
Carbonic Anhydrase IX Inhibitors as Candidates for Combination Therapy of Solid Tumors

 ,
,  , ,
, ,  and
and
Abstract
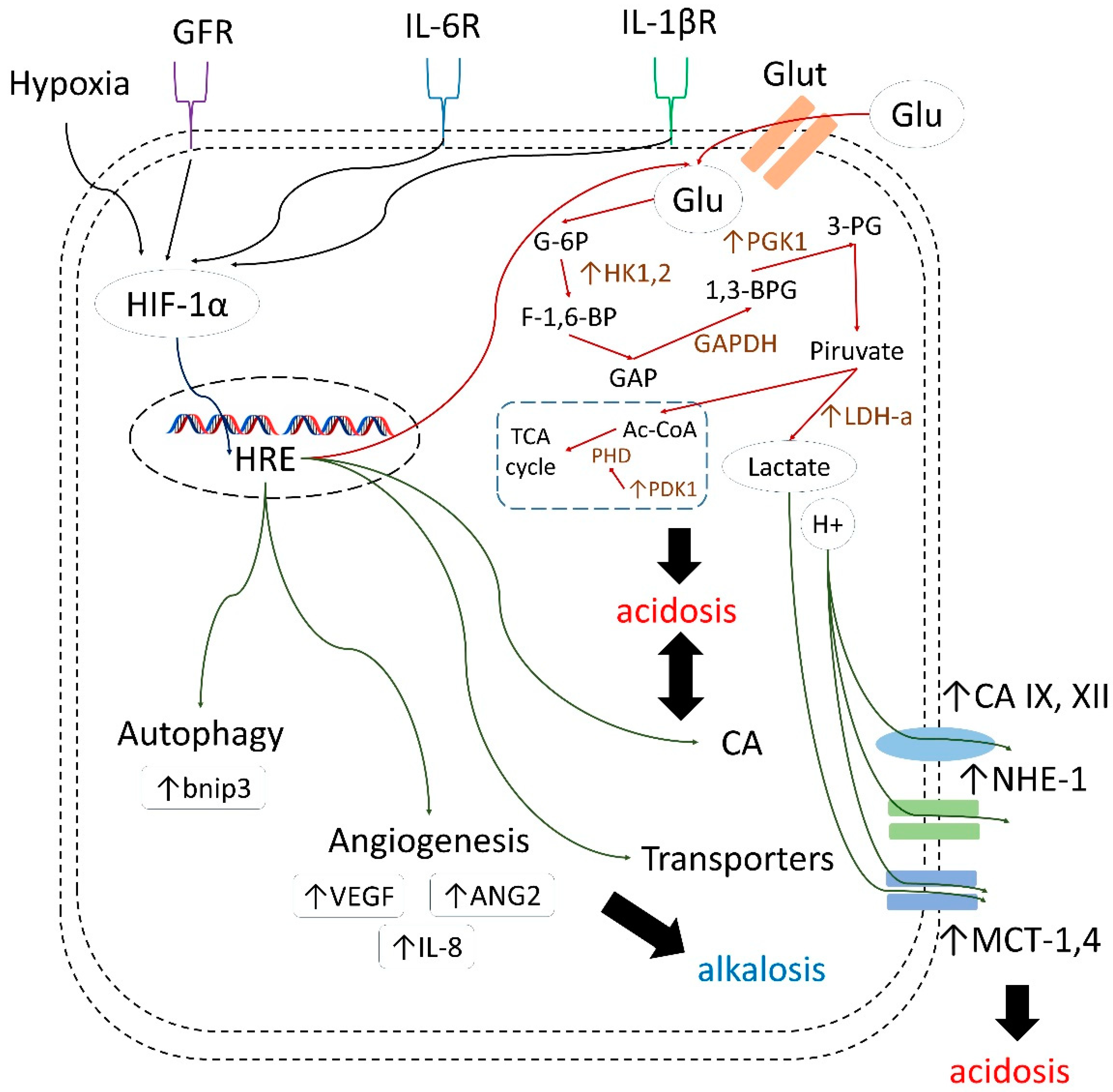
1. Introduction
- decreasing drug uptake in tumor cells due to the altered protonation state;
- selecting for highly aggressive and drug-resistant phenotypes possessing stem cell characteristics, and harboring protumorigenic mutations associated with poor treatment response;
- enhancing invasion and metastatic processes through apoptosis induction in surrounding cells, elevated secretion of proteinases, and the disruption of E-cadherin-mediated cell adhesion;
