
Single-Molecule/Cell Analyses Reveal Principles of Genome-Folding Mechanisms in the Three Domains of Life



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Overview of Genome-Folding Mechanisms
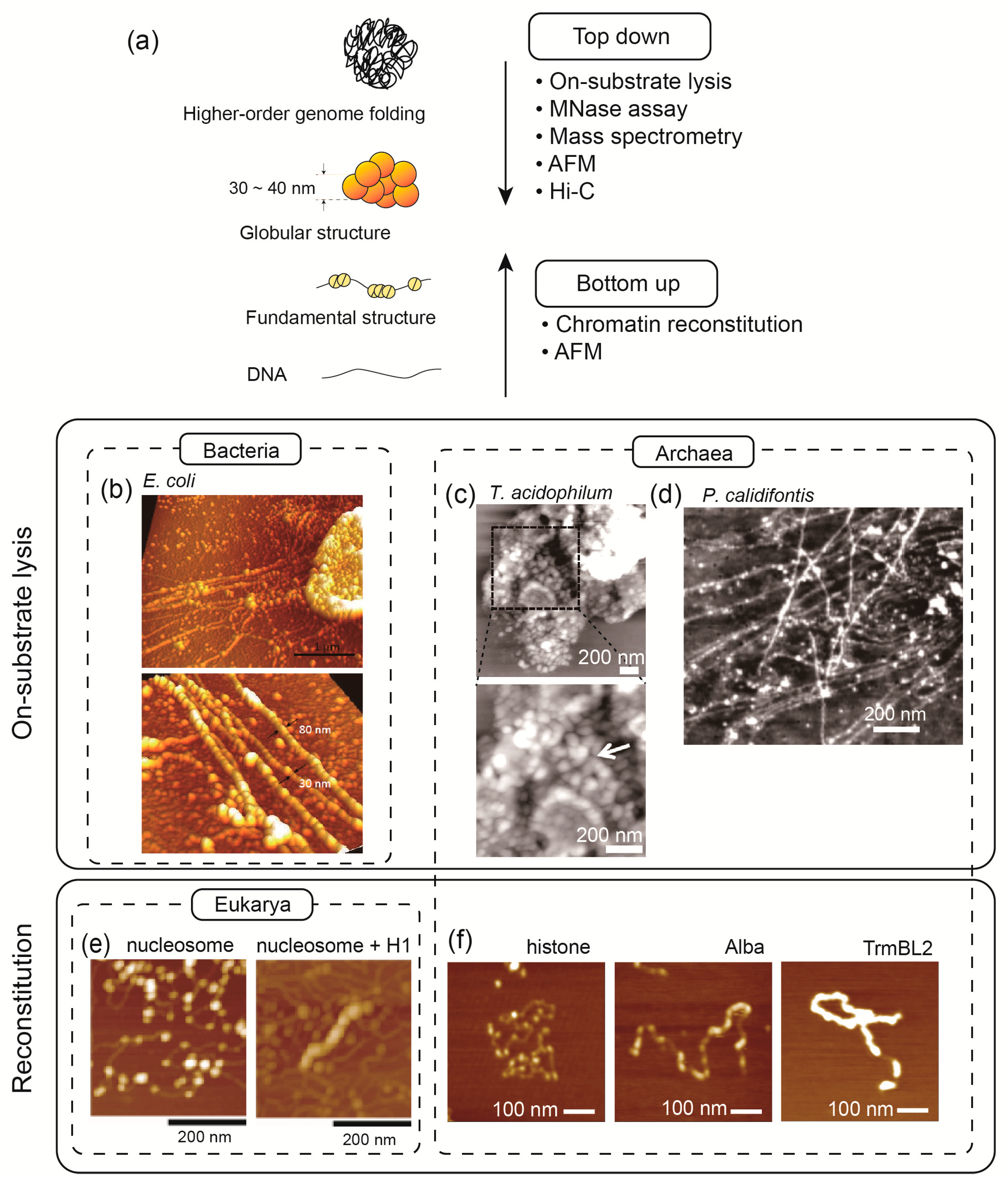
2. Strategies to Study Chromosome Architecture and Function
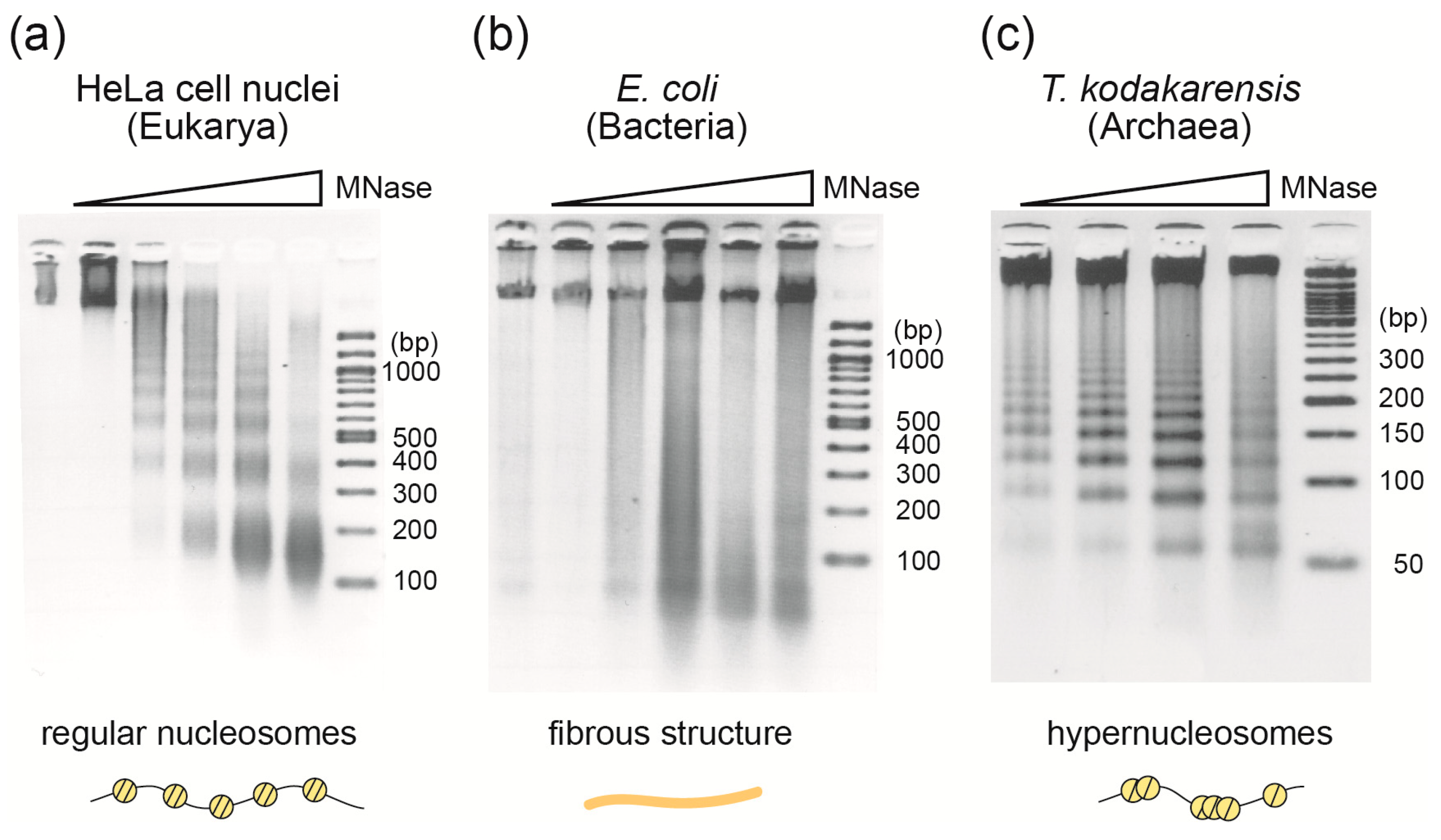
2.1. Micrococcal Nuclease Assay
2.2. Mass Spectrometry
2.3. Atomic Force Microscopy
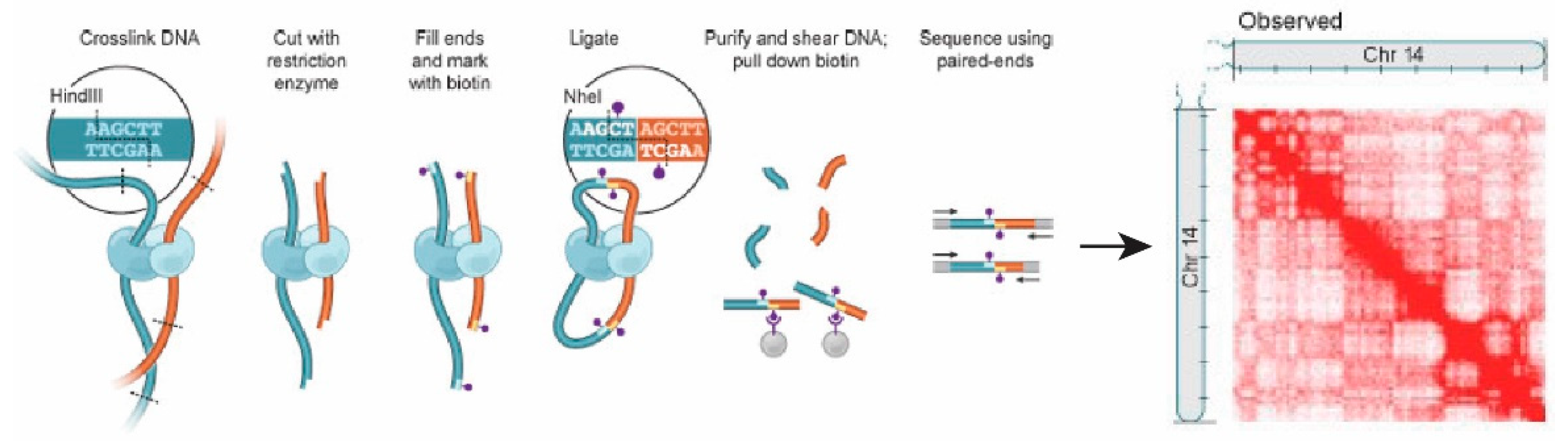
2.4. Chromosome Conformation Capture (3C) and Hi-C
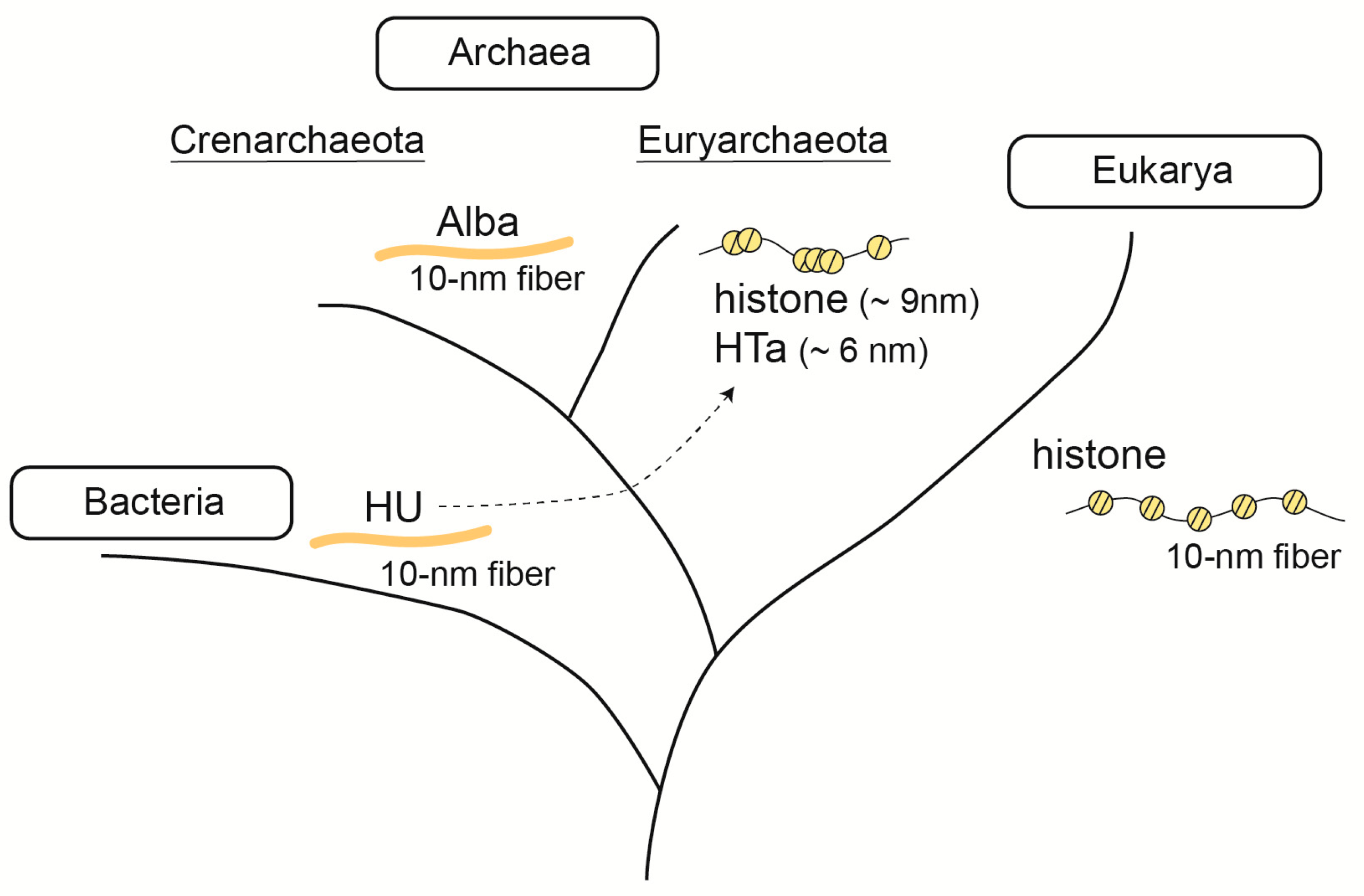
3. Fundamental Chromosomal/Nucleoid Proteins
3.1. Histone
3.2. Alba
3.3. HU
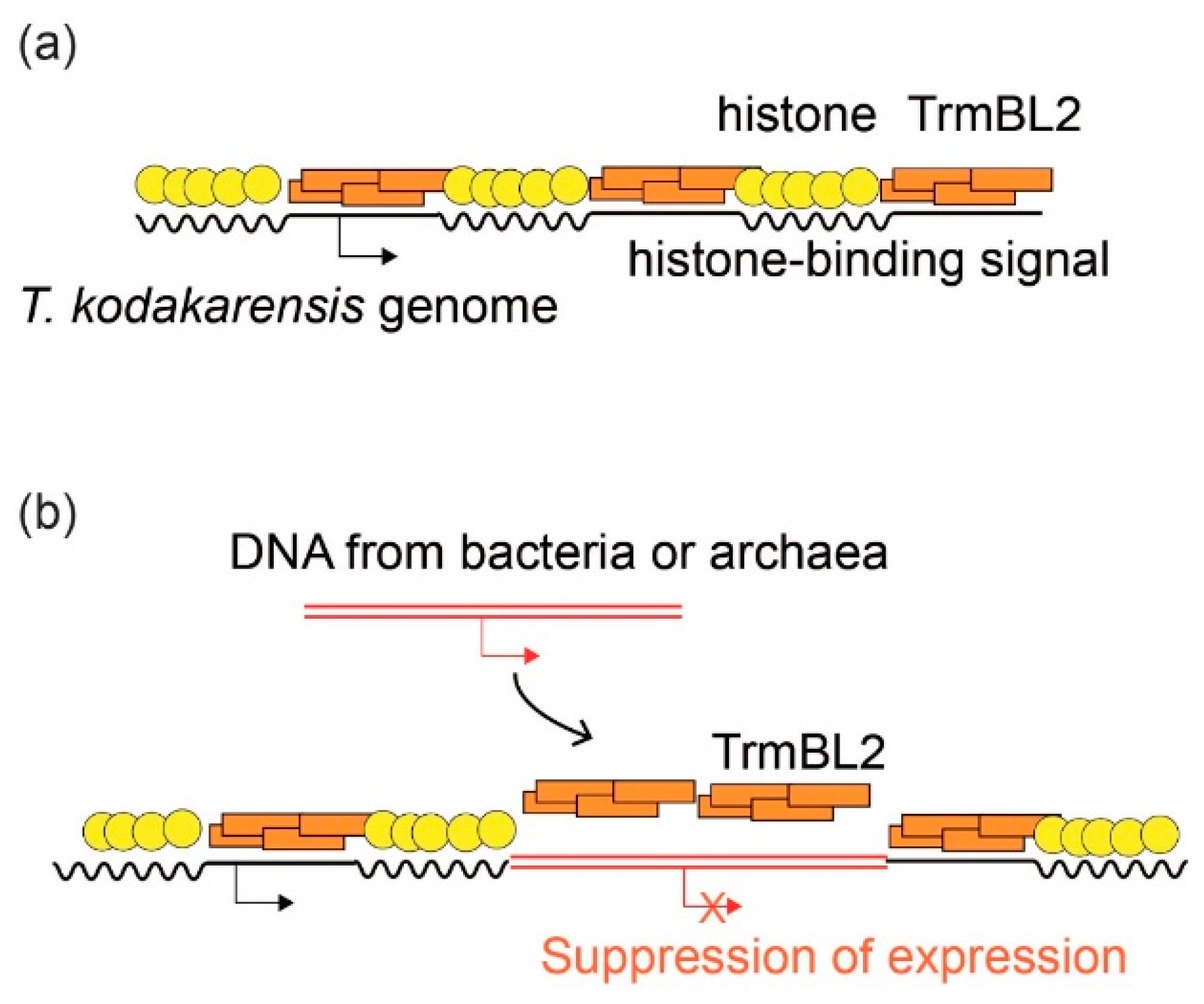
3.4. Suppression of Horizontally Transferred Genes by Global Regulatory Proteins
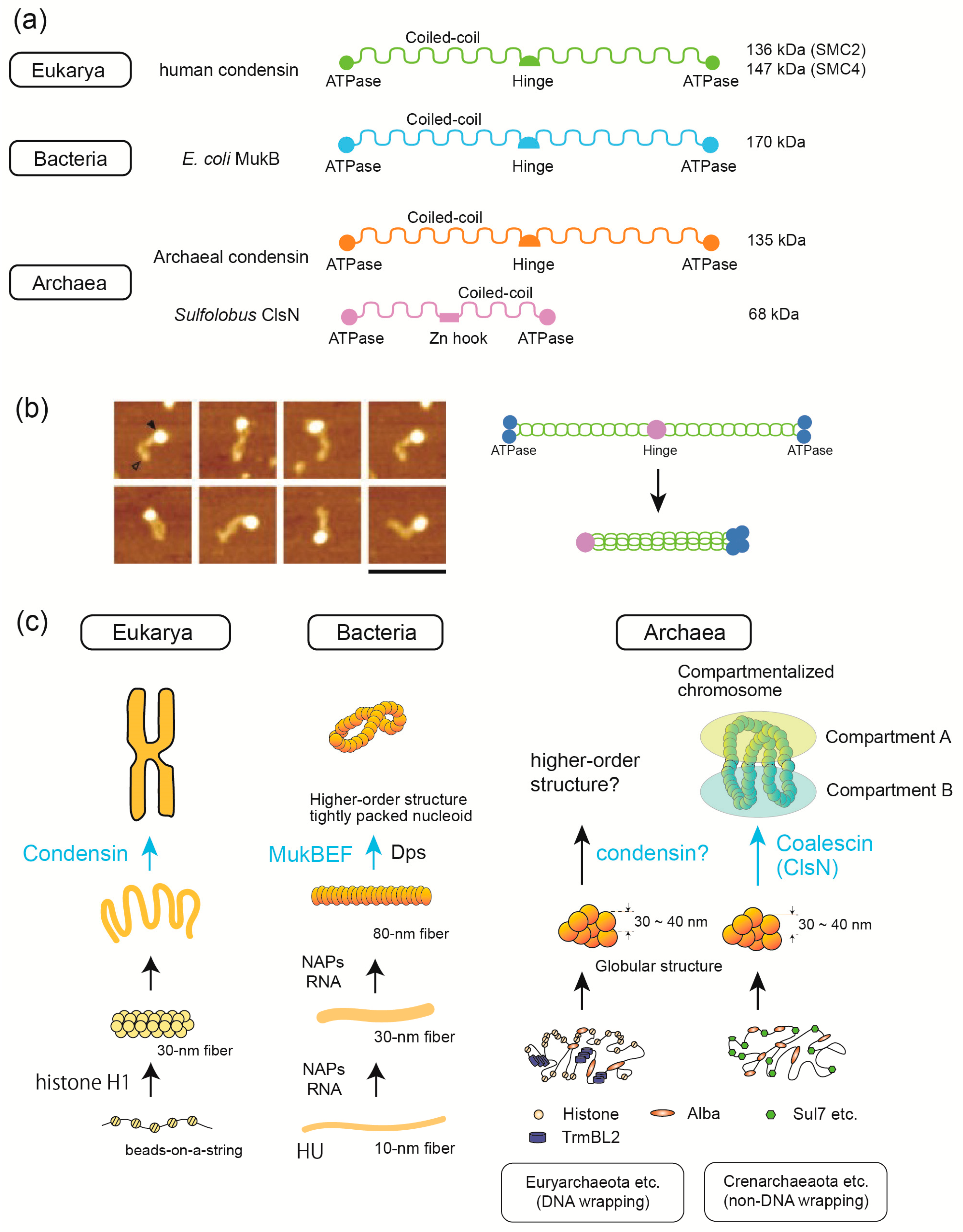
3.5. SMC Proteins Are Involved in 3D Structure Formation in the Three Domains of Life
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhou, K.; Gaullier, G.; Luger, K. Nucleosome structure and dynamics are coming of age. Nat. Struct. Mol. Biol. 2019, 26, 3–13. [Google Scholar] [CrossRef]
- McGinty, R.K.; Tan, S. Nucleosome Structure and Function. Chem. Rev. 2015, 115, 2255–2273. [Google Scholar] [CrossRef]
- Zaret, K. Micrococcal nuclease analysis of chromatin structure. Curr. Protoc. Mol. Biol. 2005, 21, 21. [Google Scholar] [CrossRef]
- Cutter, A.R.; Hayes, J.J. A brief review of nucleosome structure. FEBS Lett. 2015, 589, 2914–2922. [Google Scholar] [CrossRef]
- Morrison, O.; Thakur, J. Molecular Complexes at Euchromatin, Heterochromatin and Centromeric Chromatin. Int. J. Mol. Sci. 2021, 22, 6922. [Google Scholar] [CrossRef]
- Allshire, R.C.; Madhani, H.D. Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 2018, 19, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, F. SMC complexes: From DNA to chromosomes. Nat. Rev. Mol. Cell Biol. 2016, 17, 399–412. [Google Scholar] [CrossRef]
- Chanou, A.; Hamperl, S. Single-Molecule Techniques to Study Chromatin. Front. Cell Dev. Biol. 2021, 9, 699771. [Google Scholar] [CrossRef] [PubMed]
- Jerkovic, I.; Cavalli, G. Understanding 3D genome organization by multidisciplinary methods. Nat. Rev. Mol. Cell Biol. 2021, 22, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Hołówka, J.; Zakrzewska-Czerwińska, J. Nucleoid Associated Proteins: The Small Organizers that Help to Cope with Stress. Front. Microbiol. 2020, 11, 590. [Google Scholar] [CrossRef]
- Krogh, T.J.; Moller-Jensen, J.; Kaleta, C. Impact of Chromosomal Architecture on the Function and Evolution of Bacterial Genomes. Front. Microbiol. 2018, 9, 2019. [Google Scholar] [CrossRef]
