
Use of Zebrafish Models to Boost Research in Rare Genetic Diseases

 , , and
, , and
Abstract
1. Introduction
2. Zebrafish as a Genetic Disease Model
2.1. Genetic Approaches
2.1.1. Forward Genetics
2.1.2. Reverse Genetics
2.2. Zebrafish as a Model of Eye Diseases
2.3. Measurement of Visual Behavior in Zebrafish
2.4. Zebrafish as a Model of Deafness
2.5. Measurement of Hearing Behavior in Zebrafish
2.6. Zebrafish as a Model of Epilepsy
2.7. Measurement of Seizures in Zebrafish
2.7.1. Two-Stage Locomotor and Electrophysiological Setup
2.7.2. iZAP and Multi-Electrode Array
2.7.3. Bioluminescence and Fluorescence Calcium Imaging
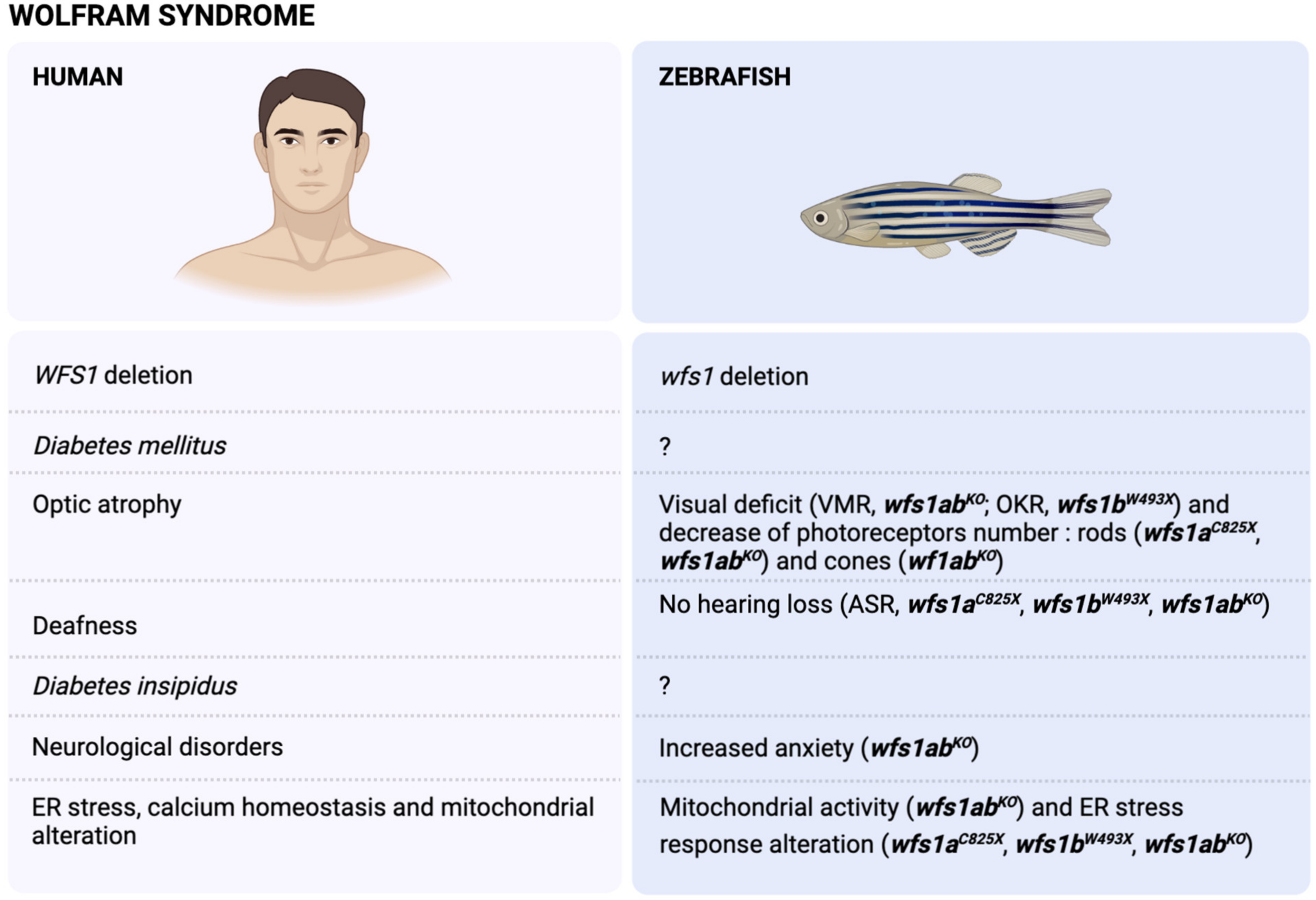
3. The Wolfram Syndrome (WS)
3.1. Physiopathology of the WS
3.2. Modelling of WS in Zebrafish
3.3. WFS1 and Stress Response
3.4. WFS1 and Ca2+ ER Homeostasis
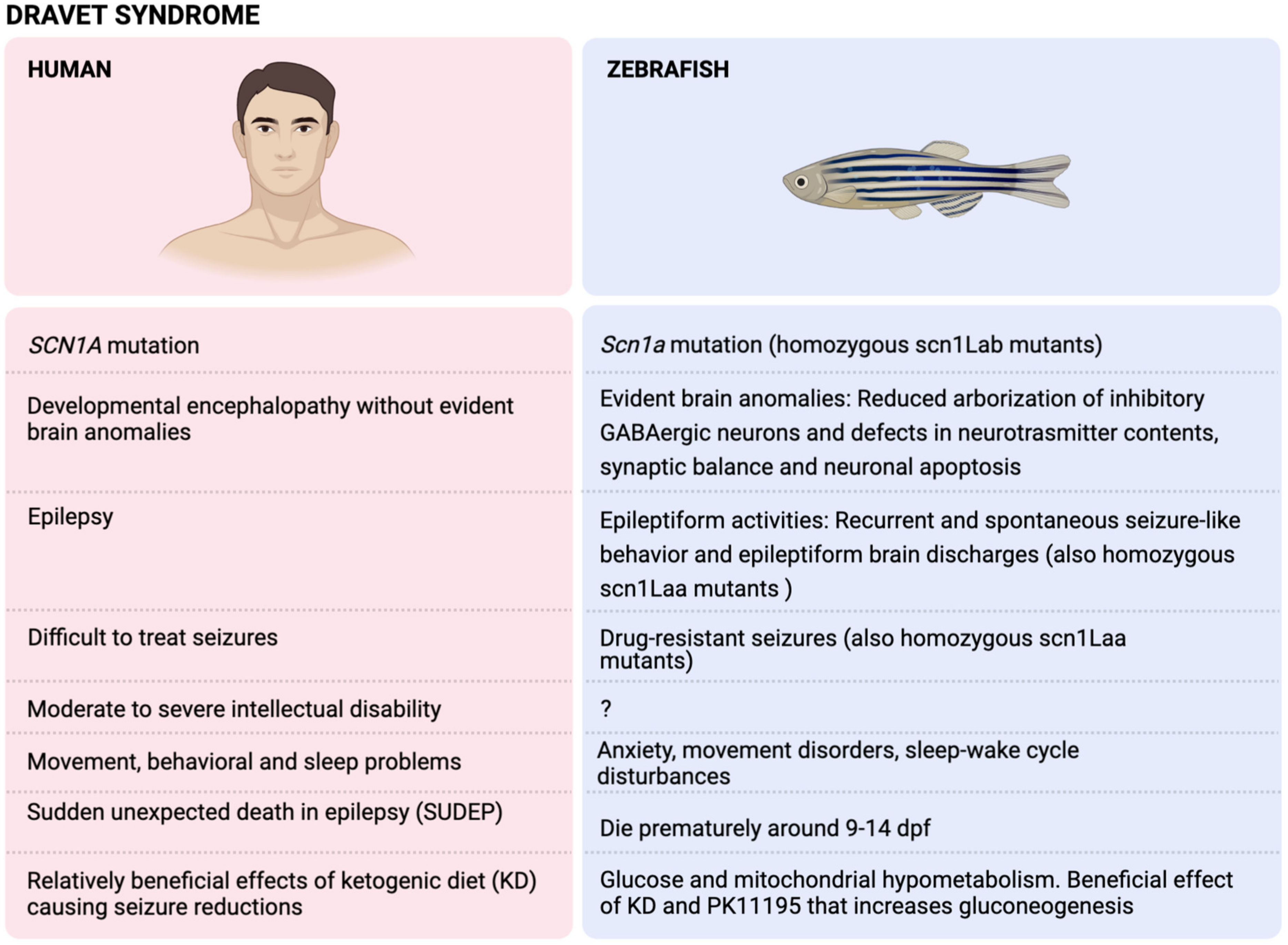
4. The Dravet Syndrome (DS)
4.1. Physiopathology of DS
4.1.1. Genetics
4.1.2. Features
4.1.3. Brain Anomalies
4.2. Modelling of DS in Zebrafish (Scn1a Mutants)
4.2.1. Genetics
4.2.2. Features
4.2.3. Brain Anomalies
4.3. DS Zebrafish Mimicking Drug-Resistant Seizures
4.4. DS Zebrafish for Drug Discovery
4.4.1. Trazodone, TCB-2 and Lisuride
4.4.2. Lorcaserin
4.4.3. Clemizole and Analogs Stimulating 5-HT2BRs
4.4.4. Fenfluramine
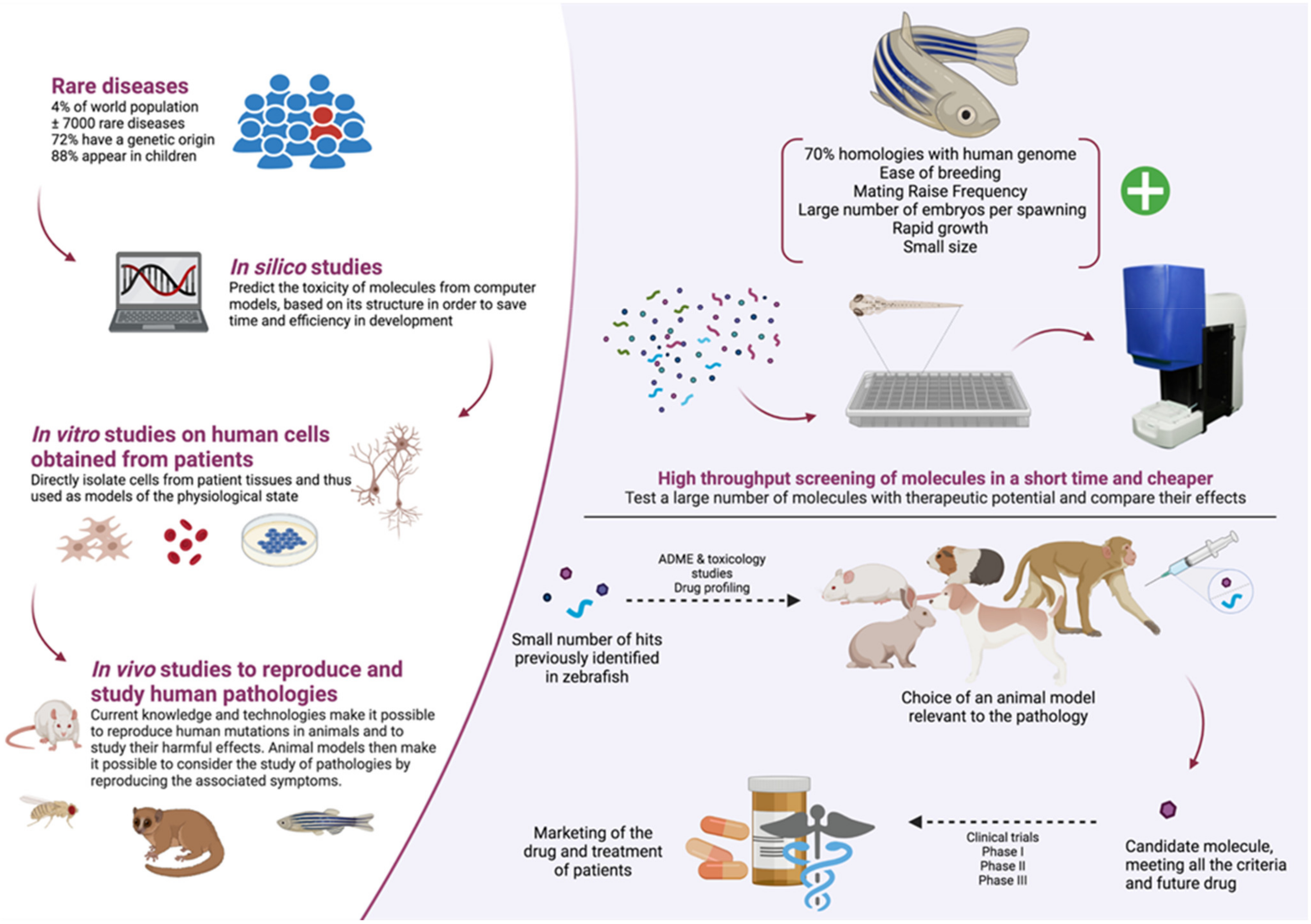
5. Pros and Cons of Zebrafish Models in a Context of Molecules High-Throughput Screening
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 5-HT | serotonin |
| 5-HTR | serotonin receptor |
| AATF | apoptosis antagonizing transcription factor |
| ASM | anti-seizure medication |
| ASR | acoustic startle response |
| ATF4 | activating transcription factor 4 |
| ATF6 | activating transcription factor 6 |
| BIP | glucose-regulated protein (GRP-78) |
| CHOP | C/EBP homologous protein |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| DEE | developmental and epileptic encephalopathies |
| DIDMOAD | diabetes insipidus, diabetes mellitus, optic atrophy, deafness |
| dpf | day post fertilization |
| DS | Dravet syndrome |
| EKP | ethyl ketopentenoate |
| EMA | European Medicines Agency |
| ENU | N-ethyl-N-nitrosourea |
| ER | endoplasmic reticulum |
| ERAD | endoplasmic reticulum-associated degradation |
| FA | fenfluramine |
| GABA | γ-aminobutyric acid |
| GCL | ganglion cell layer |
| GFP | green fluorescent protein |
| GRP75 | glucose-related protein 75 |
| ILAE | International League Against Epilepsy |
| INL | inner nuclear layer |
| IP3R | inositol 1,4,5-trisphosphate receptor |
| IRE1 | inositol-requiring enzyme 1 |
| iZAP | integrated Zebrafish Analysis Platform |
| KD | knock-down |
| KO | knock-out |
| LFP | local field potential |
| MAM | mitochondria-associated ER membrane |
| MES | maximal electroshock seizures |
| MO | morpholino oligonucleotide |
| MRI | magnetic resonance imaging |
| MTC | maximum tolerated concentration |
| NCS1 | neural calcium sensor-1 |
| OKR | optokinetic response |
| OMR | optomotor response |
| ONL | outer nuclear layer |
| ORF | open reading frame |
| PERK | protein kinase R (PKR)-like endoplasmic reticulum kinase |
| PTZ | pentylenetetrazole |
| PWE | patient with epilepsy |
| qPCR | quantitative polymerase chain reaction |
| RIDD | regulated-IRE1 dependent decay |
| RNAi | RNA interference |
| S1/2P | site 1/2 protease |
| SCN1A | sodium voltage-gated channel alpha subunit 1 |
| SUDEP | sudden unexpected death in epilepsy |
| TALEN | transcription activator-like effector nuclease |
| UPR | unfolded protein response |
| VDAC1 | voltage-dependent mitochondrial transmembrane anion channel |
| VMR | visual motor response |
| WFS1/2 | Wolfram syndrome type 1/2 |
| WS | Wolfram syndrome |
| XBP1 | X-box binding protein 1 |
References
- Hamilton, F. An Account of the Fishes Found in the River Ganges and Its Branches; Constable & Robinson Ltd.: Edinburgh, UK, 1822. [Google Scholar]
- Spence, R.; Gerlach, G.; Lawrence, C.; Smith, C. The behaviour and ecology of the zebrafish, Danio rerio. Biol. Rev. Camb. Philos. Soc. 2008, 83, 13–34. [Google Scholar] [CrossRef]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 1995, 203, 253–310. [Google Scholar] [CrossRef]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Currie, P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef]
- Phillips, J.B.; Westerfield, M. Zebrafish models in translational research: Tipping the scales toward advancements in human health. Dis. Models Mech. 2014, 7, 739–743. [Google Scholar] [CrossRef]
- White, R.M. Cross-species oncogenomics using zebrafish models of cancer. Curr. Opin. Genet. Dev. 2015, 30, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Burton, E.A. Zebrafish models of Tauopathy. Biochim. Biophys. Acta 2011, 1812, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Bandmann, O.; Burton, E.A. Genetic zebrafish models of neurodegenerative diseases. Neurobiol. Dis. 2010, 40, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Rajanikant, G.K. Huntington disease: Can a zebrafish trail leave more than a ripple? Neurosci. Biobehav. Rev. 2014, 45, 258–261. [Google Scholar] [CrossRef]
- Laird, A.S.; Mackovski, N.; Rinkwitz, S.; Becker, T.S.; Giacomotto, J. Tissue-specific models of spinal muscular atrophy confirm a critical role of SMN in motor neurons from embryonic to adult stages. Hum. Mol. Genet. 2016, 25, 1728–1738. [Google Scholar] [CrossRef]
- Boehmler, W.; Obrecht-Pflumio, S.; Canfield, V.; Thisse, C.; Thisse, B.; Levenson, R. Evolution and expression of D2 and D3 dopamine receptor genes in zebrafish. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2004, 230, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Norton, W.H.J.; Folchert, A.; Bally-Cuif, L. Comparative analysis of serotonin receptor (HTR1A/HTR1B families) and transporter (slc6a4a/b) gene expression in the zebrafish brain. J. Comp. Neurol. 2008, 511, 521–542. [Google Scholar] [CrossRef]
- Sanchez-Simon, F.M.; Rodriguez, R.E. Developmental expression and distribution of opioid receptors in zebrafish. Neuroscience 2008, 151, 129–137. [Google Scholar] [CrossRef]
- Eliceiri, B.P.; Gonzalez, A.M.; Baird, A. Zebrafish model of the blood-brain barrier: Morphological and permeability studies. Methods Mol. Biol. 2011, 686, 371–378. [Google Scholar] [CrossRef]
- Jeong, J.-Y.; Kwon, H.-B.; Ahn, J.-C.; Kang, D.; Kwon, S.-H.; Park, J.A.; Kim, K.-W. Functional and developmental analysis of the blood-brain barrier in zebrafish. Brain Res. Bull. 2008, 75, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Haffter, P.; Granato, M.; Brand, M.; Mullins, M.C.; Hammerschmidt, M.; Kane, D.A.; Odenthal, J.; van Eeden, F.J.; Jiang, Y.J.; Heisenberg, C.P.; et al. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development 1996, 123, 1–36. [Google Scholar] [CrossRef]
- Roosen-Runge, E. Observations of the early development of the zebrafish, BrachyDanio rerio. Anat. Rec. 1937, 70, 103. [Google Scholar]
- Streisinger, G.; Walker, C.; Dower, N.; Knauber, D.; Singer, F. Production of clones of homozygous diploid zebra fish (BrachyDanio rerio). Nature 1981, 291, 293–296. [Google Scholar] [CrossRef]
- Stanton, M.F. Diethylnitrosamine-induced hepatic degeneration and neoplasia in the aquarium fish, Brachydanio rerio. J. Natl. Cancer Inst. 1965, 34, 117–130. [Google Scholar] [CrossRef]
- Laale, H.W. Ethanol induced notochord and spinal cord duplications in the embryo of the zebrafish, Brachydanio rerio. J. Exp. Zool. 1971, 177, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, R.R.; Tongiorgi, E.; Anzini, P.; Schachner, M. Increased expression of specific recognition molecules by retinal ganglion cells and by optic pathway glia accompanies the successful regeneration of retinal axons in adult zebrafish. J. Comp. Neurol. 1996, 376, 253–264. [Google Scholar] [CrossRef]
- Johnson, S.L.; Weston, J.A. Temperature-sensitive mutations that cause stage-specific defects in Zebrafish fin regeneration. Genetics 1995, 141, 1583–1595. [Google Scholar] [CrossRef] [PubMed]
- White, J.A.; Boffa, M.B.; Jones, B.; Petkovich, M. A zebrafish retinoic acid receptor expressed in the regenerating caudal fin. Development 1994, 120, 1861–1872. [Google Scholar] [CrossRef]
- Lin, Y.-Y. Muscle diseases in the zebrafish. Neuromuscul. Disord. 2012, 22, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Norton, W.H.J. Toward developmental models of psychiatric disorders in zebrafish. Front. Neural Circuits 2013, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Leach, S.D. Zebrafish Models for Cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 71–93. [Google Scholar] [CrossRef]
