
Radiosensitizer Effect of β-Apopicropodophyllin against Colorectal Cancer via Induction of Reactive Oxygen Species and Apoptosis

 ,
,
Abstract
1. Introduction
2. Results
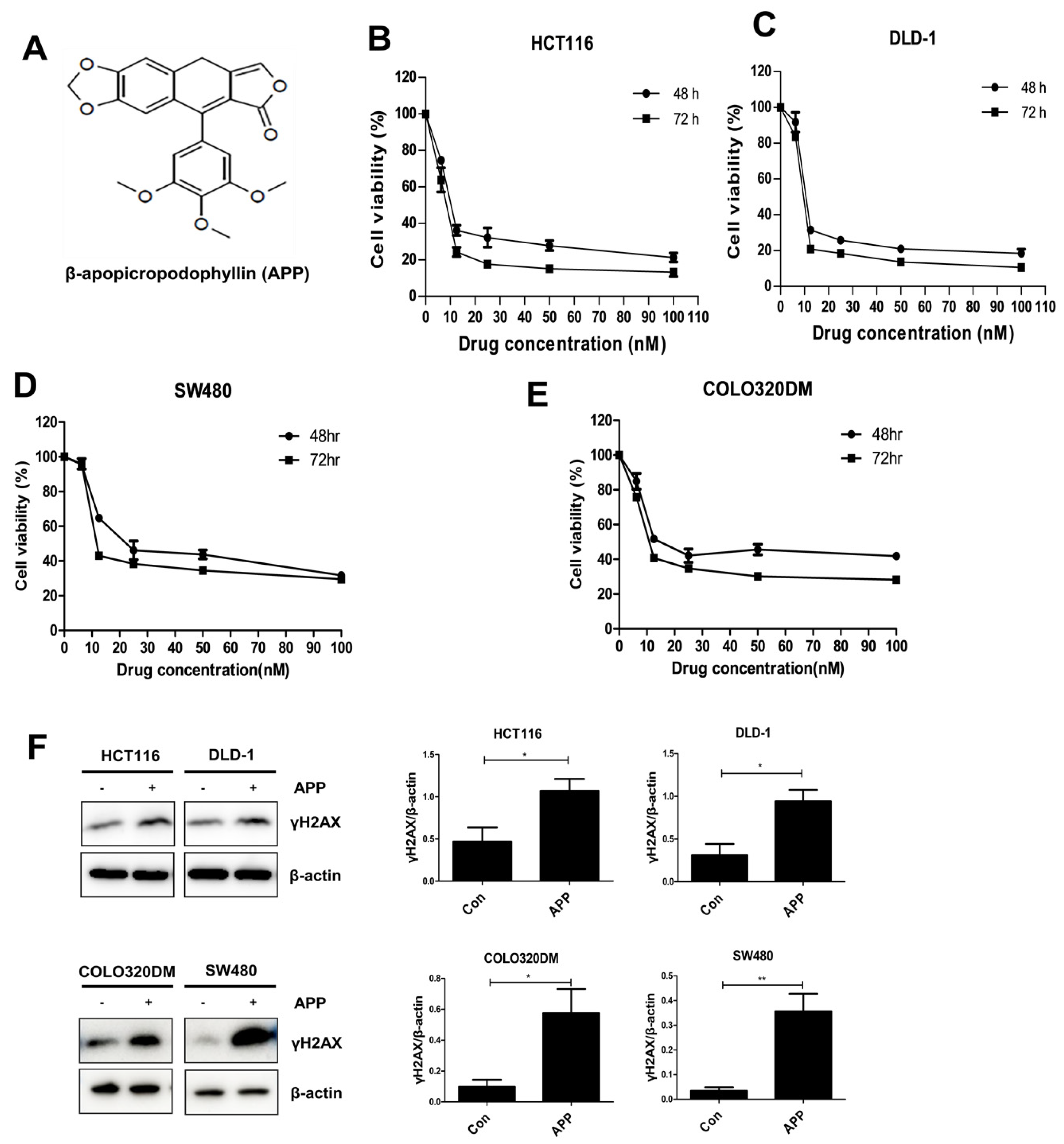
2.1. APP Induces the Death of CRC Cells by Inducing DNA Damage
2.2. APP Acts as a Radiosensitizer by Retarding Cell Growth In Vitro
2.3. Combined Treatment with APP and IR Increases Apoptosis
2.4. The Radiosensitizing Effect of APP Is Mediated by Intracellular ROS Accumulation
2.5. In Vivo Radiosensitization Effect of APP
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Chemical Reagents
4.2. MTT Assay and IC50 Determination
4.3. Clonogenic Assay
4.4. Cell Counting Assay
4.5. Immunoblot Analysis
4.6. Isolation of Mitochondrial and Cytosolic Fractions
4.7. Annexin V–Propidium Iodide Assay
4.8. ROS Detection
4.9. TUNEL Assay of Xenografts
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| APP | β-apopicropodophyllin |
| CRC | Colorectal cancer |
| IR | γ-Ionizing radiation |
| ROS | Reactive oxygen species |
| PPT | Podophyllotoxin |
| Gy | Grey |
| DER | Dose enhancement ratio |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| SD | Standard deviation |
| PI | Propidium iodide |
| NAC | N-acetylcysteine |
References
- Siegel, R.L.; Miller, K.D.; Sauer, A.G.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef]
