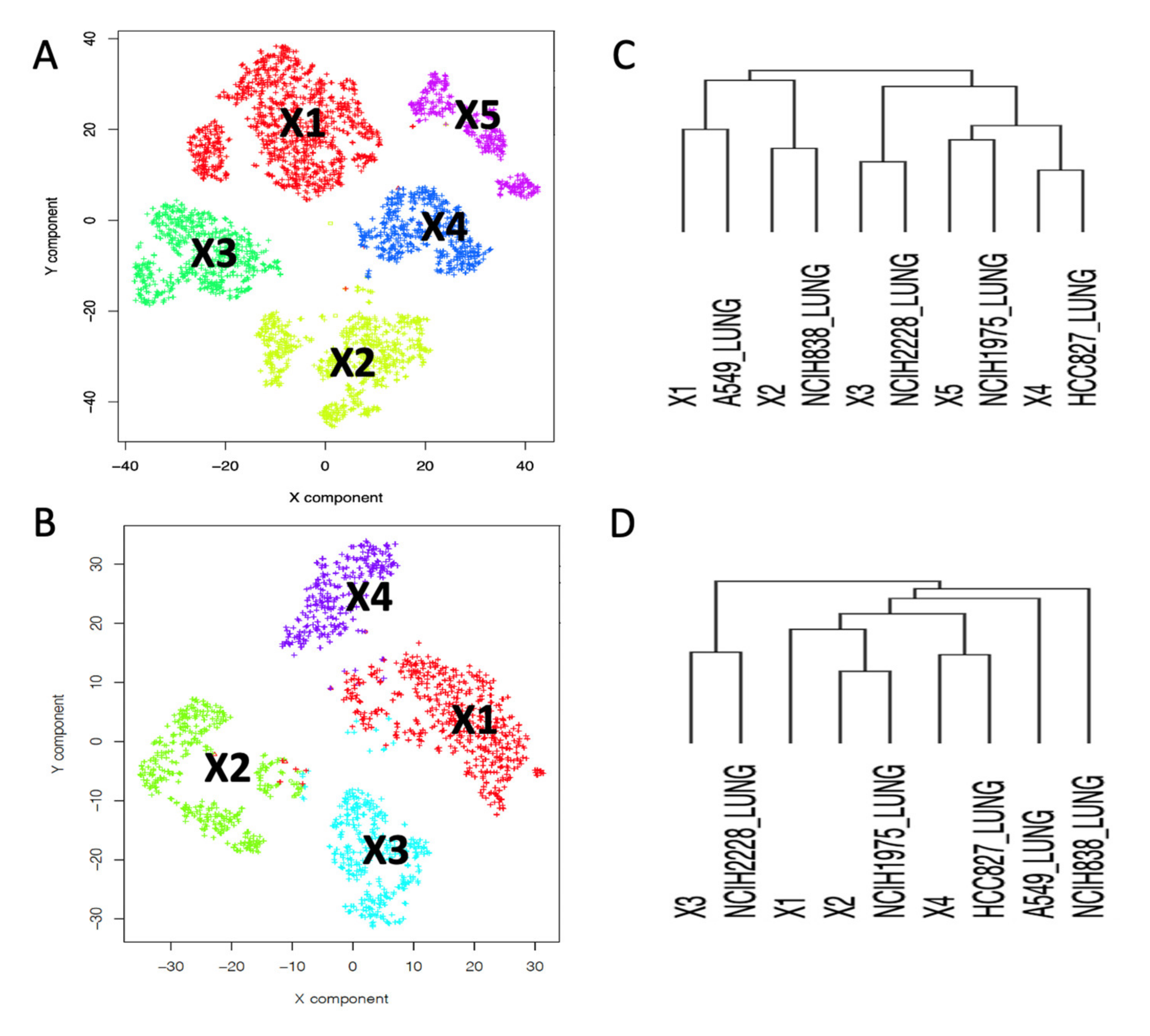
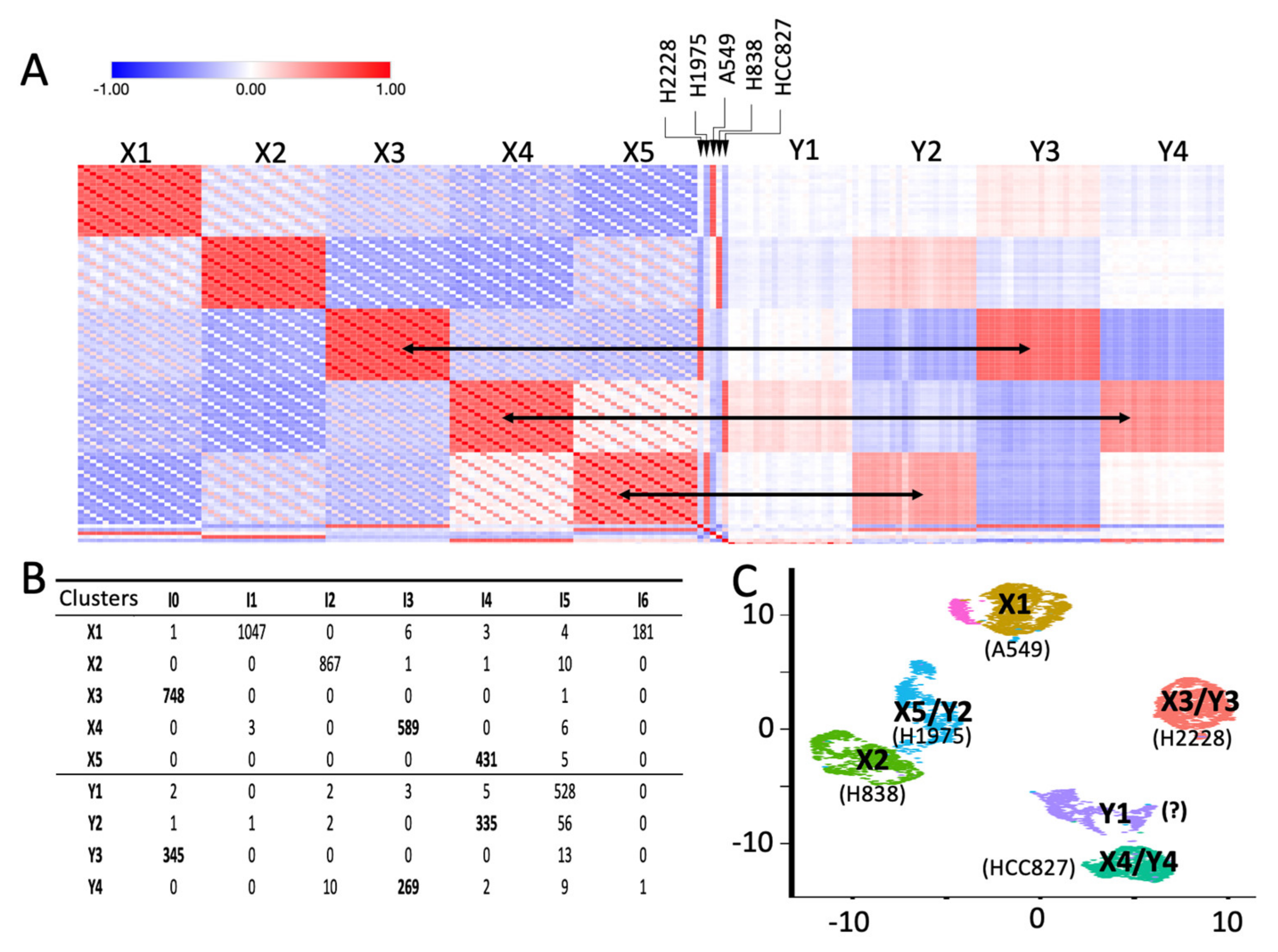
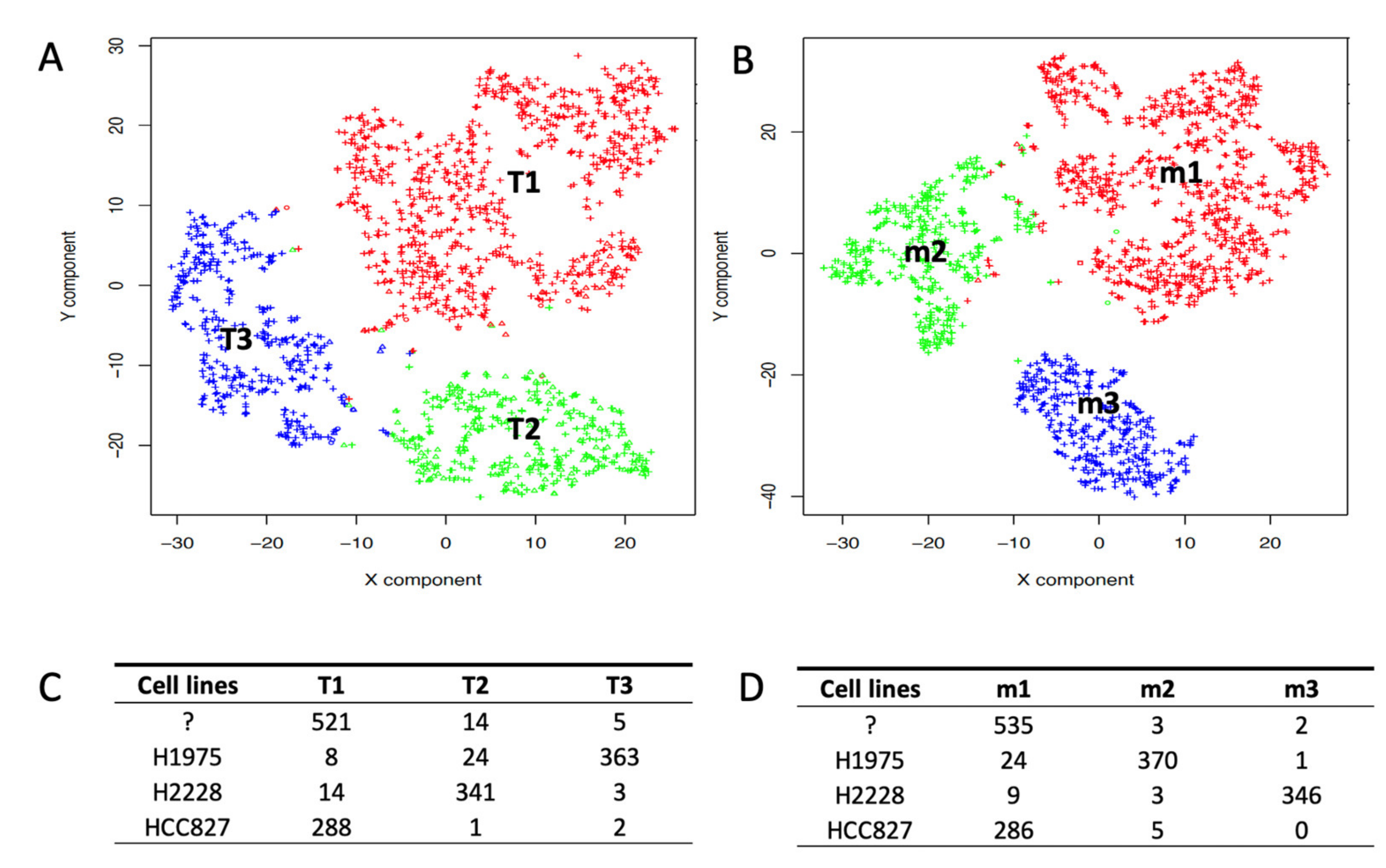
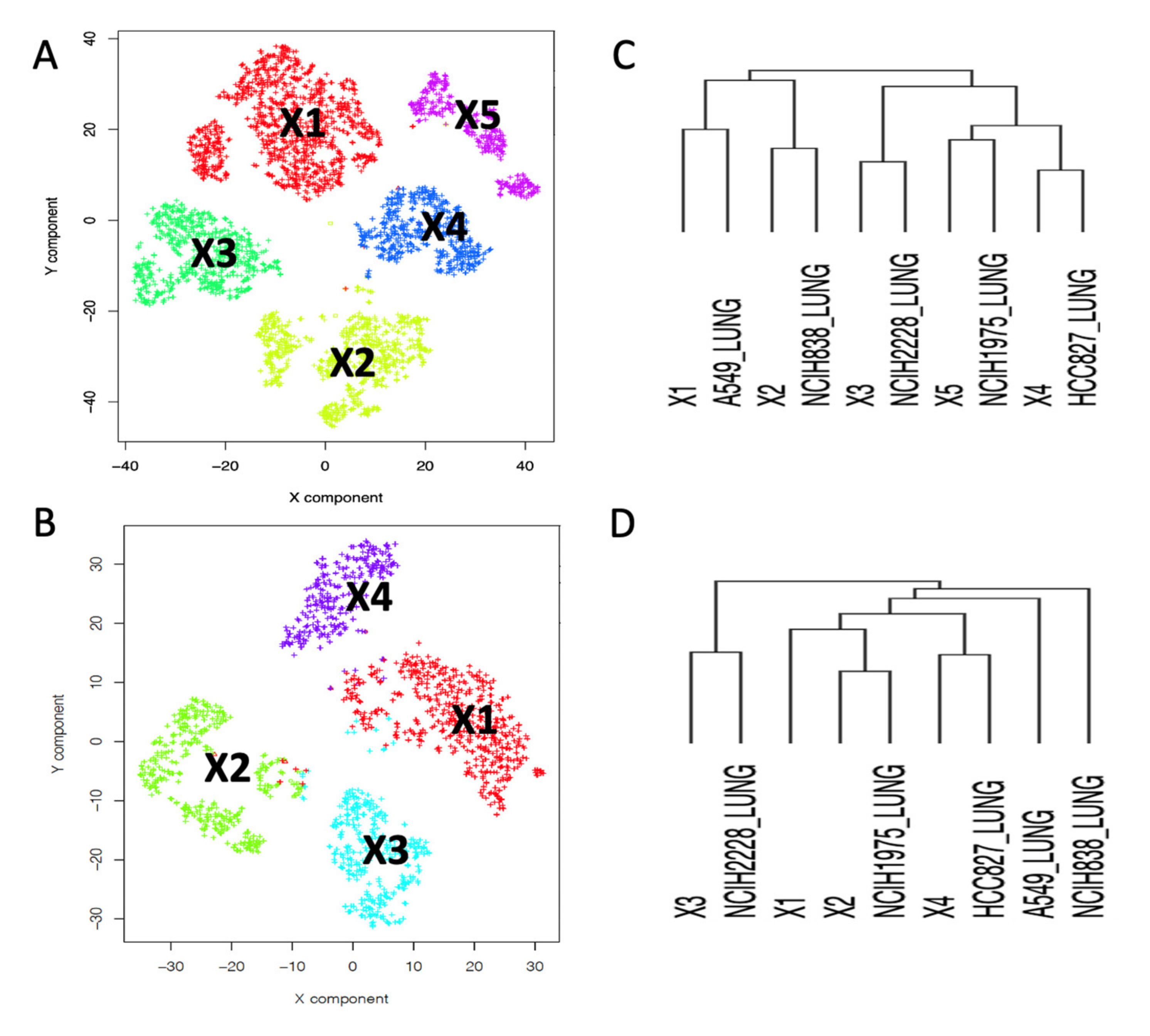
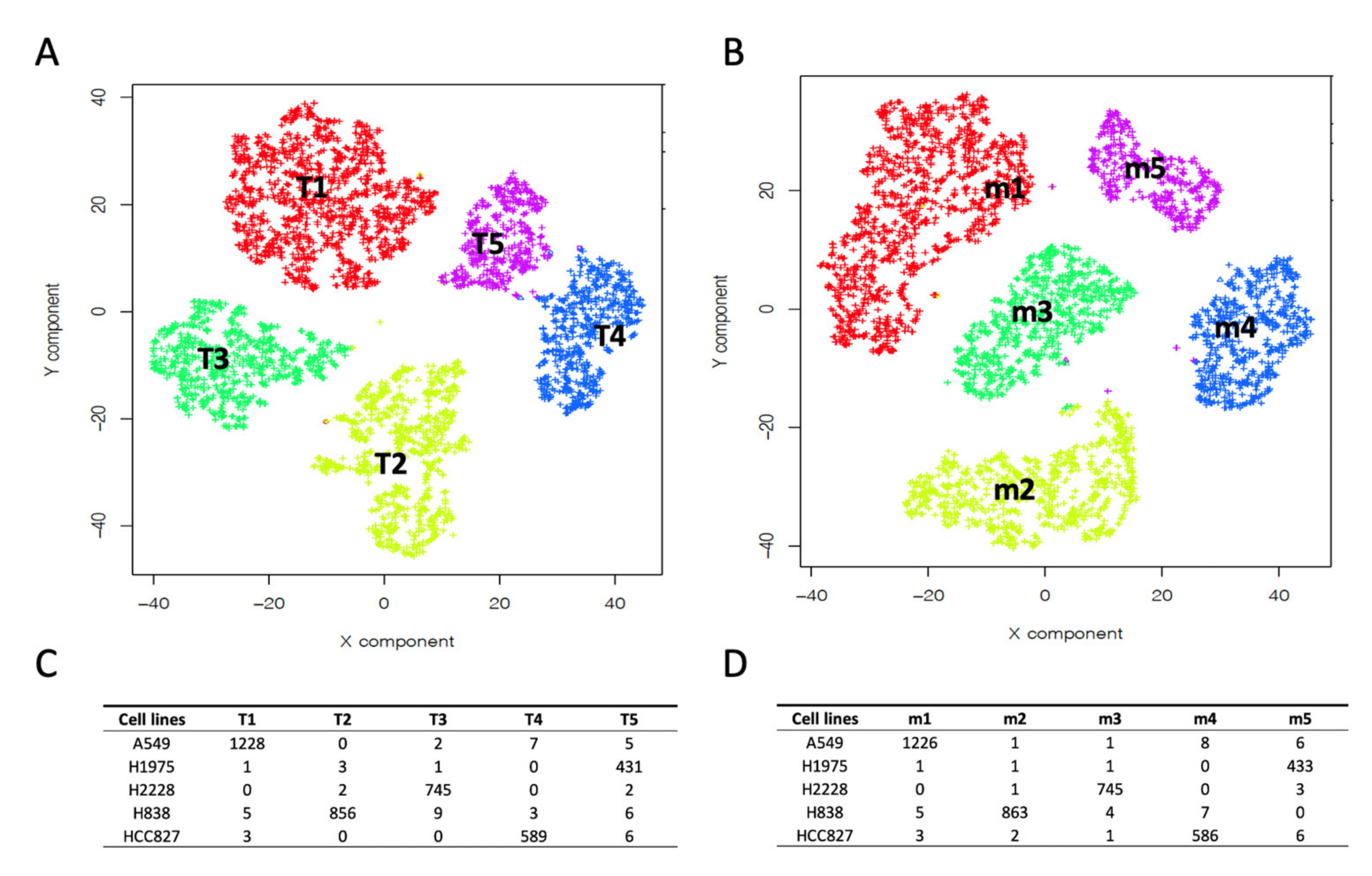
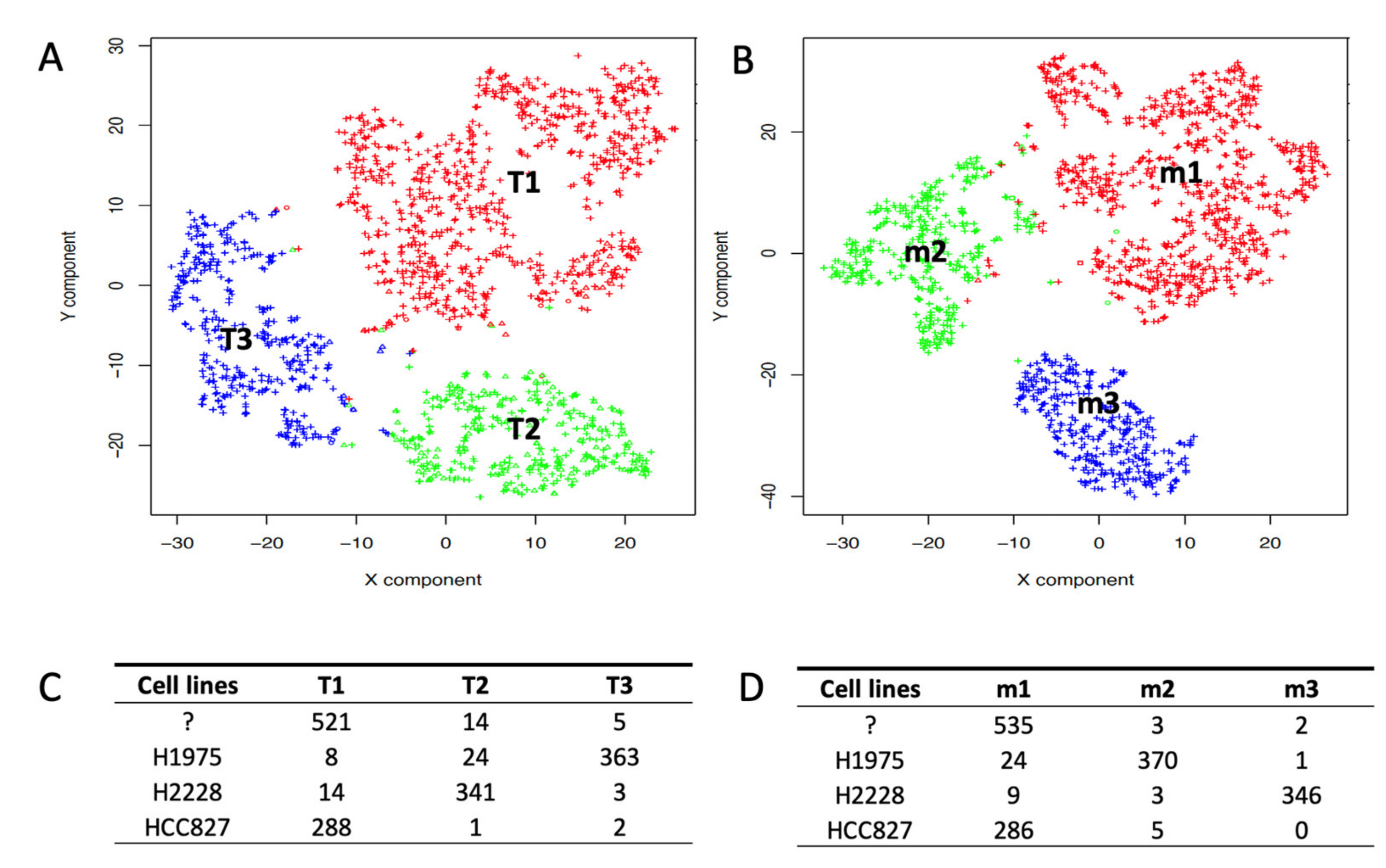
2.2. Depicting Clusters Correspondence among Independent Experiments Using SCA Pseudo-Bulks
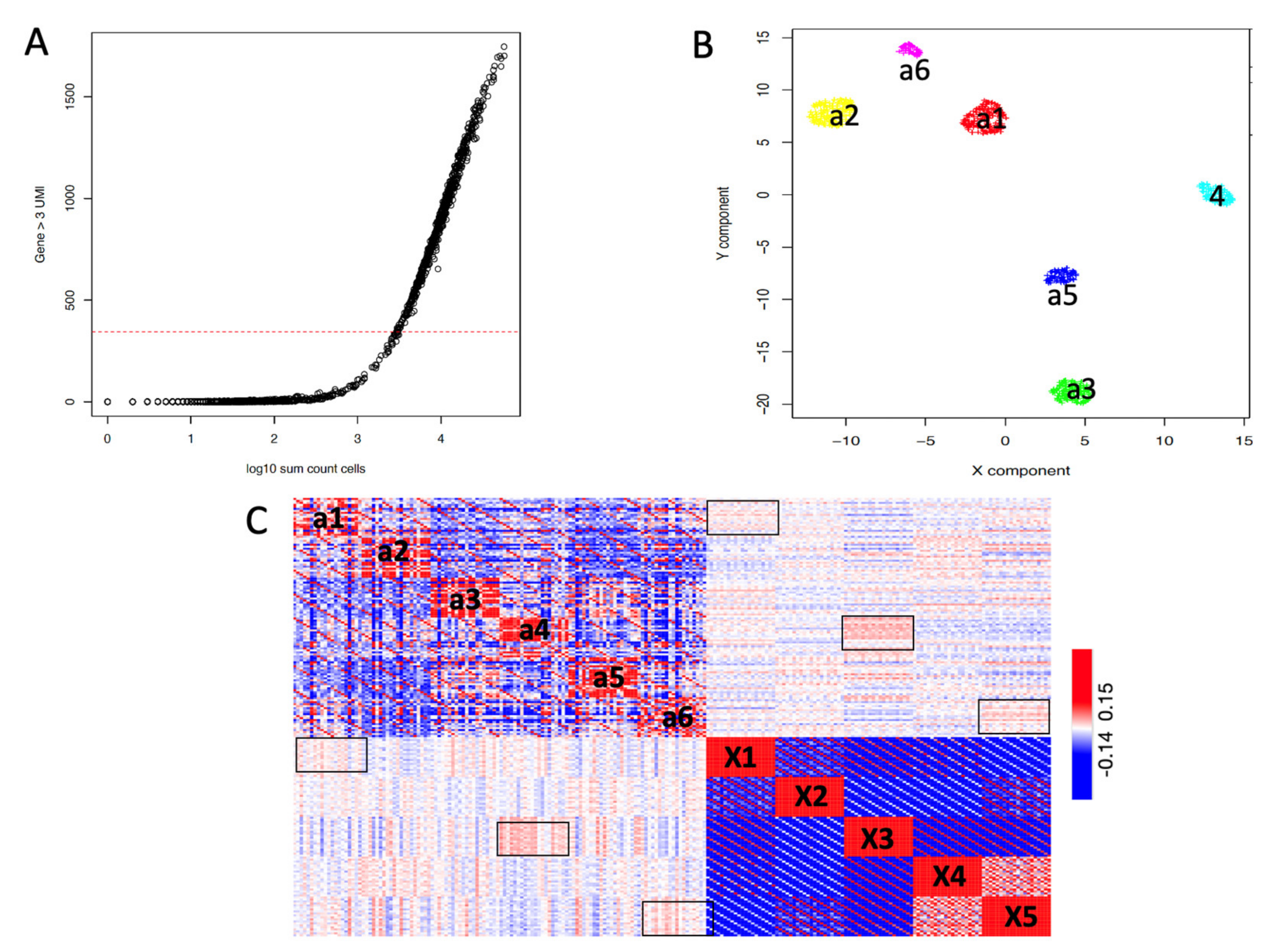
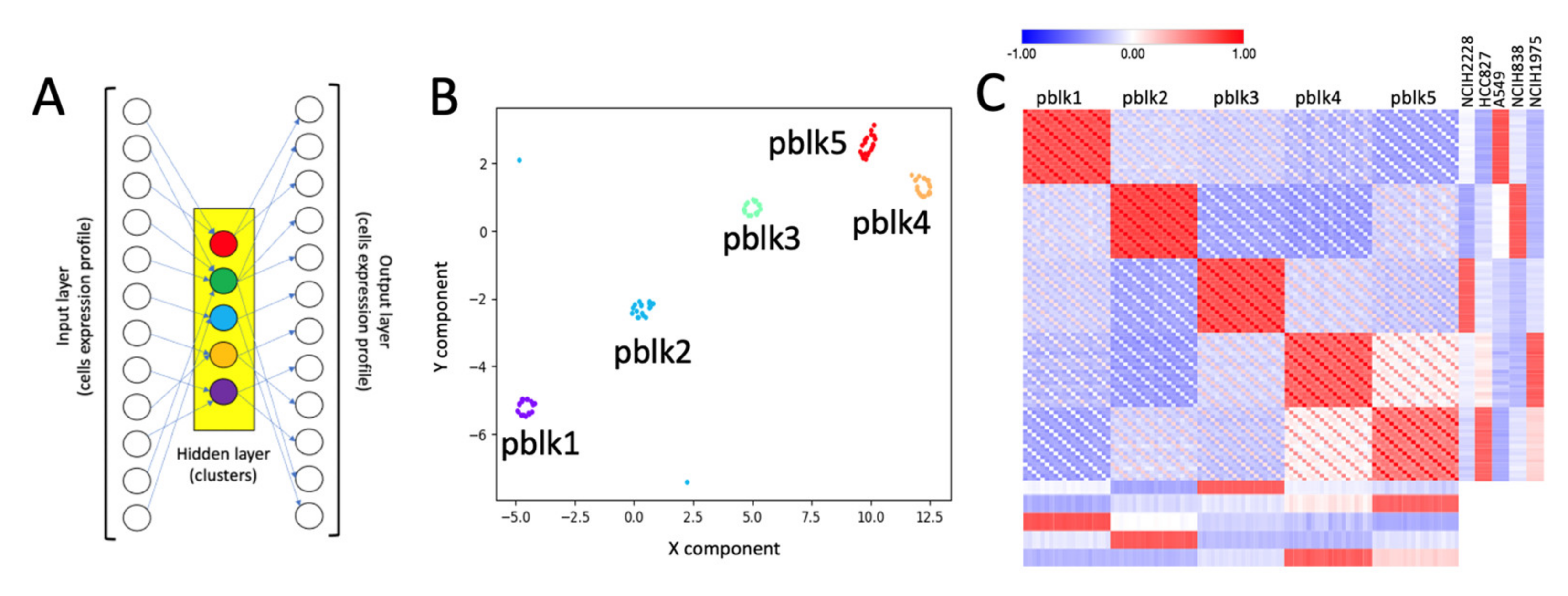
2.3. SCA Pseudo-Clusters as Tool in Multi-Modal Analysis
2.4. SCA as Tool for the Detection of Regulatory Gene Hubs
3. Discussion
4. Materials and Methods
4.1. scRNAseq Benchmark Preprocessing
4.2. scRNAseq Cell Type Association
4.3. scATACseq Benchmark Preprocessing
4.4. Software Implementation
4.5. SCA-Pseudo-Clusters Generation
4.6. Seurat Clustering
4.7. Hierarchical Clustering
4.8. Seurat Integration