- Lien, C.-L.; Harrison, M.R.; Tuan, T.-L.; Starnes, V.A. Heart repair and regeneration: Recent insights from zebrafish studies. Wound Repair Regen. 2012, 20, 638–646. [Google Scholar] [CrossRef]
- Lohi, O.; Parikka, M.; Rämet, M. The zebrafish as a model for paediatric diseases. Acta Paediatr. 2013, 102, 104–110. [Google Scholar] [CrossRef]
- Bilotta, J.; Saszik, S. The zebrafish as a model visual system. Int. J. Dev. Neurosci. 2001, 19, 621–629. [Google Scholar] [CrossRef]
- Wang, J.; Yan, Z.; Xing, Y.; Lai, K.; Wang, J.; Yu, D.; Shi, H.; Yin, S. A zebrafish model for hearing loss and regeneration induced by blast wave. J. Bio X Res. 2019, 2, 87–97. [Google Scholar] [CrossRef]
- Fuentes, R.; Letelier, J.; Tajer, B.; Valdivia, L.E.; Mullins, M.C. Fishing forward and reverse: Advances in zebrafish phenomics. Mech. Dev. 2018, 154, 296–308. [Google Scholar] [CrossRef]
- Simon, M.M.; Moresco, E.M.Y.; Bull, K.R.; Kumar, S.; Mallon, A.-M.; Beutler, B.; Potter, P.K. Current strategies for mutation detection in phenotype-driven screens utilising next generation sequencing. Mamm. Genome 2015, 26, 486–500. [Google Scholar] [CrossRef][Green Version]
- Muto, A.; Orger, M.B.; Wehman, A.M.; Smear, M.C.; Kay, J.N.; Page-McCaw, P.S.; Gahtan, E.; Xiao, T.; Nevin, L.M.; Gosse, N.J.; et al. Forward Genetic Analysis of Visual Behavior in Zebrafish. PLoS Genet. 2005, 1, e66. [Google Scholar] [CrossRef]
- Nasevicius, A.; Ekker, S.C. Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef]
- Summerton, J. Morpholino antisense oligomers: The case for an RNase H-independent structural type. Biochim. Biophys. Acta BBA Gene Struct. Expr. 1999, 1489, 141–158. [Google Scholar] [CrossRef]
- Bill, B.R.; Petzold, A.M.; Clark, K.J.; Schimmenti, L.A.; Ekker, S.C. A Primer for Morpholino Use in Zebrafish. Zebrafish 2009, 6, 69–77. [Google Scholar] [CrossRef]
- Andrews, O.E.; Cha, D.J.; Wei, C.; Patton, J.G. RNAi-Mediated Gene silencing in Zebrafish Triggered by Convergent Transcription. Sci. Rep. 2015, 4, 5222. [Google Scholar] [CrossRef] [PubMed]
- Finckbeiner, S.; Ko, P.-J.; Carrington, B.; Sood, R.; Gross, K.; Dolnick, B.; Sufrin, J.; Liu, P. Transient knockdown and overexpression reveal a developmental role for the zebrafish enosf1b gene. Cell Biosci. 2011, 1, 32. [Google Scholar] [CrossRef]
- Rosen, J.N.; Sweeney, M.F.; Mably, J.D. Microinjection of zebrafish embryos to analyze gene function. J. Vis. Exp. 2009, 1115. [Google Scholar] [CrossRef] [PubMed]
- Hruscha, A.; Schmid, B. Generation of zebrafish models by CRISPR/Cas9 genome editing. Methods Mol. Biol. 2015, 1254, 341–350. [Google Scholar] [CrossRef]
- Huang, P.; Xiao, A.; Zhou, M.; Zhu, Z.; Lin, S.; Zhang, B. Heritable gene targeting in zebrafish using customized TALENs. Nat. Biotechnol. 2011, 29, 699–700. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.-R.J.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.C.H.; Chen, Y.; Blackburn, P.R.; Ekker, S.C. TALEN-Mediated Mutagenesis and Genome Editing. Methods Mol. Biol. 2016, 1451, 17–30. [Google Scholar] [CrossRef]
- Bahadori, R.; Rinner, O.; Schonthaler, H.B.; Biehlmaier, O.; Makhankov, Y.V.; Rao, P.; Jagadeeswaran, P.; Neuhauss, S.C.F. The Zebrafish fade out mutant: A novel genetic model for Hermansky-Pudlak syndrome. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4523–4531. [Google Scholar] [CrossRef] [PubMed]
- Boyadjiev, S.A.; Fromme, J.C.; Ben, J.; Chong, S.S.; Nauta, C.; Hur, D.J.; Zhang, G.; Hamamoto, S.; Schekman, R.; Ravazzola, M.; et al. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat. Genet. 2006, 38, 1192–1197. [Google Scholar] [CrossRef]
- Hinkes, B.; Wiggins, R.C.; Gbadegesin, R.; Vlangos, C.N.; Seelow, D.; Nürnberg, G.; Garg, P.; Verma, R.; Chaib, H.; Hoskins, B.E.; et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat. Genet. 2006, 38, 1397–1405. [Google Scholar] [CrossRef]
- Shafizadeh, E.; Paw, B.H.; Foott, H.; Liao, E.C.; Barut, B.A.; Cope, J.J.; Zon, L.I.; Lin, S. Characterization of zebrafish merlot/chablis as non-mammalian vertebrate models for severe congenital anemia due to protein 4.1 deficiency. Dev. Camb. Engl. 2002, 129, 4359–4370. [Google Scholar] [CrossRef]
- Winkler, C.; Eggert, C.; Gradl, D.; Meister, G.; Giegerich, M.; Wedlich, D.; Laggerbauer, B.; Fischer, U. Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes Dev. 2005, 19, 2320–2330. [Google Scholar] [CrossRef]
- Hoshijima, K.; Jurynec, M.J.; Grunwald, D.J. Precise Editing of the Zebrafish Genome Made Simple and Efficient. Dev. Cell 2016, 36, 654–667. [Google Scholar] [CrossRef]
- Schmitt, E.A.; Dowling, J.E. Early eye morphogenesis in the zebrafish, Brachydanio rerio. J. Comp. Neurol. 1994, 344, 532–542. [Google Scholar] [CrossRef]
- Maurer, C.M.; Huang, Y.-Y.; Neuhauss, S.C.F. Application of zebrafish oculomotor behavior to model human disorders. Rev. Neurosci. 2011, 22, 5–16. [Google Scholar] [CrossRef]
- Fleisch, V.C.; Neuhauss, S.C.F. Visual behavior in zebrafish. Zebrafish 2006, 3, 191–201. [Google Scholar] [CrossRef]
- Schmitt, E.A.; Dowling, J.E. Early retinal development in the zebrafish, Danio rerio: Light and electron microscopic analyses. J. Comp. Neurol. 1999, 404, 515–536. [Google Scholar] [CrossRef]
- Chinen, A.; Hamaoka, T.; Yamada, Y.; Kawamura, S. Gene Duplication and Spectral Diversification of Cone Visual Pigments of Zebrafish. Genetics 2003, 163, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Vihtelic, T.S.; Doro, C.J.; Hyde, D.R. Cloning and characterization of six zebrafish photoreceptor opsin cDNAs and immunolocalization of their corresponding proteins. Vis. Neurosci. 1999, 16, 571–585. [Google Scholar] [CrossRef]
- Crouzier, L.; Diez, C.; Richard, E.M.; Cubedo, N.; Barbereau, C.; Rossel, M.; Delaunay, T.; Maurice, T.; Delprat, B. Loss of Pde6a Induces Rod Outer Segment Shrinkage and Visual Alterations in pde6aQ70X Mutant Zebrafish, a Relevant Model of Retinal Dystrophy. Front. Cell Dev. Biol. 2021, 9, 675517. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, P.; Mills-Henry, I.; Padmanabhan, K.R.; Pascuzzi, P.; Hassan, M.; Zhang, J.; Zhang, X.; Ma, P.; Pang, C.P.; Dowling, J.E.; et al. Rods Contribute to Visual Behavior in Larval Zebrafish. Investig. Opthalmol. Vis. Sci. 2020, 61, 11. [Google Scholar] [CrossRef] [PubMed]
- Burrill, J.D.; Easter, S.S. Development of the retinofugal projections in the embryonic and larval zebrafish (BrachyDanio rerio). J. Comp. Neurol. 1994, 346, 583–600. [Google Scholar] [CrossRef]
- Easter, S.S.; Nicola, G.N. The development of eye movements in the zebrafish (Danio rerio). Dev. Psychobiol. 1997, 31, 267–276. [Google Scholar] [CrossRef]
- Biehlmaier, O.; Neuhauss, S.C.F.; Kohler, K. Double cone dystrophy and RPE degeneration in the retina of the zebrafish gnn mutant. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1287–1298. [Google Scholar] [CrossRef]
- Li, X.-Q.; Cai, H.-C.; Zhou, S.-Y.; Yang, J.-H.; Xi, Y.-B.; Gao, X.-B.; Zhao, W.-J.; Li, P.; Zhao, G.-Y.; Tong, Y.; et al. A novel mutation impairing the tertiary structure and stability of γC-crystallin (CRYGC) leads to cataract formation in humans and zebrafish lens. Hum. Mutat. 2012, 33, 391–401. [Google Scholar] [CrossRef]
- Neuhauss, S.C.F.; Biehlmaier, O.; Seeliger, M.W.; Das, T.; Kohler, K.; Harris, W.A.; Baier, H. Genetic Disorders of Vision Revealed by a Behavioral Screen of 400 Essential Loci in Zebrafish. J. Neurosci. 1999, 19, 8603–8615. [Google Scholar] [CrossRef] [PubMed]
- Orger, M.B.; Baier, H. Channeling of red and green cone inputs to the zebrafish optomotor response. Vis. Neurosci. 2005, 22, 275–281. [Google Scholar] [CrossRef]
- Richards, F.M.; Alderton, W.K.; Kimber, G.M.; Liu, Z.; Strang, I.; Redfern, W.S.; Valentin, J.-P.; Winter, M.J.; Hutchinson, T.H. Validation of the use of zebrafish larvae in visual safety assessment. J. Pharmacol. Toxicol. Methods 2008, 58, 50–58. [Google Scholar] [CrossRef]
- Brockerhoff, S.E.; Hurley, J.B.; Janssen-Bienhold, U.; Neuhauss, S.C.; Driever, W.; Dowling, J.E. A behavioral screen for isolating zebrafish mutants with visual system defects. Proc. Natl. Acad. Sci. USA 1995, 92, 10545–10549. [Google Scholar] [CrossRef]
- Huber-Reggi, S.P.; Mueller, K.P.; Neuhauss, S.C.F. Analysis of Optokinetic Response in Zebrafish by Computer-Based Eye Tracking. In Retinal Degeneration: Methods and Protocols; Weber, B.H.F., Langmann, T., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 139–160. ISBN 978-1-62703-080-9. [Google Scholar]
- Deeti, S.; O’Farrell, S.; Kennedy, B.N. Early safety assessment of human oculotoxic drugs using the zebrafish visualmotor response. J. Pharmacol. Toxicol. Methods 2014, 69, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Emran, F.; Rihel, J.; Dowling, J.E. A behavioral assay to measure responsiveness of zebrafish to changes in light intensities. J. Vis. Exp. 2008, 20, 103. [Google Scholar] [CrossRef]