- Ohniwa, R.L.; Morikawa, K.; Takeshita, S.L.; Kim, J.; Ohta, T.; Wada, C.; Takeyasu, K. Transcription-coupled nucleoid architecture in bacteria. Genes Cells 2007, 12, 1141–1152. [Google Scholar] [CrossRef]
- Ohniwa, R.L.; Morikawa, K.; Kim, J.; Kobori, T.; Hizume, K.; Matsumi, R.; Atomi, H.; Imanaka, T.; Ohta, T.; Wada, C.; et al. Atomic Force Microscopy Dissects the Hierarchy of Genome Architectures in Eukaryote, Prokaryote, and Chloroplast. Microsc. Microanal. 2007, 13, 3–12. [Google Scholar] [CrossRef]
- Ali Azam, T.; Iwata, A.; Nishimura, A.; Ueda, S.; Ishihama, A. Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J. Bacteriol. 1999, 181, 6361–6370. [Google Scholar] [CrossRef]
- Kim, J.; Yoshimura, S.H.; Hizume, K.; Ohniwa, R.L.; Ishihama, A.; Takeyasu, K. Fundamental structural units of the Escherichia coli nucleoid revealed by atomic force microscopy. Nucleic Acids Res. 2004, 32, 1982–1992. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, K.; Ushijima, Y.; Ohniwa, R.L.; Miyakoshi, M.; Takeyasu, K. What Happens in the Staphylococcal Nucleoid under Oxidative Stress? Microorganisms 2019, 7, 631. [Google Scholar] [CrossRef]
- Fabrega, A.; Vila, J. Salmonella enterica serovar Typhimurium skills to succeed in the host: Virulence and regulation. Clin. Microbiol. Rev. 2013, 26, 308–341. [Google Scholar] [CrossRef] [PubMed]
- Ferrandiz, M.J.; Carreno, D.; Ayora, S.; de la Campa, A.G. HU of Streptococcus pneumoniae Is Essential for the Preservation of DNA Supercoiling. Front. Microbiol. 2018, 9, 493. [Google Scholar] [CrossRef]
- Priyadarshini, R.; Cugini, C.; Arndt, A.; Chen, T.; Tjokro, N.O.; Goodman, S.D.; Davey, M.E. The nucleoid-associated protein HUbeta affects global gene expression in Porphyromonas gingivalis. Microbiology 2013, 159, 219–229. [Google Scholar] [CrossRef]
- Alvarez, A.; Toledo, H. The histone-like protein HU has a role in gene expression during the acid adaptation response in Helicobacter pylori. Helicobacter 2017, 22, e12381. [Google Scholar] [CrossRef]
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef]
- Baker, B.J.; De Anda, V.; Seitz, K.W.; Dombrowski, N.; Santoro, A.E.; Lloyd, K.G. Diversity, ecology and evolution of Archaea. Nat. Microbiol. 2020, 5, 887–900. [Google Scholar] [CrossRef]
- Adam, P.S.; Borrel, G.; Brochier-Armanet, C.; Gribaldo, S. The growing tree of Archaea: New perspectives on their diversity, evolution and ecology. ISME J. 2017, 11, 2407–2425. [Google Scholar] [CrossRef] [PubMed]