- inducing angiogenesis via activating VEGF (vascular endothelial growth factor) production;
- decreasing immune infiltration of tumors.
2. Carbonic Anhydrase Inhibitors/Anticancer Agent Combinations
2.1. Acetazolamide in Combination with Conventional Cytostatic Agents
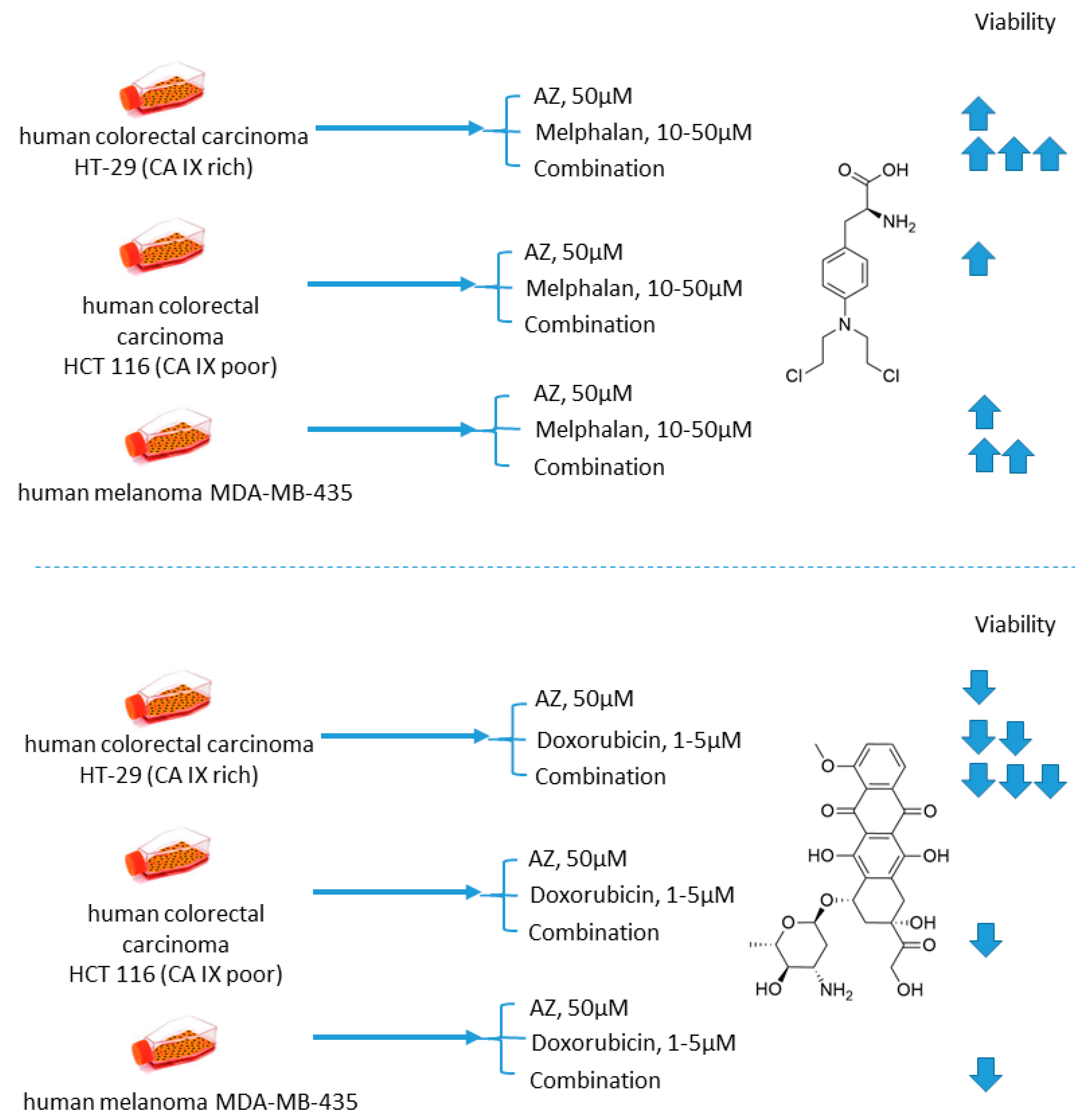
2.1.1. Acetazolamide and Doxorubicin
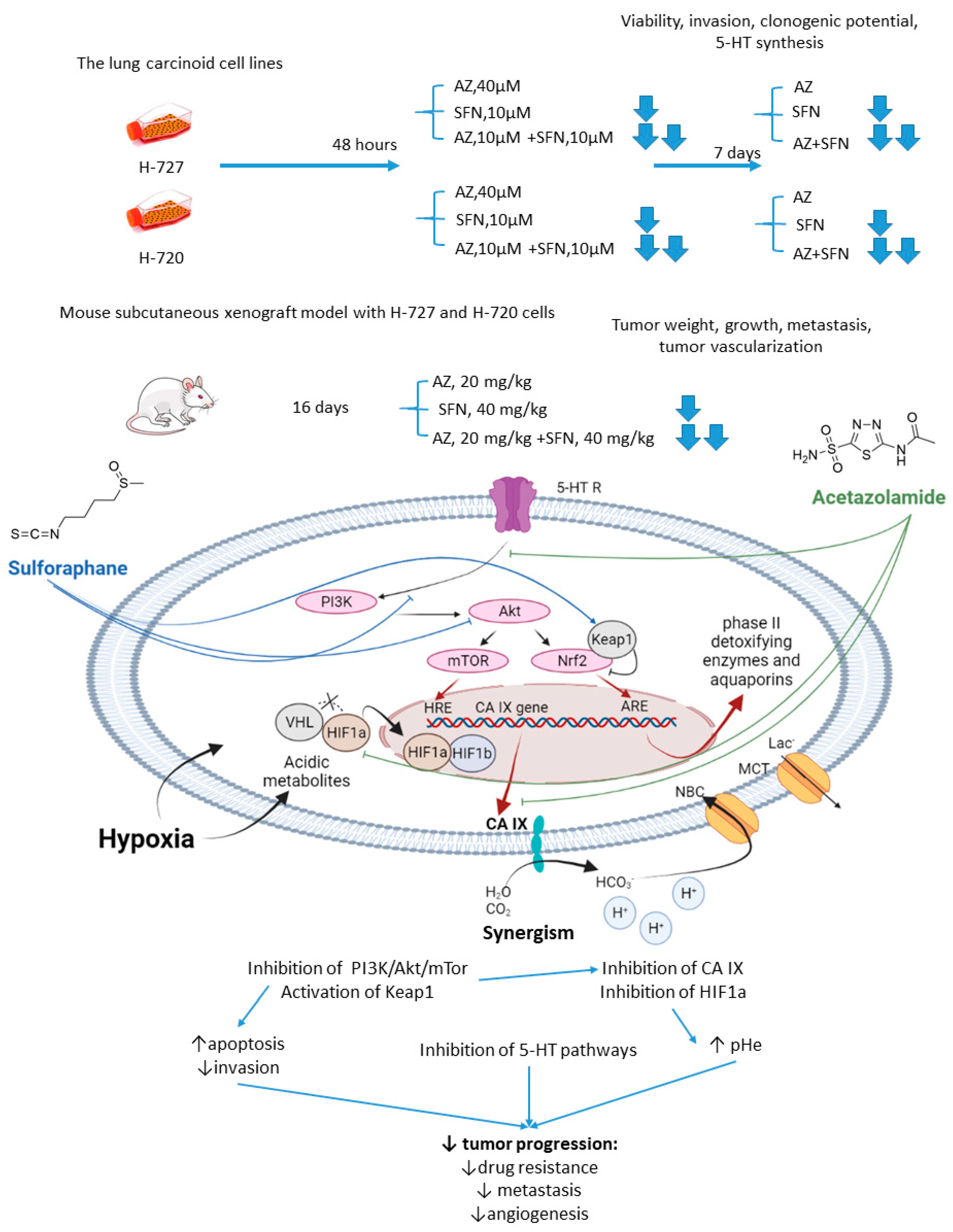
2.1.2. Acetazolamide and Sulforaphane
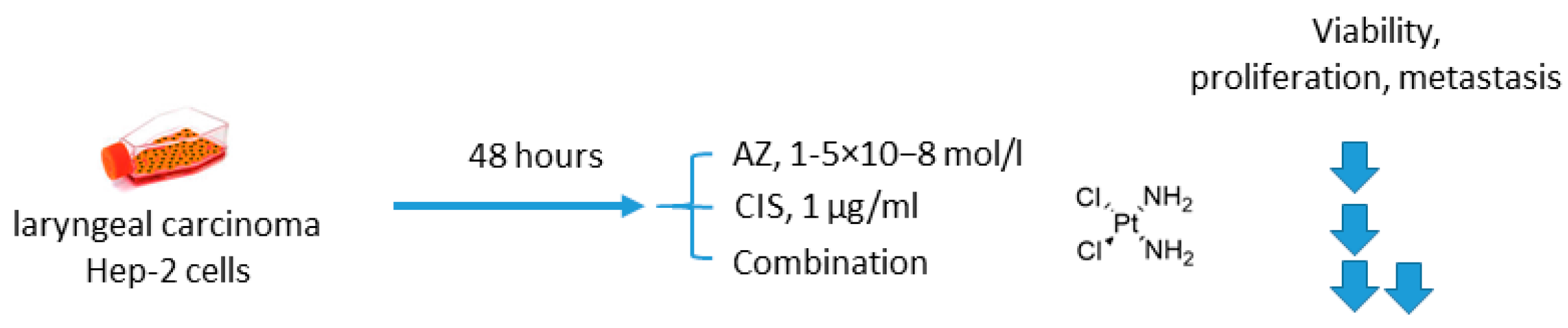
2.1.3. Acetazolamide and Cisplatin
2.2. Acetazolamide in Combination with Targeted Anticancer Drugs
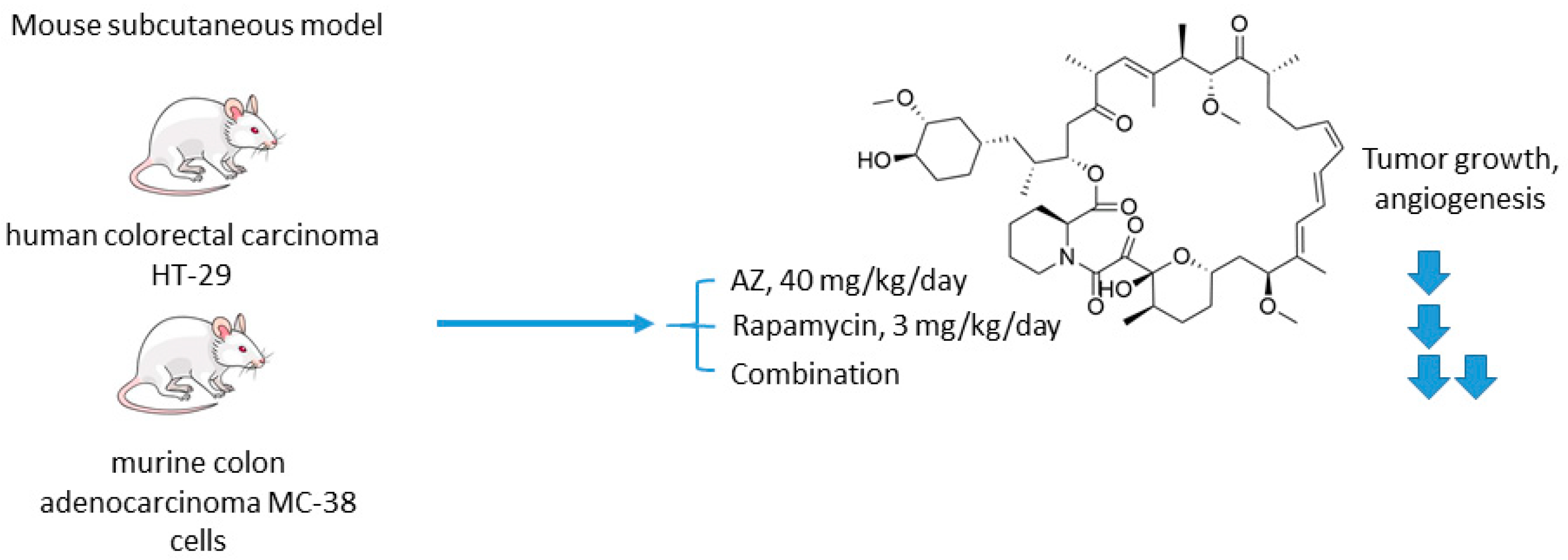
2.2.1. Acetazolamide and Rapamycin
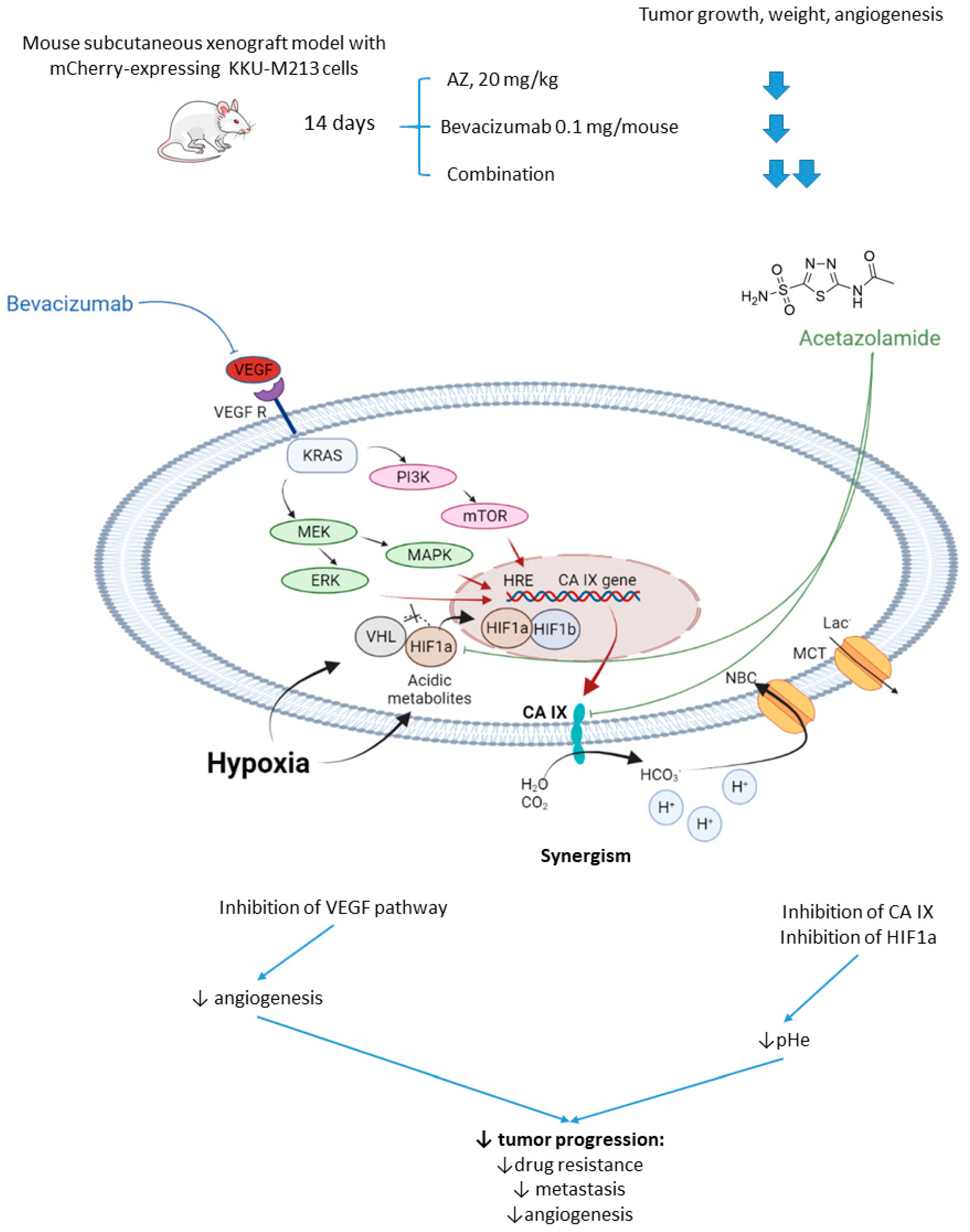
2.2.2. Acetazolamide and Bevacizumab