- Zhang, L.; Xiang, L.; Liu, Y.; Venkatraman, P.; Chong, L.; Cho, J.; Bonilla, S.; Jin, Z.-B.; Pang, C.P.; Ko, K.M.; et al. A Naturally-Derived Compound Schisandrin B Enhanced Light Sensation in the pde6c Zebrafish Model of Retinal Degeneration. PLoS ONE 2016, 11, e0149663. [Google Scholar] [CrossRef]
- Ernest, S.; Rauch, G.J.; Haffter, P.; Geisler, R.; Petit, C.; Nicolson, T. Mariner is defective in myosin VIIA: A zebrafish model for human hereditary deafness. Hum. Mol. Genet. 2000, 9, 2189–2196. [Google Scholar] [CrossRef]
- Phillips, J.B.; Blanco-Sanchez, B.; Lentz, J.J.; Tallafuss, A.; Khanobdee, K.; Sampath, S.; Jacobs, Z.G.; Han, P.F.; Mishra, M.; Titus, T.A.; et al. Harmonin (Ush1c) is required in zebrafish Müller glial cells for photoreceptor synaptic development and function. Dis. Models Mech. 2011, 4, 786–800. [Google Scholar] [CrossRef] [PubMed]
- Söllner, C.; Rauch, G.-J.; Siemens, J.; Geisler, R.; Schuster, S.C.; Müller, U.; Nicolson, T. Tübingen 2000 Screen Consortium Mutations in cadherin 23 affect tip links in zebrafish sensory hair cells. Nature 2004, 428, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Yariz, K.O.; Duman, D.; Zazo Seco, C.; Dallman, J.; Huang, M.; Peters, T.A.; Sirmaci, A.; Lu, N.; Schraders, M.; Skromne, I.; et al. Mutations in OTOGL, encoding the inner ear protein otogelin-like, cause moderate sensorineural hearing loss. Am. J. Hum. Genet. 2012, 91, 872–882. [Google Scholar] [CrossRef]
- Chiu, L.L.; Cunningham, L.L.; Raible, D.W.; Rubel, E.W.; Ou, H.C. Using the zebrafish lateral line to screen for ototoxicity. J. Assoc. Res. Otolaryngol. JARO 2008, 9, 178–190. [Google Scholar] [CrossRef]
- Coffin, A.B.; Ou, H.; Owens, K.N.; Santos, F.; Simon, J.A.; Rubel, E.W.; Raible, D.W. Chemical screening for hair cell loss and protection in the zebrafish lateral line. Zebrafish 2010, 7, 3–11. [Google Scholar] [CrossRef]
- He, Y.; Bao, B.; Li, H. Using zebrafish as a model to study the role of epigenetics in hearing loss. Expert Opin. Drug Discov. 2017, 12, 967–975. [Google Scholar] [CrossRef]
- Owens, K.N.; Coffin, A.B.; Hong, L.S.; Bennett, K.O.; Rubel, E.W.; Raible, D.W. Response of mechanosensory hair cells of the zebrafish lateral line to aminoglycosides reveals distinct cell death pathways. Hear. Res. 2009, 253, 32–41. [Google Scholar] [CrossRef]
- Wang, C.; Zhong, Z.; Sun, P.; Zhong, H.; Li, H.; Chen, F. Evaluation of the Hair Cell Regeneration in Zebrafish Larvae by Measuring and Quantifying the Startle Responses. Neural Plast. 2017, 2017, 8283075. [Google Scholar] [CrossRef]
- Baxendale, S.; Whitfield, T.T. Methods to study the development, anatomy, and function of the zebrafish inner ear across the life course. Methods Cell Biol. 2016, 134, 165–209. [Google Scholar] [PubMed]
- Whitfield, T.T.; Hammond, K.L. Axial patterning in the developing vertebrate inner ear. Int. J. Dev. Biol. 2007, 51, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Bever, M.M.; Fekete, D.M. Atlas of the developing inner ear in zebrafish. Dev. Dyn. 2002, 223, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, T. The genetics of hearing and balance in zebrafish. Annu. Rev. Genet. 2005, 39, 9–22. [Google Scholar] [CrossRef]
- Riley, B.B.; Moorman, S.J. Development of utricular otoliths, but not saccular otoliths, is necessary for vestibular function and survival in zebrafish. J. Neurobiol. 2000, 43, 329–337. [Google Scholar] [CrossRef]
- Bang, P.I.; Sewell, W.F.; Malicki, J.J. Morphology and cell type heterogeneities of the inner ear epithelia in adult and juvenile zebrafish (Danio rerio). J. Comp. Neurol. 2001, 438, 173–190. [Google Scholar] [CrossRef]
- Hudspeth, A.J. Hair-bundle mechanics and a model for mechanoelectrical transduction by hair cells. Soc. Gen. Physiol. Ser. 1992, 47, 357–370. [Google Scholar] [PubMed]
- Dijkgraaf, S. The functioning and significance of the lateral-line organs. Biol. Rev. Camb. Philos. Soc. 1963, 38, 51–105. [Google Scholar] [CrossRef]
- Kanter, M.J.; Coombs, S. Rheotaxis and prey detection in uniform currents by Lake Michigan mottled sculpin (Cottus bairdi). J. Exp. Biol. 2003, 206, 59–70. [Google Scholar] [CrossRef]
- Montgomery, J.C.; Baker, C.F.; Carton, A.G. The lateral line can mediate rheotaxis in fish. Nature 1997, 389, 960–963. [Google Scholar] [CrossRef]
- Pitcher, T.J.; Partridge, B.L.; Wardle, C.S. A blind fish can school. Science 1976, 194, 963–965. [Google Scholar] [CrossRef] [PubMed]
- Conley, R.A.; Coombs, S. Dipole source localization by mottled sculpin. III. Orientation after site-specific, unilateral denervation of the lateral line system. J. Comp. Physiol. A 1998, 183, 335–344. [Google Scholar] [CrossRef]
- Coombs, S.; Braun, C.B.; Donovan, B. The orienting response of Lake Michigan mottled sculpin is mediated by canal neuromasts. J. Exp. Biol. 2001, 204, 337–348. [Google Scholar] [CrossRef]
- Montgomery, J.C.; Coombs, S. Peripheral encoding of moving sources by the lateral line system of a sit-and-wait predator. J. Exp. Biol. 1998, 201, 91–102. [Google Scholar] [CrossRef]
- Blaxter, J.H.S.; Fuiman, L.A. Function of the Free Neuromasts of Marine Teleost Larvae. In The Mechanosensory Lateral Line; Coombs, S., Görner, P., Münz, H., Eds.; Springer: New York, NY, USA, 1989; pp. 481–499. [Google Scholar]
- Metcalfe, W.K.; Kimmel, C.B.; Schabtach, E. Anatomy of the posterior lateral line system in young larvae of the zebrafish. J. Comp. Neurol. 1985, 233, 377–389. [Google Scholar] [CrossRef]
- McHenry, M.J.; van Netten, S.M. The flexural stiffness of superficial neuromasts in the zebrafish (Danio rerio) lateral line. J. Exp. Biol. 2007, 210, 4244–4253. [Google Scholar] [CrossRef] [PubMed]
- Ghysen, A.; Dambly-Chaudière, C. Development of the zebrafish lateral line. Curr. Opin. Neurobiol. 2004, 14, 67–73. [Google Scholar] [CrossRef]
- McDermott, B.M.; Asai, Y.; Baucom, J.M.; Jani, S.D.; Castellanos, Y.; Gomez, G.; McClintock, J.M.; Hudspeth, A.J. Transgenic Labeling of Hair Cells in the Zebrafish Acousticolateralis System. Gene Expr. Patterns GEP 2010, 10, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Holder, N. Cell turnover in neuromasts of zebrafish larvae. Hear. Res. 2000, 143, 171–181. [Google Scholar] [CrossRef]
- Harris, J.A.; Cheng, A.G.; Cunningham, L.L.; MacDonald, G.; Raible, D.W.; Rubel, E.W. Neomycin-Induced Hair Cell Death and Rapid Regeneration in the Lateral Line of Zebrafish (Danio rerio). JARO J. Assoc. Res. Otolaryngol. 2003, 4, 219–234. [Google Scholar] [CrossRef]
- Ou, H.C.; Raible, D.W.; Rubel, E.W. Cisplatin-induced hair cell loss in zebrafish (Danio rerio) lateral line. Hear. Res. 2007, 233, 46–53. [Google Scholar] [CrossRef]
- Ton, C.; Parng, C. The use of zebrafish for assessing ototoxic and otoprotective agents. Hear. Res. 2005, 208, 79–88. [Google Scholar] [CrossRef]
- Domarecka, E.; Skarzynska, M.; Szczepek, A.J.; Hatzopoulos, S. Use of zebrafish larvae lateral line to study protection against cisplatin-induced ototoxicity: A scoping review. Int. J. Immunopathol. Pharmacol. 2020, 34, 205873842095955. [Google Scholar] [CrossRef]
- Vlasits, A.L.; Simon, J.A.; Raible, D.W.; Rubel, E.W.; Owens, K.N. Screen of FDA-approved drug library reveals compounds that protect hair cells from aminoglycosides and cisplatin. Hear. Res. 2012, 294, 153–165. [Google Scholar] [CrossRef]
- Vanwalleghem, G.; Heap, L.A.; Scott, E.K. A profile of auditory-responsive neurons in the larval zebrafish brain. J. Comp. Neurol. 2017, 525, 3031–3043. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B.; Patterson, J.; Kimmel, R.O. The development and behavioral characteristics of the startle response in the zebra fish. Dev. Psychobiol. 1974, 7, 47–60. [Google Scholar] [CrossRef]
- Zeddies, D.G.; Fay, R.R. Development of the acoustically evoked behavioral response in zebrafish to pure tones. J. Exp. Biol. 2005, 208, 1363–1372. [Google Scholar] [CrossRef]
- Bhandiwad, A.A.; Sisneros, J.A. Revisiting Psychoacoustic Methods for the Assessment of Fish Hearing. Adv. Exp. Med. Biol. 2016, 877, 157–184. [Google Scholar] [CrossRef]
- Liu, X.; Lin, J.; Zhang, Y.; Guo, N.; Li, Q. Sound shock response in larval zebrafish: A convenient and high-throughput assessment of auditory function. Neurotoxicol. Teratol. 2018, 66, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Trevick, S. The Epidemiology of Global Epilepsy. Neurol. Clin. 2016, 34, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef]