- Skuse, A. Constructions of Cancer in Early Modern England; Palgrave Macmillan: London, UK, 2015; pp. 20–39. [Google Scholar]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Banerjee, A.; Pathak, S.; Subramanium, V.D.; Dharanivasan, G.; Murugesan, R.; Verma, R.S. Strategies for targeted drug delivery in treatment of colon cancer: Current trends and future perspectives. Drug Discov. Today 2017, 22, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Hong, W.G.; Cho, J.H.; Kim, E.M.; Kim, J.D.; Jung, C.H.; Hwang, S.G.; Um, H.D.; Park, J.K. Podophyllotoxin acetate triggers anticancer effects against non-small cell lung cancer cells by promoting cell death via cell cycle arrest, ER stress and autophagy. Int. J. Oncol. 2015, 47, 1257–1265. [Google Scholar] [CrossRef]
- Gordaliza, M.; García, P.A.; del Corral, J.M.M.; Castro, M.A.; Gómez-Zurita, M.A. Podophyllotoxin: Distribution, sources, applications and new cytotoxic derivatives. Toxicon 2004, 44, 441–459. [Google Scholar] [CrossRef]
- Hevener, K.; Verstak, T.A.; Lutat, K.E.; Riggsbee, D.L.; Mooney, J.W. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm. Sin. B 2018, 8, 844–861. [Google Scholar] [CrossRef]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Bush, N.G.; Evans-Roberts, K.; Maxwell, A. DNA topoisomerases. Available online: https://journals.asm.org/doi/10.1128/ecosalplus.ESP-0010-2014 (accessed on 10 December 2021).