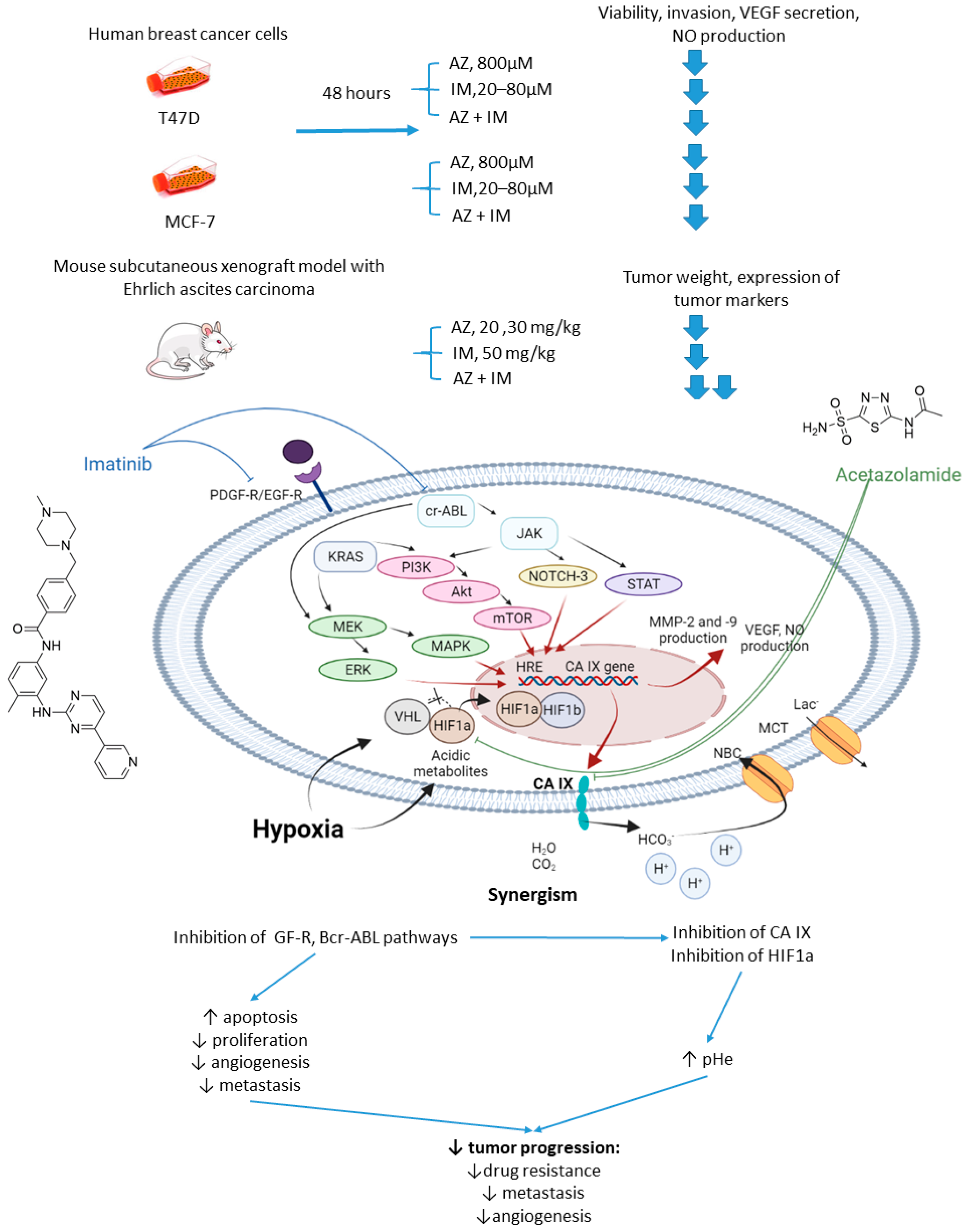
2.2.3. Acetazolamide and Imatinib
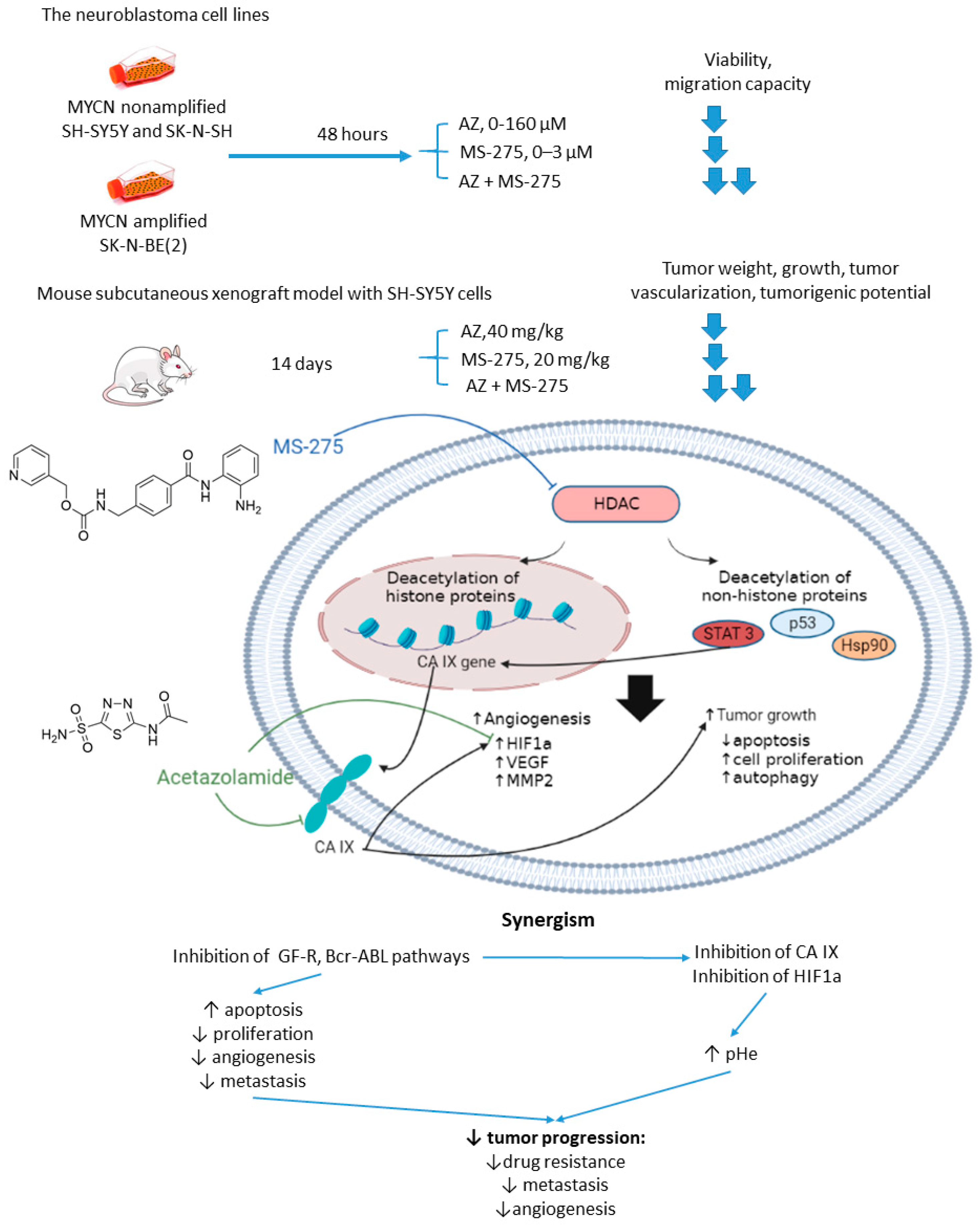
2.2.4. Acetazolamide and MS-275
2.3. Acetazolamide in Multidrug Combinations
Acetazolamide and CHOP Combination
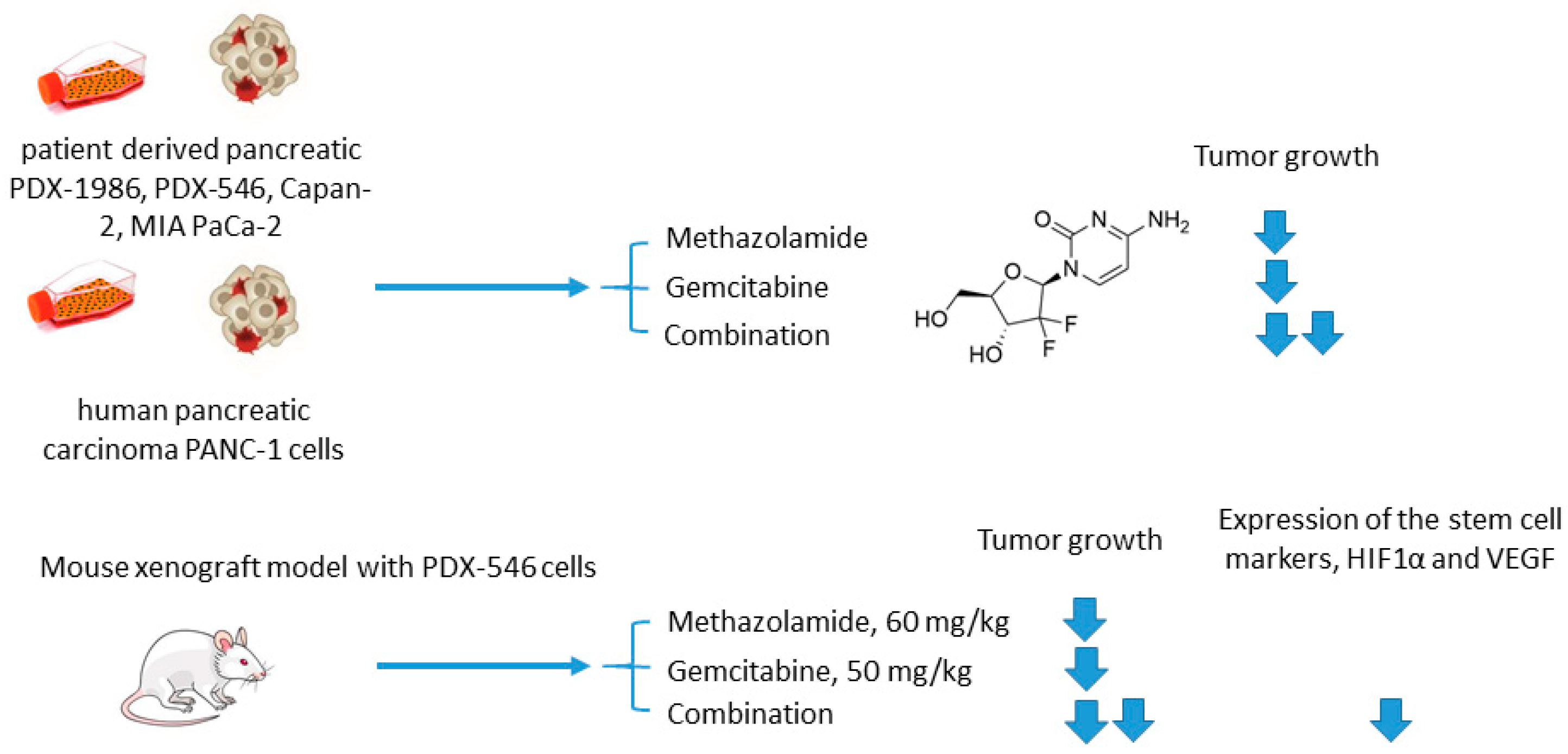
2.4. Methazolamide in Combination with Conventional Cytostatic Agents
Methazolamide and Gemcitabine
2.5. Isoform Selective Carbonic Anhydrase IX Inhibitors in Combination with Conventional Cytostatic Agents
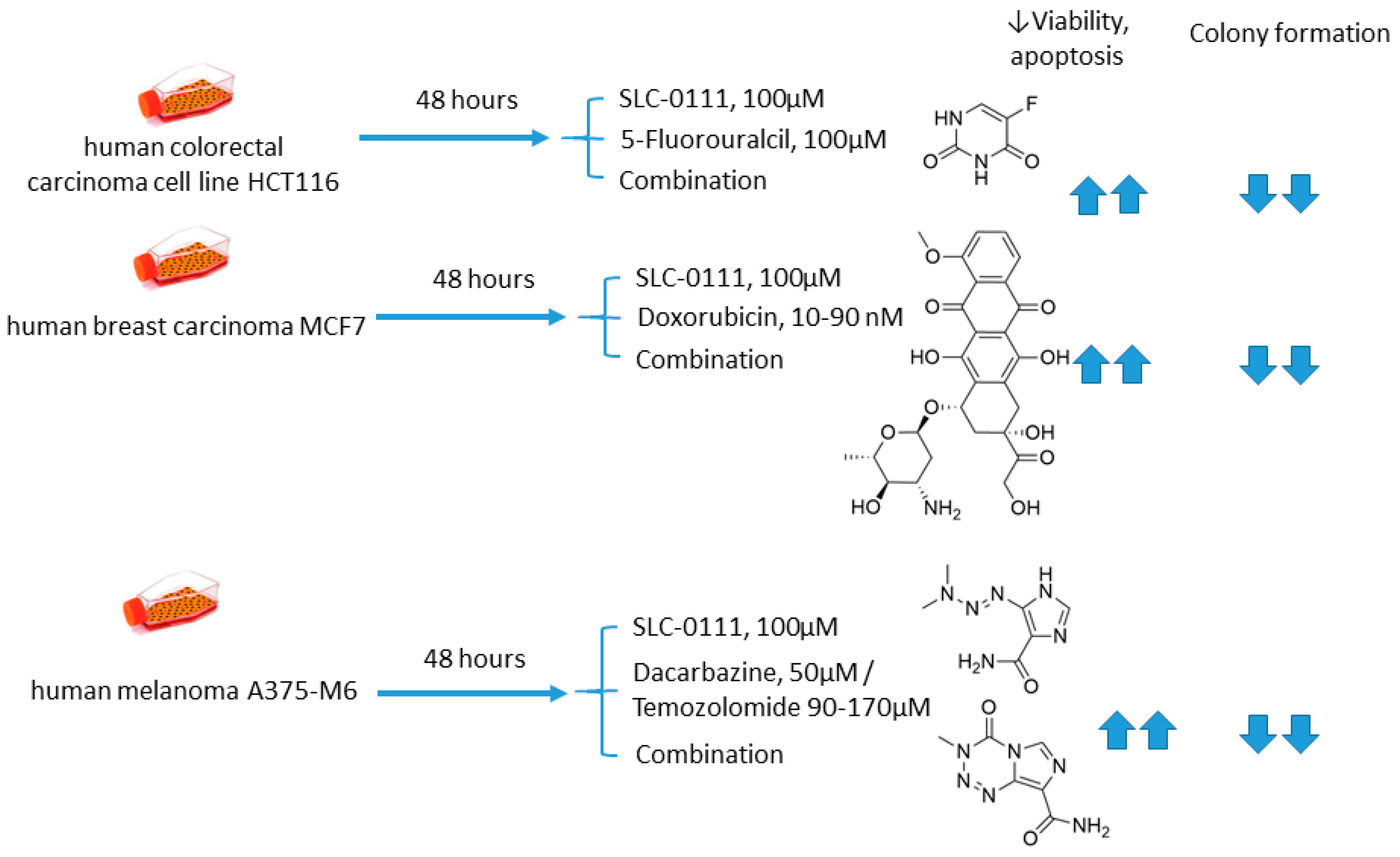
2.5.1. SLC-0111 and Dacarbazine/Temozolomide/Doxorubicin/5-Fluorouracil
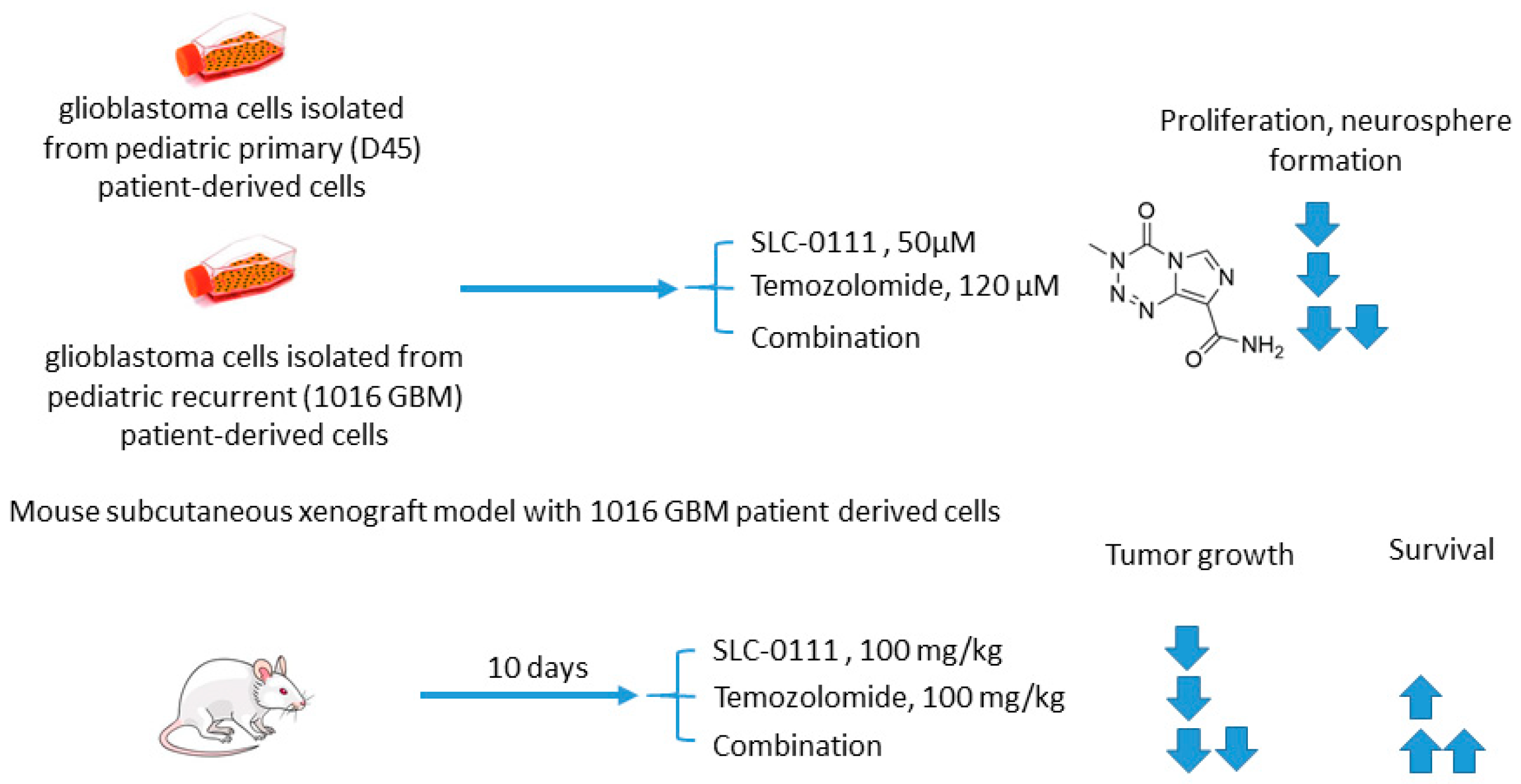
2.5.2. SLC-0111 and Temozolomide