- Yaksi, E.; Jamali, A.; Diaz Verdugo, C.; Jurisch-Yaksi, N. Past, present and future of zebrafish in epilepsy research. FEBS J. 2021. [Google Scholar] [CrossRef]
- Burrows, D.R.W.; Samarut, É.; Liu, J.; Baraban, S.C.; Richardson, M.P.; Meyer, M.P.; Rosch, R.E. Imaging epilepsy in larval zebrafish. Eur. J. Paediatr. Neurol. 2020, 24, 70–80. [Google Scholar] [CrossRef]
- Cunliffe, V.T. Building a zebrafish toolkit for investigating the pathobiology of epilepsy and identifying new treatments for epileptic seizures. J. Neurosci. Methods 2016, 260, 91–95. [Google Scholar] [CrossRef]
- Cunliffe, V.T.; Baines, R.A.; Giachello, C.N.G.; Lin, W.-H.; Morgan, A.; Reuber, M.; Russell, C.; Walker, M.C.; Williams, R.S.B. Epilepsy research methods update: Understanding the causes of epileptic seizures and identifying new treatments using non-mammalian model organisms. Seizure 2015, 24, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Ibhazehiebo, K.; Rho, J.M.; Kurrasch, D.M. Metabolism-based drug discovery in zebrafish: An emerging strategy to uncover new anti-seizure therapies. Neuropharmacology 2020, 167, 107988. [Google Scholar] [CrossRef]
- Tiraboschi, E.; Martina, S.; Ent, W.; Grzyb, K.; Gawel, K.; Cordero-Maldonado, M.L.; Poovathingal, S.K.; Heintz, S.; Satheesh, S.V.; Brattespe, J.; et al. New insights into the early mechanisms of epileptogenesis in a zebrafish model of Dravet syndrome. Epilepsia 2020, 61, 549–560. [Google Scholar] [CrossRef]
- Stafstrom, C.E.; Kossoff, E.H. Epileptic Encephalopathy in Infants and Children. Epilepsy Curr. 2016, 16, 273–279. [Google Scholar] [CrossRef]
- Ellis, C.A.; Petrovski, S.; Berkovic, S.F. Epilepsy genetics: Clinical impacts and biological insights. Lancet Neurol. 2020, 19, 93–100. [Google Scholar] [CrossRef]
- Baraban, S.C. A zebrafish-centric approach to antiepileptic drug development. Dis. Models Mech. 2021, 14, dmm049080. [Google Scholar] [CrossRef] [PubMed]
- Copmans, D.; Siekierska, A.; de Witte, P.A.M. Chapter 26 - Zebrafish Models of Epilepsy and Epileptic Seizures. In Models of Seizures and Epilepsy, 2nd ed.; Academic Press: New York, NY, USA, 2017; pp. 369–384. [Google Scholar]
- Baraban, S.C.; Taylor, M.R.; Castro, P.A.; Baier, H. Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c-fos expression. Neuroscience 2005, 131, 759–768. [Google Scholar] [CrossRef]
- Gawel, K.; Langlois, M.; Martins, T.; van der Ent, W.; Tiraboschi, E.; Jacmin, M.; Crawford, A.D.; Esguerra, C.V. Seizing the moment: Zebrafish epilepsy models. Neurosci. Biobehav. Rev. 2020, 116, 1–20. [Google Scholar] [CrossRef]
- Kumari, S.; Sharma, P.; Mazumder, A.G.; Rana, A.K.; Sharma, S.; Singh, D. Development and validation of chemical kindling in adult zebrafish: A simple and improved chronic model for screening of antiepileptic agents. J. Neurosci. Methods 2020, 346, 108916. [Google Scholar] [CrossRef] [PubMed]
- Pieróg, M.; Socała, K.; Doboszewska, U.; Wyska, E.; Guz, L.; Szopa, A.; Serefko, A.; Poleszak, E.; Wlaź, P. Effects of classic antiseizure drugs on seizure activity and anxiety-like behavior in adult zebrafish. Toxicol. Appl. Pharmacol. 2021, 415, 115429. [Google Scholar] [CrossRef]
- Sourbron, J.; Partoens, M.; Scheldeman, C.; Zhang, Y.; Lagae, L.; Witte, P. Drug repurposing for Dravet syndrome in scn1Lab−/− mutant zebrafish. Epilepsia 2019, 60, e8–e13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Vanmeert, M.; Siekierska, A.; Ny, A.; John, J.; Callewaert, G.; Lescrinier, E.; Dehaen, W.; de Witte, P.A.M.; Kaminski, R.M. Inhibition of glutamate decarboxylase (GAD) by ethyl ketopentenoate (EKP) induces treatment-resistant epileptic seizures in zebrafish. Sci. Rep. 2017, 7, 7195. [Google Scholar] [CrossRef] [PubMed]
- Duy, P.Q.; Berberoglu, M.A.; Beattie, C.E.; Hall, C.W. Cellular responses to recurrent pentylenetetrazole-induced seizures in the adult zebrafish brain. Neuroscience 2017, 349, 118–127. [Google Scholar] [CrossRef]
- Cho, S.-J.; Park, E.; Telliyan, T.; Baker, A.; Reid, A.Y. Zebrafish model of posttraumatic epilepsy. Epilepsia 2020, 61, 1774–1785. [Google Scholar] [CrossRef]
- Afrikanova, T.; Serruys, A.-S.K.; Buenafe, O.E.M.; Clinckers, R.; Smolders, I.; de Witte, P.A.M.; Crawford, A.D.; Esguerra, C.V. Validation of the zebrafish pentylenetetrazol seizure model: Locomotor versus electrographic responses to antiepileptic drugs. PLoS ONE 2013, 8, e54166. [Google Scholar] [CrossRef]
- Sourbron, J.; Schneider, H.; Kecskés, A.; Liu, Y.; Buening, E.M.; Lagae, L.; Smolders, I.; de Witte, P. Serotonergic Modulation as Effective Treatment for Dravet Syndrome in a Zebrafish Mutant Model. ACS Chem. Neurosci. 2016, 7, 588–598. [Google Scholar] [CrossRef]
- Griffin, A.; Hamling, K.R.; Knupp, K.; Hong, S.; Lee, L.P.; Baraban, S.C. Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain 2017, 140, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Buenafe, O.E.; Orellana-Paucar, A.; Maes, J.; Huang, H.; Ying, X.; De Borggraeve, W.; Crawford, A.D.; Luyten, W.; Esguerra, C.V.; de Witte, P. Tanshinone IIA Exhibits Anticonvulsant Activity in Zebrafish and Mouse Seizure Models. ACS Chem. Neurosci. 2013, 4, 1479–1487. [Google Scholar] [CrossRef]
- Orellana-Paucar, A.M.; Serruys, A.-S.K.; Afrikanova, T.; Maes, J.; De Borggraeve, W.; Alen, J.; León-Tamariz, F.; Wilches-Arizábala, I.M.; Crawford, A.D.; de Witte, P.A.M.; et al. Anticonvulsant activity of bisabolene sesquiterpenoids of Curcuma longa in zebrafish and mouse seizure models. Epilepsy Behav. 2012, 24, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Sourbron, J.; Smolders, I.; de Witte, P.; Lagae, L. Pharmacological Analysis of the Anti-epileptic Mechanisms of Fenfluramine in scn1a Mutant Zebrafish. Front. Pharmacol. 2017, 8, 191. [Google Scholar] [CrossRef]
- Liu, M.; Copmans, D.; Lu, J.-G.; Yang, M.-R.; Sourbron, J.; Ny, A.; Jiang, Z.-H.; de Witte, P.A.M.; Luyten, W. Bioassay-guided isolation of anti-seizure principles from Semen Pharbitidis using a zebrafish pentylenetetrazol seizure model. J. Ethnopharmacol. 2019, 232, 130–134. [Google Scholar] [CrossRef]
- Monach, P.A. Repeating tests: Different roles in research studies and clinical medicine. Biomark. Med. 2012, 6, 691–703. [Google Scholar] [CrossRef]
- Baraban, S.C.; Dinday, M.T.; Hortopan, G.A. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat. Commun. 2013, 4, 2410. [Google Scholar] [CrossRef]
- Baxendale, S.; Holdsworth, C.J.; Meza Santoscoy, P.L.; Harrison, M.R.M.; Fox, J.; Parkin, C.A.; Ingham, P.W.; Cunliffe, V.T. Identification of compounds with anti-convulsant properties in a zebrafish model of epileptic seizures. Dis. Models Mech. 2012, 5, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Griffin, A.L.; Jaishankar, P.; Grandjean, J.-M.; Olson, S.H.; Renslo, A.R.; Baraban, S.C. Zebrafish studies identify serotonin receptors mediating antiepileptic activity in Dravet syndrome. Brain Commun. 2019, 1, fcz008. [Google Scholar] [CrossRef]
- Diaz Verdugo, C.; Myren-Svelstad, S.; Aydin, E.; Van Hoeymissen, E.; Deneubourg, C.; Vanderhaeghe, S.; Vancraeynest, J.; Pelgrims, R.; Cosacak, M.I.; Muto, A.; et al. Glia-neuron interactions underlie state transitions to generalized seizures. Nat. Commun. 2019, 10, 3830. [Google Scholar] [CrossRef]
- Hong, S.; Lee, P.; Baraban, S.C.; Lee, L.P. A Novel Long-term, Multi-Channel and Non-invasive Electrophysiology Platform for Zebrafish. Sci. Rep. 2016, 6, 28248. [Google Scholar] [CrossRef]
- Meyer, M.; Dhamne, S.C.; LaCoursiere, C.M.; Tambunan, D.; Poduri, A.; Rotenberg, A. Microarray Noninvasive Neuronal Seizure Recordings from Intact Larval Zebrafish. PLoS ONE 2016, 11, e0156498. [Google Scholar] [CrossRef]
- Khateb, M.; Bosak, N.; Herskovitz, M. The Effect of Anti-seizure Medications on the Propagation of Epileptic Activity: A Review. Front. Neurol. 2021, 12, 674182. [Google Scholar] [CrossRef]
- Berio, A.; Piazzi, A. Activity of drugs and components of natural origin in the severe myoclonic epilepsy of infancy (Dravet syndrome). Cent. Nerv. Syst. Agents Med. Chem. 2015, 15, 95–98. [Google Scholar] [CrossRef]
- Naumann, E.A.; Kampff, A.R.; Prober, D.A.; Schier, A.F.; Engert, F. Monitoring neural activity with bioluminescence during natural behavior. Nat. Neurosci. 2010, 13, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Wolfram, D.; Wagener, H.P. Diabetes mellitus and simple optic atrophy among siblings: Report of four cases. Mayo Clin. Proc. 1938, 13, 715–718. [Google Scholar]
- Barrett, T.G.; Bundey, S.E.; Macleod, A.F. Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet Lond. Engl. 1995, 346, 1458–1463. [Google Scholar] [CrossRef]