- Pommier, Y. Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Leelaram, M.N.; Bhat, A.G.; Godbole, A.A.; Bhat, R.S.; Manjunath, R.; Nagaraja, V. Type IA topoisomerase inhibition by clampclosure. FASEB J. 2013, 27, 3030–3038. [Google Scholar] [CrossRef]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Hornyak, P.; Askwith, T.; Walker, S.; Komulainen, E.; Paradowski, M.; Pennicott, L.E.; Bartlett, E.J.; Brissett, N.C.; Raoof, A.; Watson, M.; et al. Mode of action of DNA-competitive small molecule inhibitors of tyrosyl DNA phosphodiesterase 2. Biochem. J. 2016, 473, 1869–1879. [Google Scholar] [CrossRef]
- Lindsey, R.H., Jr.; Pendleton, M.; Ashley, R.E.; Mercer, S.L.; Deweese, J.E.; Osheroff, N. Catalytic core of human topoisomerase IIα: Insightsinto enzyme-DNA interactions and drug mechanism. Biochemistry 2014, 53, 6595–6602. [Google Scholar] [CrossRef]
- Gibson, E.G.; Deweese, J.E. Covalent poisons of topoisomerase II. Curr. Top. Pharmacol. 2013, 17, 1–12. [Google Scholar]
- Olaussen, K.A.; Postel-Vinay, S. Predictors of chemotherapy efficacy in non-small-cell lung cancer: A challenging landscape. Ann. Oncol. 2016, 27, 2004–2016. [Google Scholar] [CrossRef] [PubMed]
- Meisenberg, C.; Ashour, M.E.; El-Shafie, L.; Liao, C.; Hodgson, A.; Pilborough, A.; Khurram, S.A.; Downs, J.A.; Ward, S.E.; El-Khamisy, S.F. Epigenetic changes in histone acetylation underpin resistance to the topoisomerase I inhibitor irinotecan. Nucleic Acids Res. 2017, 45, 1159–1176. [Google Scholar] [CrossRef] [PubMed]
- Saif, M.W.; Kang, S.P.; Chu, E. Treatment of metastatic colorectal cancer: From cytotoxic agents to molecular agents and multitargeted strategies. Oncology 2006, 14 (Suppl. S10), 11–19. [Google Scholar]
- Kim, J.Y.; Cho, J.H.; Kim, E.M.; Shin, H.J.; Hwang, S.G.; Song, J.Y.; Um, H.D.; Park, J.K. β-apopicropodophyllin functions as a radiosensitizer targeting ER stress in non-small cell lung cancer. Biomed. Pharmacother 2019, 113, 108769. [Google Scholar] [CrossRef]
- Kim, J.Y.; Cho, J.H.; Choi, J.R.; Shin, H.J.; Song, J.Y.; Hwang, S.G.; Um, H.D.; Yoo, Y.D.; Kim, J.; Park, J.K. A novel anti-cancer role of β-apopicropodophyllin against non-small cell lung cancer cells. Toxicol. Appl. Pharmacol. 2018, 375, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mao, Y.; Brandt-Rauf, P.W.; Williams, A.C.; Robert, L.F. Selective induction of apoptosis in mutant p53 premalignant and malignant cancer cells by PRIMA-1 through the c-Jun-NH2-kinase pathway. Mol. Cancer Ther. 2005, 4, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.; Yamamoto, H.; Ngan, C.Y.; Koi, M.; Takagi, A.; Damdinsuren, B.; Yasui, M.; Fujie, Y.; Matsuzaki, T.; Hemmi, H.; et al. Role of p21waf1/cip1 in effects of oxaliplatin in colorectal cancer cells. Mol. Cancer Ther. 2005, 4, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknæs, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Ahn, K.C.; Choi, J.Y.; Hwang, S.G.; Kim, W.J.; Um, H.D.; Park, J.K. Luteolin acts as a radiosensitizer in non-small cell lung cancer cells by enhancing apoptotic cell death through activation of a p38/ROS/caspase cascade. Int. J. Oncol. 2015, 46, 1149–1158. [Google Scholar] [CrossRef]
- Hong, W.G.; Kim, J.Y.; Cho, J.H.; Hwang, S.G.; Song, J.Y.; Lee, E.A.; Chang, T.S.; Um, H.D.; Park, J.K. AMRI-59 functions as a radiosensitizer via peroxiredoxin I-targeted ROS accumulation and apoptotic cell death induction. Oncotarget 2017, 8, 114050–114064. [Google Scholar] [CrossRef][Green Version]