- Aminov, R. Role of archaea in human disease. Front. Cell. Infect. Microbiol. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Blombach, F.; Matelska, D.; Fouqueau, T.; Cackett, G.; Werner, F. Key Concepts and Challenges in Archaeal Transcription. J. Mol. Biol. 2019, 431, 4184–4201. [Google Scholar] [CrossRef]
- Lemmens, L.; Maklad, H.R.; Bervoets, I.; Peeters, E. Transcription Regulators in Archaea: Homologies and Differences with Bacterial Regulators. J. Mol. Biol. 2019, 431, 4132–4146. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Shin, M.; Oda, T.; Matsumi, R.; Ohniwa, R.L.; Itoh, T.; Shirahige, K.; Imanaka, T.; Atomi, H.; Yoshimura, S.H.; et al. Histone and TK0471/TrmBL2 form a novel heterogeneous genome architecture in the hyperthermophilic archaeon Thermococcus kodakarensis. Mol. Biol. Cell 2011, 22, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Peeters, E.; Driessen, R.P.; Werner, F.; Dame, R.T. The interplay between nucleoid organization and transcription in archaeal genomes. Nat. Rev. Microbiol. 2015, 13, 333–341. [Google Scholar] [CrossRef]
- Takemata, N.; Bell, S.D. Multi-scale architecture of archaeal chromosomes. Mol. Cell 2021, 81, 473–487.e476. [Google Scholar] [CrossRef]
- Laursen, S.P.; Bowerman, S.; Luger, K. Archaea: The Final Frontier of Chromatin. J. Mol. Biol. 2021, 433, 166791. [Google Scholar] [CrossRef]
- Maruyama, H.; Prieto, E.I.; Nambu, T.; Mashimo, C.; Kashiwagi, K.; Okinaga, T.; Atomi, H.; Takeyasu, K. Different Proteins Mediate Step-Wise Chromosome Architectures in Thermoplasma acidophilum and Pyrobaculum calidifontis. Front. Microbiol. 2020, 11, 1247. [Google Scholar] [CrossRef]
- Hizume, K.; Yoshimura, S.H.; Takeyasu, K. Linker histone H1 per se can induce three-dimensional folding of chromatin fiber. Biochemistry 2005, 44, 12978–12989. [Google Scholar] [CrossRef]
- Clark, D.J. Nucleosome positioning, nucleosome spacing and the nucleosome code. J. Biomol. Struct. Dyn. 2010, 27, 781–793. [Google Scholar] [CrossRef]
- Rojec, M.; Hocher, A.; Stevens, K.M.; Merkenschlager, M.; Warnecke, T. Chromatinization of Escherichia coli with archaeal histones. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Harwood, J.C.; Moore, K.M.; Paszkiewicz, K.; Durley, S.C.; Fukushima, H.; Atomi, H.; Takeyasu, K.; Kent, N.A. An alternative beads-on-a-string chromatin architecture in Thermococcus kodakarensis. EMBO Rep. 2013, 14, 711–717. [Google Scholar] [CrossRef]
