
Mitochondrial Oxidative Stress—A Causative Factor and Therapeutic Target in Many Diseases

 , ,
, ,  ,
, {kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Mitochondrial Oxidative Phosphorylation (OXPHOS)—A Source of Free Radical Formation
1.2. mtDNA and nDNA—Uniqueness of mtDNA
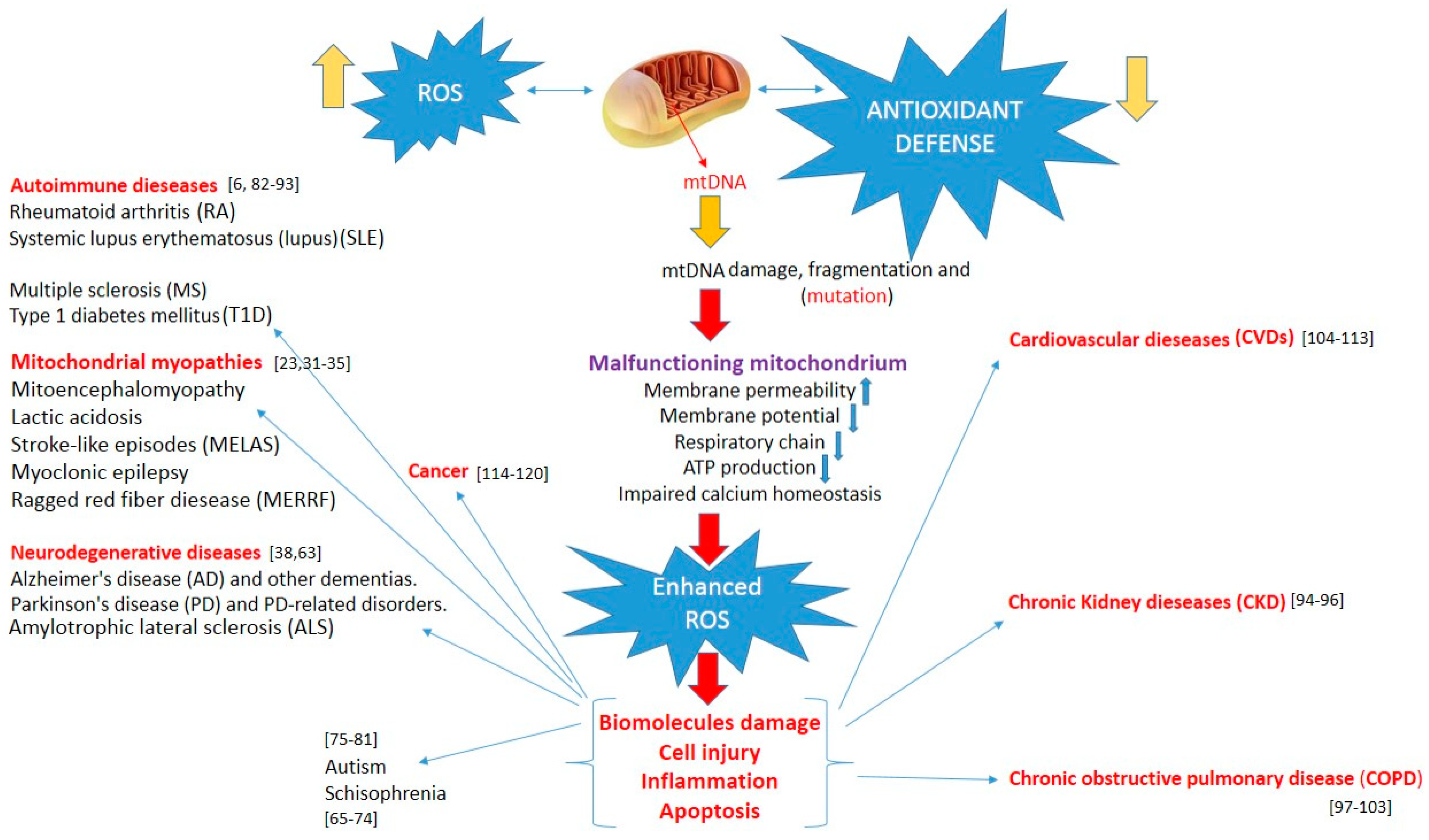
2. Mutations in mtDNA and nDNA and Mitochondrial Diseases
3. Diseases Associated with Impaired ROS Generation in Mitochondria
3.1. Neurological Diseases
3.2. Neurodevelopmental Disorders
3.3. Autoimmune Diseases
3.4. Kidney and Lung Diseases
3.5. Cardiovascular Diseases (CVDs)
3.6. Cancer
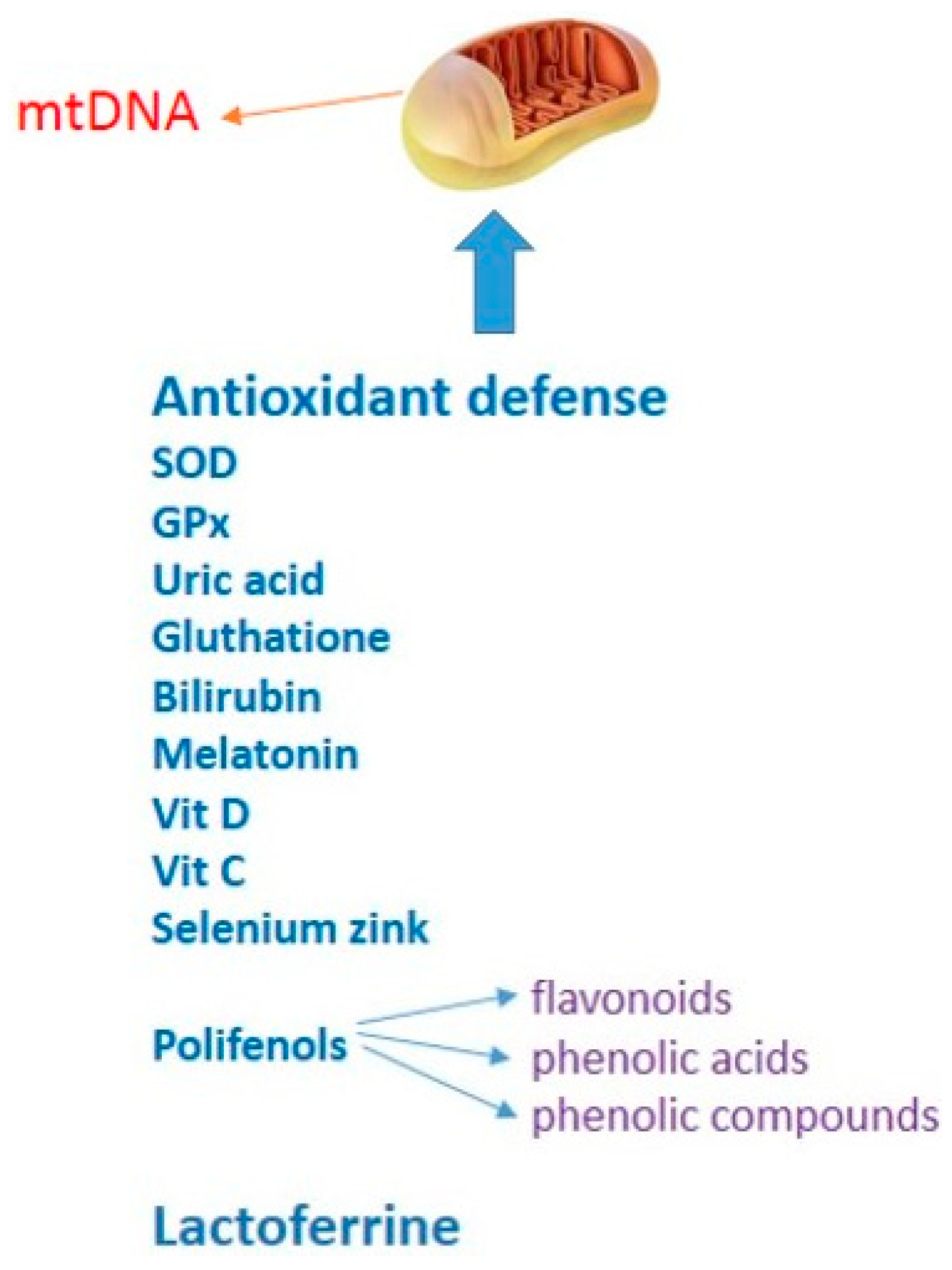
4. Oxidative Stress as a Therapeutic Target
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Wu, J.Q.; Kosten, T.R.; Zhang, X.Y. Free radicals, antioxidant defense systems, and schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 46, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Kohen, R.; Nyska, A. Oxidation of biological systems: Oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxid. Med. Cell. Longev. 2016, 2016, 7432797. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Kokkinopoulou, I.; Moutsatsou, P. Mitochondrial Glucocorticoid Receptors and Their Actions. Int. J. Mol. Sci. 2021, 22, 6054. [Google Scholar] [CrossRef]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef]
- Newmeyer, D.D.; Ferguson-Miller, S. Mitochondria: Releasing power for life and unleashing the machineries of death. Cell 2003, 112, 481–490. [Google Scholar] [CrossRef]
- Goncalves, R.L.; Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Brand, M.D. Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J. Biol. Chem. 2015, 290, 209–227. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Scialo, F.; Sriram, A.; Fernandez-Ayala, D.; Gubina, N.; Lohmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enríquez, J.A.; et al. Mitochondrial ROS Produced via reverse electron transport extend animal lifespan. Cell Metab. 2016, 23, 725–734. [Google Scholar] [CrossRef]
- Scialo, F.; Fernandez-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.G. Maintenance and expression of mammalian mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef]
- Cha, M.Y.; Kim, D.; Mook-Jung, I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp. Mol. Med. 2015, 47, e150. [Google Scholar] [CrossRef]
- Menger, K.E.; Rodríguez-Luis, A.; Chapman, J.; Nicholls, T.J. Controlling the topology of mammalian mitochondrial DNA. Open Biol. 2021, 11, 210168. [Google Scholar] [CrossRef]
- Mishmar, D.; Levin, R.; Naeem, M.M.; Sondheimer, N. Higher Order Organization of the mtDNA: Beyond Mitochondrial Transcription Factor A. Front. Genet. 2019, 10, 1285. [Google Scholar] [CrossRef] [PubMed]
- Matsui, A.; Ikeda, T.; Enomoto, K.; Hosoda, K.; Nakashima, H.; Omae, K.; Watanabe, M.; Hibi, T.; Kitajima, M. Increased Formation of Oxidative DNA Damage, 8-Hydroxy-2’-Deoxyguanosine, in Human Breast Cancer Tissue and its Relationship to GSTP1 and COMT Genotypes. Cancer Lett. 2000, 151, 87–95. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Sutovsky, P. Ubiquitin-dependent proteolysis in mammalian spermatogenesis, fertilization, and sperm quality control: Killing three birds with one stone. Microsc. Res. Tech. 2003, 61, 88–102. [Google Scholar] [CrossRef]
- Neiman, M.; Taylor, D.R. The causes of mutation accumulation in mitochondrial genomes. Proc. Biol. Sci. 2009, 276, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Zheng, X.X.; Lott, M.T.; Shoffner, J.M.; Hodge, J.A.; Kelley, R.I.; Epstein, C.M.; Hopkins, L.C. Familial mitochondrial encephalomyopathy (MERRF): Genetic, pathophysiological, and biochemical characterization of a mitochondrial DNA disease. Cell 1988, 55, 601–610. [Google Scholar] [CrossRef]
- Alston, C.L.; Rocha, M.C.; Lax, N.Z.; Turnbull, D.M.; Taylor, R.W. The genetics and pathology of mitochondrial disease. J. Pathol. 2017, 241, 236–250. [Google Scholar] [CrossRef]
- Jokinen, R.; Battersby, B.J. Insight into mammalian mitochondrial DNA segregation. Ann. Med. 2013, 45, 149–155. [Google Scholar] [CrossRef]
- Saneto, R.P. Genetics of Mitochondrial Disease. Adv. Genet. 2017, 98, 63–116. [Google Scholar] [CrossRef]
- DiMauro, S. Mitochondrial myopathies. Curr. Opin. Rheumatol. 2006, 18, 636–641. [Google Scholar] [CrossRef]