- Albino, A.P.; Huang, X.; Jorgensen, E.; Yang, J.; Gietl, D.; Traganos, F.; Darzynkiewicz, Z. Induction of H2AX phosphorylation in pulmonary cells by tobacco smoke: A new assay for carcinogens. Cell Cycle 2004, 3, 1062–1068. [Google Scholar] [CrossRef]
- Ando, M.; Yoshikawa, K.; Iwase, Y.; Ishiura, S. Usefulness of monitoring gamma-H2AX and cell cycle arrest in HepG2 cells for estimating genotoxicity using a high-content analysis system. J. Biomol. Screen. 2014, 19, 1246–1254. [Google Scholar] [CrossRef]
- Pouliliou, S.; Koukourakis, M.I. Gamma histone 2AX (γ-H2AX) as a predictive tool in radiation oncology. Biomarkers 2014, 19, 167–180. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef] [PubMed]
- Ichim, G.; Tait, S.W.G. A fate worse than death: Apoptosis as an oncogenic process. Nat. Rev. Cancer 2016, 16, 539–548. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Pennica, D.; Nedwin, G.E.; Hayflick, J.S.; Seeburg, P.H.; Derynck, R.; Palladino, M.A.; Kohr, W.J.; Aggarwal, B.B.; Goeddel, D.V. Human tumour necrosis factor: Precursor structure, expression and homology to lymphotoxin. Nature 1984, 312, 724–729. [Google Scholar] [CrossRef]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The receptor for the cytotoxic ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998, 94, 481–490. [Google Scholar] [CrossRef]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef]
- Yasui, H.; Iizuka, D.; Hiraoka, W.; Kuwabara, M.; Matsuda, A.; Inanami, O. Nucleoside analogs as a radiosensitizer modulating DNA repair, cell cycle checkpoints, and apoptosis. Nucleosides Nucleotides Nucleic Acids 2020, 39, 439–452. [Google Scholar] [CrossRef]
- Buckley, A.M.; Lynam-Lennon, N.; O’Neill, H.; O’Sullivan, J. Targeting hallmarks of cancer to enhance radiosensitivity in gastrointestinal cancers. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Mu, X.; He, H.; Zhang, X.D. Cancer Radiosensitizers. Trends Pharmacol. Sci. 2018, 39, 24–48. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, H.; De Ridder, M. Targeting antioxidant enzymes as a radiosensitizing strategy. Cancer Lett. 2018, 438, 154–164. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DER (Dose Enhancement Ratio) | |
|---|---|
| 7.5 nM | |
| HCT116 | 1.13 |
| DLD-1 | 1.31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, J.-H.; Lee, N.-G.; Kang, A.-R.; Song, J.-Y.; Hwang, S.-G.; Um, H.-D.; Kim, J.; Park, J.K. Radiosensitizer Effect of β-Apopicropodophyllin against Colorectal Cancer via Induction of Reactive Oxygen Species and Apoptosis. Int. J. Mol. Sci. 2021, 22, 13514. https://doi.org/10.3390/ijms222413514
Kwon J-H, Lee N-G, Kang A-R, Song J-Y, Hwang S-G, Um H-D, Kim J, Park JK. Radiosensitizer Effect of β-Apopicropodophyllin against Colorectal Cancer via Induction of Reactive Oxygen Species and Apoptosis. International Journal of Molecular Sciences. 2021; 22(24):13514. https://doi.org/10.3390/ijms222413514
Chicago/Turabian StyleKwon, Jin-Hee, Na-Gyeong Lee, A-Ram Kang, Jie-Young Song, Sang-Gu Hwang, Hong-Duck Um, Joon Kim, and Jong Kuk Park. 2021. "Radiosensitizer Effect of β-Apopicropodophyllin against Colorectal Cancer via Induction of Reactive Oxygen Species and Apoptosis" International Journal of Molecular Sciences 22, no. 24: 13514. https://doi.org/10.3390/ijms222413514
APA StyleKwon, J.-H., Lee, N.-G., Kang, A.-R., Song, J.-Y., Hwang, S.-G., Um, H.-D., Kim, J., & Park, J. K. (2021). Radiosensitizer Effect of β-Apopicropodophyllin against Colorectal Cancer via Induction of Reactive Oxygen Species and Apoptosis. International Journal of Molecular Sciences, 22(24), 13514. https://doi.org/10.3390/ijms222413514