4.9. SCA-Metagenes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gao, C.; Zhang, M.; Chen, L. The Comparison of Two Single-cell Sequencing Platforms: BD Rhapsody and 10x Genomics Chromium. Curr. Genomics 2020, 21, 602–609. [Google Scholar] [CrossRef]
- Zappia, L.; Theis, F.J. Over 1000 tools reveal trends in the single-cell RNA-seq analysis landscape. Genome Biol. 2021, 22, 301. [Google Scholar] [CrossRef] [PubMed]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Satpathy, A.T.; Zhou, W.; Ji, H.; Stratton, J.A.; Jaffer, A.; Bahlis, N.; Morrissy, S.; Biernaskie, J.A. Profiling Chromatin Accessibility at Single-cell Resolution. Genom. Proteom. Bioinform. 2021, 19, 172–190. [Google Scholar] [CrossRef]
- Saviano, A.; Henderson, N.C.; Baumert, T.F. Single-cell genomics and spatial transcriptomics: Discovery of novel cell states and cellular interactions in liver physiology and disease biology. J. Hepatol. 2020, 73, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Chappell, L.; Russell, A.J.C.; Voet, T. Single-Cell (Multi)omics Technologies. Annu. Rev. Genom. Hum. Genet. 2018, 19, 15–41. [Google Scholar] [CrossRef]
