
Cardiac Changes in Parkinson’s Disease: Lessons from Clinical and Experimental Evidence

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Cardiac Sympathetic Loss in PD
2.1. Neuroimaging Findings
2.2. Circulating Catecholamine Levels
3. Clinical Manifestations
4. Molecular Alterations in the Parkinsonian Heart
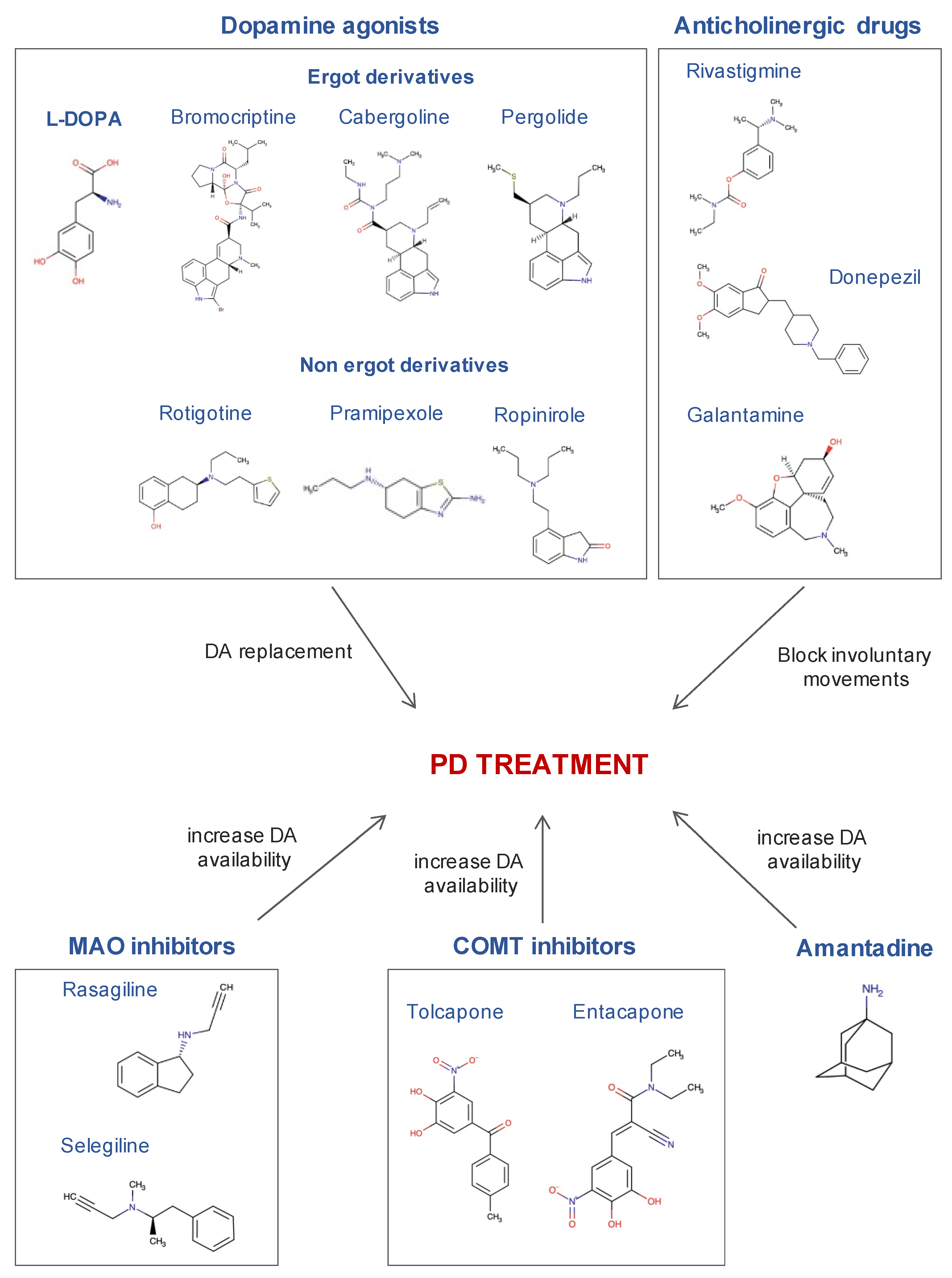
5. Effects of Antiparkinsonian Treatments on the Heart
6. Cardiac Alterations in Neurotoxin-Based Models for PD Research
6.1. Hearts in the 6-OHDA Model
6.2. Hearts in the MPTP Model
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cuenca, L.; Gil-Martinez, A.L.; Cano-Fernandez, L.; Sanchez-Rodrigo, C.; Estrada, C.; Fernandez-Villalba, E.; Herrero Ezquerro, M.T. Parkinson’s disease: A short story of 200 years. Histol. Histopathol. 2019, 34, 573–591. [Google Scholar] [CrossRef]
- Halliday, G.M.; Mccann, H. The progression of pathology in Parkinson’ s disease. Ann. N. Y. Acad. Sci. 2010, 1184, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Stamelou, M.; Goetz, C.G.; Poewe, W.; Lang, A.E.; Weintraub, D.; Burn, D.; Halliday, G.M.; Bezard, E.; Przedborski, S.; et al. Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov. Disord. 2017, 32, 1264–1310. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S. Goldstein Dysautonomia in Parkinson’s disease: Neurocardiological abnormalities. Compr. Physiol. 2014, 4, 805. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller, O.; Goss, J.E. Autonomic Deficits in Parkinson’s Syndrome. Arch. Neurol. 1971, 24, 50–57. [Google Scholar] [CrossRef]
- Takahashi, A. Autonomic Nervous System Disorders in Parkinson’s Disease. Eur. Neurol. 1991, 31, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Schnabel, R.B.; Hasenfuß, G.; Buchmann, S.; Kahl, K.G.; Aeschbacher, S.; Osswald, S.; Angermann, C.E. Heart and brain interactions. Herz 2021, 46, 138–149. [Google Scholar] [CrossRef]
- Krämer, H.H.; Lautenschläger, G.; de Azevedo, M.; Doppler, K.; Schänzer, A.; Best, C.; Oertel, W.H.; Reuter, I.; Sommer, C.; Birklein, F. Reduced central sympathetic activity in Parkinson’s disease. Brain Behav. 2019, 9, e01463. [Google Scholar] [CrossRef]
- Lamotte, G.; Holmes, C.; Wu, T.; Goldstein, D.S. Long-term trends in myocardial sympathetic innervation and function in synucleinopathies. Park. Relat. Disord. 2019, 67, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Simon, K.C.; Chen, H.; Schwarzschild, M.; Ascherio, A. Hypertension, hypercholesterolemia, diabetes, and risk of Parkinson disease. Neurology 2007, 69, 1688–1695. [Google Scholar] [CrossRef]
- Muqtadar, H.; Testai, F.D.; Gorelick, P.B. The Dementia of Cardiac Disease. Curr. Cardiol. Rep. 2012, 14, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Maluf, F.C.; Feder, D.; Carvalho, A.A.D.S. Analysis of the Relationship between Type II Diabetes Mellitus and Parkinson’s Disease: A Systematic Review. Park. Dis. 2019, 2019, 1–14. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Holmes, C.; Li, S.-T.; Bruce, S.; Metman, L.V.; Cannon, R.O. Cardiac sympathetic denervation in Parkinson disease. Ann. Intern. Med. 2000, 133, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Ruffoli, R.; Soldani, P.; Ruggieri, S.; Paparelli, A. The “Parkinsonian heart”: From novel vistas to advanced therapeutic approaches in Parkinson’s disease. Curr. Med. Chem. 2007, 14, 2421–2428. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Sharabi, Y. The heart of PD: Lewy body diseases as neurocardiologic disorders. Brain Res. 2019, 1702, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Morita, Y.; Shimizu, T.; Takahashi, K.; Suzuki, N. Cardiac parasympathetic dysfunction concurrent with cardiac sympathetic denervation in Parkinson’s disease. J. Neurol. Sci. 2009, 276, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Prell, T.; Schaller, D.; Perner, C.; Witte, O.W.; Grosskreutz, J. Sicca Symptoms in Parkinson’s Disease: Association with Other Nonmotor Symptoms and Health-Related Quality of Life. Park. Dis. 2020, 2020, 2958635. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Steur, E.N.H.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Rolland, A.-S.; Herrero, M.-T.; Garcia-Martinez, V.; Ruberg, M.; Hirsch, E.C.; François, C. Metabolic activity of cerebellar and basal ganglia-thalamic neurons is reduced in parkinsonism. Brain 2006, 130, 265–275. [Google Scholar] [CrossRef]
- Heman, P.; Barcia, C.; Gómez, A.; Ros, C.M.; Ros-Bernal, F.; Yuste, J.E.; de Pablos, V.; Fernandez-Villalba, E.; Toledo-Cárdenas, M.R.; Herrero, M.T. Nigral degeneration correlates with persistent activation of cerebellar Purkinje cells in MPTP-treated monkeys. Histol. Histopathol. 2012, 27, 89–94. [Google Scholar] [CrossRef]
- Fahn, S. The 200-year journey of Parkinson disease: Reflecting on the past and looking towards the future. Park. Relat. Disord. 2018, 46, S1–S5. [Google Scholar] [CrossRef] [PubMed]
- Buddhala, C.; Loftin, S.K.; Kuley, B.M.; Cairns, N.J.; Campbell, M.C.; Perlmutter, J.S.; Kotzbauer, P.T. Dopaminergic, serotonergic, and noradrenergic deficits in Parkinson disease. Ann. Clin. Transl. Neurol. 2015, 2, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Yao, N.; Wu, Y.; Zhou, Y.; Ju, L.; Liu, Y.; Ju, R.; Duan, D.; Xu, Q. Lesion of the locus coeruleus aggravates dopaminergic neuron degeneration by modulating microglial function in mouse models of Parkinson’s disease. Brain Res. 2015, 1625, 255–274. [Google Scholar] [CrossRef] [PubMed]