- Tekgül, S.; Oge, O.; Simşek, E.; Yordam, N.; Kendi, S. Urological manifestations of the Wolfram syndrome: Observations in 14 patients. J. Urol. 1999, 161, 616–617. [Google Scholar] [CrossRef]
- Aldenhövel, H.G.; Gallenkamp, U.; Sulemana, C.A. Juvenile onset diabetes mellitus, central diabetes insipidus and optic atrophy (Wolfram syndrome)—Neurological findings and prognostic implications. Neuropediatrics 1991, 22, 103–106. [Google Scholar] [CrossRef]
- Swift, R.G.; Sadler, D.B.; Swift, M. Psychiatric findings in Wolfram syndrome homozygotes. Lancet Lond. Engl. 1990, 336, 667–669. [Google Scholar] [CrossRef]
- Kinsley, B.T.; Swift, M.; Dumont, R.H.; Swift, R.G. Morbidity and mortality in the Wolfram syndrome. Diabetes Care 1995, 18, 1566–1570. [Google Scholar] [CrossRef]
- Kumar, S. Wolfram syndrome: Important implications for pediatricians and pediatric endocrinologists. Pediatr. Diabetes 2010, 11, 28–37. [Google Scholar] [CrossRef]
- Matsunaga, K.; Tanabe, K.; Inoue, H.; Okuya, S.; Ohta, Y.; Akiyama, M.; Taguchi, A.; Kora, Y.; Okayama, N.; Yamada, Y.; et al. Wolfram syndrome in the Japanese population; molecular analysis of WFS1 gene and characterization of clinical features. PLoS ONE 2014, 9, e106906. [Google Scholar] [CrossRef]
- Fraser, F.C.; Gunn, T. Diabetes mellitus, diabetes insipidus, and optic atrophy. An autosomal recessive syndrome? J. Med. Genet. 1977, 14, 190–193. [Google Scholar] [CrossRef]
- Medlej, R.; Wasson, J.; Baz, P.; Azar, S.; Salti, I.; Loiselet, J.; Permutt, A.; Halaby, G. Diabetes mellitus and optic atrophy: A study of Wolfram syndrome in the Lebanese population. J. Clin. Endocrinol. Metab. 2004, 89, 1656–1661. [Google Scholar] [CrossRef]
- Amr, S.; Heisey, C.; Zhang, M.; Xia, X.-J.; Shows, K.H.; Ajlouni, K.; Pandya, A.; Satin, L.S.; El-Shanti, H.; Shiang, R. A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am. J. Hum. Genet. 2007, 81, 673–683. [Google Scholar] [CrossRef]
- Inoue, H.; Tanizawa, Y.; Wasson, J.; Behn, P.; Kalidas, K.; Bernal-Mizrachi, E.; Mueckler, M.; Marshall, H.; Donis-Keller, H.; Crock, P.; et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat. Genet. 1998, 20, 143–148. [Google Scholar] [CrossRef]
- Strom, T.M.; Hörtnagel, K.; Hofmann, S.; Gekeler, F.; Scharfe, C.; Rabl, W.; Gerbitz, K.D.; Meitinger, T. Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum. Mol. Genet. 1998, 7, 2021–2028. [Google Scholar] [CrossRef] [PubMed]
- Hardy, C.; Khanim, F.; Torres, R.; Scott-Brown, M.; Seller, A.; Poulton, J.; Collier, D.; Kirk, J.; Polymeropoulos, M.; Latif, F.; et al. Clinical and molecular genetic analysis of 19 Wolfram syndrome kindreds demonstrating a wide spectrum of mutations in WFS1. Am. J. Hum. Genet. 1999, 65, 1279–1290. [Google Scholar] [CrossRef]
- Takeda, K.; Inoue, H.; Tanizawa, Y.; Matsuzaki, Y.; Oba, J.; Watanabe, Y.; Shinoda, K.; Oka, Y. WFS1 (Wolfram syndrome 1) gene product: Predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum. Mol. Genet. 2001, 10, 477–484. [Google Scholar] [CrossRef]
- Hofmann, S.; Philbrook, C.; Gerbitz, K.-D.; Bauer, M.F. Wolfram syndrome: Structural and functional analyses of mutant and wild-type wolframin, the WFS1 gene product. Hum. Mol. Genet. 2003, 12, 2003–2012. [Google Scholar] [CrossRef]
- Harding, H.P.; Calfon, M.; Urano, F.; Novoa, I.; Ron, D. Transcriptional and translational control in the Mammalian unfolded protein response. Annu. Rev. Cell Dev. Biol. 2002, 18, 575–599. [Google Scholar] [CrossRef] [PubMed]
- Patil, C.; Walter, P. Intracellular signaling from the endoplasmic reticulum to the nucleus: The unfolded protein response in yeast and mammals. Curr. Opin. Cell Biol. 2001, 13, 349–355. [Google Scholar] [CrossRef]
- Fonseca, S.G.; Fukuma, M.; Lipson, K.L.; Nguyen, L.X.; Allen, J.R.; Oka, Y.; Urano, F. WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic beta-cells. J. Biol. Chem. 2005, 280, 39609–39615. [Google Scholar] [CrossRef]
- Osman, A.A.; Saito, M.; Makepeace, C.; Permutt, M.A.; Schlesinger, P.; Mueckler, M. Wolframin expression induces novel ion channel activity in endoplasmic reticulum membranes and increases intracellular calcium. J. Biol. Chem. 2003, 278, 52755–52762. [Google Scholar] [CrossRef]
- Lu, S.; Kanekura, K.; Hara, T.; Mahadevan, J.; Spears, L.D.; Oslowski, C.M.; Martinez, R.; Yamazaki-Inoue, M.; Toyoda, M.; Neilson, A.; et al. A calcium-dependent protease as a potential therapeutic target for Wolfram syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, E5292–E5301. [Google Scholar] [CrossRef]
- Takei, D.; Ishihara, H.; Yamaguchi, S.; Yamada, T.; Tamura, A.; Katagiri, H.; Maruyama, Y.; Oka, Y. WFS1 protein modulates the free Ca2+ concentration in the endoplasmic reticulum. FEBS Lett. 2006, 580, 5635–5640. [Google Scholar] [CrossRef]
- Luuk, H.; Koks, S.; Plaas, M.; Hannibal, J.; Rehfeld, J.F.; Vasar, E. Distribution of WFS1 protein in the central nervous system of the mouse and its relation to clinical symptoms of the Wolfram syndrome. J. Comp. Neurol. 2008, 509, 642–660. [Google Scholar] [CrossRef]
- Kakiuchi, C.; Ishiwata, M.; Hayashi, A.; Kato, T. XBP1 induces WFS1 through an endoplasmic reticulum stress response element-like motif in SH-SY5Y cells. J. Neurochem. 2006, 97, 545–555. [Google Scholar] [CrossRef]
- Shang, L.; Hua, H.; Foo, K.; Martinez, H.; Watanabe, K.; Zimmer, M.; Kahler, D.J.; Freeby, M.; Chung, W.; LeDuc, C.; et al. β-Cell Dysfunction Due to Increased ER Stress in a Stem Cell Model of Wolfram Syndrome. Diabetes 2014, 63, 923–933. [Google Scholar] [CrossRef]
- Ueda, K.; Kawano, J.; Takeda, K.; Yujiri, T.; Tanabe, K.; Anno, T.; Akiyama, M.; Nozaki, J.; Yoshinaga, T.; Koizumi, A.; et al. Endoplasmic reticulum stress induces WFS1 gene expression in pancreatic beta-cells via transcriptional activation. Eur. J. Endocrinol. 2005, 153, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Ishihara, H.; Tamura, A.; Takahashi, R.; Yamaguchi, S.; Takei, D.; Tokita, A.; Satake, C.; Tashiro, F.; Katagiri, H.; et al. WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic beta-cells. Hum. Mol. Genet. 2006, 15, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Ishiwata, M.; Yamada, K.; Kasahara, T.; Kakiuchi, C.; Iwamoto, K.; Kawamura, K.; Ishihara, H.; Oka, Y. Behavioral and gene expression analyses of WFS1 knockout mice as a possible animal model of mood disorder. Neurosci. Res. 2008, 61, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Luuk, H.; Plaas, M.; Raud, S.; Innos, J.; Sütt, S.; Lasner, H.; Abramov, U.; Kurrikoff, K.; Kõks, S.; Vasar, E. WFS1-deficient mice display impaired behavioural adaptation in stressful environment. Behav. Brain Res. 2009, 198, 334–345. [Google Scholar] [CrossRef]
- Sequeira, A.; Kim, C.; Seguin, M.; Lesage, A.; Chawky, N.; Desautels, A.; Tousignant, M.; Vanier, C.; Lipp, O.; Benkelfat, C.; et al. Wolfram syndrome and suicide: Evidence for a role of WFS1 in suicidal and impulsive behavior. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2003, 119B, 108–113. [Google Scholar] [CrossRef]
- Shrestha, P.; Mousa, A.; Heintz, N. Layer 2/3 pyramidal cells in the medial prefrontal cortex moderate stress induced depressive behaviors. eLife 2015, 4, e08752. [Google Scholar] [CrossRef]
- Bonnet Wersinger, D.; Benkafadar, N.; Jagodzinska, J.; Hamel, C.; Tanizawa, Y.; Lenaers, G.; Delettre, C. Impairment of visual function and retinal ER stress activation in WFS1-deficient mice. PLoS ONE 2014, 9, e97222. [Google Scholar] [CrossRef] [PubMed]
- El-Shanti, H.; Lidral, A.C.; Jarrah, N.; Druhan, L.; Ajlouni, K. Homozygosity Mapping Identifies an Additional Locus for Wolfram Syndrome on Chromosome 4q. Am. J. Hum. Genet. 2000, 66, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Wiley, S.E.; Andreyev, A.Y.; Divakaruni, A.S.; Karisch, R.; Perkins, G.; Wall, E.A.; van der Geer, P.; Chen, Y.-F.; Tsai, T.-F.; Simon, M.I.; et al. Wolfram Syndrome protein, Miner1, regulates sulphydryl redox status, the unfolded protein response, and Ca2+ homeostasis. EMBO Mol. Med. 2013, 5, 904–918. [Google Scholar] [CrossRef] [PubMed]
- Rigoli, L.; Di Bella, C. Wolfram syndrome 1 and Wolfram syndrome 2. Curr. Opin. Pediatr. 2012, 24, 512–517. [Google Scholar] [CrossRef]
- Al-Sheyyab, M.; Jarrah, N.; Younis, E.; Shennak, M.M.; Hadidi, A.; Awidi, A.; El-Shanti, H.; Ajlouni, K. Bleeding tendency in Wolfram syndrome: A newly identified feature with phenotype genotype correlation. Eur. J. Pediatr. 2001, 160, 243–246. [Google Scholar] [CrossRef]
- Rigoli, L.; Bramanti, P.; Di Bella, C.; De Luca, F. Genetic and clinical aspects of Wolfram syndrome 1, a severe neurodegenerative disease. Pediatr. Res. 2018, 83, 921–929. [Google Scholar] [CrossRef]
- Urano, F. Wolfram Syndrome: Diagnosis, Management, and Treatment. Curr. Diab. Rep. 2016, 16, 6. [Google Scholar] [CrossRef]