- Miao, Z.; Humphreys, B.D.; McMahon, A.P.; Kim, J. Multi-omics integration in the age of million single-cell data. Nat. Rev. Nephrol. 2021, 17, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Lotfollahi, M.; Wolf, F.A.; Theis, F.J. Machine Learning for perturbational single-cell omics. Cell Syst. 2021, 12, 522–537. [Google Scholar] [CrossRef]
- Yan, R.; Fan, C.; Yin, Z.; Wang, T.; Chen, X. Potential applications of Deep Learning in single-cell RNA sequencing analysis for cell therapy and regenerative medicine. Stem Cells 2021, 39, 511–521. [Google Scholar] [CrossRef]
- Chougoni, K.K.; Grossman, S.R. Extraction of high-quality RNA from mouse pancreatic tumors. MethodsX 2020, 7, 101163. [Google Scholar] [CrossRef]
- Tran, D.; Nguyen, H.; Tran, B.; La Vecchia, C.; Luu, H.N.; Nguyen, T. Fast and precise single-cell data analysis using a hierarchical autoencoder. Nat. Commun. 2021, 12, 1029. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Greene, C.S. Parameter tuning is a key part of dimensionality reduction via deep variational autoencoders for single cell RNA transcriptomics. Pac. Symp. Biocomput. 2019, 24, 362–373. [Google Scholar]
- Gold, M.P.; LeNail, A.; Fraenkel, E. Shallow Sparsely-Connected Autoencoders for Gene Set Projection. Pac. Symp. Biocomput. 2019, 24, 374–385. [Google Scholar]
- Alessandri, L.; Cordero, F.; Beccuti, M.; Licheri, N.; Arigoni, M.; Olivero, M.; Di Renzo, M.F.; Sapino, A.; Calogero, R. Sparsely-connected autoencoder (SCA) for single cell RNAseq data mining. NPJ Syst. Biol. Appl. 2021, 7, 1. [Google Scholar] [CrossRef]
- Tian, L.; Dong, X.; Freytag, S.; Le Cao, K.A.; Su, S.; JalalAbadi, A.; Amann-Zalcenstein, D.; Weber, T.S.; Seidi, A.; Jabbari, J.S.; et al. Benchmarking single cell RNA-sequencing analysis pipelines using mixture control experiments. Nat. Methods 2019, 16, 479–487. [Google Scholar] [CrossRef]