- Chan-Palay, V.L.; Jentsch, B. Chapter 18 Galanin tuberomammillary neurons in the hypothalamus in Alzheimer’s and Parkinson’s diseases. Prog. Brain Res. 1992, 93, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Neuropathological Staging of Brain Pathology in Sporadic Parkinson’s disease: Separating the Wheat from the Chaff. J. Park. Dis. 2017, 7, S71–S85. [Google Scholar] [CrossRef] [PubMed]
- Cersosimo, M.G.; Benarroch, E.E. Central Control of Autonomic Function and Involvement in Neurodegenerative Disorders, 1st ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2013; Volume 117. [Google Scholar]
- Donadio, V.; Incensi, A.; Del Sorbo, F.; Rizzo, G.; Infante, R.; Scaglione, C.; Modugno, N.; Fileccia, E.; Elia, A.E.; Cencini, F.; et al. Skin Nerve Phosphorylated α-Synuclein Deposits in Parkinson Disease With Orthostatic Hypotension. J. Neuropathol. Exp. Neurol. 2018, 77, 942–949. [Google Scholar] [CrossRef]
- Sakakibara, R.; Tateno, F.; Aiba, Y.; Ogata, T.; Kishi, M.; Terada, H.; Inaoka, T.; Nakatsuka, T.; Matsuoka, K. MIBG Myocardial Scintigraphy Identifies Premotor PD/DLB During a Negative DAT Scan Period: Second Report. Mov. Disord. Clin. Pract. 2018, 6, 46–50. [Google Scholar] [CrossRef]
- Gonçalves, V.C.; Cuenca-Bermejo, L.; Fernandez-Villalba, E.; Martin-Balbuena, S.; da Silva Fernandes, M.J.; Scorza, C.A.; Herrero, M.-T. Heart Matters: Cardiac Dysfunction and Other Autonomic Changes in Parkinson’s Disease. Neuroscience 2021, 15, 1073858421990000. [Google Scholar] [CrossRef] [PubMed]
- Scorza, F.A.; Fiorini, A.C.; Scorza, C.A.; Finsterer, J. Cardiac abnormalities in Parkinson’s disease and Parkinsonism. J. Clin. Neurosci. 2018, 53, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Taki, J.; Nakajima, K.; Hwang, E.-H.; Matsunari, I.; Komai, K.; Yoshita, M.; Sakajiri, K.; Tonami, N. Peripheral sympathetic dysfunction in patients with Parkinson’s disease without autonomic failure is heart selective and disease specific. Eur. J. Nucl. Med. Mol. Imaging 2000, 27, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S.; Holmes, C.S.; Dendi, R.; Bruce, S.R.; Li, S.-T. Orthostatic hypotension from sympathetic denervation in Parkinson’s disease. Neurology 2002, 58, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-T.; Dendi, R.; Holmes, C.; Goldstein, D.S. Progressive loss of cardiac sympathetic innervation in Parkinson’s disease. Ann. Neurol. 2002, 52, 220–223. [Google Scholar] [CrossRef]
- Braune, S.; Reinhardt, M.J.; Bathmann, J.; Krause, T.; Lehmann, M.J.; Lucking, C.H. Impaired cardiac uptake of meta-[123I]iodobenzylguanidine in Parkinson’s disease with autonomic failure. Acta Neurol. Scand. 1998, 97, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Yoshita, M.; Hayashi, M.; Hirai, S. Decreased myocardial accumulation of 123i-meta-iodobenzyl guanidine in parkinson’s disease. Nucl. Med. Commun. 1998, 19, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Takatsu, H.; Nishida, H.; Matsuo, H.; Watanabe, S.; Nagashima, K.; Wada, H.; Noda, T.; Nishigaki, K.; Fujiwara, H. Cardiac sympathetic denervation from the early stage of Parkinson’s disease: Clinical and experimental studies with radiolabeled MIBG. J. Nucl. Med. 2000, 41, 71–77. [Google Scholar] [PubMed]
- Senard, P.J.-M.; Brefel-Courbon, C.; Rascol, O.; Montastruc, J.-L. Orthostatic Hypotension in Patients with Parkinson’s Disease: Pathophysiology and management. Drugs Aging 2001, 18, 495–505. [Google Scholar] [CrossRef]
- Saiki, S.; Hirose, G.; Sakai, K.; Kataoka, S.; Hori, A.; Saiki, M.; Kaito, M.; Higashi, K.; Taki, S.; Kakeshita, K.; et al. Cardiac 123I-MIBG scintigraphy can assess the disease severity and phenotype of PD. J. Neurol. Sci. 2004, 220, 105–111. [Google Scholar] [CrossRef]
- Orimo, S.; Ozawa, E.; Oka, T.; Nakade, S.; Tsuchiya, K.; Yoshimoto, M.; Wakabayashi, K.; Takahashi, H. Different histopathology accounting for a decrease in myocardial MIBG uptake in PD and MSA. Neurology 2001, 57, 1140–1141. [Google Scholar] [CrossRef]
- Orimo, S.; Oka, T.; Miura, H.; Tsuchiya, K.; Mori, F.; Wakabayashi, K.; Nagao, T.; Yokochi, M. Sympathetic cardiac denervation in Parkinson’s disease and pure autonomic failure but not in multiple system atrophy. J. Neurol. Neurosurg. Psychiatry 2002, 73, 776. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Ghebremedhin, E.; Del Tredici, K.; Langston, J.W.; Braak, H. Diminished tyrosine hydroxylase immunoreactivity in the cardiac conduction system and myocardium in Parkinson’s disease: An anatomical study. Acta Neuropathol. 2009, 118, 777–784. [Google Scholar] [CrossRef]
- Metzger, J.M.; Matsoff, H.N.; Zinnen, A.D.; Fleddermann, R.A.; Bondarenko, V.; Simmons, H.A.; Mejia, A.; Moore, C.F.; Emborg, M.E. Post mortem evaluation of inflammation, oxidative stress, and PPARγ activation in a nonhuman primate model of cardiac sympathetic neurodegeneration. PLoS ONE 2020, 15, e0226999. [Google Scholar] [CrossRef] [PubMed]
- Joers, V.; Emborg, M.E. Modeling and imaging cardiac sympathetic neurodegeneration in Parkinson’s disease. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 125–159. [Google Scholar] [PubMed]