- Crouzier, L.; Richard, E.M.; Diez, C.; Alzaeem, H.; Denus, M.; Cubedo, N.; Delaunay, T.; Glendenning, E.; Baxendale, S.; Whitfield, T.T.; et al. Morphological, behavioral and cellular analyses revealed different phenotype in 2 Wolfram syndrome WFS1a and WFS1b zebrafish mutant lines. Human Mol. Genet. 2021. Submitted. [Google Scholar]
- Crouzier, L.; Richard, E.M.; Diez, C.; Alzaeem, H.; Denus, M.; Cubedo, N.; Maurice, T.; Delprat, B. NCS1 overexpression restored mitochondrial activity and behavioral anomalies in a novel zebrafish model of Wolfram syndrome. Molec. Ther. 2021. Submitted. [Google Scholar]
- Kaufman, R.J.; Scheuner, D.; Schröder, M.; Shen, X.; Lee, K.; Liu, C.Y.; Arnold, S.M. The unfolded protein response in nutrient sensing and differentiation. Nat. Rev. Mol. Cell Biol. 2002, 3, 411–421. [Google Scholar] [CrossRef]
- Mori, K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 2000, 101, 451–454. [Google Scholar] [CrossRef]
- Ariyasu, D.; Yoshida, H.; Hasegawa, Y. Endoplasmic Reticulum (ER) Stress and Endocrine Disorders. Int. J. Mol. Sci. 2017, 18, 382. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.-P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Kaufman, R.J. That which does not kill me makes me stronger: Adapting to chronic ER stress. Trends Biochem. Sci. 2007, 32, 469–476. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 Activated by Proteolysis Binds in the Presence of NF-Y (CBF) Directly to the cis-Acting Element Responsible for the Mammalian Unfolded Protein Response. Mol. Cell. Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef]
- Coelho, D.S.; Domingos, P.M. Physiological roles of regulated Ire1 dependent decay. Front. Genet. 2014, 5, 76. [Google Scholar] [CrossRef]
- Martindale, J.J.; Fernandez, R.; Thuerauf, D.; Whittaker, R.; Gude, N.; Sussman, M.A.; Glembotski, C.C. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ. Res. 2006, 98, 1186–1193. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufman, R.J. Divergent Roles of IRE1α and PERK in the Unfolded Protein Response. Available online: https://www.ingentaconnect.com/content/ben/cmm/2006/00000006/00000001/art00002 (accessed on 27 April 2020).
- Lu, P.D.; Harding, H.P.; Ron, D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 2004, 167, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress–mediated diabetes. J. Clin. Investig. 2002, 109, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef]
- Bruno, T.; De Nicola, F.; Iezzi, S.; Lecis, D.; D’Angelo, C.; Di Padova, M.; Corbi, N.; Dimiziani, L.; Zannini, L.; Jekimovs, C.; et al. Che-1 phosphorylation by ATM/ATR and Chk2 kinases activates p53 transcription and the G2/M checkpoint. Cancer Cell 2006, 10, 473–486. [Google Scholar] [CrossRef]
- Bruno, T.; Iezzi, S.; De Nicola, F.; Di Padova, M.; Desantis, A.; Scarsella, M.; Di Certo, M.G.; Leonetti, C.; Floridi, A.; Passananti, C.; et al. Che-1 activates XIAP expression in response to DNA damage. Cell Death Differ. 2008, 15, 515–520. [Google Scholar] [CrossRef]
- Ishigaki, S.; Fonseca, S.G.; Oslowski, C.M.; Jurczyk, A.; Shearstone, J.R.; Zhu, L.J.; Permutt, M.A.; Greiner, D.L.; Bortell, R.; Urano, F. AATF mediates anti-apoptotic effect of the unfolded protein response through transcriptional regulation of AKT1. Cell Death Differ. 2010, 17, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Chen, X.; Karnovsky, A.; Sans, M.D.; Andrews, P.C.; Williams, J.A. Molecular characterization of the endoplasmic reticulum: Insights from proteomic studies. Proteomics 2010, 10, 4040–4052. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yoshida, H.; Kokame, K.; Kaufman, R.J.; Mori, K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 2004, 136, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Fonseca, S.G.; Ishigaki, S.; Oslowski, C.M.; Lu, S.; Lipson, K.L.; Ghosh, R.; Hayashi, E.; Ishihara, H.; Oka, Y.; Permutt, M.A.; et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J. Clin. Investig. 2010, 120, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. The binary switch that controls the life and death decisions of ER stressed beta cells. Curr. Opin. Cell Biol. 2011, 23, 207–215. [Google Scholar] [CrossRef]
- Azim, M.; Surani, H. Glycoprotein synthesis and inhibition of glycosylation by tunicamycin in preimplantation mouse embryos: Compaction and trophoblast adhesion. Cell 1979, 18, 217–227. [Google Scholar] [CrossRef]
- Hakulinen, J.K.; Hering, J.; Brändén, G.; Chen, H.; Snijder, A.; Ek, M.; Johansson, P. MraY–antibiotic complex reveals details of tunicamycin mode of action. Nat. Chem. Biol. 2017, 13, 265–267. [Google Scholar] [CrossRef]
- Yoo, J.; Mashalidis, E.H.; Kuk, A.C.Y.; Yamamoto, K.; Kaeser, B.; Ichikawa, S.; Lee, S.-Y. GlcNAc-1-P-transferase–tunicamycin complex structure reveals basis for inhibition of N-glycosylation. Nat. Struct. Mol. Biol. 2018, 25, 217–224. [Google Scholar] [CrossRef]
- Li, J.; Chen, Z.; Gao, L.-Y.; Colorni, A.; Ucko, M.; Fang, S.; Du, S.J. A transgenic zebrafish model for monitoring xbp1 splicing and endoplasmic reticulum stress in vivo. Mech. Dev. 2015, 137, 33–44. [Google Scholar] [CrossRef]
- Clark, E.M.; Nonarath, H.J.T.; Bostrom, J.R.; Link, B.A. Establishment and validation of an endoplasmic reticulum stress reporter to monitor zebrafish ATF6 activity in development and disease. Dis. Models Mech. 2019, 13, dmm041426. [Google Scholar] [CrossRef]
- Lee, H.-C.; Chen, Y.-J.; Liu, Y.-W.; Lin, K.-Y.; Chen, S.-W.; Lin, C.-Y.; Lu, Y.-C.; Hsu, P.-C.; Lee, S.-C.; Tsai, H.-J. Transgenic zebrafish model to study translational control mediated by upstream open reading frame of human chop gene. Nucleic Acids Res. 2011, 39, e139. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Joshi, A.U.; Kornfeld, O.S.; Mochly-Rosen, D. The Entangled ER-Mitochondrial axis as a potential therapeutic strategy in Neurodegeneration: A Tangled Duo Unchained. Cell Calcium 2016, 60, 218–234. [Google Scholar] [CrossRef]
- Krols, M.; van Isterdael, G.; Asselbergh, B.; Kremer, A.; Lippens, S.; Timmerman, V.; Janssens, S. Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol. 2016, 131, 505–523. [Google Scholar] [CrossRef] [PubMed]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.H.W.; Mueller, S.; Miller, T.; Miller, C.C.J. There’s Something Wrong with my MAM; the ER-Mitochondria Axis and Neurodegenerative Diseases. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef]
- Ruby, J.R.; Dyer, R.F.; Skalko, R.G. Continuities between mitochondria and endoplasmic reticulum in the mammalian ovary. Z. Zellforsch. Mikrosk. Anat. 1969, 97, 30–37. [Google Scholar] [CrossRef]
- Delprat, B.; Maurice, T.; Delettre, C. Wolfram syndrome: MAMs’ connection? Cell Death Dis. 2018, 9, 364. [Google Scholar] [CrossRef]
- Colombini, M. The VDAC channel: Molecular basis for selectivity. Biochim. Biophys. Acta BBA Mol. Cell Res. 2016, 1863, 2498–2502. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Nakamura, T.Y.; Jeromin, A.; Mikoshiba, K.; Wakabayashi, S. Neuronal calcium sensor-1 promotes immature heart function and hypertrophy by enhancing Ca2+ signals. Circ. Res. 2011, 109, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Pongs, O.; Lindemeier, J.; Zhu, X.R.; Theil, T.; Engelkamp, D.; Krah-Jentgens, I.; Lambrecht, H.G.; Koch, K.W.; Schwemer, J.; Rivosecchi, R. Frequenin—A novel calcium-binding protein that modulates synaptic efficacy in the Drosophila nervous system. Neuron 1993, 11, 15–28. [Google Scholar] [CrossRef]
- Saab, B.J.; Georgiou, J.; Nath, A.; Lee, F.J.S.; Wang, M.; Michalon, A.; Liu, F.; Mansuy, I.M.; Roder, J.C. NCS-1 in the dentate gyrus promotes exploration, synaptic plasticity, and rapid acquisition of spatial memory. Neuron 2009, 63, 643–656. [Google Scholar] [CrossRef]
- Sippy, T.; Cruz-Martín, A.; Jeromin, A.; Schweizer, F.E. Acute changes in short-term plasticity at synapses with elevated levels of neuronal calcium sensor-1. Nat. Neurosci. 2003, 6, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Drumond, L.E.; Mourão, F.A.G.; Leite, H.R.; Abreu, R.V.; Reis, H.J.; Moraes, M.F.D.; Pereira, G.S.; Massensini, A.R. Differential effects of swimming training on neuronal calcium sensor-1 expression in rat hippocampus/cortex and in object recognition memory tasks. Brain Res. Bull. 2012, 88, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.; Fei, G.-H.; Saab, B.J.; Su, J.; Roder, J.C.; Feng, Z.-P. Neuronal calcium sensor-1 modulation of optimal calcium level for neurite outgrowth. Dev. Camb. Engl. 2007, 134, 4479–4489. [Google Scholar] [CrossRef]