- D’Errico, M.; Parlanti, E.; Pascucci, B.; Filomeni, G.; Mastroberardino, P.G.; Dogliotti, E. The interplay between mitochondrial functionality and genome integrity in the prevention of human neurologic diseases. Arch. Biochem. Biophys. 2021, 23, 108977. [Google Scholar] [CrossRef]
- Mancuso, M.; Orsucci, D.; Siciliano, G.; Murri, L. Mitochondria, mitochondrial DNA and Alzheimer’s disease. What comes first? Curr. Alzheimer Res. 2008, 5, 457–468. [Google Scholar] [CrossRef]
- Butterfield, D.A. β-amyloid-associated free radical oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. Chem. Res. Toxicol. 1997, 10, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef]
- Sharma, C.; Kim, S.; Nam, Y.; Jung, U.J.; Kim, S.R. Mitochondrial Dysfunction as a Driver of Cognitive Impairment in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4850. [Google Scholar] [CrossRef]
- Reddy, P.H. Amyloid precursor protein-mediated free radicals and oxidative damage: Implications for the development and progression of Alzheimer’s disease. J. Neurochem. 2006, 96, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Knight, W.C.; Xu, J. Striatal oxidative damages and neuroinflammation correlate with progression and survival of Lewy body and Alzheimer diseases. Neural Regen. Res. 2022, 17, 867–874. [Google Scholar] [CrossRef]
- Caito, S.W.; Aschner, M. Mitochondrial redox dysfunction and environmental exposures. Antioxid. Redox Signal. 2015, 23, 578–595. [Google Scholar] [CrossRef] [PubMed]
- Davie, C.A. A review of Parkinson’s disease. Br. Med. Bull. 2008, 80, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef]
- Pyle, A.; Anugrha, H.; Kurzawa-Akanbi, M.; Yarnall, A.; Burn, D.; Hudson, G. Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol. Aging 2016, 38, e7–e216. [Google Scholar] [CrossRef]
- Grünewald, A.; Rygiel, K.A.; Hepplewhite, P.D.; Morris, C.M.; Picardm, M.; Turnbullm, D.M. Mitochondrial DNA Depletion in Respiratory Chain-Deficient Parkinson Disease Neurons. Ann. Neurol. 2016, 79, 366–378. [Google Scholar] [CrossRef]
- Dölle, C.; Flønes, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 22, 13548. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef]
- Silva, R.; Domingues, H.S.; Salgado, A.J.; Teixeira, F.G. From regenerative strategies to pharmacological approaches: Can we fine-tune treatment for Parkinson’s disease? Neural Regen. Res. 2022, 17, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, B.; Adamczyk-Sowa, M. New Insights into the Role of Oxidative Stress Mechanisms in the Pathophysiology and Treatment of Multiple Sclerosis. Oxid. Med. Cell. Longev. 2016, 2016, 1973834. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef] [PubMed]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef]
- Simpson, E.P.; Henry, Y.K.; Henkel, J.S.; Smith, R.G.; Appel, S.H. Increased lipid peroxidation in sera of ALS patients: A potential biomarker of disease burden. Neurology 2004, 62, 1758–1765. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef]
- Wiedau-Pazos, M.; Goto, J.J.; Rabizadeh, S.; Gralla, E.B.; Roe, J.A.; Lee, M.K.; Valentine, J.S.; Bredesen, D.E. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science 1996, 271, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Petrozziello, T.; Bordt, E.A.; Mills, A.N.; Kim, S.E.; Sapp, E.A.; Devlin, B.A.; Obeng-Marnu, A.A.; Farhan, S.M.K.; Amaral, A.C.; Dujardin, S.; et al. Targeting Tau Mitigates Mitochondrial Fragmentation and Oxidative Stress in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Carrera-Juliá, S.; Moreno, M.L.; Barrios, C.; de la Rubia Ortí, J.E.; Drehmer, E. Antioxidant alternatives in the treatment of amyotrophic lateral sclerosis: A comprehensive review. Front. Physiol. 2020, 11, 63. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Liu, J.; Li, M.; Song, Y.; Zhang, C.; Zhai, Z.; Gao, Y. Neuroprotective Effects of Shenqi Fuzheng Injection in a Transgenic SOD1-G93A Mouse Model of Amyotrophic Lateral Sclerosis. Front. Pharmacol. 2021, 19, 701886. [Google Scholar] [CrossRef]
- Murray, A.J.; Rogers, J.C.; Katshu, M.Z.U.H.; Liddle, P.F.; Upthegrove, R. Oxidative Stress and the Pathophysiology and Symptom Profile of Schizophrenia Spectrum Disorders. Front. Psychiatry 2021, 12, 703452. [Google Scholar] [CrossRef]
