
Vascular Calcification: Key Roles of Phosphate and Pyrophosphate


Abstract
1. Introduction
2. Role of Phosphate
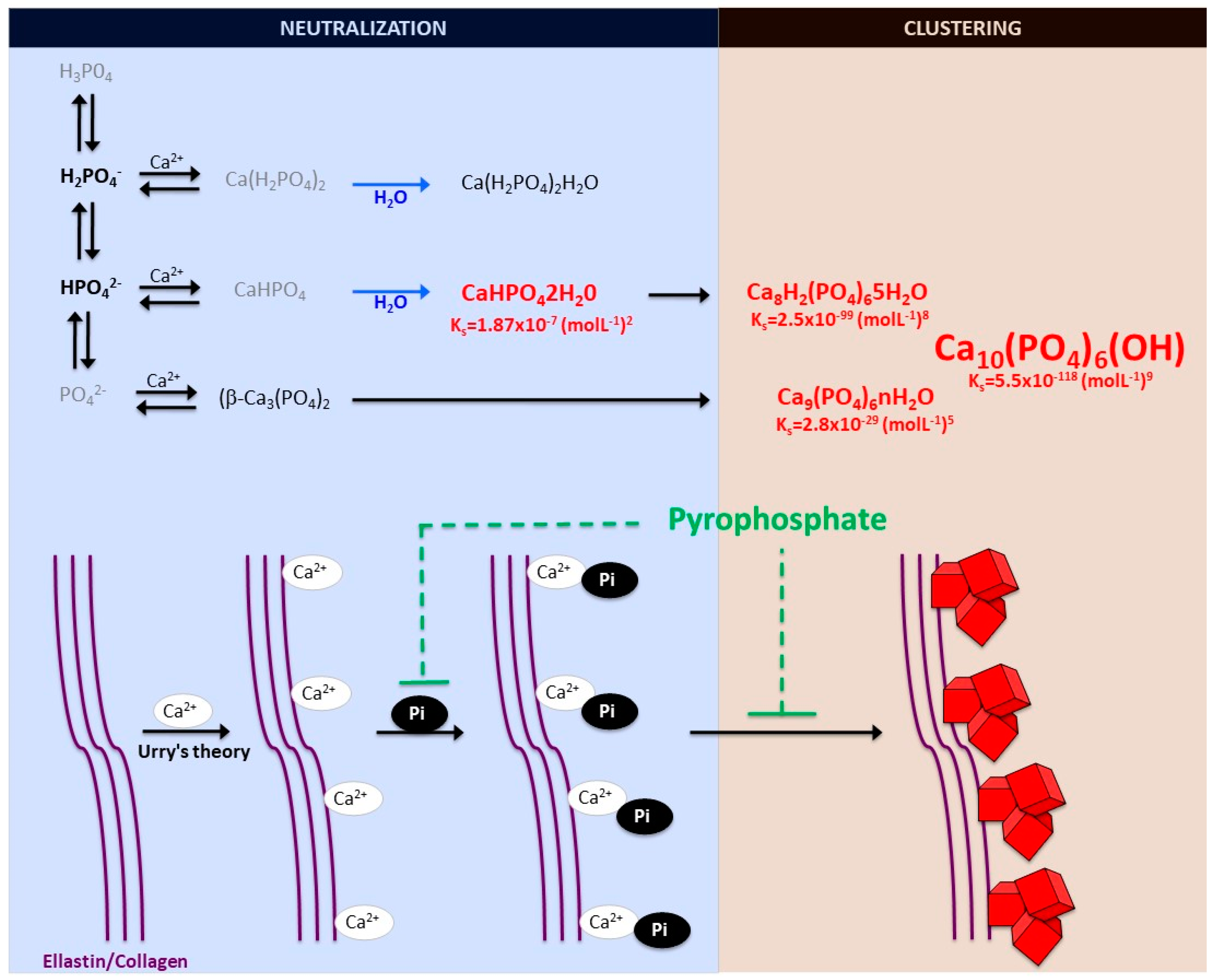
2.1. Biomineralization Process
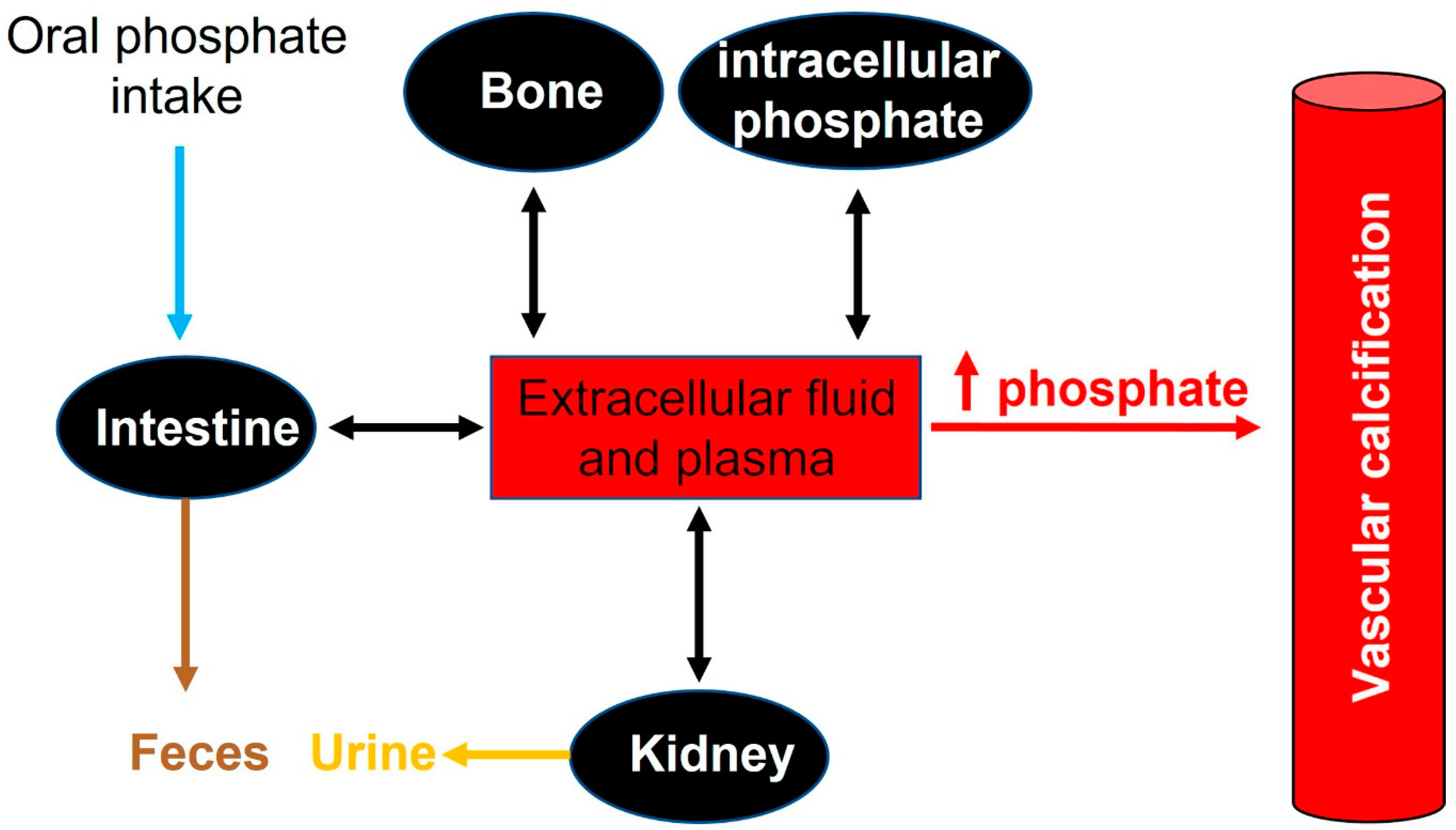
2.2. Phosphate Homeostasis
2.3. Phosphate Transporters
3. Role of Pyrophosphate
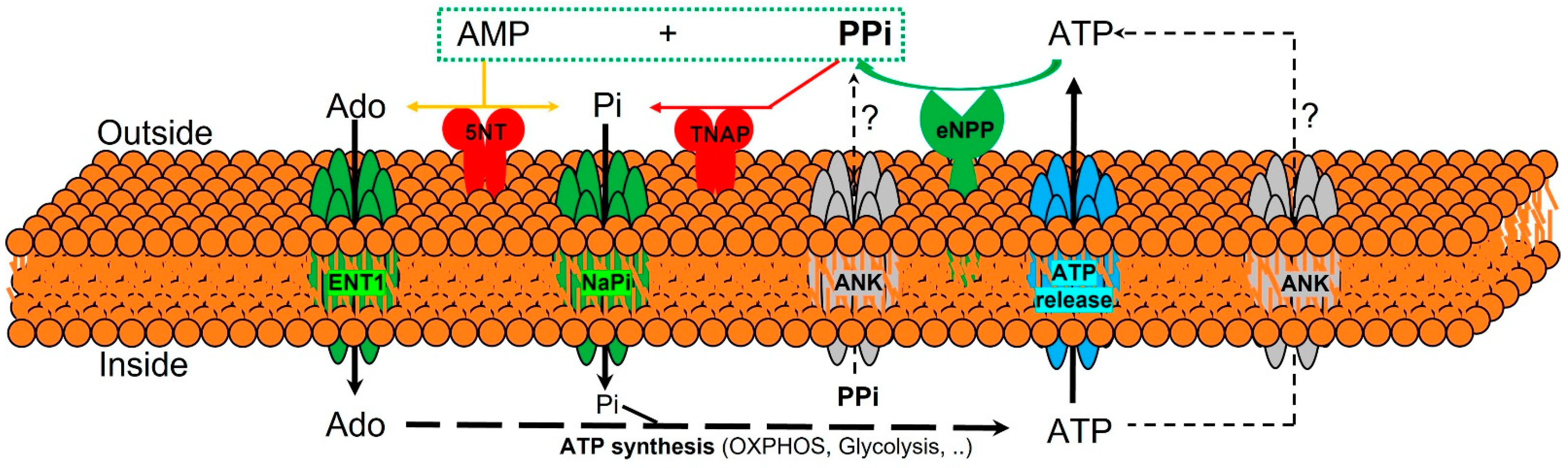
3.1. Extracelular Pyrophosphate Metabolism
3.2. Extracelular Pyrophosphate Metabolism in the Aortic Wall
4. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Rutsch, F.; Nitschke, Y.; Terkeltaub, R. Genetics in arterial calcification: Pieces of a puzzle and cogs in a wheel. Circ. Res. 2011, 109, 578–592. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Singh, K.J.; Zeller, T.; Jaff, M.R. Peripheral arterial calcification: Prevalence, mechanism, detection, and clinical implications. Catheter. Cardiovasc. Interv. 2014, 83, E212–E220. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R. New insights into endogenous mechanisms of protection against arterial calcification. Atherosclerosis 2020, 306, 68–74. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Millan, A.; Sorribas, V. Role of calcium-phosphate deposition in vascular smooth muscle cell calcification. Am. J. Physiol. Cell Physiol. 2011, 300, C210–C220. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R. Synthesis of Extracellular Pyrophosphate Increases in Vascular Smooth Muscle Cells During Phosphate-Induced Calcification. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2137–2147. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M.; Speer, M.Y.; Li, X.; Rajachar, R.M.; Yang, H. Regulation of vascular calcification: Roles of phosphate and osteopontin. Circ. Res. 2005, 96, 717–722. [Google Scholar] [CrossRef]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, E10–E17. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.-Y.; Giachelli, C.M. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ. Res. 2006, 98, 905–912. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Bogaert, Y.E.; Levi, M.; Sorribas, V. Characterization of phosphate transport in rat vascular smooth muscle cells: Implications for vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1030–1036. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Sorribas, V. Calcium phosphate deposition with normal phosphate concentration. -Role of pyrophosphate-. Circ. J. 2011, 75, 2705–2710. [Google Scholar] [CrossRef] [PubMed]