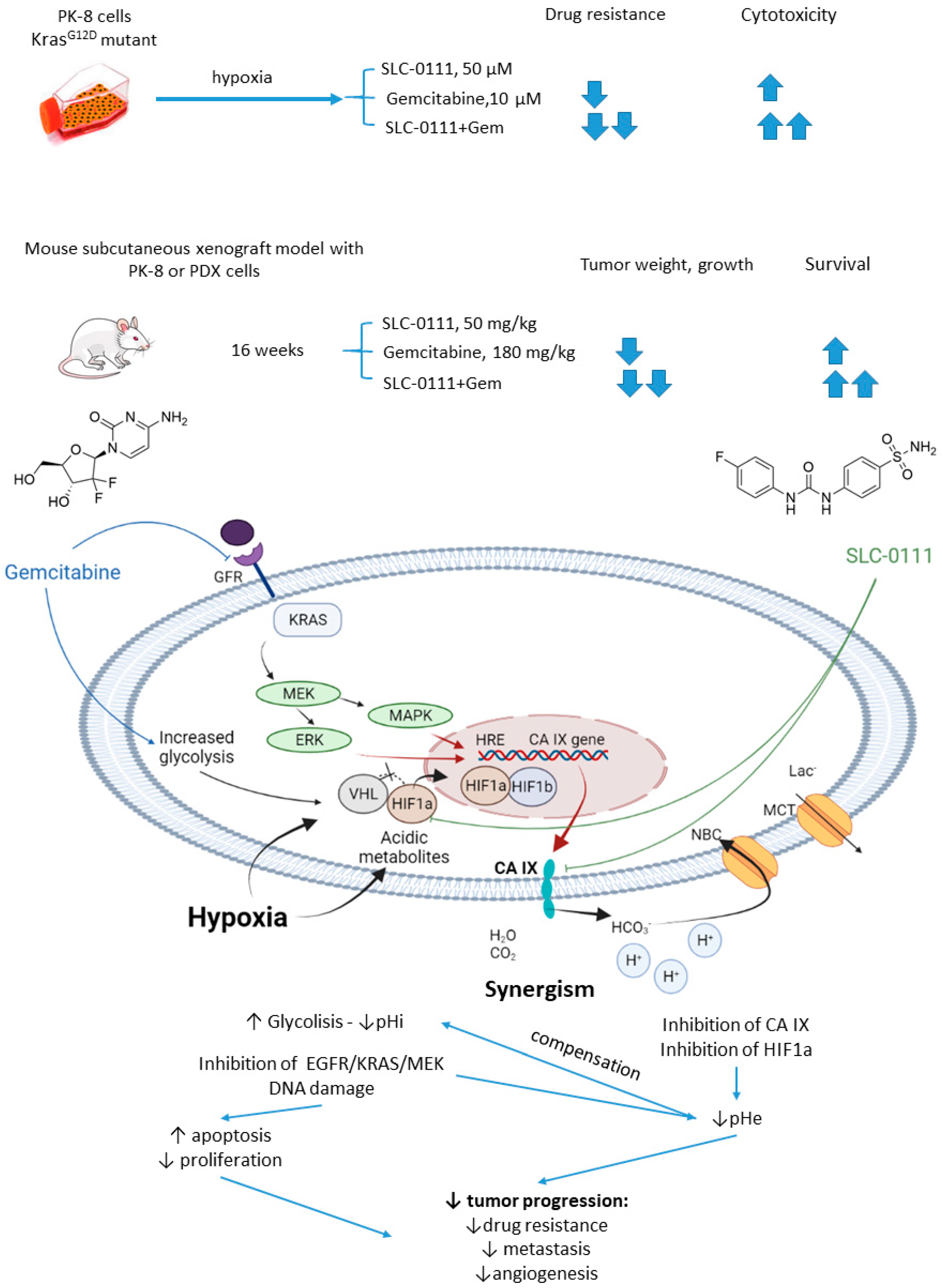
2.5.3. SLC-0111 and Gemcitabine
2.5.4. S4 and Doxorubicin
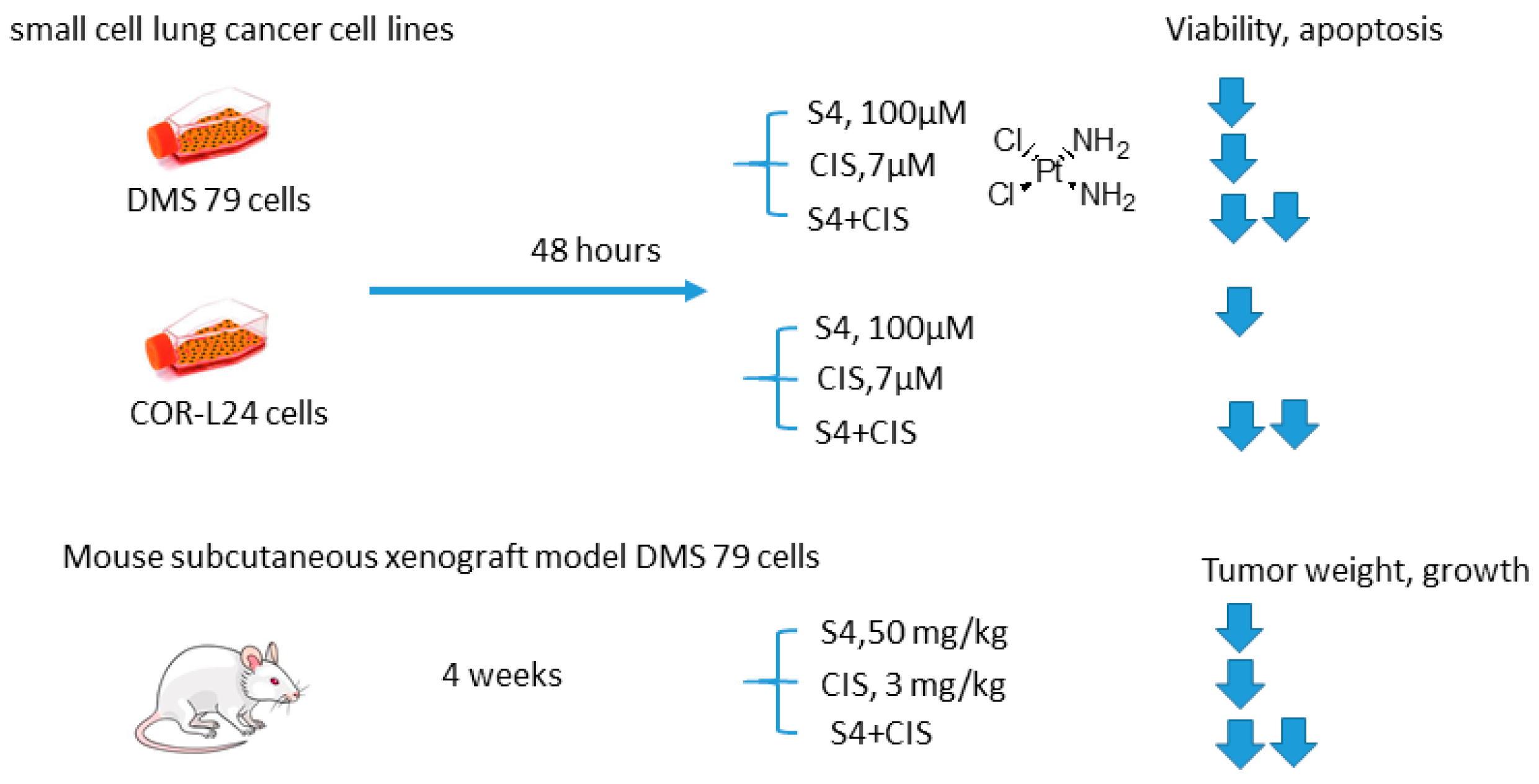
2.5.5. S4 and Cisplatin
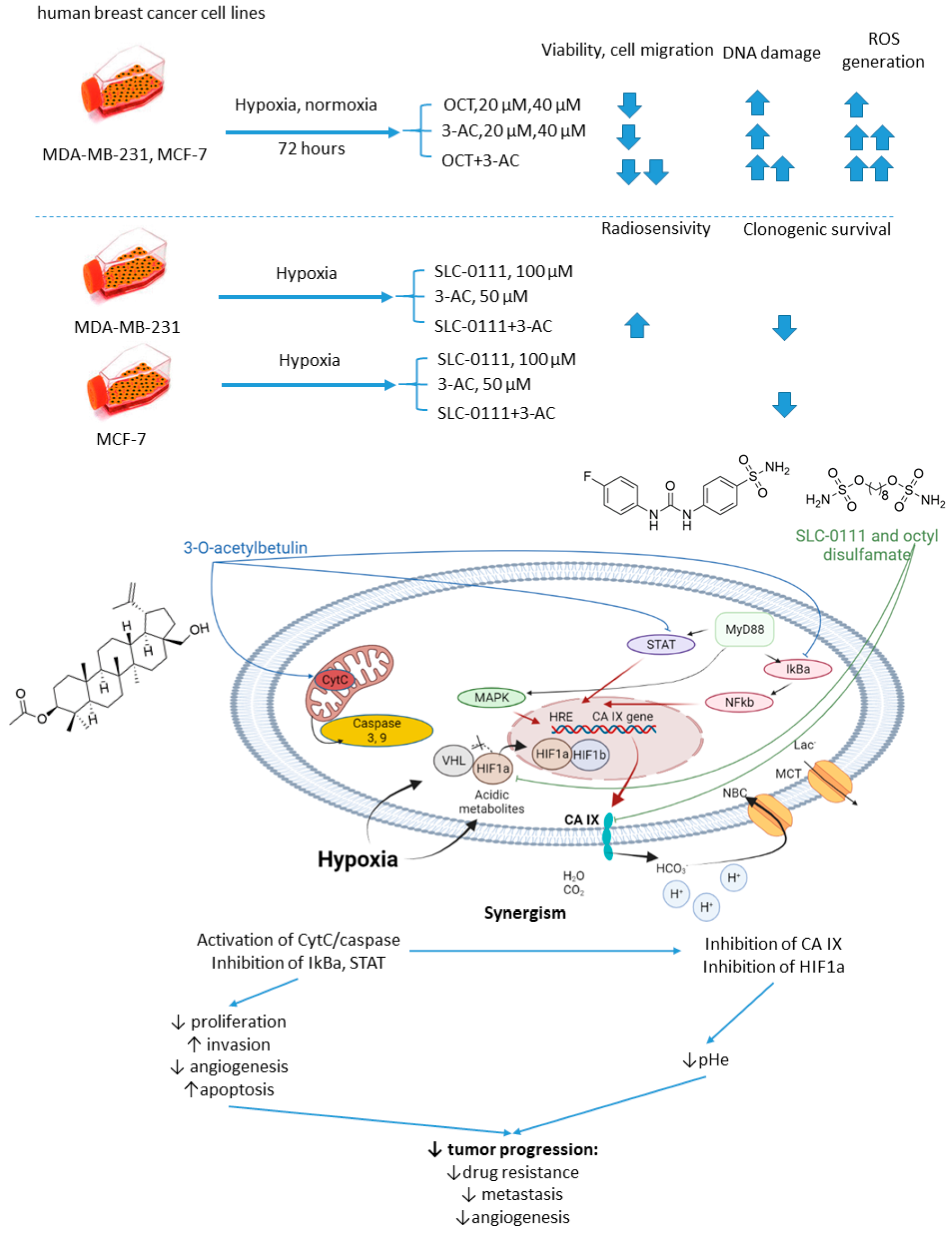
2.5.6. n-Octyl Disulfamate/SCL-0111 and 3-O-acetylbetulin
2.6. Isoform Selective Carbonic Anhydrase IX Inhibitors in Combination with Targeted Anticancer Drugs
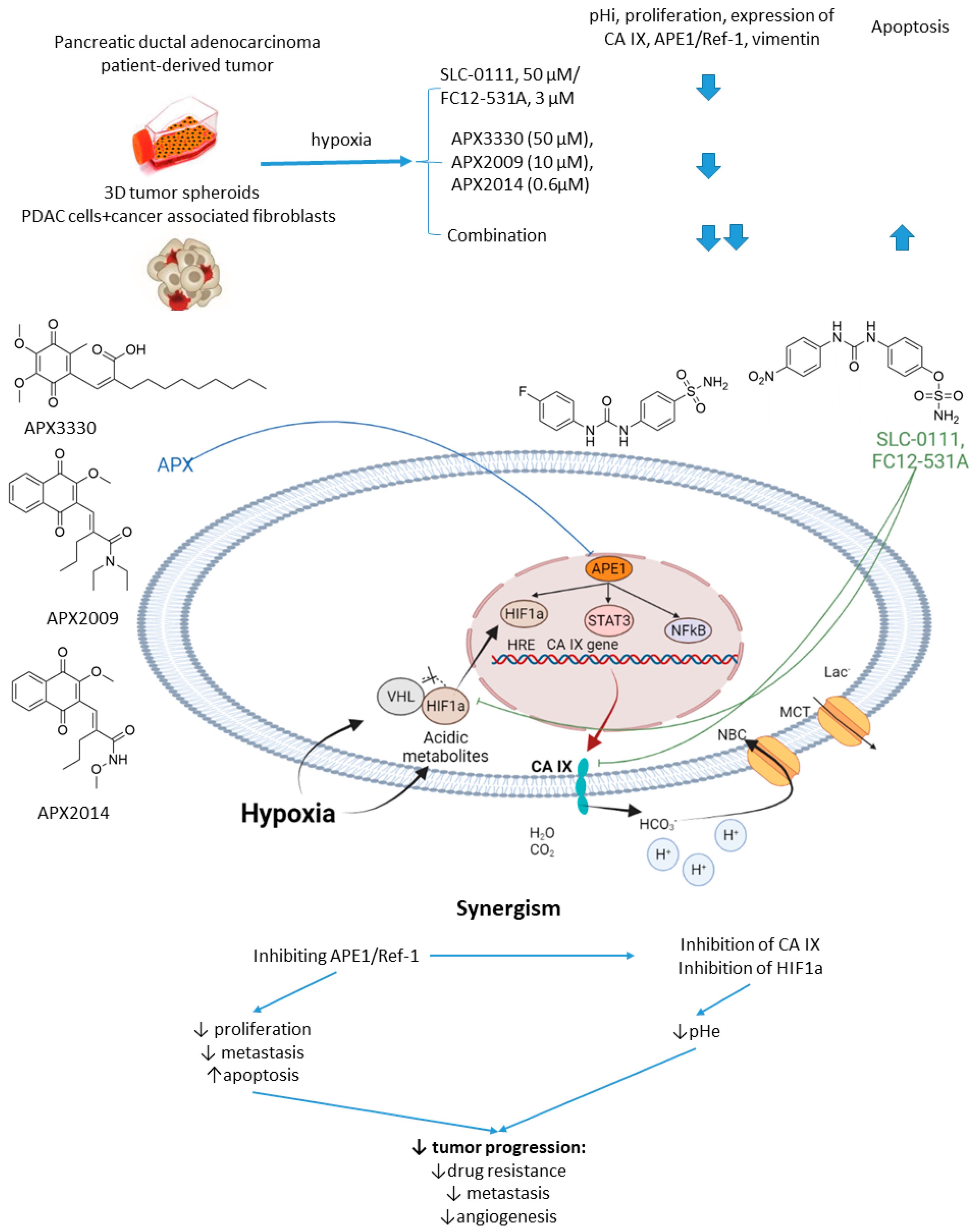
2.6.1. SCL-0111 and APE1/Ref-1 Inhibitors
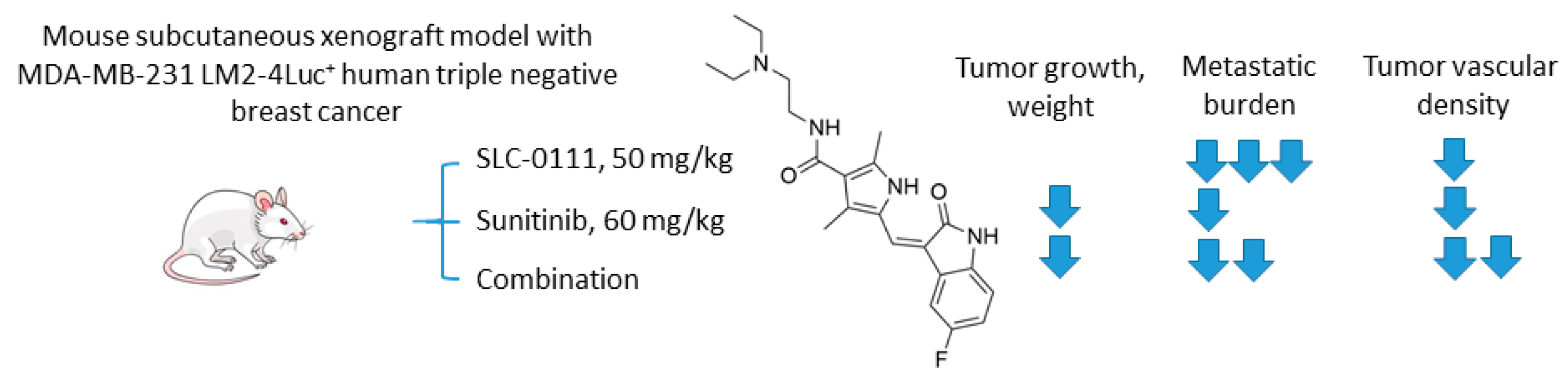
2.6.2. SCL-0111 and Sunitinib
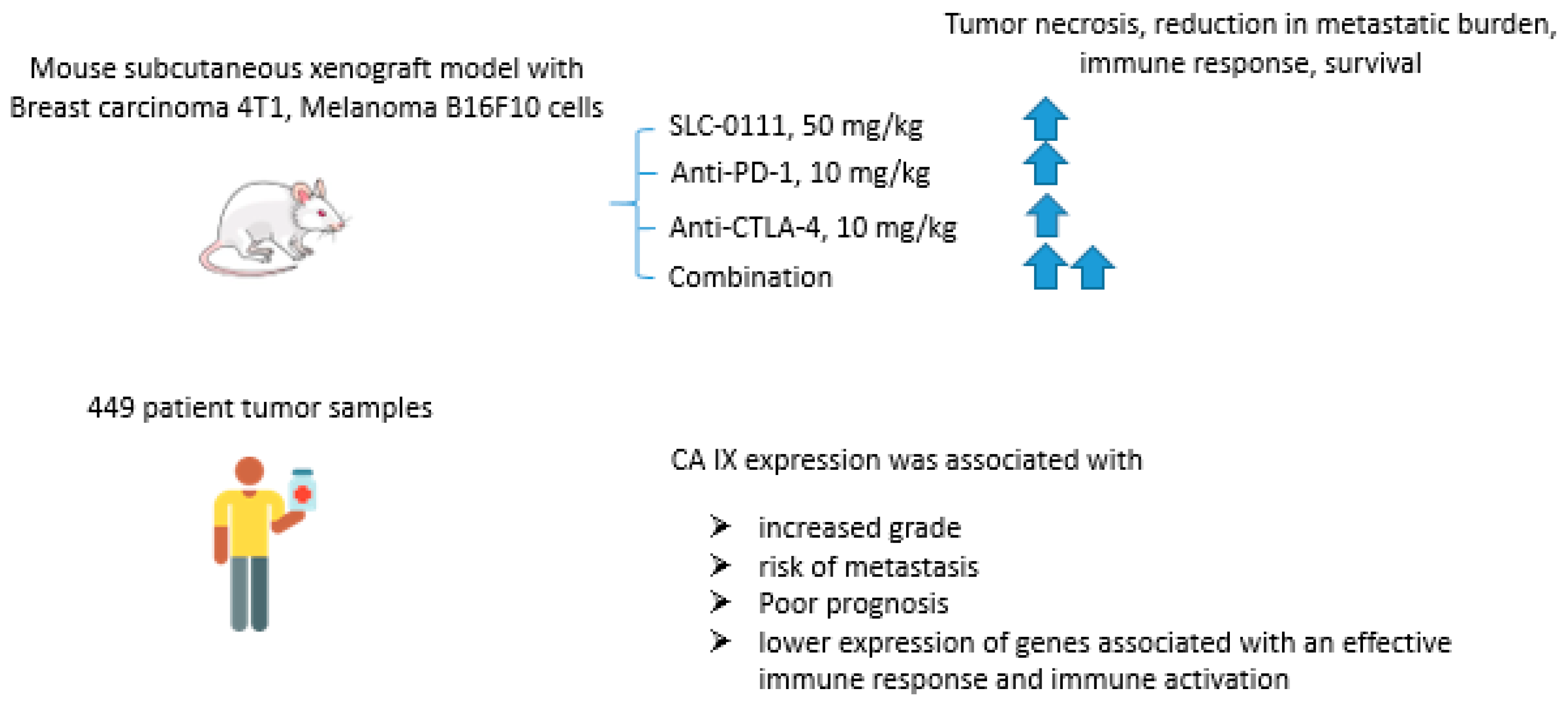
2.6.3. SCL-0111 and Immune Checkpoint Blockade Inhibitors
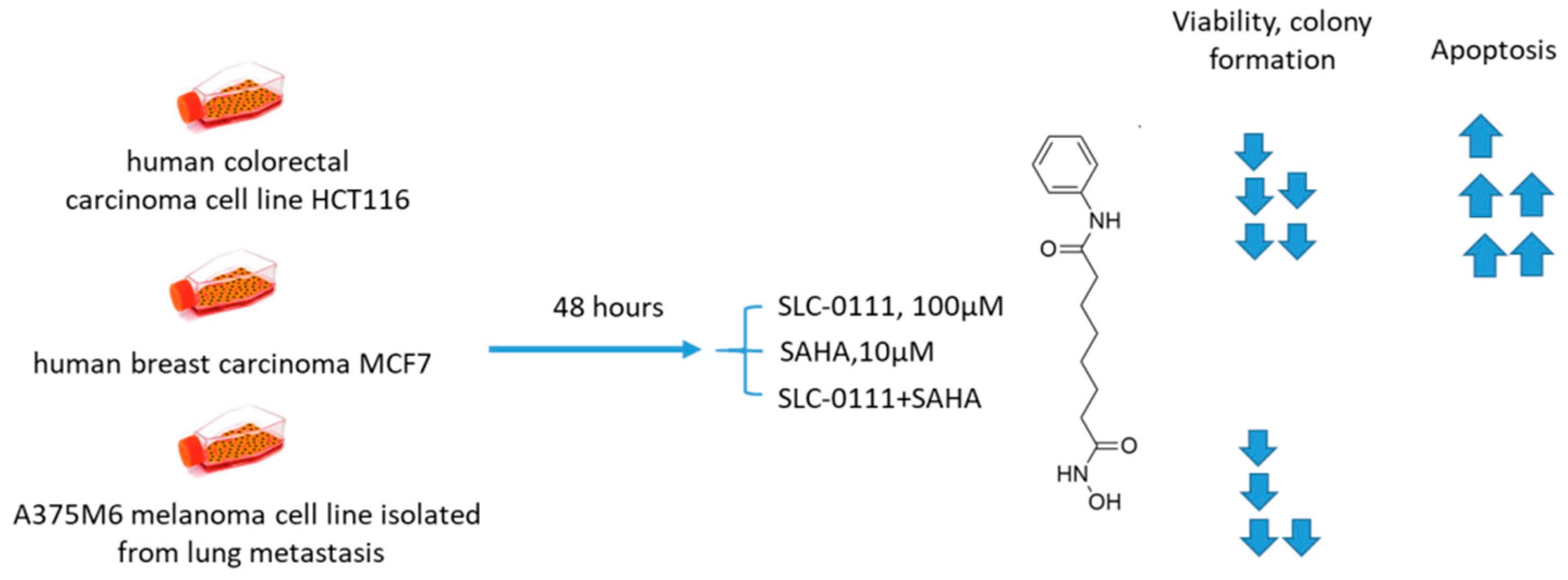