- Raffa, M.; Atig, F.; Mhalla, A.; Kerkeni, A.; Mechri, A. Decreased glutathione levels and impaired antioxidant enzyme activities in drug-naive first-episode schizophrenic patients. BMC Psychiatry 2011, 11, 124. [Google Scholar] [CrossRef]
- Gunes, M.; Altindag, A.; Bulut, M.; Demir, S.; Ibiloglu, A.O.; Kaya, M.C.; Atli, A.; Aksoy, N. Oxidative metabolism may be associated with negative symptoms in schizophrenia. Psychiatry Clin. Psychopharmacol. 2017, 27, 54–61. [Google Scholar] [CrossRef]
- Solberg, D.K.; Refsum, H.; Andreassen, O.A.; Bentsen, H. A five-year follow-up study of antioxidants, oxidative stress and polyunsaturated fatty acids in schizophrenia. Acta Neuropsychiatr. 2019, 31, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Dietrich-Muszalska, A.; Kwiatkowska, A. Generation of superoxide anion radicals and platelet glutathione peroxidase activity in patients with schizophrenia. Neuropsych. Dis. Treat. 2014, 10, 703–709. [Google Scholar] [CrossRef]
- Do, K.Q.; Trabesinger, A.H.; Kirsten-Kruger, M.; Lauer, C.J.; Dydak, U.; Hell, D.; Holsboer, F.; Boesiger, P.; Cuénod, M. Schizophrenia: Glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur. J. Neurosci. 2000, 12, 3721–3728. [Google Scholar] [CrossRef]
- Gawryluk, J.W.; Wang, J.F.; Andreazza, A.C.; Shao, L.; Young, L.T. Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. Int. J. Neuropsychopharmacol. 2011, 14, 123–130. [Google Scholar] [CrossRef]
- Yao, J.K.; Keshavan, M.S. Antioxidants, redox signaling, and pathophysiology in schizophrenia: An integrative view. Antioxid. Redox Signal. 2011, 15, 2011–2035. [Google Scholar] [CrossRef] [PubMed]
- Cuenod, M.; Steullet, P.; Cabungcal, J.H.; Dwir, D.; Khadimallah, I.; Klauser, P.; Conus, P.; Do, K.Q. Caught in vicious circles: A perspective on dynamic feed-forward loops driving oxidative stress in schizophrenia. Mol. Psychiatry 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Toyoshima, M.; Hisano, Y.; Balan, S.; Iwayama, Y.; Aono, H.; Futamura, Y.; Osada, H.; Owada, Y.; Yoshikawa, T. Glyoxalase I disruption and external carbonyl stress impair mitochondrial function in human induced pluripotent stem cells and derived neurons. Transl. Psychiatry 2021, 11. [Google Scholar] [CrossRef]
- Michels, S.; Wöhr, M.; Schwarting, R.K.; Culmsee, C. Psychiatric risk gene cacna1c determines mitochondrial resilience against oxidative stress in neurons. Cell Death Dis. 2018, 9, 645. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Kanda, Y.; Sone, H.; Aoyama, H. Oxidative Stress as a Common Key Event in Developmental Neurotoxicity. Oxid. Med. Cell. Longev. 2021, 19, 6685204. [Google Scholar] [CrossRef] [PubMed]
- Balachandar, V.; Rajagopalan, K.; Jayaramayya, K.; Jeevanandam, M.; Iyer, M. Mitochondrial dysfunction: A hidden trigger of autism? Genes Dis. 2020, 8, 629–639. [Google Scholar] [CrossRef]
- Toscano, C.V.A.; Barros, L.; Lima, A.B.; Nunes, T.; Carvalho, H.M.; Gaspar, J.M. Neuroinflammation in autism spectrum disorders: Exercise as a “pharmacological” tool. Neurosci. Biobehav. Rev. 2021, 129, 63–74. [Google Scholar] [CrossRef]
- Omotosho, I.O.; Akinade, A.O.; Lagunju, I.A.; Yakubu, M.A. Oxidative stress indices in ASD children in Sub-Sahara Africa. J. Neurodev. Disord. 2021, 13, 50. [Google Scholar] [CrossRef]
- Zawadzka, A.; Cieślik, M.; Adamczyk, A. The Role of Maternal Immune Activation in the Pathogenesis of Autism: A Review of the Evidence, Proposed Mechanisms and Implications for Treatment. Int. J. Mol. Sci. 2021, 22, 11516. [Google Scholar] [CrossRef]
- Hardan, A.Y.; Fung, L.K.; Libove, R.A.; Obukhanych, T.V.; Nair, S.; Herzenberg, L.A.; Frazier, T.W.; Tirouvanziam, R. A randomized controlled pilot trial of oral N-acetylcysteine in children with autism. Biol. Psychiatry 2012, 71, 956–961. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Z.; Du, Y.; Shi, S.; Cheng, Y. Antioxidant interventions in autism spectrum disorders: A meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 113, 110476. [Google Scholar] [CrossRef]
- Chavez, M.D.; Tse, H.M. Targeting Mitochondrial-Derived Reactive Oxygen Species in T Cell Mediated Autoimmune Diseases. Front. Immunol. 2021, 12, 703972. [Google Scholar] [CrossRef] [PubMed]