- Schinke, T.; Karsenty, G. Vascular calcification—A passive process in need of inhibitors. Nephrol. Dial. Transplant. 2000, 15, 1272–1274. [Google Scholar] [CrossRef][Green Version]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.C.; McNair, R.; Figg, N.; Skepper, J.N.; Schurgers, L.; Gupta, A.; Hiorns, M.; Donald, A.E.; Deanfield, J.; Rees, L.; et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation 2008, 118, 1748–1757. [Google Scholar] [CrossRef]
- Mansfield, K.; Teixeira, C.C.; Adams, C.S.; Shapiro, I.M. Phosphate ions mediate chondrocyte apoptosis through a plasma membrane transporter mechanism. Bone 2001, 28, 1–8. [Google Scholar] [CrossRef]
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.Y.; Haynes, P.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth muscle cell phenotypic transition associated with calcification: Upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ. Res. 2001, 89, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, H.-Y.; Giachelli, C.M. BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis 2008, 199, 271–277. [Google Scholar] [CrossRef]
- Speer, M.Y.; Li, X.; Hiremath, P.G.; Giachelli, C.M. Runx2/Cbfa1, but not loss of myocardin, is required for smooth muscle cell lineage reprogramming toward osteochondrogenesis. J. Cell. Biochem. 2010, 110, 935–947. [Google Scholar] [CrossRef]
- Lei, Y.; Sinha, A.; Nosoudi, N.; Grover, A.; Vyavahare, N. Hydroxyapatite and calcified elastin induce osteoblast-like differentiation in rat aortic smooth muscle cells. Exp. Cell Res. 2014, 323, 198–208. [Google Scholar] [CrossRef]
- Sage, A.P.; Lu, J.; Tintut, Y.; Demer, L.L. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 2011, 79, 414–422. [Google Scholar] [CrossRef]
- Villa-Bellosta, R. Vascular Calcification Revisited: A New Perspective for Phosphate Transport. Curr. Cardiol. Rev. 2015, 11, 341–351. [Google Scholar] [CrossRef]
- Kanzaki, N.; Treboux, G.; Onuma, K.; Tsutsumi, S.; Ito, A. Calcium phosphate clusters. Biomaterials 2001, 22, 2921–2929. [Google Scholar] [CrossRef]
- Johnsson, M.S.; Nancollas, G.H. The role of brushite and octacalcium phosphate in apatite formation. Crit. Rev. Oral Biol. Med. 1992, 3, 61–82. [Google Scholar] [CrossRef]
- P’ng, C.H.; Boadle, R.; Horton, M.; Bilous, M.; Bonar, F. Magnesium whitlockite of the aorta. Pathology 2008, 40, 539–540. [Google Scholar] [CrossRef]
- Reid, J.D.; Andersen, M.E. Medial calcification (whitlockite) in the aorta. Atherosclerosis 1993, 101, 213–224. [Google Scholar] [CrossRef]
- Kay, M.I.; Young, R.A.; Posner, A.S. Crystal structure of hydroxyapatite. Nature 1964, 204, 1050–1052. [Google Scholar] [CrossRef]
- Posner, A.S. The structure of bone apatite surfaces. J. Biomed. Mater. Res. 1985, 19, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Posner, A.S.; Beebe, R.A. The surface chemistry of bone mineral and related calcium phosphates. Semin. Arthritis Rheum. 1975, 4, 267–291. [Google Scholar] [CrossRef]