- Nakamura, T.Y.; Jeromin, A.; Smith, G.; Kurushima, H.; Koga, H.; Nakabeppu, Y.; Wakabayashi, S.; Nabekura, J. Novel role of neuronal Ca2+ sensor-1 as a survival factor up-regulated in injured neurons. J. Cell Biol. 2006, 172, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Angebault, C.; Fauconnier, J.; Patergnani, S.; Rieusset, J.; Danese, A.; Affortit, C.A.; Jagodzinska, J.; Mégy, C.; Quiles, M.; Cazevieille, C.; et al. ER-mitochondria cross-talk is regulated by the Ca2+ sensor NCS1 and is impaired in Wolfram syndrome. Sci. Signal. 2018, 11, eaaq1380. [Google Scholar] [CrossRef] [PubMed]
- Delprat, B.; Rieusset, J.; Delettre, C. Defective endoplasmic reticulum–mitochondria 2270 connection is a hallmark of Wolfram syndrome. Contact 2019, 2, 1–5. [Google Scholar] [CrossRef]
- Ashworth, R. Approaches to measuring calcium in zebrafish: Focus on neuronal development. Cell Calcium 2004, 35, 393–402. [Google Scholar] [CrossRef]
- Ahrens, M.B.; Orger, M.B.; Robson, D.N.; Li, J.M.; Keller, P.J. Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat. Methods 2013, 10, 413–420. [Google Scholar] [CrossRef]
- Panier, T.; Romano, S.A.; Olive, R.; Pietri, T.; Sumbre, G.; Candelier, R.; Debrégeas, G. Fast functional imaging of multiple brain regions in intact zebrafish larvae using selective plane illumination microscopy. Front. Neural Circuits 2013, 7, 65. [Google Scholar] [CrossRef]
- Yang, W.; Miller, J.K.; Carrillo-Reid, L.; Pnevmatikakis, E.; Paninski, L.; Yuste, R.; Peterka, D.S. Simultaneous Multi-plane Imaging of Neural Circuits. Neuron 2016, 89, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Mukamel, E.A.; Nimmerjahn, A.; Schnitzer, M.J. Automated Analysis of Cellular Signals from Large-Scale Calcium Imaging Data. Neuron 2009, 63, 747–760. [Google Scholar] [CrossRef]
- Pnevmatikakis, E.A.; Soudry, D.; Gao, Y.; Machado, T.A.; Merel, J.; Pfau, D.; Reardon, T.; Mu, Y.; Lacefield, C.; Yang, W.; et al. Simultaneous Denoising, Deconvolution, and Demixing of Calcium Imaging Data. Neuron 2016, 89, 285–299. [Google Scholar] [CrossRef]
- Chen, T.-W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295–300. [Google Scholar] [CrossRef]
- Keller, P.J.; Ahrens, M.B. Visualizing Whole-Brain Activity and Development at the Single-Cell Level Using Light-Sheet Microscopy. Neuron 2015, 85, 462–483. [Google Scholar] [CrossRef]
- Muto, A.; Ohkura, M.; Kotani, T.; Higashijima, S.-I.; Nakai, J.; Kawakami, K. Genetic visualization with an improved GCaMP calcium indicator reveals spatiotemporal activation of the spinal motor neurons in zebrafish. Proc. Natl. Acad. Sci. USA 2011, 108, 5425–5430. [Google Scholar] [CrossRef]
- Liu, J.; Baraban, S.C. Network Properties Revealed during Multi-Scale Calcium Imaging of Seizure Activity in Zebrafish. eNeuro 2019, 6, ENEURO.0041-19.2019. [Google Scholar] [CrossRef] [PubMed]
- Muto, A.; Kawakami, K. Imaging functional neural circuits in zebrafish with a new GCaMP and the Gal4FF-UAS system. Commun. Integr. Biol. 2011, 4, 566–568. [Google Scholar] [CrossRef]
- Esterberg, R.; Hailey, D.W.; Rubel, E.W.; Raible, D.W. ER-Mitochondrial Calcium Flow Underlies Vulnerability of Mechanosensory Hair Cells to Damage. J. Neurosci. 2014, 34, 9703–9719. [Google Scholar] [CrossRef] [PubMed]
- Lagae, L. Dravet syndrome. Curr. Opin. Neurol. 2021, 34, 213–218. [Google Scholar] [CrossRef]
- Rink, E.; Wullimann, M.F. The teleostean (zebrafish) dopaminergic system ascending to the subpallium (striatum) is located in the basal diencephalon (posterior tuberculum). Brain Res. 2001, 889, 316–330. [Google Scholar] [CrossRef]
- Brunklaus, A.; Zuberi, S.M. Dravet syndrome—From epileptic encephalopathy to channelopathy. Epilepsia 2014, 55, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Bayat, A.; Hjalgrim, H.; Møller, R.S. The incidence of SCN1A-related Dravet syndrome in Denmark is 1:22,000: A population-based study from 2004 to 2009. Epilepsia 2015, 56, e36–e39. [Google Scholar] [CrossRef] [PubMed]
- Connolly, M.B. Dravet Syndrome: Diagnosis and Long-Term Course. Can. J. Neurol. Sci. 2016, 43, S3–S8. [Google Scholar] [CrossRef]
- Darra, F.; Battaglia, D.; Dravet, C.; Patrini, M.; Offredi, F.; Chieffo, D.; Piazza, E.; Fontana, E.; Olivieri, G.; Turrini, I.; et al. Dravet syndrome: Early electroclinical findings and long-term outcome in adolescents and adults. Epilepsia 2019, 60, S49–S58. [Google Scholar] [CrossRef]
- Li, W.; Schneider, A.L.; Scheffer, I.E. Defining Dravet syndrome: An essential pre-requisite for precision medicine trials. Epilepsia 2021, 62, 2205–2217. [Google Scholar] [CrossRef]
- Shmuely, S.; Sisodiya, S.M.; Gunning, W.B.; Sander, J.W.; Thijs, R.D. Mortality in Dravet syndrome: A review. Epilepsy Behav. 2016, 64, 69–74. [Google Scholar] [CrossRef]
- Al-Baradie, R.S. Dravet syndrome, what is new? Neurosciences 2013, 18, 11–17. [Google Scholar]
- Scheffer, I.E.; Nabbout, R. SCN1A-related phenotypes: Epilepsy and beyond. Epilepsia 2019, 60, S17–S24. [Google Scholar] [CrossRef]
- Guerrini, R.; Striano, P.; Catarino, C.; Sisodiya, S.M. Neuroimaging and neuropathology of Dravet syndrome: Neuroimaging and Neuropathology of Dravet Syndrome. Epilepsia 2011, 52, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Yum, M.-S.; Kim, M.-J.; Shim, W.-H.; Yoon, H.M.; Yoo, I.H.; Lee, J.; Lim, B.C.; Kim, K.J.; Ko, T.-S. Large-scale structural alteration of brain in epileptic children with SCN1A mutation. NeuroImage Clin. 2017, 15, 594–600. [Google Scholar] [CrossRef]
- Schoonheim, P.J.; Arrenberg, A.B.; Del Bene, F.; Baier, H. Optogenetic Localization and Genetic Perturbation of Saccade-Generating Neurons in Zebrafish. J. Neurosci. 2010, 30, 7111–7120. [Google Scholar] [CrossRef]
- Dinday, M.T.; Baraban, S.C. Large-Scale Phenotype-Based Antiepileptic Drug Screening in a Zebrafish Model of Dravet Syndrome. eNeuro 2015, 2, ENEURO.0068-15.2015. [Google Scholar] [CrossRef] [PubMed]
- Eimon, P.M.; Ghannad-Rezaie, M.; De Rienzo, G.; Allalou, A.; Wu, Y.; Gao, M.; Roy, A.; Skolnick, J.; Yanik, M.F. Brain activity patterns in high-throughput electrophysiology screen predict both drug efficacies and side effects. Nat. Commun. 2018, 9, 219. [Google Scholar] [CrossRef] [PubMed]
- Ademuwagun, I.A.; Rotimi, S.O.; Syrbe, S.; Ajamma, Y.U.; Adebiyi, E. Voltage Gated Sodium Channel Genes in Epilepsy: Mutations, Functional Studies, and Treatment Dimensions. Front. Neurol. 2021, 12, 600050. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.E.; Taylor, A.D.; Pineda, R.H.; Lasda, E.L.; Wright, M.A.; Ribera, A.B. Embryonic and larval expression of zebrafish voltage-gated sodium channel α-subunit genes. Dev. Dyn. 2006, 235, 1962–1973. [Google Scholar] [CrossRef]
- Zhang, Y.; Kecskés, A.; Copmans, D.; Langlois, M.; Crawford, A.D.; Ceulemans, B.; Lagae, L.; de Witte, P.A.M.; Esguerra, C.V. Pharmacological Characterization of an Antisense Knockdown Zebrafish Model of Dravet Syndrome: Inhibition of Epileptic Seizures by the Serotonin Agonist Fenfluramine. PLoS ONE 2015, 10, e0125898. [Google Scholar] [CrossRef]
- Schoonjans, A.-S.; Lagae, L.; Ceulemans, B. Low-dose fenfluramine in the treatment of neurologic disorders: Experience in Dravet syndrome. Ther. Adv. Neurol. Disord. 2015, 8, 328–338. [Google Scholar] [CrossRef]
- Brenet, A.; Hassan-Abdi, R.; Somkhit, J.; Yanicostas, C.; Soussi-Yanicostas, N. Defective Excitatory/Inhibitory Synaptic Balance and Increased Neuron Apoptosis in a Zebrafish Model of Dravet Syndrome. Cells 2019, 8, 1199. [Google Scholar] [CrossRef]
- Grone, B.P.; Qu, T.; Baraban, S.C. Behavioral Comorbidities and Drug Treatments in a Zebrafish scn1lab Model of Dravet Syndrome. eNeuro 2017, 4, ENEURO.0066-17.2017. [Google Scholar] [CrossRef]
- Stahl, S.M. Mechanism of Action of Trazodone: A Multifunctional Drug. CNS Spectr. 2009, 14, 536–546. [Google Scholar] [CrossRef]
- Cacabelos, R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef] [PubMed]
- Griffin, A.; Hamling, K.R.; Hong, S.; Anvar, M.; Lee, L.P.; Baraban, S.C. Preclinical Animal Models for Dravet Syndrome: Seizure Phenotypes, Comorbidities and Drug Screening. Front. Pharmacol. 2018, 9, 573. [Google Scholar] [CrossRef]
- Banerji, R.; Huynh, C.; Figueroa, F.; Dinday, M.T.; Baraban, S.C.; Patel, M. Enhancing glucose metabolism via gluconeogenesis is therapeutic in a zebrafish model of Dravet syndrome. Brain Commun. 2021, 3, fcab004. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.G.; Rowley, S.; Fulton, R.; Dinday, M.T.; Baraban, S.C.; Patel, M. Altered Glycolysis and Mitochondrial Respiration in a Zebrafish Model of Dravet Syndrome. eNeuro 2016, 3, ENEURO.0008-16.2016. [Google Scholar] [CrossRef]