- Clayton, S.A.; MacDonald, L.; Kurowska-Stolarska, M.; Clark, A.R. Mitochondria as Key Players in the Pathogenesis and Treatment of Rheumatoid Arthritis. Front. Immunol. 2021, 12, 673916. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Stimpson, S.E.; Fernandez-Bueno, G.A.; Mathews, C.E. Mitochondrial Reactive Oxygen Species and Type 1 Diabetes. Antioxid. Redox Signal. 2018, 29, 1361–1372. [Google Scholar] [CrossRef]
- Chatterjee, S. Chapter Two—Oxidative Stress, Inflammation, and Disease. In Oxidative Stress and Biomaterials; Dziubla, T., Butterfield, D.A., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 35–58. [Google Scholar] [CrossRef]
- Singh, V.; Ubaid, S. Role of Silent Information Regulator 1 (SIRT1) in Regulating Oxidative Stress and Inflammation. Inflammation 2020, 43, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Geto, Z.; Molla, M.D.; Challa, F.; Belay, Y.; Getahun, T. Mitochondrial Dynamic Dysfunction as a Main Triggering Factor for Inflammation Associated Chronic Non-Communicable Diseases. J. Inflamm. Res. 2020, 13, 97–107. [Google Scholar] [CrossRef]
- Wincup, C.; Radziszewska, A. Abnormal Mitochondrial Physiology in the Pathogenesis of Systemic Lupus Erythematosus. Rheum. Dis. Clin. 2021, 47, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Mistry, N.; Wood, C.; Herbert, K.; Lunec, J. Immunogenicity of DNA damaged by reactive oxygen species–implications for anti-DNA antibodies in lupus. Free Radic. Biol. Med. 1997, 22, 151–159. [Google Scholar] [CrossRef]
- Hebert, S.L.; Nair, K.S. Protein and energy metabolism in type 1 diabetes. Clin. Nutr. 2010, 29, 13–17. [Google Scholar] [CrossRef]
- Domínguez, C.; Ruiz, E.; Gussinye, M.; Carrascosa, A. Oxidative stress at onset and in early stages of type 1 diabetes in children and adolescents. Diabetes Care 1998, 21, 1736–1742. [Google Scholar] [CrossRef]
- Gurgul-Convey, E.; Mehmeti, I.; Lortz, S.; Lenzen, S. Cytokine toxicity in insulin-producing cells is mediated by nitro-oxidative stress-induced hydroxyl radical formation in mitochondria. J. Mol. Med. 2011, 89, 785–798. [Google Scholar] [CrossRef]
- Quiñonez-Flores, C.M.; González-Chávez, S.A.; Del Río Nájera, D.; Pacheco-Tena, C. Oxidative Stress Relevance in the Pathogenesis of the Rheumatoid Arthritis: A Systematic Review. BioMed Res. Int. 2016, 2016, 6097417. [Google Scholar] [CrossRef] [PubMed]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Front. Physiol. 2021, 12, 627837. [Google Scholar] [CrossRef]
- Ling, X.C.; Kuo, K. Oxidative stress in chronic kidney disease. Ren. Replace. Ther. 2018, 4, 53. [Google Scholar] [CrossRef]
- Picard, M.; McEwen, B.S.; Epel, E.S.; Sandi, C. An energetic view of stress: Focus on mitochondria. Front. Neuroendocr. 2018, 49, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.L.; Yao, H. Mitochondrial dysfunction in inflammatory responses and cellular senescence: Pathogenesis and pharmacological targets for chronic lung diseases. Br. J. Pharmacol. 2016, 173, 2305–2318. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020, 33, 101544. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Rowe, J.H.; Garcia-de-Alba, C.; Kim, C.F.; Sharpe, A.H.; Haigis, M.C. The aging lung: Physiology, disease, and 465 immunity. Cell 2021, 184, 1990–2019. [Google Scholar] [CrossRef]
- Rahman, I.; Biswas, S.K.; Kode, A. Oxidant and antioxidant balance in the airways and airway diseases. Eur. J. Pharmacol. 2006, 533, 222–239. [Google Scholar] [CrossRef] [PubMed]
- Wiegman, C.H.; Michaeloudes, C.; Haji, G.; Narang, P.; Clarke, C.J.; Russell, K.E.; Bao, W.; Pavlidis, S.; Barnes, P.J.; Kanerva, J.; et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015, 136, 769–780. [Google Scholar] [CrossRef]
- Haji, G.; Wiegman, C.H.; Michaeloudes, C.; Patel, M.S.; Curtis, K.; Bhavsar, P.; Polkey, M.I.; Adcock, I.M.; Chung, K.F. Mitochondrial dysfunction in airways and quadriceps muscle of patients with chronic obstructive pulmonary disease. Respir. Res. 2020, 21, 262. [Google Scholar] [CrossRef]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220. [Google Scholar] [CrossRef] [PubMed]
- Burtenshaw, D.; Kitching, M.; Redmond, E.M.; Megson, I.L.; Cahill, P.A. Reactive oxygen species (ROS), intimal thickening, and subclinical atherosclerotic disease. Front. Cardiovasc. Med. 2019, 6, 89. [Google Scholar] [CrossRef]