- Posner, A.S.; Betts, F.; Blumenthal, N.C. Role of ATP and Mg in the stabilization of biological and synthetic amorphous calcium phosphates. Calcif. Tissue Res. 1977, 22, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, N.C.; Betts, F.; Posner, A.S. Stabilization of amorphous calcium phosphate by Mg and ATP. Calcif. Tissue Res. 1977, 23, 245–250. [Google Scholar] [CrossRef]
- Urry, D.W. Neutral sites for calcium ion binding to elastin and collagen: A charge neutralization theory for calcification and its relationship to atherosclerosis. Proc. Natl. Acad. Sci. USA 1971, 68, 810–814. [Google Scholar] [CrossRef]
- Khavandgar, Z.; Roman, H.; Li, J.; Lee, S.; Vali, H.; Brinckmann, J.; Davis, E.C.; Murshed, M. Elastin haploinsufficiency impedes the progression of arterial calcification in MGP-deficient mice. J. Bone Miner. Res. 2014, 29, 327–337. [Google Scholar] [CrossRef]
- Hosaka, N.; Mizobuchi, M.; Ogata, H.; Kumata, C.; Kondo, F.; Koiwa, F.; Kinugasa, E.; Akizawa, T. Elastin degradation accelerates phosphate-induced mineralization of vascular smooth muscle cells. Calcif. Tissue Int. 2009, 85, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Azpiazu, D.; González-Parra, E.; Egido, J.; Villa-Bellosta, R. Hydrolysis of Extracellular Pyrophosphate increases in post-hemodialysis plasma. Sci. Rep. 2018, 8, 11089. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Egido, J. Phosphate, pyrophosphate, and vascular calcification: A question of balance. Eur. Heart J. 2017, 38, 1801–1804. [Google Scholar] [CrossRef]
- Manghat, P.; Sodi, R.; Swaminathan, R. Phosphate homeostasis and disorders. Ann. Clin. Biochem. 2014, 51 Pt 6, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Christov, M.; Jüppner, H. Phosphate homeostasis disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 685–706. [Google Scholar] [CrossRef]
- Bergwitz, C.; Jüppner, H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu. Rev. Med. 2010, 61, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, S.; Villa-Bellosta, R. The role of sodium phosphate cotransporters in ectopic calcification. Endokrynol. Pol. 2019, 70, 496–503. [Google Scholar] [CrossRef]
- Forster, I.C.; Hernando, N.; Biber, J.; Murer, H. Phosphate transporters of the SLC20 and SLC34 families. Mol. Asp. Med. 2013, 34, 386–395. [Google Scholar] [CrossRef]
- Forster, I.C.; Hernando, N.; Biber, J.; Murer, H. Phosphate transport kinetics and structure-function relationships of SLC34 and SLC20 proteins. Curr. Top. Membr. 2012, 70, 313–356. [Google Scholar]