2.6.4. SCL-0111 and Suberoylanilide Hydroxamic Acid
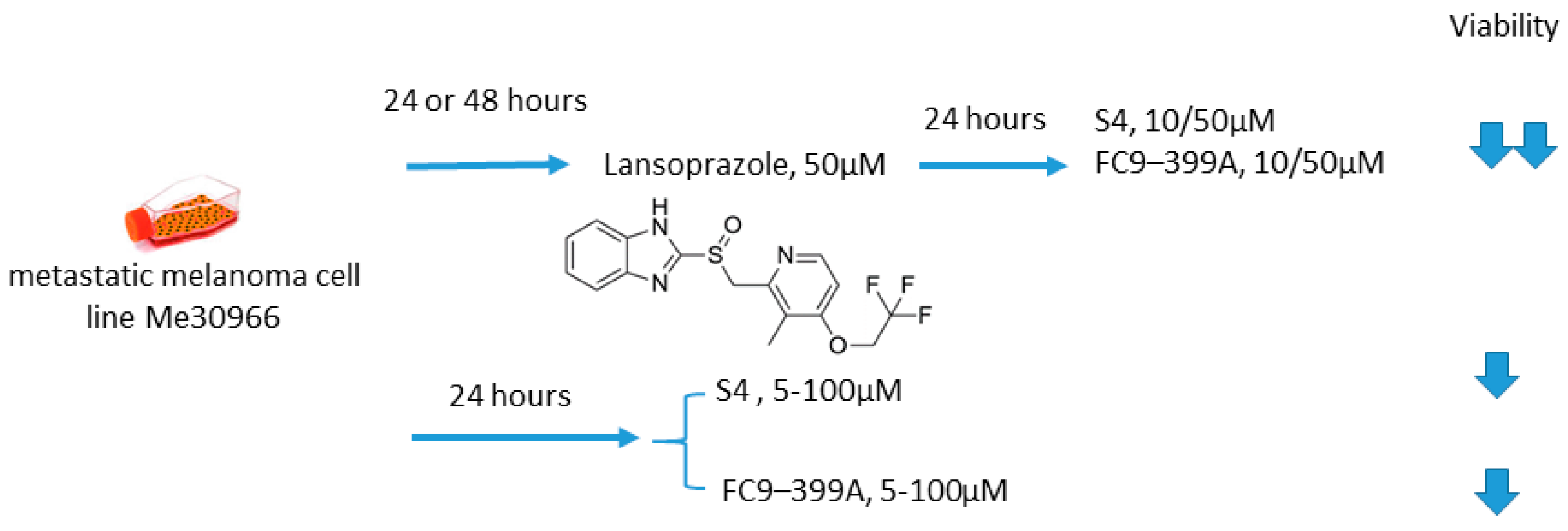
2.6.5. S4/FC9–399A and Proton Pump Inhibitors
3. Discussion and Perspectives
- the cytoskeleton;
- cell cycle and mitosis, ribosome biogenesis, RNA processing, DNA damage repair, and nucleic acid metabolism;
- mitochondrial organization;
- redox homeostasis: thioredoxin, glutathione S-transferases, glutathione reductase, and the molybdenum cofactor and iron-sulfur cluster biogenesis: the cysteine desulfurase (NFS1) and iron-sulfur cluster scaffold protein;
- iron-sulfur cluster adapter protein, adenosine 5′-triphosphate–binding cassette subfamily B member 7, and glutaredoxin 5 [103].