- Wakabayashi, K.; Takakashi, H. Neuropathology of Autonomic Nervous System in Parkinson’s Disease. Eur. Neurol. 1997, 38, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.-Y.; Liu, G.-L.; Wang, D.-X.; Zhang, M.-M.; Kou, W.-Y.; Feng, T. Alpha-Synuclein in Peripheral Tissues in Parkinson’s Disease. ACS Chem. Neurosci. 2019, 10, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Fujishiro, H.; Frigerio, R.; Burnett, M.; Klos, K.J.; Josephs, K.A.; Delledonne, A.; Parisi, J.E.; Ahlskog, J.E.; Dickson, D.W. Cardiac sympathetic denervation correlates with clinical and pathologic stages of Parkinson’s disease. Mov. Disord. 2008, 23, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Amino, T.; Orimo, S.; Itoh, Y.; Takahashi, A.; Uchihara, T.; Mizusawa, H. Profound Cardiac Sympathetic Denervation Occurs in Parkinson Disease. Brain Pathol. 2006, 15, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Leys, F.; Fanciulli, A.; Ndayisaba, J.-P.; Granata, R.; Struhal, W.; Wenning, G.K. Cardiovascular autonomic function testing in multiple system atrophy and Parkinson’s disease: An expert-based blinded evaluation. Clin. Auton. Res. 2020, 30, 255–263. [Google Scholar] [CrossRef]
- Lenka, A.; Lamotte, G.; Goldstein, D.S. Cardiac 18 F-Dopamine PET Distinguishes PD with Orthostatic Hypotension from Parkinsonian MSA. Mov. Disord. Clin. Pract. 2021, 8, 582–586. [Google Scholar] [CrossRef]
- Reinhardt, M.J.; Jüngling, F.D.; Krause, T.M.; Braune, S. Scintigraphic differentiation between two forms of primary dysautonomia early after onset of autonomic dysfunction: Value of cardiac and pulmonary iodine-123 MIBG uptake. Eur. J. Nucl. Med. Mol. Imaging 2000, 27, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, Y.; Imrich, R.; Holmes, C.; Rn, S.P.; Goldstein, D.S. Generalized and neurotransmitter-selective noradrenergic denervation in Parkinson’s disease with orthostatic hypotension. Mov. Disord. 2008, 23, 1725–1732. [Google Scholar] [CrossRef] [PubMed]
- Gilman, S.; Wenning, G.K.; Low, P.A.; Brooks, D.; Mathias, C.J.; Trojanowski, J.Q.; Wood, N.; Colosimo, C.; Durr, A.; Fowler, C.J.; et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008, 71, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Hague, K.; Lento, P.; Morgello, S.; Caro, S.; Kaufmann, H. The distribution of Lewy bodies in pure autonomic failure: Autopsy findings and review of the literature. Acta Neuropathol. 1997, 94, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Riachi, N.J.; Harik, S.I.; Kalaria, R.N.; Sayre, L.M. On the mechanisms underlying 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. II. Susceptibility among mammalian species correlates with the toxin’s metabolic patterns in brain microvessels and liver. J. Pharmacol. Exp. Ther. 1988, 244, 443–448. [Google Scholar] [PubMed]
- Torabi, P.; Ricci, F.; Hamrefors, V.; Sutton, R.; Fedorowski, A. Classical and Delayed Orthostatic Hypotension in Patients With Unexplained Syncope and Severe Orthostatic Intolerance. Front. Cardiovasc. Med. 2020, 7, 21. [Google Scholar] [CrossRef]
- Smit, A.A.J.; Halliwill, J.R.; Low, P.A.; Wieling, W. Pathophysiological basis of orthostatic hypotension in autonomic failure. J. Physiol. 1999, 519, 1–10. [Google Scholar] [CrossRef]
- Turkka, J.T. Correlation of the Severity of Autonomic Dysfunction to Cardiovascular Reflexes and to Plasma Noradrenaline Levels in Parkinson’s Disease. Eur. Neurol. 1987, 26, 203–210. [Google Scholar] [CrossRef]
- Senard, J.M.; Valet, P.; Durrieu, G.; Berlan, M.; Tran, M.A.; Montastruc, J.L.; Rascol, A.; Montastruc, P. Adrenergic supersensitivity in Parkinsonians with orthostatic hypotension. Eur. J. Clin. Investig. 2008, 20, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Galinier, M.; Senard, J.M.; Valet, P.; Doazan, J.P.; Durrieu, G.; Tran, M.A.; Monstastruc, J.L.; Bounhoure, J.P. Relationship Between Arterial Blood Pressure Disturbances and Alpha Adrenoceptor Density. Clin. Exp. Hypertens. 1994, 16, 373–389. [Google Scholar] [CrossRef]
- Niimi, Y.; Ieda, T.; Hirayama, M.; Koike, Y.; Sobue, G.; Hasegawa, Y.; Takahashi, A. Clinical and physiological characteristics of autonomic failure with Parkinson’s disease. Clin. Auton. Res. 1999, 9, 139–144. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Pechnik, S.; Holmes, C.; Eldadah, B.; Sharabi, Y. Association Between Supine Hypertension and Orthostatic Hypotension in Autonomic Failure. Hypertension 2003, 42, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Fanciulli, A.; Göbel, G.; Ndayisaba, J.P.; Granata, R.; Duerr, S.; Strano, S.; Colosimo, C.; Poewe, W.; Pontieri, F.E.; Wenning, G.K. Supine hypertension in Parkinson’s disease and multiple system atrophy. Clin. Auton. Res. 2016, 26, 97–105. [Google Scholar] [CrossRef]
- D’Andrea, G.; Pizzolato, G.; Gucciardi, A.; Stocchero, M.; Giordano, G.; Baraldi, E.; Leon, A. Different Circulating Trace Amine Profiles in De Novo and Treated Parkinson’s Disease Patients. Sci. Rep. 2019, 9, 6151. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Zaragoza, J.; Cuenca-Bermejo, L.; Almela, P.; Laorden, M.-L.; Herrero, M.-T. Could Small Heat Shock Protein HSP27 Be a First-Line Target for Preventing Protein Aggregation in Parkinson’s Disease? Int. J. Mol. Sci. 2021, 22, 3038. [Google Scholar] [CrossRef]