- Reimer, R.J. SLC17: A functionally diverse family of organic anion transporters. Mol. Asp. Med. 2013, 34, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.A.; Hernando, N.; Forster, I.C.; Biber, J. The SLC34 family of sodium-dependent phosphate transporters. Pflugers Arch. 2014, 466, 139–153. [Google Scholar] [CrossRef]
- Silverstein, D.M.; Barac-Nieto, M.; Murer, H.; Spitzer, A. A putative growth-related renal Na(+)-Pi cotransporter. Am. J. Physiol. 1997, 273 Pt 2, R928–R933. [Google Scholar] [CrossRef] [PubMed]
- Segawa, H.; Kaneko, I.; Takahashi, A.; Kuwahata, M.; Ito, M.; Ohkido, I.; Tatsumi, S.; Miyamoto, K.-I. Growth-related renal type II Na/Pi cotransporter. J. Biol. Chem. 2002, 277, 19665–19672. [Google Scholar] [CrossRef]
- Collins, J.F.; Bai, L.; Ghishan, F.K. The SLC20 family of proteins: Dual functions as sodium-phosphate cotransporters and viral receptors. Pflugers Arch. 2004, 447, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, G.; Svanberg, E.; Dadar, M.; Card, D.J.; Chirumbolo, S.; Harrington, D.J.; Aaseth, J. The Role of Matrix Gla Protein (MGP) in Vascular Calcification. Curr. Med. Chem. 2020, 27, 1647–1660. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Emoto, M.; Inaba, M. Fetuin-A: A multifunctional protein. Recent Pat. Endocr. Metab. Immune Drug Discov. 2011, 5, 124–146. [Google Scholar] [CrossRef]
- Jahnen-Dechent, W.; Schinke, T.; Trindl, A.; Müller-Esterl, W.; Sablitzky, F.; Kaiser, S.; Blessing, M. Cloning and targeted deletion of the mouse fetuin gene. J. Biol. Chem. 1997, 272, 31496–31503. [Google Scholar] [CrossRef]
- Schäfer, C.; Heiss, A.; Schwarz, A.; Westenfeld, R.; Ketteler, M.; Floege, J.; Müller-Esterl, W.; Schinke, T.; Jahnen-Dechent, W. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J. Clin. Investig. 2003, 112, 357–366. [Google Scholar] [CrossRef]
- Ulutas, O.; Taskapan, M.C.; Dogan, A.; Baysal, T.; Taskapan, H. Vascular calcification is not related to serum fetuin-A and osteopontin levels in hemodialysis patients. Int. Urol. Nephrol. 2018, 50, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Franzén, A.; Heinegård, D. Isolation and characterization of two sialoproteins present only in bone calcified matrix. Biochem. J. 1985, 232, 715–724. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.R.; Garvin, M.R.; Stewart, D.K.; Hinohara, T.; Simpson, J.B.; Schwartz, S.M.; Giachelli, C.M. Osteopontin is synthesized by macrophage, smooth muscle, and endothelial cells in primary and restenotic human coronary atherosclerotic plaques. Arterioscler. Thromb. J. Vasc. Biol. 1994, 14, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Shirasawa, T.; Esaki, Y.; Yoshiki, S.; Hirokawa, K. Osteopontin mRNA is expressed by smooth muscle-derived foam cells in human atherosclerotic lesions of the aorta. J. Clin. Investig. 