- Alessandri, L.; Arigoni, M.; Calogero, R. Differential Expression Analysis in Single-Cell Transcriptomics. Methods Mol. Biol. 2019, 1979, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Alessandri, L.; Cordero, F.; Beccuti, M.; Arigoni, M.; Olivero, M.; Romano, G.; Rabellino, S.; Licheri, N.; De Libero, G.; Pace, L.; et al. rCASC: Reproducible classification analysis of single-cell sequencing data. Gigascience 2019, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Das, P.; McCord, R.P. SMILE: Mutual Information Learning for Integration of Single-cell Omics Data. Bioinformatics 2021, btab706. [Google Scholar] [CrossRef]
- Dong, J.; Zhou, P.; Wu, Y.; Chen, Y.; Xie, H.; Gao, Y.; Lu, J.; Yang, J.; Zhang, X.; Wen, L.; et al. Integrating single-cell datasets with ambiguous batch information by incorporating molecular network features. Brief. Bioinform. 2021. [CrossRef]
- Tran, H.T.N.; Ang, K.S.; Chevrier, M.; Zhang, X.; Lee, N.Y.S.; Goh, M.; Chen, J. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 2020, 21, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaney, C.; Schnell, A.; Cammarata, L.V.; Yao-Smith, A.; Regev, A.; Kuchroo, V.K.; Singer, M. Combinatorial prediction of marker panels from single-cell transcriptomic data. Mol. Syst. Biol. 2019, 15, e9005. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Xia, J. OmicsNet: A web-based tool for creation and visual analysis of biological networks in 3D space. Nucleic Acids Res. 2018, 46, W514–W522. [Google Scholar] [CrossRef] [Green Version]
- Foulkes, W.D.; Flanders, T.Y.; Pollock, P.M.; Hayward, N.K. The CDKN2A (p16) gene and human cancer. Mol. Med. 1997, 3, 5–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierson, E.; Yau, C. ZIFA: Dimensionality reduction for zero-inflated single-cell gene expression analysis. Genome Biol. 2015, 16, 241. [Google Scholar] [CrossRef] [Green Version]