- Boi, L.; Pisanu, A.; Palmas, M.F.; Fusco, G.; Carboni, E.; Casu, M.A.; Satta, V.; Scherma, M.; Janda, E.; Mocci, I.; et al. Modeling Parkinson’s Disease Neuropathology and Symptoms by Intranigral Inoculation of Preformed Human α-Synuclein Oligomers. Int. J. Mol. Sci. 2020, 21, 8535. [Google Scholar] [CrossRef] [PubMed]
- Velseboer, D.C.; de Haan, R.J.; Wieling, W.; Goldstein, D.S.; de Bie, R.M.A. Prevalence of orthostatic hypotension in Parkinson’s disease: A systematic review and meta-analysis. Park. Relat. Disord. 2011, 17, 724–729. [Google Scholar] [CrossRef]
- Szili-Török, T.; Kálmán, J.; Paprika, D.; Dibó, G.; Rózsa, Z.; Rudas, L. Depressed baroreflex sensitivity in patients with Alzheimer’s and Parkinson’s disease. Neurobiol. Aging 2001, 22, 435–438. [Google Scholar] [CrossRef]
- Ariza, D.; Lopes, F.N.C.; Crestani, C.C.; Martins-Pinge, M.C. Chemoreflex and baroreflex alterations in Parkinsonism induced by 6-OHDA in unanesthetized rats. Neurosci. Lett. 2015, 607, 77–82. [Google Scholar] [CrossRef]
- Palma, J.-A.; Kaufmann, H. Autonomic disorders predicting Parkinson’s disease. Park. Relat. Disord. 2014, 20, S94–S98. [Google Scholar] [CrossRef]
- Umehara, T.; Toyoda, C.; Oka, H. Postprandial hypotension in de novo Parkinson’s disease: A comparison with orthostatic hypotension. Park. Relat. Disord. 2014, 20, 573–577. [Google Scholar] [CrossRef]
- Solla, P.; Cadeddu, C.; Cannas, A.; Deidda, M.; Mura, N.; Mercuro, G.; Marrosu, F. Heart rate variability shows different cardiovascular modulation in Parkinson’s disease patients with tremor dominant subtype compared to those with akinetic rigid dominant subtype. J. Neural Transm. 2015, 122, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Kanegusuku, H.; Silva-Batista, C.; Peçanha, T.; Silva-Junior, N.; Queiroz, A.; Costa, L.; Mello, M.; Piemonte, M.; Ugrinowitsch, C.; Forjaz, C. Patients with Parkinson disease present high ambulatory blood pressure variability. Clin. Physiol. Funct. Imaging 2016, 37, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Sommer, S.; Aral-Becher, B.; Jost, W. Nondipping in Parkinson’s Disease. Park. Dis. 2011, 2011, 897586. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, A.; Atmis, V.; Cengiz, O.K.; Cinar, E.; Aras, S.; Varli, M.; Atli, T. Evaluation of Cardiac Autonomic Functions in Older Parkinson’s Disease Patients: A Cross-Sectional Study. Aging Dis. 2016, 7, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Zesiewicz, T.A.; Strom, J.A.; Borenstein, A.R.; Hauser, R.A.; Cimino, C.R.; Fontanet, H.L.; Cintron, G.B.; Staffetti, J.F.; Dunne, P.B.; Sullivan, K.L. Heart failure in Parkinson’s disease: Analysis of the United States medicare current beneficiary survey. Park. Relat. Disord. 2004, 10, 417–420. [Google Scholar] [CrossRef]
- Pennington, S.; Snell, K.; Lee, M.; Walker, R. The cause of death in idiopathic Parkinson’s disease. Park. Relat. Disord. 2010, 16, 434–437. [Google Scholar] [CrossRef]
- Fernandez, H.H.; Lapane, K.L. Predictors of mortality among nursing home residents with a diagnosis of Parkinson’s disease. Med Sci. Monit. 2002, 8, 241–247. [Google Scholar]
- Piqueras-Flores, J.; López-García, A.; Moreno-Reig, Á.; Gonzalez-Martinez, A.; Hernández-González, A.; Vaamonde-Gamo, J.; Jurado-Román, A. Structural and functional alterations of the heart in Parkinson’s disease. Neurol. Res. 2017, 40, 53–61. [Google Scholar] [CrossRef]
- Malik, M.; Andreas, J.-O.; Hnatkova, K.; Hoeckendorff, J.; Cawello, W.; Middle, M.; Horstmann, R.; Braun, M. Thorough QT/QTc Study in Patients With Advanced Parkinson’s Disease: Cardiac Safety of Rotigotine. Clin. Pharmacol. Ther. 2008, 84, 595–603. [Google Scholar] [CrossRef]
- Çanga, Y.; Emre, A.; Yüksel, G.A.; Karataş, M.B.; Yelgeç, N.S.; Gürkan, U.; Çalık, A.N.; Tireli, H.; Terzi, S. Assessment of Atrial Conduction Times in Patients with Newly Diagnosed Parkinson’s Disease. Park. Dis. 2018, 2018, 1–5. [Google Scholar] [CrossRef]
- Xu, J.; Bu, L.; Huang, L.; Yang, Y.; Yu, M.; Liu, J.; Wang, P.; Huang, D.; Bai, X.; Ma, Y.; et al. Heart failure having little effect on the progression of Parkinson’s disease: Direct evidence from mouse model. Int. J. Cardiol. 2014, 177, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Cunnington, A.-L.; Hood, K.; White, L. Outcomes of screening Parkinson’s patients for QTc prolongation. Park. Relat. Disord. 2013, 19, 1000–1003. [Google Scholar] [CrossRef] [PubMed]
- Nataraj, A.; Rajput, A.H. Parkinson’s disease, stroke, and related epidemiology. Mov. Disord. 2005, 20, 1476–1480. [Google Scholar] [CrossRef] [PubMed]
- Renoux, C.; Dell’Aniello, S.; Khairy, P.; Marras, C.; Bugden, S.; Turin, T.C.; Blais, L.; Tamim, H.; Evans, C.; Steele, R.; et al. Ventricular tachyarrhythmia and sudden cardiac death with domperidone use in Parkinson’s disease. Br. J. Clin. Pharmacol. 2016, 82, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Scorza, F.A.; do Carmo, A.C.; Fiorini, A.C.; Nejm, M.B.; Scorza, C.A.; Finsterer, J.; Ferraz, H.B. Sudden unexpected death in Parkinson’s disease (SUDPAR): A review of publications since the decade of the brain. Clinics 2017, 72, 649–651. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W., II. PINK1-Phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, U.A.; Ong, S.-B.; Hausenloy, D.J. Parkinson’s disease proteins: Novel mitochondrial targets for cardioprotection. Pharmacol. Ther. 2015, 156, 34–43. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Trilck-Winkler, M.; Borsche, M.; König, I.R.; Balck, A.; Lenz, I.; Kasten, M.; Lohmann, K.; Brockmann, K.; Valente, E.M.; Klein, C.; et al. Parkin Deficiency Appears Not to Be Associated with Cardiac Damage in Parkinson’s Disease. Mov. Disord. 2021, 36, 271–273. [Google Scholar] [CrossRef]