1993, 92, 2814–2820. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D.; Skepper, J.N.; Shanahan, C.M.; Weissberg, P.L. Calcification of human vascular cells in vitro is correlated with high levels of matrix Gla protein and low levels of osteopontin expression. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 379–388. [Google Scholar] [CrossRef]
- Jono, S.; Peinado, C.; Giachelli, C.M. Phosphorylation of osteopontin is required for inhibition of vascular smooth muscle cell calcification. J. Biol. Chem. 2000, 275, 20197–20203. [Google Scholar] [CrossRef] [PubMed]
- Schibler, D.; Russell, R.G.; Fleisch, H. Inhibition by pyrophosphate and polyphosphate of aortic calcification induced by vitamin D3 in rats. Clin. Sci. 1968, 35, 363–372. [Google Scholar]
- Lomashvili, K.A.; Khawandi, W.; O’Neill, W.C. Reduced plasma pyrophosphate levels in hemodialysis patients. J. Am. Soc. Nephrol. 2005, 16, 2495–2500. [Google Scholar] [CrossRef]
- O’Neill, W.C.; Lomashvili, K.A.; Malluche, H.H.; Faugere, M.-C.; Riser, B.L. Treatment with pyrophosphate inhibits uremic vascular calcification. Kidney Int. 2011, 79, 512–517. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Rivera-Torres, J.; Osorio, F.G.; Acín-Pérez, R.; Enriquez, J.A.; López-Otín, C.; Andrés, V. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation 2013, 127, 2442–2451. [Google Scholar] [CrossRef] [PubMed]
- Riser, B.L.; Barreto, F.C.; Rezg, R.; Valaitis, P.W.; Cook, C.S.; White, J.A.; Gass, J.H.; Maizel, J.; Louvet, L.; Drueke, T.B.; et al. Daily peritoneal administration of sodium pyrophosphate in a dialysis solution prevents the development of vascular calcification in a mouse model of uraemia. Nephrol. Dial. Transplant. 2011, 26, 3349–3357. [Google Scholar] [CrossRef]
- Villa-Bellosta, R. ATP-based therapy prevents vascular calcification and extends longevity in a mouse model of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 23698–23704. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R. Dietary magnesium supplementation improves lifespan in a mouse model of progeria. EMBO Mol. Med. 2020, 12, e12423. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; O’Neill, W.C. Pyrophosphate deficiency in vascular calcification. Kidney Int. 2018, 93, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Zebisch, M.; Sträter, N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef]