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 1,3-BPG | 1,3-Bisphosphoglycerate |
| 3-PG | 3-phosphoglyceric acid |
| Ac-CoA | acetyl coenzyme A |
| Akt (PKB) | protein kinase B |
| ANG2 | angiopoietin 2 |
| APE1/Ref-1 | apurinic/apyrimidinic endonuclease-1/redox effector factor 1 |
| AZ | acetazolamide |
| Bnip3 | Bcl-2/adenovirus EIB 19-kD interacting protein 3 |
| BTIC | brain tumor initiating cell |
| CA | carboanhydrase |
| CA IX | carbonic anhydrase IX |
| CAIs | CA inhibitors |
| CHOP | cyclophosphamide, doxorubicin hydrochloride hydroxydaunorubicin, vincristine sulfate Oncovin, and prednisone |
| CIS | cisplatin |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| DOX | doxorubicin |
| EAC | Ehrlich ascites carcinoma |
| EGFR | epidermal growth factor receptor |
| ERK | extracellular signal-regulated kinase |
| EV1 | empty vector |
| F-1,6-BP | fructose-1,6-bisphosphate |
| G-6P | glucose -6 phosphate |
| GAP | glyceraldehyde-3-phosphate |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| GFR | growth factor receptor |
| GLU | glucose |
| GLUT | glucose transporter |
| GSH | glutathione |
| HDAC | histone deacetylases |
| HIF-1a | hypoxia-inducible factor 1a |
| HK | hexokinase |
| HRE | hypoxia response element |
| HUVEC | human umbilical vein endothelial cells |
| IL | interleukin |
| IM | imatinib |
| JAK | Janus kinase |
| KEAP1 | Kelch Like ECH Associated Protein 1 |
| KRAS | Kirsten rat sarcoma |
| LDH-a | lactic dehydrogenase-a |
| MAPK | mitogen activated protein kinase |
| MCT | monocarboxylate transporters |
| MEK | MAPK/ERK kinase |
| MMP | matrix metalloproteinases |
| mTOR | mechanistic target of rapamycin |
| mTORC | mTOR complexe |
| MyD88 | myeloid differentiation primary response gene (88) |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NFS1 | iron-sulfur cluster biogenesis: the cysteine desulfurase |
| NHE | Na+,H+-exchanger |
| OCT | n-octyl disulfamate |
| PARP | poly(ADP-ribose) polymerase |
| PD-1 | programmed cell death protein 1 |
| PDGFR | platelet-derived growth factor receptor |
| PDK1 | pyruvate dehydrogenase kinase 1 |
| PGK1 | phosphoglycerate kinase 1 |
| PHD | prolyl hydroxylases |
| pHe | extracellular pH |
| pHi | intracellular pH |
| PI3K | phosphatidylinositol-3 kinase |
| PPIs | proton pump inhibitors |
| ROS | reactive oxygen species |
| SAHA | suberoylanilide hydroxamic acid |
| SFN | sulforaphane |
| STAT | signal transducer and activator of transcription proteins |
| TCA | tricarboxylic acid |
| Th17 | T-helper 17 cells |
| TIMP-1 | tissue inhibitor of metalloprotease-1 |
| TNBC | triple-negative breast cancer |
| Tregs | T regulatory cells |
| VEGF | vascular endothelial growth factor |
| VHL | Von Hippel-Lindau |
References
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The different mechanisms of cancer drug resistance: A brief review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting tumor microenvironment for cancer therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef]
- Wojtkowiak, J.W.; Verduzco, D.; Schramm, K.J.; Gillies, R.J. Drug resistance and cellular adaptation to tumor acidic pH microenvironment. Mol. Pharm. 2011, 8, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.R.; Damaghi, M.; Marunaka, Y.; Spugnini, E.P.; Fais, S.; Gillies, R.J. Causes, consequences, and therapy of tumors acidosis. Cancer Metastasis Rev. 2019, 38, 205–222. [Google Scholar] [CrossRef]
- Ibrahim-Hashim, A.; Estrella, V. Acidosis and cancer: From mechanism to neutralization. Cancer Metastasis Rev. 2019, 38, 149–155. [Google Scholar] [CrossRef]
- Pouysségur, J.; Chambard, J.C.; Franchi, A.; Paris, S.; Van Obberghen-Schilling, E. Growth factor activation of an amiloride-sensitive Na+/H+ exchange system in quiescent fibroblasts: Coupling to ribosomal protein S6 phosphorylation. Proc. Natl. Acad. Sci. USA 1982, 79, 3935–3939. [Google Scholar] [CrossRef] [PubMed]
- Benej, M.; Pastorekova, S.; Pastorek, J. Carbonic Anhydrase IX: Regulation and Role in Cancer. In Carbonic Anhydrase: Mechanism, Regulation, Links to Disease, and Industrial Applications; Frost, S., McKenna, R., Eds.; Subcellular Biochemistry, Vol 75; Springer: Dordrecht, The Netherlands, 2014; Chapter 11; pp. 199–219. ISBN 9789400773592. [Google Scholar]
- Swietach, P.; Patiar, S.; Supuran, C.T.; Harris, A.L.; Vaughan-Jones, R.D. The role of carbonic anhydrase 9 in regulating extracellular and intracellular pH in three-dimensional tumor cell growths. J. Biol. Chem. 2009, 284, 20299–20310. [Google Scholar] [CrossRef]
- Chiche, J.; Ilc, K.; Laferrière, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouysségur, J. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009, 69, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Mishra, C.B.; Tiwari, M.; Supuran, C.T. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: Where are we today? Med. Res. Rev. 2020, 40, 2485–2565. [Google Scholar] [CrossRef]
- Singh, S.; Lomelino, C.L.; Mboge, M.Y.; Frost, S.C.; McKenna, R. Cancer drug development of carbonic anhydrase inhibitors beyond the active site. Molecules 2018, 23, 1045. [Google Scholar] [CrossRef]
- Vullo, D.; Franchi, M.; Gallori, E.; Pastorek, J.; Scozzafava, A.; Pastorekova, S.; Supuran, C.T. Carbonic anhydrase inhibitors: Inhibition of the tumor-associated isozyme IX with aromatic and heterocyclic sulfonamides. Bioorg. Med. Chem. Lett. 2003, 13, 1005–1009. [Google Scholar] [CrossRef]
- Nishimori, I.; Vullo, D.; Innocenti, A.; Scozzafava, A.; Mastrolorenzo, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Inhibition of the transmembrane isozyme XIV with sulfonamides. Bioorganic Med. Chem. Lett. 2005, 15, 3828–3833. [Google Scholar] [CrossRef]
- Carta, F.; Vullo, D.; Osman, S.M.; AlOthman, Z.; Supuran, C.T. Synthesis and carbonic anhydrase inhibition of a series of SLC-0111 analogs. Bioorganic Med. Chem. 2017, 25, 2569–2576. [Google Scholar] [CrossRef]
- Winum, J.Y.; Pastorekova, S.; Jakubickova, L.; Montero, J.L.; Scozzafava, A.; Pastorek, J.; Vullo, D.; Innocenti, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis and inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II, and IX with bis-sulfamates. Bioorganic Med. Chem. Lett. 2005, 15, 579–584. [Google Scholar] [CrossRef]
- Federici, C.; Lugini, L.; Marino, M.L.; Carta, F.; Iessi, E.; Azzarito, T.; Supuran, C.T.; Fais, S. Lansoprazole and carbonic anhydrase IX inhibitors sinergize against human melanoma cells. J. Enzym. Inhib. Med. Chem. 2016, 31, 119–125. [Google Scholar] [CrossRef]
- Oosterwijk-Wakka, J.C.; Boerman, O.C.; Mulders, P.F.A.M.; Oosterwijk, E. Application of monoclonal antibody G250 recognizing carbonic anhydrase IX in renal cell carcinoma. Int. J. Mol. Sci. 2013, 14, 11402–11423. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.; Lin, K.S.; Bénard, F. Past, present, and future: Development of theranostic agents targeting carbonic anhydrase IX. Theranostics 2017, 7, 4322–4339. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Cui, Y.; Xu, S.; Wu, X.; Huang, Y.; Zhou, W.; Wang, S.; Fu, Z.; Xie, H. Altered glycolysis results in drug-resistant in clinical tumor therapy. Oncol. Lett. 2021, 21, 369. [Google Scholar] [CrossRef]
- Dubois, L.; Peeters, S.G.J.A.; Van Kuijk, S.J.A.; Yaromina, A.; Lieuwes, N.G.; Saraya, R.; Biemans, R.; Rami, M.; Parvathaneni, N.K.; Vullo, D.; et al. Targeting carbonic anhydrase IX by nitroimidazole based sulfamides enhances the therapeutic effect of tumor irradiation: A new concept of dual targeting drugs. Radiother. Oncol. 2013, 108, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.; Peeters, S.; Lieuwes, N.G.; Geusens, N.; Thiry, A.; Wigfield, S.; Carta, F.; McIntyre, A.; Scozzafava, A.; Dogné, J.M.; et al. Specific inhibition of carbonic anhydrase IX activity enhances the in vivo therapeutic effect of tumor irradiation. Radiother. Oncol. 2011, 99, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Inhibition of CArbonic Anhydrase in Combination with Platinum and Etoposide-based Radiochemotherapy in Patients with Localized Small Cell Lung Cancer—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03467360 (accessed on 16 October 2021).