- Zhang, S.-X.; Zhuang, L.-L.; Liu, J.; Jing, Y.-Y.; Sun, J.; Gong, L.; Liu, X.-Y. The role of Parkin protein in cardiac function and ventricular remodeling in myocardial infarction rats. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5004–5013. [Google Scholar] [CrossRef]
- Sun, Y.; Yao, X.; Zhang, Q.-J.; Zhu, M.; Liu, Z.-P.; Ci, B.; Xie, Y.; Carlson, D.; Rothermel, B.A.; Sun, Y.; et al. Beclin-1-Dependent Autophagy Protects the Heart During Sepsis. Circulation 2018, 138, 2247–2262. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Kolwicz, S.C.; Wang, P.; Roe, N.D.; Villet, O.; Nishi, K.; Hsu, Y.-W.A.; Flint, G.V.; Caudal, A.; Wang, W.; et al. Increasing Fatty Acid Oxidation Prevents High Fat Diet Induced Cardiomyopathy through Regulating Parkin Mediated Mitophagy. Circulation 2020, 142. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ouyang, Y.; Yang, L.; Beal, M.F.; McQuibban, A.; Vogel, O.H.; Lu, B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc. Natl. Acad. Sci. USA 2008, 105, 7070–7075. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Wu, Y.E.; Wang, W.; Zhang, S.; Liu, D.; Liu, H. Decreased dynamin-related protein 1-related mitophagy induces myocardial apoptosis in the aging heart. Acta Biochim. et Biophys. Sin. 2021, 53, 1354–1366. [Google Scholar] [CrossRef]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. USA 2011, 108, 9572–9577. [Google Scholar] [CrossRef]
- Siddall, H.K.; Yellon, D.M.; Ong, S.-B.; Mukherjee, U.A.; Burke, N.; Hall, A.R.; Angelova, P.R.; Ludtmann, M.H.; Deas, E.; Davidson, S.M.; et al. Loss of PINK1 Increases the Heart’s Vulnerability to Ischemia-Reperfusion Injury. PLoS ONE 2013, 8, e62400. [Google Scholar] [CrossRef]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef]
- Matsuda, N.; Kimura, M.; Queliconi, B.B.; Kojima, W.; Mishima, M.; Takagi, K.; Koyano, F.; Yamano, K.; Mizushima, T.; Ito, Y.; et al. Parkinson’s disease-related DJ-1 functions in thiol quality control against aldehyde attack in vitro. Sci. Rep. 2017, 7, 12816. [Google Scholar] [CrossRef]
- Yu, H.-H.; Xu, Q.; Chen, H.-P.; Wang, S.; Huang, X.-S.; Huang, Q.-R.; He, M. Stable overexpression of DJ-1 protects H9c2 cells against oxidative stress under a hypoxia condition. Cell Biochem. Funct. 2013, 31, 643–651. [Google Scholar] [CrossRef]
- Dongworth, R.K.; Mukherjee, U.A.; Hall, A.R.; Astin, R.; Ong, S.-B.; Yao, Z.; Dyson, A.; Szabadkai, G.; Davidson, S.M.; Yellon, D.M.; et al. DJ-1 protects against cell death following acute cardiac ischemia-reperfusion injury. Cell Death Dis. 2014, 5, e1082. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-S.; Chen, H.-P.; Wang, S.; Yu, H.-H.; Huang, X.-S.; Huang, Q.-R.; He, M. Hypoxic preconditioning up-regulates DJ-1 protein expression in rat heart-derived H9c2 cells through the activation of extracellular-regulated kinase 1/2 pathway. Mol. Cell. Biochem. 2012, 370, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Billia, F.; Hauck, L.; Grothe, D.; Konecny, F.; Rao, V.; Kim, R.H.; Mak, T.W. Parkinson-susceptibility gene DJ-1/PARK7 protects the murine heart from oxidative damage in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 6085–6090. [Google Scholar] [CrossRef]
- Naranjo, C.C.; Marras, C.; Visanji, N.; Cornforth, D.J.; Sanchez-Rodriguez, L.; Schuele, B.; Goldman, S.; Estévez, M.; Stein, P.K.; Lang, A.E.; et al. Increased markers of cardiac vagal activity in leucine-rich repeat kinase 2-associated Parkinson’s disease. Clin. Auton. Res. 2019, 29, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Otano, J.; Gelpi, E.; Mestres, C.A.; Quintana, E.; Rauek, S.; Ribalta, T.; Santiago, V.; Tolosa, E. Alpha-synuclein aggregates in epicardial fat tissue in living subjects without parkinsonism. Park. Relat. Disord. 2013, 19, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Pickrell, A.M. Hidden phenotypes of PINK1/Parkin knockout mice. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129871. [Google Scholar] [CrossRef]
- Gao, L.; Liu, Y.; Guo, S.; Yao, R.; Wu, L.; Xiao, L.; Wang, Z.; Liu, Y.; Zhang, Y. Circulating Long Noncoding RNA HOTAIR is an Essential Mediator of Acute Myocardial Infarction. Cell. Physiol. Biochem. 2017, 44, 1497–1508. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, X.; Guo, Y.; Rong, H.; Liu, T. The long noncoding RNA HOTAIR promotes Parkinson’s disease by upregulating LRRK2 expression. Oncotarget 2017, 8, 24449–24456. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Ni, J.; Shu, Y.; Wang, H.; Hu, T. MiR-124 attenuates doxorubicin-induced cardiac injury via inhibiting p66Shc-mediated oxidative stress. Biochem. Biophys. Res. Commun. 2019, 521, 420–426. [Google Scholar] [CrossRef]