- Zimmermann, H. Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch. Pharmacol. 2000, 362, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Rutsch, F.; Ruf, N.; Vaingankar, S.; Toliat, M.R.; Suk, A.; Höhne, W.; Schauer, G.; Lehmann, M.; Roscioli, T.; Schnabel, D.; et al. Mutations in ENPP1 are associated with «idiopathic» infantile arterial calcification. Nat. Genet. 2003, 34, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Wang, X.; Millán, J.L.; Dubyak, G.R.; O’Neill, W.C. Extracellular pyrophosphate metabolism and calcification in vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H61–H68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dyment, N.A.; Rowe, D.W.; Siu, S.Y.; Sundberg, J.P.; Uitto, J.; Li, Q. Ectopic mineralization of cartilage and collagen-rich tendons and ligaments in Enpp1asj-2J mice. Oncotarget 2016, 7, 12000–12009. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Sorribas, V. Prevention of vascular calcification by polyphosphates and nucleotides-role of ATP. Circ. J. 2013, 77, 2145–2151. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Sorribas, V. Phosphonoformic acid prevents vascular smooth muscle cell calcification by inhibiting calcium-phosphate deposition. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Lomashvili, K.A.; Cobbs, S.; Hennigar, R.A.; Hardcastle, K.I.; O’Neill, W.C. Phosphate-induced vascular calcification: Role of pyrophosphate and osteopontin. J. Am. Soc. Nephrol. 2004, 15, 1392–1401. [Google Scholar] [CrossRef]
- Lomashvili, K.A.; Garg, P.; Narisawa, S.; Millan, J.L.; O’Neill, W.C. Upregulation of alkaline phosphatase and pyrophosphate hydrolysis: Potential mechanism for uremic vascular calcification. Kidney Int. 2008, 73, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Narisawa, S.; Harmey, D.; Yadav, M.C.; O’Neill, W.C.; Hoylaerts, M.F.; Millán, J.L. Novel inhibitors of alkaline phosphatase suppress vascular smooth muscle cell calcification. J. Bone Miner. Res. 2007, 22, 1700–1710. [Google Scholar] [CrossRef]
- Azpiazu, D.; Gonzalo, S.; Villa-Bellosta, R. Tissue Non-Specific Alkaline Phosphatase and Vascular Calcification: A Potential Therapeutic Target. Curr. Cardiol. Rev. 2019, 15, 91–95. [Google Scholar] [CrossRef] [PubMed]
- St Hilaire, C.; Ziegler, S.G.; Markello, T.C.; Brusco, A.; Groden, C.; Gill, F.; Carlson-Donohoe, H.; Lederman, R.J.; Chen, M.Y.; Yang, D.; et al. NT5E mutations and arterial calcifications. N. Engl. J. Med. 2011, 364, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Warraich, S.; Bone, D.; Quinonez, D.; Ii, H.; Choi, D.