- Mattiroli, F.; Bhattacharyya, S.; Dyer, P.N.; White, A.E.; Sandman, K.; Burkhart, B.W.; Byrne, K.R.; Lee, T.; Ahn, N.G.; Santangelo, T.J.; et al. Structure of histone-based chromatin in Archaea. Science 2017, 357, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Hocher, A.; Rojec, M.; Swadling, J.B.; Esin, A.; Warnecke, T. The DNA-binding protein HTa from Thermoplasma acidophilum is an archaeal histone analog. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Zivanovic, Y.; Armengaud, J.; Lagorce, A.; Leplat, C.; Guerin, P.; Dutertre, M.; Anthouard, V.; Forterre, P.; Wincker, P.; Confalonieri, F. Genome analysis and genome-wide proteomics of Thermococcus gammatolerans, the most radioresistant organism known amongst the Archaea. Genome Biol. 2009, 10, R70. [Google Scholar] [CrossRef]
- Hocher, A.; Borrel, G.; Fadhlaoui, K.; Brugère, J.-F.; Gribaldo, S.; Warnecke, T. Growth temperature is the principal driver of chromatinization in archaea. bioRxiv 2021. [Google Scholar] [CrossRef]
- McCarthy, R.L.; Kaeding, K.E.; Keller, S.H.; Zhong, Y.; Xu, L.; Hsieh, A.; Hou, Y.; Donahue, G.; Becker, J.S.; Alberto, O.; et al. Diverse heterochromatin-associated proteins repress distinct classes of genes and repetitive elements. Nat. Cell Biol. 2021, 23, 905–914. [Google Scholar] [CrossRef]
- Yoshimura, S.H.; Kim, J.; Takeyasu, K. On-substrate lysis treatment combined with scanning probe microscopy revealed chromosome structures in eukaryotes and prokaryotes. J. Electron. Microsc. 2003, 52, 415–423. [Google Scholar] [CrossRef]
- Hizume, K.; Yoshimura, S.H.; Maruyama, H.; Kim, J.; Wada, H.; Takeyasu, K. Chromatin reconstitution: Development of a salt-dialysis method monitored by nano-technology. Arch. Histol. Cytol. 2002, 65, 405–413. [Google Scholar] [CrossRef][Green Version]
- Okimune, K.I.; Nagy, S.K.; Hataya, S.; Endo, Y.; Takasuka, T.E. Reconstitution of Drosophila and human chromatins by wheat germ cell-free co-expression system. BMC Biotechnol. 2020, 20, 62. [Google Scholar] [CrossRef] [PubMed]
- Hizume, K.; Araki, S.; Yoshikawa, K.; Takeyasu, K. Topoisomerase II, scaffold component, promotes chromatin compaction in vitro in a linker-histone H1-dependent manner. Nucleic Acids Res. 2007, 35, 2787–2799. [Google Scholar] [CrossRef]
- Efremov, A.K.; Qu, Y.; Maruyama, H.; Lim, C.J.; Takeyasu, K.; Yan, J. Transcriptional Repressor TrmBL2 from Thermococcus kodakarensis Forms Filamentous Nucleoprotein Structures and Competes with Histones for DNA Binding in a Salt- and DNA Supercoiling-dependent Manner. J. Biol. Chem. 2015, 290, 15770–15784. [Google Scholar] [CrossRef] [PubMed]
- Birnie, A.; Dekker, C. Genome-in-a-Box: Building a Chromosome from the Bottom Up. ACS Nano 2021, 15, 111–124. [Google Scholar] [CrossRef]
- Jerkovic, I.; Szabo, Q.; Bantignies, F.; Cavalli, G. Higher-Order Chromosomal Structures Mediate Genome Function. J. Mol. Biol. 2020, 432, 676–681. [Google Scholar] [CrossRef]