- Zhang, L.-M.; Wang, M.-H.; Yang, H.-C.; Tian, T.; Sun, G.-F.; Ji, Y.-F.; Hu, W.-T.; Liu, X.; Wang, J.-P.; Lu, H. Dopaminergic neuron injury in Parkinson’s disease is mitigated by interfering lncRNA SNHG14 expression to regulate the miR-133b/ α-synuclein pathway. Aging 2019, 11, 9264–9279. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Gong, Z.; Jin, X.; Zhao, P.; Zhang, Y.; Wang, Z. LncRNA MALAT1 targeting miR-124-3p regulates DAPK1 expression contributes to cell apoptosis in Parkinson’s Disease. J. Cell. Biochem. 2020, 121, 4838–4848. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Salgado-Somoza, A.; Stefanizzi, F.M.; Lumley, A.I.; Zhang, L.; Glaab, E.; May, P.; Devaux, Y. Non-Coding RNAs in the Brain-Heart Axis: The Case of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 6513. [Google Scholar] [CrossRef] [PubMed]
- Noack, C.; Schroeder, C.; Heusser, K.; Lipp, A. Cardiovascular effects of levodopa in Parkinson’s disease. Park. Relat. Disord. 2014, 20, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Heranval, A.; Lefaucheur, R.; Fetter, D.; Rouillé, A.; Le Goff, F.; Maltête, D. Drugs with potential cardiac adverse effects: Retrospective study in a large cohort of parkinsonian patients. Rev. Neurol. 2016, 172, 318–323. [Google Scholar] [CrossRef]
- Perez-Lloret, S.; Rey, M.V.; Crispo, J.; Krewski, D.; Lapeyre-Mestre, M.; Montastruc, J.-L.; Rascol, O. Risk of heart failure following treatment with dopamine agonists in Parkinson’s disease patients. Expert Opin. Drug Saf. 2014, 13, 351–360. [Google Scholar] [CrossRef]
- Günaydın, Z.Y.; Özer, F.F.; Karagöz, A.; Bektaş, O.; Karataş, M.B.; Vural, A.; Bayramoğlu, A.; Çelik, A.; Yaman, M. Evaluation of cardiovascular risk in patients with Parkinson disease under levodopa treatment. J. Geriatr. Cardiol. 2016, 13, 75–80. [Google Scholar] [CrossRef]
- Almela, P.; Bermejo, L.C.; Yuste, J.E.; Estrada, C.; De Pablos, V.; Bautista-Hernandez, V.; Fernández-Villalba, E.; Laorden, M.; Herrero, M.-T. Cardiac Noradrenaline Turnover and Heat Shock Protein 27 Phosphorylation in Dyskinetic Monkeys. Mov. Disord. 2019, 35, 698–703. [Google Scholar] [CrossRef]
- Van Camp, G.; Flamez, A.; Cosyns, B.; Goldstein, J.; Perdaens, C.; Schoors, D. Heart valvular disease in patients with Parkinson’s disease treated with high-dose pergolide. Neurology 2003, 61, 859–861. [Google Scholar] [CrossRef]
- Van Camp, G.; Flamez, A.; Cosyns, B.; Weytjens, C.; Muyldermans, L.; Van Zandijcke, M.; De Sutter, J.; Santens, P.; Decoodt, P.; Moerman, C.; et al. Treatment of Parkinson’s disease with pergolide and relation to restrictive valvular heart disease. Lancet 2004, 363, 1179–1183. [Google Scholar] [CrossRef]
- Horvath, J.; Fross, R.D.; Kleiner-Fisman, G.; Lerch, R.; Stalder, H.; Liaudat, S.; Raskoff, W.J.; Flachsbart, K.D.; Rakowski, H.; Pache, J.-C.; et al. Severe multivalvular heart disease: A new complication of the ergot derivative dopamine agonists. Mov. Disord. 2004, 19, 656–662. [Google Scholar] [CrossRef]
- Schade, R.; Andersohn, F.; Suissa, S.; Haverkamp, W.G. Dopamine Agonists and the Risk of Cardiac-Valve Regurgitation. Surv. Anesthesiol. 2007, 51, 273–274. [Google Scholar] [CrossRef]
- Zanetti, R.; Antonini, A.; Gatto, G.; Gentile, R.; Tesei, S.; Pezzoli, G. Valvular Heart Disease and the Use of Dopamine Agonists for Parkinson’s Disease. Surv. Anesthesiol. 2007, 51, 274–275. [Google Scholar] [CrossRef]
- Roth, B.L. Drugs and Valvular Heart Disease. N. Engl. J. Med. 2007, 356, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.; Brophy, J.M.; Suissa, S.; Renoux, C. Risks of Cardiac Valve Regurgitation and Heart Failure Associated with Ergot- and Non-Ergot-Derived Dopamine Agonist Use in Patients with Parkinson’s Disease: A Systematic Review of Observational Studies. CNS Drugs 2015, 29, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Apraxine, M.; Pasquet, A.; Jeanjean, A. Pramipexole-Induced Reversible Heart Failure. Mov. Disord. Clin. Pract. 2014, 1, 381–382. [Google Scholar] [CrossRef]
- Finberg, J.P.M.; Gross, A.; Bar-Am, O.; Friedman, R.; Loboda, Y.; Youdim, M.B.H. Cardiovascular responses to combined treatment with selective monoamine oxidase type B inhibitors and L-DOPA in the rat. Br. J. Pharmacol. 2006, 149, 647–656. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stryjer, R.; Treves, T.A.; Klein, C.; Rabey, J.M. The effects of acute loading with levodopa and levodopa with selegiline on blood pressure and plasma norepinephrine levels in chronic Parkinson’s disease patients. Acta Neurol. Scand. 2005, 111, 89–94. [Google Scholar] [CrossRef]
- Grünig, D.; Felser, A.; Bouitbir, J.; Krähenbühl, S. The catechol-O-methyltransferase inhibitors tolcapone and entacapone uncouple and inhibit the mitochondrial respiratory chain in HepaRG cells. Toxicol. Vitr. 2017, 42, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Meco, G.; Vanacore, N.; Locuratolo, N.; Bonifati, V.V.; Vella, C.; Giovani, A.; Tubani, L.; Baratta, L.; Mastrocola, C. Heart rate variability in Parkinson’s disease patients treated with tolcapone. Park. Relat. Disord. 2000, 6, 223–227. [Google Scholar] [CrossRef]
- Patel, P.; Karch, J. Regulation of Cell Death in the CARDIOVASCULAR System, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; Volume 353, ISBN 9780128201350. [Google Scholar]