-S.; Holdsworth, D.; Drangova, M.; Dixon, S.J.; Séguin, C.A.; Hammond, J. Loss of equilibrative nucleoside transporter 1 in mice leads to progressive ectopic mineralization of spinal tissues resembling diffuse idiopathic skeletal hyperostosis in humans. J. Bone Miner. Res. 2013, 28, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.M.; Johnson, M.D.; Kingsley, D.M. Role of the mouse ank gene in control of tissue calcification and arthritis. Science 2000, 289, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Nürnberg, P.; Thiele, H.; Chandler, D.; Höhne, W.; Cunningham, M.L.; Ritter, H.; Leschik, G.; Uhlmann, K.; Mischung, C.; Harrop, K.; et al. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat. Genet. 2001, 28, 37–41. [Google Scholar] [CrossRef]
- Reichenberger, E.; Tiziani, V.; Watanabe, S.; Park, L.; Ueki, Y.; Santanna, C.; Baur, S.T.; Shiang, R.; Grange, D.K.; Beighton, P.; et al. Autosomal dominant craniometaphyseal dysplasia is caused by mutations in the transmembrane protein ANK. Am. J. Hum. Genet. 2001, 68, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, A.; Johnson, M.D.; Hughes, A.; Gurley, K.A.; Ho, A.M.; Doherty, M.; Dixey, J.; Gillet, P.; Loeuille, D.; McGrath, R.; et al. Mutations in ANKH cause chondrocalcinosis. Am. J. Hum. Genet. 2002, 71, 933–940. [Google Scholar] [CrossRef]
- Williams, C.J.; Zhang, Y.; Timms, A.; Bonavita, G.; Caeiro, F.; Broxholme, J.; Cuthbertson, J.; Jones, Y.; Marchegiani, R.; Reginato, A.; et al. Autosomal dominant familial calcium pyrophosphate dihydrate deposition disease is caused by mutation in the transmembrane protein ANKH. Am. J. Hum. Genet. 2002, 71, 985–991. [Google Scholar] [CrossRef]
- Williams, C.J.; Pendleton, A.; Bonavita, G.; Reginato, A.J.; Hughes, A.E.; Peariso, S.; Doherty, M.; Mccarty, D.J.; Ryan, L.M. Mutations in the amino terminus of ANKH in two US families with calcium pyrophosphate dihydrate crystal deposition disease. Arthritis Rheum. 2003, 48, 2627–2631. [Google Scholar] [CrossRef]
- Le Saux, O.; Urban, Z.; Tschuch, C.; Csiszar, K.; Bacchelli, B.; Quaglino, D.; Pasquali-Ronchetti, I.; Pope, F.M.; Richards, A.; Terry, S.; et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 223–227. [Google Scholar] [CrossRef]
- Bergen, A.A.; Plomp, A.S.; Schuurman, E.J.; Terry, S.F.; Breuning, M.H.; Dauwerse, H.G.; Swart, J.; Kool, M.; Van Soest, S.; Baas, F.; et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [PubMed]
- Colin, S.; Chinetti-Gbaguidi, G.; Staels, B. Macrophage phenotypes in atherosclerosis. Immunol. Rev. 2014, 262, 153–166. [Google Scholar] [CrossRef]