- Peng, W.; Cai, G.; Xia, Y.; Chen, J.; Wu, P.; Wang, Z.; Li, G.; Wei, D. Mitochondrial Dysfunction in Atherosclerosis. DNA Cell Biol. 2019, 38, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Shemiakova, T.; Ivanova, E.; Grechko, A.V.; Gerasimova, E.V.; Sobenin, I.A.; Orekhov, A.N. Mitochondrial Dysfunction and DNA Damage in the Context of Pathogenesis of Atherosclerosis. Biomedicines 2020, 18, 166. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, Y.; Li, Z.; Song, Y.; Cai, X.; Liu, Y.; Zhang, T.; Yang, L.; Li, L.; Gao, S.; et al. Identification of essential hypertension biomarkers in human urine by non-targeted metabolomics based on UPLC-Q-TOF/MS. Clin. Chim. Acta 2018, 486, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Pinzón-Díaz, C.E.; Calderón-Salinas, J.V.; Rosas-Flores, M.M.; Hernández, G.; López-Betancourt, A.; Quintanar-Escorza, M.A. Eryptosis and oxidative damage in hypertensive and dyslipidemic patients. Mol. Cell. Biochem. 2018, 440, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Kiyuna, L.A.; Albuquerque, R.P.E.; Chen, C.H.; Mochly-Rosen, D.; Ferreira, J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic. Biol. Med. 2018, 129, 155–168. [Google Scholar] [CrossRef]
- Sánchez-Rodríguez, M.A.; Mendoza-Núñez, V.M. Oxidative stress indexes for diagnosis of health or disease in humans. Oxid. Med. Cell. Long. 2019, 2019, 4128152. [Google Scholar] [CrossRef]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- Li, A.; Zheng, N.; Ding, X. Mitochondrial abnormalities: A hub in metabolic syndrome-related cardiac dysfunction caused by oxidative stress. Heart Fail. Rev. 2021, 1–8. [Google Scholar] [CrossRef]
- Xiang, D.; Liu, Y.; Zhou, S.; Zhou, E.; Wang, Y. Protective Effects of Estrogen on Cardiovascular Disease Mediated by Oxidative Stress. Oxid. Med. Cell. Longev. 2021, 28, 5523516. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Sato, K. Maternal inheritance of mitochondrial DNA by diverse mechanisms to eliminate paternal mitochondrial DNA. Biochim. Biophys. 2013, 1833, 1979–1984. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Singh, K.K. Genetic insights into OXPHOS defect and its role in cancer. Biochim. Biophys. Acta 2011, 1807, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Di Emidio, G.; Falone, S.; Artini, P.G.; Amicarelli, F.; D’Alessandro, A.M.; Tatone, C. Mitochondrial Sirtuins in Reproduction. Antioxidants 2021, 10, 1047. [Google Scholar] [CrossRef] [PubMed]
- Zahra, K.; Lefter, R.; Ali, A.; Abdellah, E.C.; Trus, C.; Ciobica, A.; Timofte, D. The Involvement of the Oxidative Stress Status in Cancer Pathology: A Double View on the Role of the Antioxidants. Oxid. Med. Cell. Longev. 2021, 5, 9965916. [Google Scholar] [CrossRef]
- Harper, M.E.; Bevilacqua, L.; Hagopian, K.; Weindruch, R.; Ramsey, J.J. Ageing, oxidative stress, and mitochondrial uncoupling. Acta Physiol. Scand. 2004, 182, 321–331. [Google Scholar] [CrossRef]
- Moncada, S.; Palmer, R.M.J.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Chan, P.H.; Kawase, M.; Murakami, K.; Chen, S.F.; Li, Y.; Calagui, B.; Reola, L.; Carlson, E.; Epstein, C.J. Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J. Neurosci. 1998, 18, 8292–8299. [Google Scholar] [CrossRef]
- Isogawa, A.; Yamakado, M.; Yano, M.; Shiba, T. Serum superoxide dismutase activity correlates with the components of metabolic syndrome or carotid artery intima-media thickness. Diabetes Res. Clin. Pract. 2009, 86, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Soto, M.E.; Soria-Castro, E.; Lans, V.G.; Ontiveros, E.M.; Mejía, B.I.; Hernandez, H.J.; García, R.B.; Herrera, V.; Pérez-Torres, I. Analysis of oxidative stress enzymes and structural and functional proteins on human aortic tissue from different aortopathies. Oxid. Med. Cell. Longev. 2014, 760694. [Google Scholar] [CrossRef]
- Gupta, R.K.; Patel, A.K.; Kumari, R.; Chugh, S.; Shrivastav, C.; Mehra, S.; Sharma, A.N. Interactions between oxidative stress, lipid profile and antioxidants in breast cancer: A case control study. Asian Pac. J. Cancer Prev. 2012, 13, 6295–6298. [Google Scholar] [CrossRef] [PubMed]