- Kandiah, N.; Pai, M.-C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: The advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin. Interv. Aging 2017, 12, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Alsabbagh, M.W. Comparative risk of cardiac arrhythmias associated with acetylcholinesterase inhibitors used in treatment of dementias—A narrative review. Pharmacol. Res. Perspect. 2020, 8, e00622. [Google Scholar] [CrossRef]
- Chang, C.; Ramphul, K. Amantadine. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kin, K.; Yasuhara, T.; Kameda, M.; Date, I. Animal Models for Parkinson’s Disease Research: Trends in the 2000s. Int. J. Mol. Sci. 2019, 20, 5402. [Google Scholar] [CrossRef]
- Barcia, C.; Ros, F.; Annese, V.; Gómez, C.M.R.; Bernal, F.R.; Aguado-Llera, D.; Pagán, M.E.M.; De Pablos, V.; Villalba, E.F.; Herrero, M.T. IFN-γ signaling, with the synergistic contribution of TNF-α, mediates cell specific microglial and astroglial activation in experimental models of Parkinson’s disease. Cell Death Dis. 2011, 2, e142. [Google Scholar] [CrossRef]
- Annese, V.; Herrero, M.T.; Di Pentima, M.; Gomez, A.; Lombardi, L.; Ros, C.M.; De Pablos, V.; Fernandez-Villalba, E.; De Stefano, M.E. Metalloproteinase-9 contributes to inflammatory glia activation and nigro-striatal pathway degeneration in both mouse and monkey models of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism. Brain Struct. Funct. 2014, 220, 703–727. [Google Scholar] [CrossRef] [PubMed]
- Gil-Martínez, A.L.; Cuenca, L.; Sánchez-Rodrigo, C.; Estrada, C.; Fernández-Villalba, E.; Herrero, M.T. Effect of NAC treatment and physical activity on neuroinflammation in subchronic Parkinsonism; is physical activity essential ? J. Neuroinflamm. 2018, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.J.; Tan, E.-K.; Chao, Y.-X. Historical Perspective: Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2464. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, P.; Nadeau, R.; de Champlain, J. Acute and Chronic Cardiovascular Effects of 6-Hydroxydopamine in Dogs. Circ. Res. 1972, 31, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Joers, V.; Seneczko, K.; Goecks, N.C.; Kamp, T.J.; Hacker, T.A.; Brunner, K.G.; Engle, J.W.; Barnhart, T.E.; Nickles, R.J.; Holden, J.E.; et al. Nonuniform Cardiac Denervation Observed by 11C-meta-Hydroxyephedrine PET in 6-OHDA-Treated Monkeys. PLoS ONE 2012, 7, e35371. [Google Scholar] [CrossRef]
- Laverty, R.; Sharman, D.F. Modification by drugs of the metabolism of 3,4-dihydroxyphenylethylamine, noradrenaline and 5-hydroxytryptamine in the brain. Br. J. Pharmacol. Chemother. 1965, 24, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S.; Grossman, E.; Tamrat, M.; Chang, P.C.; Eisenhofer, G.; Bacher, J.; Kirk, K.L.; Bacharach, S.; Kopin, I.J. Positron emission imaging of cardiac sympathetic innervation and function using 18F-6-fluorodopamine: Effects of chemical sympathectomy by 6-hydroxydopamine. J. Hypertens. 1991, 9, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Anzai, T.; Yoshikawa, T.; Baba, A.; Nishimura, H.; Shiraki, H.; Nagami, K.; Suzuki, M.; Wainai, Y.; Ogawa, S. Myocardial sympathetic denervation prevents chamber-specific alteration of beta-adrenergic transmembrane signaling in rabbits with heart failure. J. Am. Coll. Cardiol. 1996, 28, 1314–1322. [Google Scholar] [CrossRef][Green Version]
- Nomura, Y.; Matsunari, I.; Takamatsu, H.; Murakami, Y.; Matsuya, T.; Taki, J.; Nakajima, K.; Nekolla, S.G.; Chen, W.-P.; Kajinami, K. Quantitation of cardiac sympathetic innervation in rabbits using 11C-hydroxyephedrine PET: Relation to 123I-MIBG uptake. Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.C.; Totaro, J.A.; Stone, C.A. Effect of 6-hydroxydopamine and some other compounds on the concentration of norepinephrine in the hearts of mice. J. Pharmacol. Exp. Ther. 1963, 140, 308–316. [Google Scholar] [PubMed]
- Arbab, A.S.; Koizumi, K.; Araki, T. Uptake and washout of I-123-MIBG in neuronal and non-neuronal sites in rat hearts: Relationship to renal clearance. Ann. Nucl. Med. 1996, 10, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Joers, V.; Dilley, K.; Rahman, S.; Jones, C.; Shultz, J.; Simmons, H.; Emborg, M.E. Cardiac Sympathetic Denervation in 6-OHDA-Treated Nonhuman Primates. PLoS ONE 2014, 9, e104850. [Google Scholar] [CrossRef]
- Slack, K.; Billing, R.; Matthews, S.; Allbutt, H.N.; Einstein, R.; Henderson, J.M. Subtle Cardiovascular Dysfunction in the Unilateral 6-Hydroxydopamine-Lesioned Rat. Park. Dis. 2010, 2010, 1–10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ariza, D.; Sisdeli, L.; Crestani, C.C.; Fazan, R.; Martins-Pinge, M.C. Dysautonomias in Parkinson’s disease: Cardiovascular changes and autonomic modulation in conscious rats after infusion of bilateral 6-OHDA in substantia nigra. Am. J. Physiol. Circ. Physiol. 2015, 308, H250–H257. [Google Scholar] [CrossRef]
- de Campos, B.H.; de Jager, L.; Reginato, G.S.; Pereira, R.S.; Crestani, C.C.; Pinge-Filho, P.; Martins-Pinge, M.C. Cardiovascular evaluation of female rats with 6-OHDA-induced parkinsonism: Possible protection by ovarian hormones and participation of nitric oxide. Life Sci. 2020, 259, 118259. [Google Scholar] [CrossRef]