- Tintut, Y.; Patel, J.; Territo, M.; Saini, T.; Parhami, F.; Demer, L.L. Monocyte/macrophage regulation of vascular calcification in vitro. Circulation 2002, 105, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Hamczyk, M.R.; Andrés, V. Alternatively activated macrophages exhibit an anticalcifying activity dependent on extracellular ATP/pyrophosphate metabolism. Am. J. Physiol. Cell Physiol. 2016, 310, C788–C799. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Speer, M.Y.; Yang, H.; Bergen, J.; Giachelli, C.M. Vitamin D receptor activators induce an anticalcific paracrine program in macrophages: Requirement of osteopontin. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Villa-Bellosta, R.; Hamczyk, M.R.; Andrés, V. Novel phosphate-activated macrophages prevent ectopic calcification by increasing extracellular ATP and pyrophosphate. PLoS ONE 2017, 12, e0174998. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, Y.; Shi, L.; Ren, J.; Patti, M.; Wang, T.; De Oliveira, J.R.M.; Sobrido, M.-J.; Quintáns, B.; Baquero, M.; et al. Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat. Genet. 2012, 44, 254–256. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genetic Disease | Ectopic Calcification | Protein Affected | Main Reference | Role |
|---|---|---|---|---|
| Generalized Arterial Calcification of Infancy | Medial Arterial | eNPP1 | Rutsch et al., 2003 | Synthesis of pyrophosphate |
| Medial Arterial and Periarticular | 5NT | St Hilaire et al., 2011 | Hydrolysis of AMP | |
| Idiopatic Skeletal Hypertosis | Spinal Tissues | ENT1 | Warraich et al., 2013 | Ado Transporter |
| Familial Idiopathic basal Ganglia Calcification | Basal Ganglia and cortex | Pit-2 | Wang et al., 2012 | Phosphate Transporter |
| Pseudoxanthoma ellasticum | Elastic fibers in skin, arteries and retine. | ABCC6 | La Seux et al., 2000 Bergen et al., 2000 | ATP transporter |
| Craniometaphyseal dysplasia | Craniofacial Bones | ANK | Nürnberg et al., 2001 | ? |
| Condrocalcinosis | Articular cartilage | ANK | Pendleton et al., 2002 | ? |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villa-Bellosta, R. Vascular Calcification: Key Roles of Phosphate and Pyrophosphate. Int. J. Mol. Sci. 2021, 22, 13536. https://doi.org/10.3390/ijms222413536
Villa-Bellosta R. Vascular Calcification: Key Roles of Phosphate and Pyrophosphate. International Journal of Molecular Sciences. 2021; 22(24):13536. https://doi.org/10.3390/ijms222413536
Chicago/Turabian StyleVilla-Bellosta, Ricardo. 2021. "Vascular Calcification: Key Roles of Phosphate and Pyrophosphate" International Journal of Molecular Sciences 22, no. 24: 13536. https://doi.org/10.3390/ijms222413536
APA StyleVilla-Bellosta, R. (2021). Vascular Calcification: Key Roles of Phosphate and Pyrophosphate. International Journal of Molecular Sciences, 22(24), 13536. https://doi.org/10.3390/ijms222413536