- Omar, R.A.; Chyan, Y.J.; Andorn, A.C.; Poeggeler, B.; Robakis, N.K.; Pappolla, M.A. Increased Expression but Reduced Activity of Antioxidant Enzymes in Alzheimer’s Disease. J. Alzheimers Dis. 1999, 1, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Pappolla, M.A.; Chyan, Y.-J.; Omar, R.; Hsiao, K.; Perry, G.; Smith, M.A.; Bozner, P. Evidence of oxidative stress and in vivo neurotoxicity of β- amyloid in a transgenic mouse model of Alzheimer’s disease. Am. J. Pathol. 1998, 152, 871–877. [Google Scholar]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Onodera, K.; Patterson, D. Structure of human chromosome 21-understanding of genetic diseases including Down’s syndrome. Biosci. Biotechnol. Bioch. 1997, 61, 403–409. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Yulyana, Y.; Tovmasyan, A.; Ho, I.A.; Sia, K.C.; Newman, J.P.; Ng, W.; Guo, C.M.; Hui, K.M.; Batinic-Haberle, I.; Lam, P.Y.P. Redox-active mn porphyrin-based potent SOD mimic, mnTnBuOE-2-pyP(5+), enhances carbenoxolone-mediated TRAIL-induced apoptosis in glioblastoma multiforme. Stem Cell Rev. 2016, 12, 140–155. [Google Scholar] [CrossRef]
- Aston, K.; Rath, N.; Naik, A.; Slomczynska, U.; Schall, O.F.; Riley, D.P. Computer-aided design (CAD) of Mn(II) complexes: Superoxide dismutase mimetics with catalytic activity exceeding the native enzyme. Inorg. Chem. 2001, 40, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Heer, C.D.; Davis, A.B.; Riffe, D.B.; Wagner, B.A.; Falls, K.C.; Allen, B.G.; Buettner, G.R.; Beardsley, R.A.; Riley, D.P.; Spitz, D.R. Superoxide Dismutase Mimetic GC4419 Enhances the Oxidation of Pharmacological Ascorbate and Its Anticancer Effects in an H₂O₂-Dependent Manner. Antioxidants 2018, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Salvemini, D.; Mazzon, E.; Dugo, L.; Riley, D.P.; Serraino, I.; Caputi, A.P.; Cuzzocrea, S. Pharmacological manipulation of the inflammatory cascade by the superoxide dismutase mimetic, M40403. Br. J. Pharmacol. 2001, 132, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Kakaroubas, N.; Brennan, S.; Keon, M.; Saksena, N.K. Pathomechanisms of Blood-Brain Barrier Disruption in ALS. Neurosci. J. 2019, 2019, 2537698. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Murawaki, Y.; Tsuchiya, H.; Kanbe, T.; Harada, K.; Yashima, K.; Nozaka, K.; Tanida, O.; Kohno, M.; Mukoyama, T.; Nishimuki, E.; et al. Aberrant expression of selenoproteins in the progression of colorectal cancer. Cancer Lett. 2008, 259, 218–230. [Google Scholar] [CrossRef]
- Baliga, M.S.; Wang, H.; Zhuo, P.; Schwartz, J.L.; Diamond, A.M. Selenium and GPx-1 over expression protect mammalian cells against UV-induced DNA damage. Biol. Trace Elem. Res. 2007, 115, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Ito, D. Promise of Nucleic Acid Therapeutics for Amyotrophic Lateral Sclerosis. Ann. Neurol. 2021. [Google Scholar] [CrossRef]
- Hosler, B.A.; Brown, R.H. Copper/Zinc superoxide dismutase mutations and free radical damage in amyotrophic lateral sclerosis. Adv. Neurol. 1995, 68, 41–46. [Google Scholar]
- Gaya-Bover, A.; Hernández-López, R.; Alorda-Clara, M.; Ibarra de la Rosa, J.M.; Falcó, E.; Fernández, T.; Company, M.M.; Torrens-Mas, M.; Roca, P.; Oliver, J.; et al. Antioxidant enzymes change in different non-metastatic stages in tumoral and peritumoral tissues of colorectal cancer. Int. J. Biochem. Cell Biol. 2020, 120, 105698. [Google Scholar] [CrossRef]
- Dhar, S.K.; Tangpong, J.; Chaiswing, L.; Oberley, T.D.; St Clair, D.K. Manganese superoxide dismutase is a P53-regulated gene that switches cancers between early and advanced stages. Cancer Res. 2011, 71, 6684–6695. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.K.; St Clair, D.K. Manganese superoxide dismutase regulation and cancer. Free Radic. Biol. Med. 2012, 52, 2209–2222. [Google Scholar] [CrossRef]
- Gouaze, V.; Mirault, M.E.; Carpentier, S.; Salvayre, R.; Levade, T.; Andrieu-Abadie, N. Glutathione peroxidase-1 overexpression prevents ceramide production and partially inhibits apoptosis in doxorubicin-treated human breast carcinoma cells. Mol. Pharmacol. 2001, 60, 488–496. [Google Scholar]
- Sanchez-Cespedes, M.; Parrella, P.; Esteller, M.; Nomoto, S.; Trink, B.; Engles, J.M.; Westra, W.H.; Herman, J.G.; Sidransky, D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002, 62, 3659–3662. [Google Scholar] [PubMed]