- de Wit, E.; de Laat, W. A decade of 3C technologies: Insights into nuclear organization. Genes Dev. 2012, 26, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Gibcus, J.H.; Dekker, J. The hierarchy of the 3D genome. Mol. Cell 2013, 49, 773–782. [Google Scholar] [CrossRef]
- Davis, S.Z.; Hollin, T.; Lenz, T.; Le Roch, K.G. Three-dimensional chromatin in infectious disease-A role for gene regulation and pathogenicity? PLoS Pathog. 2021, 17, e1009207. [Google Scholar] [CrossRef]
- Nagano, T.; Lubling, Y.; Stevens, T.J.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 2013, 502, 59–64. [Google Scholar] [CrossRef]
- Yu, M.; Abnousi, A.; Zhang, Y.; Li, G.; Lee, L.; Chen, Z.; Fang, R.; Lagler, T.M.; Yang, Y.; Wen, J.; et al. SnapHiC: A computational pipeline to identify chromatin loops from single-cell Hi-C data. Nat. Methods 2021, 18, 1056–1059. [Google Scholar] [CrossRef]
- Zhou, T.; Zhang, R.; Ma, J. The 3D Genome Structure of Single Cells. Annu Rev. Biomed. Data Sci. 2021, 4, 21–41. [Google Scholar] [CrossRef]
- Lioy, V.S.; Cournac, A.; Marbouty, M.; Duigou, S.; Mozziconacci, J.; Espeli, O.; Boccard, F.; Koszul, R. Multiscale Structuring of the E. coli Chromosome by Nucleoid-Associated and Condensin Proteins. Cell 2018, 172, 771–783.e718. [Google Scholar] [CrossRef]
- Takemata, N.; Samson, R.Y.; Bell, S.D. Physical and Functional Compartmentalization of Archaeal Chromosomes. Cell 2019, 179, 165–179.e118. [Google Scholar] [CrossRef]
- van Berkum, N.L.; Lieberman-Aiden, E.; Williams, L.; Imakaev, M.; Gnirke, A.; Mirny, L.A.; Dekker, J.; Lander, E.S. Hi-C: A method to study the three-dimensional architecture of genomes. J. Vis. Exp. 2010, e1869. [Google Scholar] [CrossRef]
- Sandman, K.; Krzycki, J.A.; Dobrinski, B.; Lurz, R.; Reeve, J.N. HMf, a DNA-binding protein isolated from the hyperthermophilic archaeon Methanothermus fervidus, is most closely related to histones. Proc. Natl. Acad. Sci. USA 1990, 87, 5788–5791. [Google Scholar] [CrossRef] [PubMed]
- Henneman, B.; van Emmerik, C.; van Ingen, H.; Dame, R.T. Structure and function of archaeal histones. PLoS Genet. 2018, 14, e1007582. [Google Scholar] [CrossRef]
- Stevens, K.M.; Swadling, J.B.; Hocher, A.; Bang, C.; Gribaldo, S.; Schmitz, R.A.; Warnecke, T. Histone variants in archaea and the evolution of combinatorial chromatin complexity. Proc. Natl. Acad. Sci. USA 2020, 117, 33384–33395. [Google Scholar] [CrossRef]