- Huber, V.; De Milito, A.; Harguindey, S.; Reshkin, S.J.; Wahl, M.L.; Rauch, C.; Chiesi, A.; Pouysségur, J.; Gatenby, R.A.; Rivoltini, L.; et al. Proton dynamics in cancer. J. Transl. Med. 2010, 8, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Švastová, E.; Hulíková, A.; Rafajová, M.; Zat’Ovičová, M.; Gibadulinová, A.; Casini, A.; Cecchi, A.; Scozzafava, A.; Supuran, C.T.; Pastorek, J.; et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 2004, 577, 439–445. [Google Scholar] [CrossRef]
- Vullo, D.; Franchi, M.; Gallori, E.; Pastorek, J.; Scozzafava, A.; Pastorekova, S.; Supuran, C.T. Carbonic anhydrase inhibitors. Inhibition of cytosolic isozymes I and II and transmembrane, cancer-associated isozyme IX with anions. J. Enzym. Inhib. Med. Chem. 2003, 18, 403–406. [Google Scholar] [CrossRef]
- Mboge, M.Y.; Mahon, B.P.; McKenna, R.; Frost, S.C. Carbonic anhydrases: Role in pH control and cancer. Metabolites 2018, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Cuffaro, D.; Nuti, E.; Rossello, A. An overview of carbohydrate-based carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2020, 35, 1906–1922. [Google Scholar] [CrossRef]
- Mahoney, B.P.; Raghunand, N.; Baggett, B.; Gillies, R.J. Tumor acidity, ion trapping and chemotherapeutics: I. Acid pH affects the distribution of chemotherapeutic agents in vitro. Biochem. Pharmacol. 2003, 66, 1207–1218. [Google Scholar] [CrossRef]
- Becker, H.M. Carbonic anhydrase IX and acid transport in cancer. Br. J. Cancer 2020, 122, 157–167. [Google Scholar] [CrossRef]
- Gieling, R.G.; Parker, C.A.; De Costa, L.A.; Robertson, N.; Harris, A.L.; Stratford, I.J.; Williams, K.J. Inhibition of carbonic anhydrase activity modifies the toxicity of doxorubicin and melphalan in tumour cells in vitro. J. Enzym. Inhib. Med. Chem. 2012, 28, 360–369. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Kumar, S.; Islam, S.S.; Yazdanpanah, M.; Adeli, K.; Cutz, E.; Yeger, H. Combination of carbonic anhydrase inhibitor, acetazolamide, and sulforaphane, reduces the viability and growth of bronchial carcinoid cell lines. BMC Cancer 2013, 13, 378. [Google Scholar] [CrossRef]
- Su, X.; Jiang, X.; Meng, L.; Dong, X.; Shen, Y.; Xin, Y. Anticancer activity of sulforaphane: The epigenetic mechanisms and the Nrf2 signaling pathway. Oxid. Med. Cell. Longev. 2018, 2018, 5438179. [Google Scholar] [CrossRef] [PubMed]
- Bernkopf, D.B.; Daum, G.; Brückner, M.; Behrens, J. Sulforaphane inhibits growth and blocks Wnt/β-catenin signaling of colorectal cancer cells. Oncotarget 2018, 9, 33982–33994. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Essel, K.G.; Benbrook, D.M.; Garland, J.; Zhao, Y.D.; Chandra, V. Preclinical efficacy and involvement of AKT, mTOR, and ERK kinases in the mechanism of sulforaphane against endometrial cancer. Cancers 2020, 12, 1273. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Dashwood, W.M.; Li, L.; Kang, Y.; Kim, E.; Johnson, G.; Fischer, K.A.; Löhr, C.V.; Williams, D.E.; Ho, E.; et al. Nrf2 status affects tumor growth, HDAC3 gene promoter associations, and the response to sulforaphane in the colon. Clin. Epigenetics 2015, 7, 102. [Google Scholar] [CrossRef]
- Tian, Y.; Wu, K.; Liu, Q.; Han, N.; Zhang, L.; Chu, Q.; Chen, Y. Modification of platinum sensitivity by KEAP1/NRF2 signals in non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 83. [Google Scholar] [CrossRef]
- Zhang, P.; Singh, A.; Yegnasubramanian, S.; Esopi, D.; Kombairaju, P.; Bodas, M.; Wu, H.; Bova, S.G.; Biswal, S. Loss of kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol. Cancer Ther. 2010, 9, 336–346. [Google Scholar] [CrossRef]
- Mastrangelo, L.; Cassidy, A.; Mulholland, F.; Wang, W.; Bao, Y. Serotonin receptors, novel targets of sulforaphane identified by proteomic analysis in Caco-2 cells. Cancer Res. 2008, 68, 5487–5491. [Google Scholar] [CrossRef][Green Version]
- Kamal, M.M.; Akter, S.; Lin, C.N.; Nazzal, S. Sulforaphane as an anticancer molecule: Mechanisms of action, synergistic effects, enhancement of drug safety, and delivery systems. Arch. Pharm. Res. 2020, 43, 371–384. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Sparaneo, A.; Muscarella, L.A.; Zhao, S.; Cheng, H.L.; Das, B.; Yeger, H. Human bronchial carcinoid tumor initiating cells are targeted by the combination of acetazolamide and sulforaphane. BMC Cancer 2019, 19, 864. [Google Scholar] [CrossRef]
- Islam, S.S.; Mokhtari, R.B.; Akbari, P.; Hatina, J.; Yeger, H.; Farhat, W.A. Simultaneous Targeting of Bladder Tumor Growth, Survival, and Epithelial-to-Mesenchymal Transition with a Novel Therapeutic Combination of Acetazolamide (AZ) and Sulforaphane (SFN). Target. Oncol. 2016, 11, 209–227. [Google Scholar] [CrossRef]
- Gao, H.; Dong, H.; Li, G.; Jin, H. Combined treatment with acetazolamide and cisplatin enhances chemosensitivity in laryngeal carcinoma Hep-2 cells. Oncol. Lett. 2018, 15, 9299–9306. [Google Scholar] [CrossRef]
- Nathan, N.; Keppler-Noreuil, K.M.; Biesecker, L.G.; Moss, J.; Darling, T.N. Mosaic Disorders of the PI3K/PTEN/AKT/TSC/mTORC1 Signaling Pathway. Dermatol. Clin. 2017, 35, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, M.D.; Hopkins, B.D.; Cantley, L.C. Phosphatidylinositol 3-Kinase, Growth Disorders, and Cancer. N. Engl. J. Med. 2018, 379, 2052–2062. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Luo, Y.; Huang, S. Updates of mTOR Inhibitors. Anticancer. Agents Med. Chem. 2012, 10, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Kelly, K.R.; Nawrocki, S.T. Mechanisms of mTOR inhibitor resistance in cancer therapy. Target. Oncol. 2011, 6, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Faes, S.; Planche, A.; Uldry, E.; Santoro, T.; Pythoud, C.; Stehle, J.C.; Horlbeck, J.; Letovanec, I.; Riggi, N.; Datta, D.; et al. Targeting carbonic anhydrase IX improves the anti-cancer efficacy of mTOR inhibitors. Oncotarget 2016, 7, 36666–36680. [Google Scholar] [CrossRef]
- Vaeteewoottacharn, K.; Kariya, R.; Dana, P.; Fujikawa, S.; Matsuda, K.; Ohkuma, K.; Kudo, E.; Kraiklang, R.; Wongkham, C.; Wongkham, S.; et al. Inhibition of carbonic anhydrase potentiates bevacizumab treatment in cholangiocarcinoma. Tumor Biol. 2016, 37, 9023–9035. [Google Scholar] [CrossRef]
- Yoshikawa, D.; Ojima, H.; Iwasaki, M.; Hiraoka, N.; Kosuge, T.; Kasai, S.; Hirohashi, S.; Shibata, T. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br. J. Cancer 2008, 98, 418–425. [Google Scholar] [CrossRef]
- Morine, Y.; Shimada, M.; Utsunomiya, T.; Imura, S.; Ikemoto, T.; Mori, H.; Hanaoka, J.; Kanamoto, M.; Iwahashi, S.; Miyake, H. Hypoxia inducible factor expression in intrahepatic cholangiocarcinoma. Hepatogastroenterology. 2011, 58, 1439–1444. [Google Scholar] [CrossRef] [PubMed]
- Abd-El Fattah, A.A.; Darwish, H.A.; Fathy, N.; Shouman, S.A. Carbonic anhydrase inhibition boosts the antitumor effects of Imatinib mesylate via potentiating the antiangiogenic and antimetastatic machineries. Toxicol. Appl. Pharmacol. 2017, 316, 123–138. [Google Scholar] [CrossRef]
- Metibemu, D.S.; Akinloye, O.A.; Akamo, A.J.; Ojo, D.A.; Okeowo, O.T.; Omotuyi, I.O. Exploring receptor tyrosine kinases-inhibitors in Cancer treatments. Egypt. J. Med. Hum. Genet. 2019, 20, 35. [Google Scholar] [CrossRef]
- Singh, A.K.; Bishayee, A.; Pandey, A.K. Targeting Histone Deacetylases with Natural and Synthetic Agents: An Emerging Anticancer Strategy. Nutrients 2018, 10, 731. [Google Scholar] [CrossRef]
- Halsall, J.A.; Turner, B.M. Histone deacetylase inhibitors for cancer therapy: An evolutionarily ancient resistance response may explain their limited success. BioEssays 2016, 38, 1102–1110. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: Achieving the full therapeutic potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef]
- Witt, O.; Deubzer, H.; Lodrini, M.; Milde, T.; Oehme, I. Targeting Histone Deacetylases in Neuroblastoma. Curr. Pharm. Des. 2009, 15, 436–447. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Baluch, N.; Ka Hon Tsui, M.; Kumar, S.; Homayouni, T.S.; Aitken, K.; Das, B.; Baruchel, S.; Yeger, H. Acetazolamide potentiates the anti-tumor potential of HDACi, MS-275, in neuroblastoma. BMC Cancer 2017, 17, 156. [Google Scholar] [CrossRef] [PubMed]
- Lue, J.K.; O’Connor, O.A. A perspective on improving the R-CHOP regimen: From Mega-CHOP to ROBUST R-CHOP, the PHOENIX is yet to rise. Lancet Haematol. 2020, 7, e838–e850. [Google Scholar] [CrossRef]
- Hauke, R.J.; Armitage, J.O. Treatment of non-Hodgkin lymphoma. Curr. Opin. Oncol. 2000, 12, 412–418. [Google Scholar] [CrossRef]
- Senjo, H.; Hirata, K.; Izumiyama, K.; Minauchi, K.; Tsukamoto, E.; Itoh, K.; Kanaya, M.; Mori, A.; Ota, S.; Hashimoto, D.; et al. High metabolic heterogeneity on baseline 18FDG-PET/CT scan as a poor prognostic factor for newly diagnosed diffuse large B-cell lymphoma. Blood Adv. 2020, 4, 2286–2296. [Google Scholar] [CrossRef]
- Klener, P. Drug Resistance in Non-Hodgkin Lymphomas. Int. J. Mol. Sci. 2020, 21, 2081. [Google Scholar] [CrossRef] [PubMed]
- Méhes, G.; Matolay, O.; Beke, L.; Czenke, M.; Pórszász, R.; Mikó, E.; Bai, P.; Berényi, E.; Trencsényi, G. Carbonic anhydrase inhibitor acetazolamide enhances chop treatment response and stimulates effector t-cell infiltration in a20/balbc murine b-cell lymphoma. Int. J. Mol. Sci. 2020, 21, 5001. [Google Scholar] [CrossRef] [PubMed]
- Bascuas, T.; Moreno, M.; Mónaco, A.; Reyes, L.; Paolino, A.; Oliver, P.; Kramer, M.G.; Engler, H.; Pacheco, J.P.; Grille, S.; et al. A novel non-Hodgkin lymphoma murine model closer to the standard clinical scenario. J. Transl. Med. 2016, 14, 323. [Google Scholar] [CrossRef]
- Torring, M.S.; Holmgaard, K.; Hessellund, A.; Aalkjaer, C.; Bek, T. The vasodilating effect of acetazolamide and dorzolamide involves mechanisms other than carbonic anhydrase inhibition. Investig. Ophthalmol. Vis. Sci. 2009, 50, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Joshi, K.; Ghosalkar, J.D.; Sonawane, V.R.; Raut, S.R.; Malhotra, G. Abstract 2013: A carbonic anhydrase inhibitor methazolamide potentiates efficacy of gemcitabine by modulating anti-proliferative activity and cancer stem cell markers in pancreatic carcinoma. In Proceedings of the AACR Annual Meeting 2019, Atlanta, GA, USA, 29 March–3 April 2019; Volume 79. [Google Scholar] [CrossRef]
- Andreucci, E.; Ruzzolini, J.; Peppicelli, S.; Bianchini, F.; Laurenzana, A.; Carta, F.; Supuran, C.T.; Calorini, L. The carbonic anhydrase IX inhibitor SLC-0111 sensitises cancer cells to conventional chemotherapy. J. Enzym. Inhib. Med. Chem. 2019, 34, 117–123. [Google Scholar] [CrossRef]