- Nikolayeva, O.; Robinson, M.D. edgeR for differential RNA-seq and ChIP-seq analysis: An application to stem cell biology. Methods Mol. Biol. 2014, 1150, 45–79. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Muto, Y.; Wilson, P.C.; Ledru, N.; Wu, H.; Dimke, H.; Waikar, S.S.; Humphreys, B.D. Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. Nat. Commun. 2021, 12, 2190. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wang, J.; Torre, E.; Dueck, H.; Shaffer, S.; Bonasio, R.; Murray, J.I.; Raj, A.; Li, M.; Zhang, N.R. SAVER: Gene expression recovery for single-cell RNA sequencing. Nat. Methods 2018, 15, 539–542. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alessandri, L.; Ratto, M.L.; Contaldo, S.G.; Beccuti, M.; Cordero, F.; Arigoni, M.; Calogero, R.A. Sparsely Connected Autoencoders: A Multi-Purpose Tool for Single Cell omics Analysis. Int. J. Mol. Sci. 2021, 22, 12755. https://doi.org/10.3390/ijms222312755
Alessandri L, Ratto ML, Contaldo SG, Beccuti M, Cordero F, Arigoni M, Calogero RA. Sparsely Connected Autoencoders: A Multi-Purpose Tool for Single Cell omics Analysis. International Journal of Molecular Sciences. 2021; 22(23):12755. https://doi.org/10.3390/ijms222312755
Chicago/Turabian StyleAlessandri, Luca, Maria Luisa Ratto, Sandro Gepiro Contaldo, Marco Beccuti, Francesca Cordero, Maddalena Arigoni, and Raffaele A. Calogero. 2021. "Sparsely Connected Autoencoders: A Multi-Purpose Tool for Single Cell omics Analysis" International Journal of Molecular Sciences 22, no. 23: 12755. https://doi.org/10.3390/ijms222312755
APA StyleAlessandri, L., Ratto, M. L., Contaldo, S. G., Beccuti, M., Cordero, F., Arigoni, M., & Calogero, R. A. (2021). Sparsely Connected Autoencoders: A Multi-Purpose Tool for Single Cell omics Analysis. International Journal of Molecular Sciences, 22(23), 12755. https://doi.org/10.3390/ijms222312755