- De Jager, L.; Amorim, E.D.T.; Lucchetti, B.F.C.; Lopes, F.N.C.; Crestani, C.C.; Pinge-Filho, P.; Martins-Pinge, M.C. Nitric oxide alterations in cardiovascular system of rats with Parkinsonism induced by 6-OHDA and submitted to previous exercise. Life Sci. 2018, 204, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.S.; Ariza, D.; Dias, D.P.M.; Crestani, C.C.; Martins-Pinge, M.C. Cardiovascular and autonomic alterations in rats with Parkinsonism induced by 6-OHDA and treated with L-DOPA. Life Sci. 2015, 127, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.D.; Oliveira, L.F.; Shinoda, L.; Scorza, C.A.; Faber, J.; Ferraz, H.B.; Britto, L.R.G.; Scorza, F.A. Cardiovascular alterations in rats with Parkinsonism induced by 6-OHDA and treated with Domperidone. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Langston, J.W. The MPTP Story. J. Park. Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef]
- Takatsu, H.; Wada, H.; Maekawa, N.; Takemura, M.; Saito, K.; Fujiwara, H. Significant reduction of 125I-meta-iodobenzylguanidine accumulation directly caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydroxypyridine, a toxic agent for inducing experimental Parkinson’s disease. Nucl. Med. Commun. 2002, 23, 161–166. [Google Scholar] [CrossRef]
- Fukumitsu, N.; Suzuki, M.; Fukuda, T.; Kiyono, Y. Multipoint analysis of reduced 125I-meta-iodobenzylguanidine uptake and norepinephrine turnover in the hearts of mice with 1-methyl-4-phenyl-1,2,3,6-tetrahydroxypyridine-induced parkinsonism. Nucl. Med. Biol. 2009, 36, 623–629. [Google Scholar] [CrossRef]
- Algeri, S.; Ambrosio, S.; Garofalo, P.; Gerli, P. Peripheral effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and its main metabolite 1-methyl-4-phenylpyridinium ion (MPP+) in the rat. Eur. J. Pharmacol. 1987, 141, 309–312. [Google Scholar] [CrossRef]
- Carmona-Abellan, M.; Martínez-Valbuena, I.; DiCaudo, C.; Marcilla, I.; Luquin, M.R. Cardiac sympathetic innervation in the MPTP non-human primate model of Parkinson disease. Clin. Auton. Res. 2019, 29, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Fuller, R.W.; Steranka, L.R., VIII. Central and peripheral catecholamine depletion by 1-methyl-4-phenyl-tetrahydropyridine (MPTP) in rodents. Life Sci. 1985, 36, 243–247. [Google Scholar] [CrossRef]
- Goldstein, D.S.; Li, S.-T.; Holmes, C.; Bankiewicz, K. Sympathetic Innervation in the 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine Primate Model of Parkinson’s Disease. J. Pharmacol. Exp. Ther. 2003, 306, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Amino, T.; Uchihara, T.; Tsunekawa, H.; Takahata, K.; Shimazu, S.; Mizusawa, H.; Orimo, S. Myocardial nerve fibers are preserved in MPTP-treated mice, despite cardiac sympathetic dysfunction. Neurosci. Res. 2008, 60, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wei, B.; Bi, Q.; Sun, Q.; Li, L.; He, J.; Weng, Y.; Zhang, S.; Mao, G.; Bao, Y.; et al. MPTP-Induced Impairment of Cardiovascular Function. Neurotox. Res. 2020, 38, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Fukumitsu, N.; Suzuki, M.; Fukuda, T.; Kiyono, Y.; Kajiyama, S.; Saji, H. Reduced 125I-meta-iodobenzylguanidine uptake and norepinephrine transporter density in the hearts of mice with MPTP-induced parkinsonism. Nucl. Med. Biol. 2006, 33, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Cuenca-Bermejo, L.; Almela, P.; Gallo-Soljancic, P.; Yuste, J.E.; de Pablos, V.; Bautista-Hernández, V.; Fernández-Villalba, E.; Laorden, M.-L.; Herrero, M.-T. Cardiac tyrosine hydroxylase activation and MB-COMT in dyskinetic monkeys. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cano-Jaimez, M.; Sánchez, F.P.; Milán, M.; Buendía, P.; Ambrosio, S.; Fariñas, I. Vulnerability of peripheral catecholaminergic neurons to MPTP is not regulated by α-synuclein. Neurobiol. Dis. 2010, 38, 92–103. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuenca-Bermejo, L.; Almela, P.; Navarro-Zaragoza, J.; Fernández Villalba, E.; González-Cuello, A.-M.; Laorden, M.-L.; Herrero, M.-T. Cardiac Changes in Parkinson’s Disease: Lessons from Clinical and Experimental Evidence. Int. J. Mol. Sci. 2021, 22, 13488. https://doi.org/10.3390/ijms222413488
Cuenca-Bermejo L, Almela P, Navarro-Zaragoza J, Fernández Villalba E, González-Cuello A-M, Laorden M-L, Herrero M-T. Cardiac Changes in Parkinson’s Disease: Lessons from Clinical and Experimental Evidence. International Journal of Molecular Sciences. 2021; 22(24):13488. https://doi.org/10.3390/ijms222413488
Chicago/Turabian StyleCuenca-Bermejo, Lorena, Pilar Almela, Javier Navarro-Zaragoza, Emiliano Fernández Villalba, Ana-María González-Cuello, María-Luisa Laorden, and María-Trinidad Herrero. 2021. "Cardiac Changes in Parkinson’s Disease: Lessons from Clinical and Experimental Evidence" International Journal of Molecular Sciences 22, no. 24: 13488. https://doi.org/10.3390/ijms222413488
APA StyleCuenca-Bermejo, L., Almela, P., Navarro-Zaragoza, J., Fernández Villalba, E., González-Cuello, A.-M., Laorden, M.-L., & Herrero, M.-T. (2021). Cardiac Changes in Parkinson’s Disease: Lessons from Clinical and Experimental Evidence. International Journal of Molecular Sciences, 22(24), 13488. https://doi.org/10.3390/ijms222413488