- Boyd, N.H.; Walker, K.; Fried, J.; Hackney, J.R.; McDonald, P.C.; Benavides, G.A.; Spina, R.; Audia, A.; Scott, S.E.; Libby, C.J.; et al. Addition of carbonic anhydrase 9 inhibitor SLC-0111 to temozolomide treatment delays glioblastoma growth in vivo. JCI Insight 2017, 2, 16022. [Google Scholar] [CrossRef]
- McDonald, P.C.; Chafe, S.C.; Brown, W.S.; Saberi, S.; Swayampakula, M.; Venkateswaran, G.; Nemirovsky, O.; Gillespie, J.A.; Karasinska, J.M.; Kalloger, S.E.; et al. Regulation of pH by Carbonic Anhydrase 9 Mediates Survival of Pancreatic Cancer Cells with Activated KRAS in Response to Hypoxia. Gastroenterology 2019, 157, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef]
- Lee, K.E.; Spata, M.; Bayne, L.J.; Buza, E.L.; Durham, A.C.; Allman, D.; Vonderheide, R.H.; Simon, M.C. Hif1a deletion reveals pro-neoplastic function of B cells in pancreatic neoplasia. Cancer Discov. 2016, 6, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Van Kuijk, S.J.A.; Gieling, R.G.; Niemans, R.; Lieuwes, N.G.; Biemans, R.; Telfer, B.A.; Haenen, G.R.M.M.; Yaromina, A.; Lambin, P.; Dubois, L.J.; et al. The sulfamate small molecule CAIX inhibitor S4 modulates doxorubicin efficacy. PLoS ONE 2016, 11, e161040. [Google Scholar] [CrossRef] [PubMed]
- Winum, J.Y.; Carta, F.; Ward, C.; Mullen, P.; Harrison, D.; Langdon, S.P.; Cecchi, A.; Scozzafava, A.; Kunkler, I.; Supuran, C.T. Ureido-substituted sulfamates show potent carbonic anhydrase IX inhibitory and antiproliferative activities against breast cancer cell lines. Bioorganic Med. Chem. Lett. 2012, 22, 4681–4685. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Meehan, J.; Mullen, P.; Supuran, C.; Michael Dixon, J.; Thomas, J.S.; Winum, J.Y.; Lambin, P.; Dubois, L.; Pavathaneni, N.K.; et al. Evaluation of carbonic anhydrase IX as a therapeutic target for inhibition of breast cancer invasion and metastasis using a series of in vitro breast cancer models. Oncotarget 2015, 6, 24856–24870. [Google Scholar] [CrossRef]
- Gieling, R.G.; Babur, M.; Mamnani, L.; Burrows, N.; Telfer, B.A.; Carta, F.; Winum, J.Y.; Scozzafava, A.; Supuran, C.T.; Williams, K.J. Antimetastatic effect of sulfamate carbonic anhydrase IX inhibitors in breast carcinoma xenografts. J. Med. Chem. 2012, 55, 5591–5600. [Google Scholar] [CrossRef]
- Bryant, J.L.; Gieling, R.G.; Meredith, S.L.; Allen, T.-J.; Walker, L.; Telfer, B.A. Novel carbonic anhydrase IX-targeted therapy enhances the anti-tumour effects of cisplatin in small cell lung cancer. Int. J. Cancer 2017, 142, 191–201. [Google Scholar] [CrossRef]
- Petrenko, M.; Güttler, A.; Funtan, A.; Keßler, J.; Emmerich, D.; Paschke, R.; Vordermark, D.; Bache, M. Combined 3-O-acetylbetulin treatment and carbonic anhydrase IX inhibition results in additive effects on human breast cancer cells. Chem. Biol. Interact. 2021, 333, 109326. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.R.; Logsdon, D.; Fishel, M.L. Targeting DNA repair pathways for cancer treatment: What’s new? Futur. Oncol. 2014, 10, 1215–1237. [Google Scholar] [CrossRef]
- Tell, G.; Quadrifoglio, F.; Tiribelli, C.; Kelley, M.R. The many functions of APE1/Ref-1: Not only a DNA repair enzyme. Antioxid. Redox Signal. 2009, 11, 601–619. [Google Scholar] [CrossRef]
- Fishel, M.L.; Jiang, Y.; Rajeshkumar, N.V.; Scandura, G.; Sinn, A.L.; He, Y.; Shen, C.; Jones, D.R.; Pollok, K.E.; Ivan, M.; et al. Impact of APE1/ref-1 redox inhibition on pancreatic tumor growth. Mol. Cancer Ther. 2011, 10, 1698–1708. [Google Scholar] [CrossRef]
- Logsdon, D.P.; Grimard, M.; Luo, M.; Shahda, S.; Jiang, Y.; Tong, Y. Regulation of HIF1 α under Hypoxia by APE1/Ref-1 Impacts CA9 Expression: Dual-Targeting in Patient-Derived 3D Pancreatic Cancer Models. Mol. Cancer Ther. 2016, 15, 2722–2732. [Google Scholar] [CrossRef]
- Logsdon, D.P.; Shah, F.; Carta, F.; Supuran, C.T.; Kamocka, M.; Jacobsen, M.H.; Sandusky, G.E.; Kelley, M.R.; Fishel, M.L. Blocking HIF signaling via novel inhibitors of CA9 and APE1/Ref-1 dramatically affects pancreatic cancer cell survival. Sci. Rep. 2018, 8, 13759. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, E.M.E.; McDonald, P.C.; Nemirovsky, O.; Awrey, S.; Jensen, L.D.E.; Dedhar, S. Harnessing induced essentiality: Targeting carbonic anhydrase IX and angiogenesis reduces lung metastasis of triple negative breast cancer xenografts. Cancers 2019, 11, 1002. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: Mechanisms of action, efficacy, and limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Chafe, S.C.; McDonald, P.C.; Saberi, S.; Nemirovsky, O.; Venkateswaran, G.; Burugu, S.; Gao, D.; Delaidelli, A.; Kyle, A.H.; Baker, J.H.E.; et al. Targeting hypoxia-induced carbonic anhydrase IX enhances immune-checkpoint blockade locally and systemically. Cancer Immunol. Res. 2019, 7, 1064–1078. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Ruzzolini, J.; Laurenzana, A.; Andreucci, E.; Peppicelli, S.; Bianchini, F.; Carta, F.; Supuran, C.T.; Romanelli, M.N.; Nediani, C.; Calorini, L. A potentiated cooperation of carbonic anhydrase IX and histone deacetylase inhibitors against cancer. J. Enzym. Inhib. Med. Chem. 2020, 35, 391–397. [Google Scholar] [CrossRef]
- Luciani, F.; Spada, M.; De Milito, A.; Molinari, A.; Rivoltini, L.; Montinaro, A.; Marra, M.; Lugini, L.; Logozzi, M.; Lozupone, F.; et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J. Natl. Cancer Inst. 2004, 96, 1702–1713. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Huang, S.L.; Zhang, X.Q.; Zhang, B.; Zhu, H.; Yang, V.W.; Zou, X.P. Reversal effects of pantoprazole on multidrug resistance in human gastric adenocarcinoma cells by down-regulating the V-ATPases/mTOR/HIF-1α/P-gp and MRP1 signaling pathway in vitro and in vivo. J. Cell. Biochem. 2012, 113, 2474–2487. [Google Scholar] [CrossRef]
- Uchiyama, A.A.T.; Silva, P.A.I.A.; Lopes, M.S.M.; Yen, C.T.; Ricardo, E.D.; Mutão, T.; Pimenta, J.R.; Machado, L.M.; Shimba, D.S.; Peixoto, R.D. Proton pump inhibitors and oncologic treatment efficacy: A practical review of the literature for oncologists. Curr. Oncol. 2021, 28, 783-799. [Google Scholar] [CrossRef]
- Fais, S. Evidence-based support for the use of proton pump inhibitors in cancer therapy. J. Transl. Med. 2015, 13, 368. [Google Scholar] [CrossRef] [PubMed]
- Iessi, E.; Logozzi, M.; Mizzoni, D.; Di Raimo, R.; Supuran, C.T.; Fais, S. Rethinking the combination of proton exchanger inhibitors in cancer therapy. Metabolites 2018, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.C.; Chia, S.; Bedard, P.L.; Chu, Q.; Lyle, M.; Tang, L.; Singh, M.; Zhang, Z.; Supuran, C.T.; Renouf, D.J.; et al. A Phase 1 Study of SLC-0111, a Novel Inhibitor of Carbonic Anhydrase IX, in Patients with Advanced Solid Tumors. Am. J. Clin. Oncol. Cancer Clin. Trials 2020, 43, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Ducreux, M.; Seufferlein, T.; Van Laethem, J.L.; Laurent-Puig, P.; Smolenschi, C.; Malka, D.; Boige, V.; Hollebecque, A.; Conroy, T. Systemic treatment of pancreatic cancer revisited. Semin. Oncol. 2019, 46, 28–38. [Google Scholar] [CrossRef]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef]
- Su, J.; Chen, X.; Kanekura, T. A CD147-targeting siRNA inhibits the proliferation, invasiveness, and VEGF production of human malignant melanoma cells by down-regulating glycolysis. Cancer Lett. 2009, 273, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, L.A.; Fallon, M.; Pisarcik, S.; Wang, J.; Semenza, G.L. HIF-1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am. J. Physiology-Lung Cell. Mol. Physiol. 2006, 291, L941–L949. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Chen, J.L.Y.; Lucas, J.E.; Schroeder, T.; Mori, S.; Wu, J.; Nevins, J.; Dewhirst, M.; West, M.; Chi, J.T. The genomic analysis of lactic acidosis and acidosis response in human cancers. PLoS Genet. 2008, 4, e1000293. [Google Scholar] [CrossRef]
- Chafe, S.C.; Vizeacoumar, F.S.; Venkateswaran, G.; Nemirovsky, O.; Awrey, S.; Brown, W.S.; McDonald, P.C.; Carta, F.; Metcalfe, A.; Karasinska, J.M.; et al. Genome-wide synthetic lethal screen unveils novel CAIX-NFS1/xCT axis as a targetable vulnerability in hypoxic solid tumors. Sci. Adv. 2021, 7, eabj0364. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase inhibitors: An update on experimental agents for the treatment and imaging of hypoxic tumors. Expert. Opin. Investig. Drugs 2021, 27, 963–970. [Google Scholar] [CrossRef] [PubMed]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
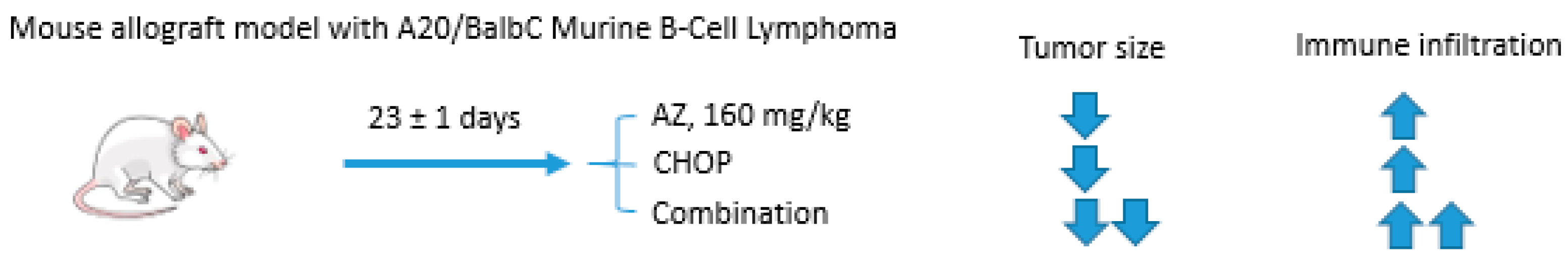{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug/Drug Candidate | Chemical Structure | Ki (nM) | Current Status | Ref | |
|---|---|---|---|---|---|
| Acetazolamide (AZ) |  | CA I | 250 | systemic CAI clinically used to treat glaucoma, edema, certain types of epilepsy | [13,14] |
| CA II | 12 | ||||
| CA IX | 25 | ||||
| CA XII | 5.7 | ||||
| Methazolamide |  | CA I | 50 | systemic CAI clinically used to treat glaucoma | [13,14] |
| CA II | 14 | ||||
| CA IX | 27 | ||||
| CA XII | 3.4 | ||||
| SLC-0111 |  | CA I | 5080 | in clinical trials as adjuvant agent in advanced solid tumorsPhase Ib/II | [15] |
| CA II | 960 | ||||
| CA IX | 45 | ||||
| CA XII | 4.5 | ||||
| n-Octyl sulfamate |  | CA I | 378 | rarely used tool compound for in vitro experiments | [16] |
| CA II | 15 | ||||
| CA IX | 4 | ||||
| CA XII | - | ||||
| S4 |  | CA I | 5600 | in preclinical studies | [17] |
| CA II | 546 | ||||
| CA IX | 7 | ||||
| CA XII | 2 | ||||
| FC-531A |  | CA I | 1230 | in preclinical studies | [17] |
| CA II | 450 | ||||
| CA IX | 6 | ||||
| CA XII | 4 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalinin, S.; Malkova, A.; Sharonova, T.; Sharoyko, V.; Bunev, A.; Supuran, C.T.; Krasavin, M. Carbonic Anhydrase IX Inhibitors as Candidates for Combination Therapy of Solid Tumors. Int. J. Mol. Sci. 2021, 22, 13405. https://doi.org/10.3390/ijms222413405
Kalinin S, Malkova A, Sharonova T, Sharoyko V, Bunev A, Supuran CT, Krasavin M. Carbonic Anhydrase IX Inhibitors as Candidates for Combination Therapy of Solid Tumors. International Journal of Molecular Sciences. 2021; 22(24):13405. https://doi.org/10.3390/ijms222413405
Chicago/Turabian StyleKalinin, Stanislav, Anna Malkova, Tatiana Sharonova, Vladimir Sharoyko, Alexander Bunev, Claudiu T. Supuran, and Mikhail Krasavin. 2021. "Carbonic Anhydrase IX Inhibitors as Candidates for Combination Therapy of Solid Tumors" International Journal of Molecular Sciences 22, no. 24: 13405. https://doi.org/10.3390/ijms222413405
APA StyleKalinin, S., Malkova, A., Sharonova, T., Sharoyko, V., Bunev, A., Supuran, C. T., & Krasavin, M. (2021). Carbonic Anhydrase IX Inhibitors as Candidates for Combination Therapy of Solid Tumors. International Journal of Molecular Sciences, 22(24), 13405. https://doi.org/10.3390/ijms222413405