- Alpha-Bazin, B.; Gorlas, A.; Lagorce, A.; Joulie, D.; Boyer, J.B.; Dutertre, M.; Gaillard, J.C.; Lopes, A.; Zivanovic, Y.; Dedieu, A.; et al. Lysine-specific acetylated proteome from the archaeon Thermococcus gammatolerans reveals the presence of acetylated histones. J. Proteom. 2021, 232, 104044. [Google Scholar] [CrossRef]
- Bowerman, S.; Wereszczynski, J.; Luger, K. Archaeal chromatin ‘slinkies’ are inherently dynamic complexes with deflected DNA wrapping pathways. Elife 2021, 10. [Google Scholar] [CrossRef]
- Sanders, T.J.; Ullah, F.; Gehring, A.M.; Burkhart, B.W.; Vickerman, R.L.; Fernando, S.; Gardner, A.F.; Ben-Hur, A.; Santangelo, T.J. Extended Archaeal Histone-Based Chromatin Structure Regulates Global Gene Expression in Thermococcus kodakarensis. Front. Microbiol. 2021, 12, 681150. [Google Scholar] [CrossRef]
- Henneman, B.; Brouwer, T.B.; Erkelens, A.M.; Kuijntjes, G.-J.; van Emmerik, C.; van der Valk, R.A.; Timmer, M.; Kirolos, N.C.S.; van Ingen, H.; van Noort, J.; et al. Mechanical and structural properties of archaeal hypernucleosomes. Nucleic Acids Res. 2020, 49, 4338–4349. [Google Scholar] [CrossRef]
- Koyama, M.; Kurumizaka, H. Structural diversity of the nucleosome. J. Biochem. 2018, 163, 85–95. [Google Scholar] [CrossRef]
- Goyal, M.; Banerjee, C.; Nag, S.; Bandyopadhyay, U. The Alba protein family: Structure and function. Biochim. Biophys. Acta 2016, 1864, 570–583. [Google Scholar] [CrossRef]
- Laurens, N.; Driessen, R.P.; Heller, I.; Vorselen, D.; Noom, M.C.; Hol, F.J.; White, M.F.; Dame, R.T.; Wuite, G.J. Alba shapes the archaeal genome using a delicate balance of bridging and stiffening the DNA. Nat. Commun. 2012, 3, 1328. [Google Scholar] [CrossRef] [PubMed]
- Ohniwa, R.L.; Muchaku, H.; Saito, S.; Wada, C.; Morikawa, K. Atomic Force Microscopy Analysis of the Role of Major DNA-Binding Proteins in Organization of the Nucleoid in Escherichia coli. PLoS ONE 2013, 8, e72954. [Google Scholar] [CrossRef] [PubMed]
- Stojkova, P.; Spidlova, P.; Stulik, J. Nucleoid-Associated Protein HU: A Lilliputian in Gene Regulation of Bacterial Virulence. Front. Cell Infect. Microbiol. 2019, 9, 159. [Google Scholar] [CrossRef]
- Bettridge, K.; Verma, S.; Weng, X.; Adhya, S.; Xiao, J. Single-molecule tracking reveals that the nucleoid-associated protein HU plays a dual role in maintaining proper nucleoid volume through differential interactions with chromosomal DNA. Mol. Microbiol. 2021, 115, 12–27. [Google Scholar] [CrossRef]
- Searcy, D.G.; Stein, D.B. Nucleoprotein subunit structure in an unusual prokaryotic organism: Thermoplasma acidophilum. Biochim. Biophys. Acta 1980, 609, 180–195. [Google Scholar] [CrossRef]