- McCarty, M.F.; Di Nicolantonio, J.J. An increased need for dietary cysteine in support of glutathione synthesis may underlie the increased risk for mortality associated with low protein intake in the elderly. AGE 2015, 37, 96. [Google Scholar] [CrossRef]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s Disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Vina, J.; Lloret, A.; Ortí, R.; Alonso, D. Molecular bases of the treatment of Alzheimer’s Disease with antioxidants: Prevention of oxidative stress. Mol. Asp. Med. 2004, 25, 117–123. [Google Scholar] [CrossRef]
- Kwon, D.H.; Cha, H.J.; Lee, H.; Hong, S.H.; Park, C.; Park, S.H.; Kim, G.Y.; Kim, S.; Kim, H.S.; Hwang, H.J.; et al. Protective Effect of Glutathione against Oxidative Stress-induced Cytotoxicity in RAW 264.7 Macrophages through Activating the Nuclear Factor Erythroid 2-Related Factor-2/Heme Oxygenase-1 Pathway. Antioxidants 2019, 8, 82. [Google Scholar] [CrossRef]
- Hagen, T.M.; Brown, L.A.; Jones, D.P. Protection against paraquat-induced injury by exogenous GSH in pulmonary alveolar type II cells. Biochem. Pharmacol. 1986, 35, 4537–4542. [Google Scholar] [CrossRef]
- Babson, J.R.; Abell, N.S.; Reed, D.J. Protective role of the glutathione redox cycle against adriamycin-mediated toxicity in isolated hepatocytes. Biochem. Pharmacol. 1981, 30, 2299–2304. [Google Scholar] [CrossRef]
- Dumaswala, U.J.; Wilson, M.J.; Wu, Y.L.; Wykle, J.; Zhuo, L.; Douglass, L.M.; Daleke, D.L. Glutathione loading prevents free radical injury in red blood cells after storage. Free Radic. Res. 2000, 33, 517–529. [Google Scholar] [CrossRef] [PubMed]
- van de Wetering, C.; Elko, E.; Berg, M.; Schiffers, C.H.J.; Stylianidis, V.; van den Berge, M.; Nawijn, M.C.; Wouters, E.F.M.; Janssen-Heininger, Y.M.W.; Reynaert, N.L. Glutathione S-transferases and their implications in the lung diseases asthma and chronic obstructive pulmonary disease: Early life susceptibility? Redox Biol. 2021, 43, 101995. [Google Scholar] [CrossRef]
- Meredith, M.J.; Reed, D.J. Depletion in vitro of mitochondrial glutathione in rat hepatocytes and enhancement of lipid peroxidation by adriamycin and 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU). Biochem. Pharmacol. 1983, 32, 1383–1388. [Google Scholar] [CrossRef]
- Hooper, D.C.; Spitsin, S.; Kean, R.B.; Champion, J.M.; Dickson, G.M.; Chaudhry, I.; Koprowski, H. Uric acid, a natural scavenger of peroxynitrite, in experimental allergic encephalomyelitis and multiple sclerosis. Proc. Natl. Acad. Sci. USA 1998, 95, 675–680. [Google Scholar] [CrossRef]
- Mazza, A.; Pessina, A.C.; Pavei, A.; Scarpa, R.; Tikhonoff, V.; Casiglia, E. Predictors of stroke mortality in elderly people from the general population. The cardiovascular study in the elderly. Eur. J. Epidemiol. 2001, 17, 1097–1104. [Google Scholar] [CrossRef]
- Sautin, Y.Y.; Nakagawa, T.; Zharikov, S.; Johnson, R.J. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am. J. Physiol. Cell Physiol. 2007, 293, C584–C596. [Google Scholar] [CrossRef]
- Sautin, Y.Y.; Johnson, R.J. Uric acid: The oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids 2008, 27, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef]
- Ollinger, R.; Kogler, P.; Troppmair, J.; Hermann, M.; Wurm, M.; Drasche, A.; Konigsrainer, I.; Amberger, A.; Weiss, H.; Ofner, D.; et al. Bilirubin inhibits tumor cell growth via activation of ERK. Cell Cycle 2007, 6, 3078–3085. [Google Scholar] [CrossRef]
- Comai, S.; Gobbi, G. Unveiling the role of melatonin MT2 receptors in sleep, anxiety and other neuropsychiatric diseases: A novel target in psychopharmacology. J. Psychiatry Neurosci. 2014, 39, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Onaolapo, O.J.; Onaolapo, A.Y. Melatonin, adolescence, and the brain: An insight into the period-specific influences of a multifunctional signaling molecule. Birth Defects Res. 2017, 109, 1659–1671. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, V. Melatonin oxidative stress and neurodegenerative diseases. Indian J. Exp. Biol. 2002, 40, 668–679. [Google Scholar] [PubMed]
- Hardeland, R. Antioxidative protection by melatonin—Multiplicity of mechanisms from radical detoxification to radical avoidance. Endocrine 2005, 27, 119–130. [Google Scholar] [CrossRef]