- López-García, P.; Zivanovic, Y.; Deschamps, P.; Moreira, D. Bacterial gene import and mesophilic adaptation in archaea. Nat. Rev. Microbiol. 2015, 13, 447–456. [Google Scholar] [CrossRef]
- Wagner, A.; Whitaker, R.J.; Krause, D.J.; Heilers, J.H.; van Wolferen, M.; van der Does, C.; Albers, S.V. Mechanisms of gene flow in archaea. Nat. Rev. Microbiol. 2017, 15, 492–501. [Google Scholar] [CrossRef]
- Lim, C.J.; Lee, S.Y.; Kenney, L.J.; Yan, J. Nucleoprotein filament formation is the structural basis for bacterial protein H-NS gene silencing. Sci. Rep. 2012, 2, 509. [Google Scholar] [CrossRef]
- Dorman, C.J. H-NS, the genome sentinel. Nat. Rev. Microbiol. 2007, 5, 157–161. [Google Scholar] [CrossRef]
- Wiedenbeck, J.; Cohan, F.M. Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol. Rev. 2011, 35, 957–976. [Google Scholar] [CrossRef]
- Maruyama, H.; Kent, N.A.; Nishida, H.; Oshima, T. Functions of Archaeal Nucleoid Proteins: Archaeal Silencers are Still Missing. In DNA Traffic in the Environment; Springer: Amsterdam, The Netherlands, 2019; pp. 29–45. [Google Scholar]
- Nalabothula, N.; Xi, L.; Bhattacharyya, S.; Widom, J.; Wang, J.P.; Reeve, J.N.; Santangelo, T.J.; Fondufe-Mittendorf, Y.N. Archaeal nucleosome positioning in vivo and in vitro is directed by primary sequence motifs. BMC Genom. 2013, 14, 391. [Google Scholar] [CrossRef]
- Husnik, F.; McCutcheon, J.P. Functional horizontal gene transfer from bacteria to eukaryotes. Nat. Rev. Microbiol. 2018, 16, 67–79. [Google Scholar] [CrossRef]
- Etheridge, T.J.; Villahermosa, D.; Campillo-Funollet, E.; Herbert, A.D.; Irmisch, A.; Watson, A.T.; Dang, H.Q.; Osborne, M.A.; Oliver, A.W.; Carr, A.M.; et al. Live-cell single-molecule tracking highlights requirements for stable Smc5/6 chromatin association in vivo. eLife 2021, 10, e68579. [Google Scholar] [CrossRef]
- Yoshimura, S.H.; Hizume, K.; Murakami, A.; Sutani, T.; Takeyasu, K.; Yanagida, M. Condensin architecture and interaction with DNA: Regulatory non-SMC subunits bind to the head of SMC heterodimer. Curr. Biol. 2002, 12, 508–513. [Google Scholar] [CrossRef]
- Sakai, A.; Hizume, K.; Sutani, T.; Takeyasu, K.; Yanagida, M. Condensin but not cohesin SMC heterodimer induces DNA reannealing through protein-protein assembly. EMBO J. 2003, 22, 2764–2775. [Google Scholar] [CrossRef]
- Davidson, I.F.; Peters, J.M. Genome folding through loop extrusion by SMC complexes. Nat. Rev. Mol. Cell Biol. 2021, 22, 445–464. [Google Scholar] [CrossRef]
- Bauer, B.W.; Davidson, I.F.; Canena, D.; Wutz, G.; Tang, W.; Litos, G.; Horn, S.; Hinterdorfer, P.; Peters, J.M. Cohesin mediates DNA loop extrusion by a “swing and clamp” mechanism. Cell 2021, 184, 5448–5464.e5422. [Google Scholar] [CrossRef]
- Nolivos, S.; Sherratt, D. The bacterial chromosome: Architecture and action of bacterial SMC and SMC-like complexes. FEMS Microbiol. Rev. 2014, 38, 380–392. [Google Scholar] [CrossRef]
- Schibany, S.; Kleine Borgmann, L.A.K.; Rösch, T.C.; Knust, T.; Ulbrich, M.H.; Graumann, P.L. Single molecule tracking reveals that the bacterial SMC complex moves slowly relative to the diffusion of the chromosome. Nucleic Acids Res. 2018, 46, 7805–7819. [Google Scholar] [CrossRef]
- Schibany, S.; Hinrichs, R.; Hernández-Tamayo, R.; Graumann, P.L.; Ellermeier, C.D. The Major Chromosome Condensation Factors Smc, HBsu, and Gyrase in Bacillus subtilis Operate via Strikingly Different Patterns of Motion. mSphere 2020, 5, e00817–e00820. [Google Scholar] [CrossRef]
- Burmann, F.; Funke, L.F.H.; Chin, J.W.; Lowe, J. Cryo-EM structure of MukBEF reveals DNA loop entrapment at chromosomal unloading sites. Mol. Cell 2021, 81, 4891–4906.e4898. [Google Scholar] [CrossRef]
- Hirano, T. Condensin-Based Chromosome Organization from Bacteria to Vertebrates. Cell 2016, 164, 847–857. [Google Scholar] [CrossRef]
- Cockram, C.; Thierry, A.; Gorlas, A.; Lestini, R.; Koszul, R. Euryarchaeal genomes are folded into SMC-dependent loops and domains, but lack transcription-mediated compartmentalization. Mol. Cell 2021, 81, 459–472.e410. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maruyama, H.; Nambu, T.; Mashimo, C.; Okinaga, T.; Takeyasu, K. Single-Molecule/Cell Analyses Reveal Principles of Genome-Folding Mechanisms in the Three Domains of Life. Int. J. Mol. Sci. 2021, 22, 13432. https://doi.org/10.3390/ijms222413432
Maruyama H, Nambu T, Mashimo C, Okinaga T, Takeyasu K. Single-Molecule/Cell Analyses Reveal Principles of Genome-Folding Mechanisms in the Three Domains of Life. International Journal of Molecular Sciences. 2021; 22(24):13432. https://doi.org/10.3390/ijms222413432
Chicago/Turabian StyleMaruyama, Hugo, Takayuki Nambu, Chiho Mashimo, Toshinori Okinaga, and Kunio Takeyasu. 2021. "Single-Molecule/Cell Analyses Reveal Principles of Genome-Folding Mechanisms in the Three Domains of Life" International Journal of Molecular Sciences 22, no. 24: 13432. https://doi.org/10.3390/ijms222413432
APA StyleMaruyama, H., Nambu, T., Mashimo, C., Okinaga, T., & Takeyasu, K. (2021). Single-Molecule/Cell Analyses Reveal Principles of Genome-Folding Mechanisms in the Three Domains of Life. International Journal of Molecular Sciences, 22(24), 13432. https://doi.org/10.3390/ijms222413432