- Srinivasan, V.; Pandi-Perumal, S.; Cardinali, D.; Poeggeler, B.; Hardeland, R. Melatonin in Alzheimer’s Disease and other neurodegenerative disorders. Behav. Brain Funct. 2006, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. A review of research trends in physiological abnormalities in autism spectrum disorders: Immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol. Psychiatry 2012, 17, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, T.; Ali, T.; Ikram, M.; Khan, A.; Alam, S.I.; Kim, M.O. Melatonin Rescue Oxidative Stress-Mediated Neuroinflammation/ Neurodegeneration and Memory Impairment in Scopolamine-Induced Amnesia Mice Model. J. Neuroimmune Pharmacol. 2019, 14, 278–294. [Google Scholar] [CrossRef]
- Pappolla, M.A.; Sos, M.; Omar, R.A.; Bick, R.J.; Hickson-Bick, D.L.; Reiter, R.J.; Efthimiopoulos, S.; Robakis, N.K. Melatonin prevents death of neuroblastoma cells exposed to the Alzheimer amyloid peptide. J. Neurosci. 1997, 17, 1683–1690. [Google Scholar] [CrossRef]
- Pappolla, M.; Bozner, P.; Soto, C.; Shao, H.; Robakis, N.K.; Zagorski, M.; Frangione, B.; Ghiso, J. Inhibition of Alzheimer β-fibrillogenesis by melatonin. J. Biol. Chem. 1998, 273, 7185–7188. [Google Scholar] [CrossRef]
- Andersen, L.P.H.; Gögenur, I.; Rosenberg, J. The Safety of Melatonin in Humans. Clin. Drug Investig. 2016, 36, 169–175. [Google Scholar] [CrossRef]
- Cegolon, L.; Mirandola, M.; Salaris, C.; Salvati, M.; Mastrangelo, G.; Salata, C. Hypothiocyanite and Hypothiocyanite/Lactoferrin Mixture Exhibit Virucidal Activity In Vitro against SARS-CoV-2. Pathogens 2021, 10, 233. [Google Scholar] [CrossRef]
- Habib, H.M.; Ibrahim, S.; Zaim, A.; Ibrahim, W.H. The role of iron in the pathogenesis of COVID-18 and possible treatment with lactoferrin and other iron chelators. Biomed. Pharmacother. 2021, 136, 111228. [Google Scholar] [CrossRef]
- Moreno-Expósito, L.; Illescas-Montes, R.; Melguizo-Rodríguez, L.; Ruiz, C.; Ramos-Torrecillas, J.; de Luna-Bertos, E. Multifunctional capacity and therapeutic potential of lactoferrin. Life Sci. 2018, 195, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Serrano, G.; Kochergina, I.; Albors, A.; Diaz, E.; Oroval, M.; Hueso, G.; Serrano, J.M. Liposomal Lactoferrin as Potential Preventative and Cure for COVID-19. Int. J. Res. Health Sci. 2020, 8, 8–15. [Google Scholar] [CrossRef]
- Mrityunjaya, M.; Pavithra, V.; Neelam, R.; Janhavi, P.; Halami, P.M.; Ravindra, P.V. Immune-Boosting, Antioxidant and Antiinflammatory Food Supplements Targeting Pathogenesis of COVID-19. Front. Immunol. 2020, 11, 570122. [Google Scholar] [CrossRef]
- Bukowska-Ośko, I.; Popiel, M.; Kowalczyk, P. The Immunological Role of the Placenta in SARS-CoV-2 Infection—Viral Transmission, Immune Regulation, and Lactoferrin Activity. Int. J. Mol. Sci. 2021, 22, 5799. [Google Scholar] [CrossRef] [PubMed]
- Konieczka, P.; Barszcz, M.; Kowalczyk, P.; Szlis, M.; Jankowski, J. The potential of acetylsalicylic acid and vitamin E in modulating inflammatory cascades in chickens under lipopolysaccharide-induced inflammation. Vet. Res. 2019, 50, 65. [Google Scholar] [CrossRef] [PubMed]
- Młyczyńska, E.; Myszka, M.; Kurowska, P.; Dawid, M.; Milewicz, T.; Bałajewicz-Nowak, M.; Kowalczyk, P.; Rak, A. Anti-Apoptotic Effect of Apelin in Human Placenta: Studies on BeWo Cells and Villous Explants from Third-Trimester Human Pregnancy. Int. J. Mol. Sci. 2021, 22, 2760. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial Oxidative Stress—A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. https://doi.org/10.3390/ijms222413384
Kowalczyk P, Sulejczak D, Kleczkowska P, Bukowska-Ośko I, Kucia M, Popiel M, Wietrak E, Kramkowski K, Wrzosek K, Kaczyńska K. Mitochondrial Oxidative Stress—A Causative Factor and Therapeutic Target in Many Diseases. International Journal of Molecular Sciences. 2021; 22(24):13384. https://doi.org/10.3390/ijms222413384
Chicago/Turabian StyleKowalczyk, Paweł, Dorota Sulejczak, Patrycja Kleczkowska, Iwona Bukowska-Ośko, Marzena Kucia, Marta Popiel, Ewa Wietrak, Karol Kramkowski, Karol Wrzosek, and Katarzyna Kaczyńska. 2021. "Mitochondrial Oxidative Stress—A Causative Factor and Therapeutic Target in Many Diseases" International Journal of Molecular Sciences 22, no. 24: 13384. https://doi.org/10.3390/ijms222413384
APA StyleKowalczyk, P., Sulejczak, D., Kleczkowska, P., Bukowska-Ośko, I., Kucia, M., Popiel, M., Wietrak, E., Kramkowski, K., Wrzosek, K., & Kaczyńska, K. (2021). Mitochondrial Oxidative Stress—A Causative Factor and Therapeutic Target in Many Diseases. International Journal of Molecular Sciences, 22(24), 13384. https://doi.org/10.3390/ijms222413384