- Weuring, W.J.; Singh, S.; Volkers, L.; Rook, M.B.; van’t Slot, R.H.; Bosma, M.; Inserra, M.; Vetter, I.; Verhoeven-Duif, N.M.; Braun, K.P.J.; et al. NaV1.1 and NaV1.6 selective compounds reduce the behavior phenotype and epileptiform activity in a novel zebrafish model for Dravet Syndrome. PLoS ONE 2020, 15, e0219106. [Google Scholar] [CrossRef] [PubMed]
- Nabbout, R.; Mistry, A.; Zuberi, S.; Villeneuve, N.; Gil-Nagel, A.; Sanchez-Carpintero, R.; Stephani, U.; Laux, L.; Wirrell, E.; Knupp, K.; et al. Fenfluramine for Treatment-Resistant Seizures in Patients with Dravet Syndrome Receiving Stiripentol-Inclusive Regimens: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 300. [Google Scholar] [CrossRef]
- Lagae, L.; Schoonjans, A.-S.; Gammaitoni, A.R.; Galer, B.S.; Ceulemans, B. A pilot, open-label study of the effectiveness and tolerability of low-dose ZX008 (fenfluramine HCl) in Lennox-Gastaut syndrome. Epilepsia 2018, 59, 1881–1888. [Google Scholar] [CrossRef]
- Geenen, K.R.; Doshi, S.P.; Patel, S.; Sourbron, J.; Falk, A.; Morgan, A.; Vu, U.; Bruno, P.L.; Thiele, E.A. Fenfluramine for seizures associated with Sunflower syndrome. Dev. Med. Child Neurol. 2021, 63, 1427–1432. [Google Scholar] [CrossRef]
- Poyurovsky, M.; Weizman, A. Treatment of Antipsychotic-Induced Akathisia: Role of Serotonin 5-HT2a Receptor Antagonists. Drugs 2020, 80, 871–882. [Google Scholar] [CrossRef]
- Jarema, M.; Dudek, D.; Landowski, J.; Heitzman, J.; Rabe-Jabłońska, J.; Rybakowski, J. Trazodon—The antidepressant: Mechanism of action and its position in the treatment of depression. Psychiatr. Pol. 2011, 45, 611–625. [Google Scholar]
- Karaki, S.; Becamel, C.; Murat, S.; la Cour, C.M.; Millan, M.J.; Prézeau, L.; Bockaert, J.; Marin, P.; Vandermoere, F. Quantitative phosphoproteomics unravels biased phosphorylation of serotonin 2A receptor at Ser280 by hallucinogenic versus nonhallucinogenic agonists. Mol. Cell. Proteom. MCP 2014, 13, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, E.; Poggioni, M.; Michelacci, S. Lisuride as a migraine prophylactic in children: An open clinical trial. Int. J. Clin. Pharmacol. Res. 1983, 3, 137–141. [Google Scholar]
- Obeso, J.A.; Rothwell, J.C.; Quinn, N.P.; Lang, A.E.; Thompson, C.; Marsden, C.D. Cortical Reflex Myoclonus Responds to Intravenous Lisuride. Clin. Neuropharmacol. 1983, 6, 231–240. [Google Scholar] [CrossRef]
- Hofmann, C.; Penner, U.; Dorow, R.; Pertz, H.H.; Jähnichen, S.; Horowski, R.; Latté, K.P.; Palla, D.; Schurad, B. Lisuride, a dopamine receptor agonist with 5-HT2B receptor antagonist properties: Absence of cardiac valvulopathy adverse drug reaction reports supports the concept of a crucial role for 5-HT2B receptor agonism in cardiac valvular fibrosis. Clin. Neuropharmacol. 2006, 29, 80–86. [Google Scholar] [CrossRef]
- Catterall, W.A. Dravet syndrome: A sodium channel interneuronopathy. Curr. Opin. Physiol. 2018, 2, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Tai, C.; Abe, Y.; Westenbroek, R.E.; Scheuer, T.; Catterall, W.A. Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, E3139–E3148. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.-Y.; Andrade, R. 5-Hydroxytryptamine2 Receptor Facilitates GABAergic Neurotransmission in Rat Hippocampus. J. Pharmacol. Exp. Ther. 1998, 285, 805–812. [Google Scholar] [PubMed]
- Guiard, B.P.; Giovanni, G.D. Central serotonin-2A (5-HT2A) receptor dysfunction in depression and epilepsy: The missing link? Front. Pharmacol. 2015, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Svob Strac, D.; Pivac, N.; Smolders, I.J.; Fogel, W.A.; De Deurwaerdere, P.; Di Giovanni, G. Monoaminergic Mechanisms in Epilepsy May Offer Innovative Therapeutic Opportunity for Monoaminergic Multi-Target Drugs. Front. Neurosci. 2016, 10, 492. [Google Scholar] [CrossRef]
- Deidda, G.; Crunelli, V.; Di Giovanni, G. 5-HT/GABA interaction in epilepsy. Prog. Brain Res. 2021, 259, 265–286. [Google Scholar] [CrossRef]
- Buchanan, G.F.; Murray, N.M.; Hajek, M.A.; Richerson, G.B. Serotonin neurones have anti-convulsant effects and reduce seizure-induced mortality. J. Physiol. 2014, 592, 4395–4410. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, G.F.; Smith, H.R.; MacAskill, A.; Richerson, G.B. 5-HT 2A receptor activation is necessary for CO2-induced arousal. J. Neurophysiol. 2015, 114, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Petrucci, A.N.; Joyal, K.G.; Purnell, B.S.; Buchanan, G.F. Serotonin and sudden unexpected death in epilepsy. Exp. Neurol. 2020, 325, 113145. [Google Scholar] [CrossRef]
- De Witte, P.; Lagae, L. Serotonergic modulation as a pharmacological modality in the treatment of Dravet syndrome. Brain 2017, 140, e35. [Google Scholar] [CrossRef][Green Version]
- Brennan, T.J.; Seeley, W.W.; Kilgard, M.; Schreiner, C.E.; Tecott, L.H. Sound-Induced Seizures in Serotonin 5-HT2c Receptor Mutant Mice. Nat. Genet. 1997, 16, 387–390. [Google Scholar] [CrossRef]
- Di Giovanni, G.; De Deurwaerdère, P. New therapeutic opportunities for 5-HT2C receptor ligands in neuropsychiatric disorders. Pharmacol. Ther. 2016, 157, 125–162. [Google Scholar] [CrossRef] [PubMed]
- Isaac, M. Serotonergic 5-HT2C Receptors as a Potential Therapeutic Target for the Design Antiepileptic Drugs. Curr. Top. Med. Chem. 2005, 5, 59–67. [Google Scholar] [CrossRef][Green Version]
- Silenieks, L.B.; Carroll, N.K.; Van Niekerk, A.; Van Niekerk, E.; Taylor, C.; Upton, N.; Higgins, G.A. Evaluation of Selective 5-HT2C Agonists in Acute Seizure Models. ACS Chem. Neurosci. 2019, 10, 3284–3295. [Google Scholar] [CrossRef]
- Panczyk, K.; Golda, S.; Waszkielewicz, A.; Zelaszczyk, D.; Gunia-Krzyzak, A.; Marona, H. Serotonergic system and its role in epilepsy and neuropathic pain treatment: A review based on receptor ligands. Curr. Pharm. Des. 2015, 21, 1723–1740. [Google Scholar] [CrossRef]
- Cheng, J.; Kozikowski, A.P. We Need 2C but Not 2B: Developing Serotonin 2C (5-HT2C) Receptor Agonists for the Treatment of CNS Disorders. ChemMedChem 2015, 10, 1963–1967. [Google Scholar] [CrossRef]
- Elangbam, C.S. Drug-induced Valvulopathy: An Update. Toxicol. Pathol. 2010, 38, 837–848. [Google Scholar] [CrossRef]
- Rothman, R.B.; Baumann, M.H. Serotonergic drugs and valvular heart disease. Expert Opin. Drug Saf. 2009, 8, 317–329. [Google Scholar] [CrossRef]
- Rothman, R.B.; Baumann, M.H.; Savage, J.E.; Rauser, L.; McBride, A.; Hufeisen, S.J.; Roth, B.L. Evidence for possible involvement of 5-HT2B receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation 2000, 102, 2836–2841. [Google Scholar] [CrossRef]
- Schoonjans, A.; Ceulemans, B. An Old Drug for a New Indication: Repurposing Fenfluramine from an Anorexigen to an Antiepileptic Drug. Clin. Pharmacol. Ther. 2019, 106, 929–932. [Google Scholar] [CrossRef]
- Li, R.; Serdula, M.K.; Williamson, D.F.; Bowman, B.A.; Graham, D.J.; Green, L. Dose-effect of fenfluramine use on the severity of valvular heart disease among fen-phen patients with valvulopathy. Int. J. Obes. 1999, 23, 926–928. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, A.-S.; Ceulemans, B. A critical evaluation of fenfluramine hydrochloride for the treatment of Dravet syndrome. Expert Rev. Neurother. 2021, 1–14, in press. [Google Scholar] [CrossRef]
- Hatini, P.G.; Commons, K.G. A 5-HT1D -receptor agonist protects Dravet syndrome mice from seizure and early death. Eur. J. Neurosci. 2020, 52, 4370–4374. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Muñoz, M.; Sánchez-Blázquez, P.; Garzón, J. Fenfluramine diminishes NMDA receptor-mediated seizures via its mixed activity at serotonin 5HT2A and type 1 sigma receptors. Oncotarget 2018, 9, 23373–23389. [Google Scholar] [CrossRef]
- Tupal, S.; Faingold, C.L. Fenfluramine, a serotonin-releasing drug, prevents seizure-induced respiratory arrest and is anticonvulsant in the DBA/1 mouse model of SUDEP. Epilepsia 2019, 60, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Tupal, S.; Faingold, C.L. Serotonin 5-HT4 receptors play a critical role in the action of fenfluramine to block seizure-induced sudden death in a mouse model of SUDEP. Epilepsy Res. 2021, 177, 106777. [Google Scholar] [CrossRef]
- Gogou, M.; Cross, J.H. Fenfluramine as antiseizure medication for epilepsy. Dev. Med. Child Neurol. 2021, 63, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; de Witte, P.A.M.; Maurice, T.; Gammaitoni, A.; Farfel, G.; Galer, B. Fenfluramine acts as a positive modulator of sigma-1 receptors. Epilepsy Behav. 2020, 105, 106989. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Reeder, T.; Sourbron, J.; de Witte, P.A.M.; Gammaitoni, A.R.; Galer, B.S. An Emerging Role for Sigma-1 Receptors in the Treatment of Developmental and Epileptic Encephalopathies. Int. J. Mol. Sci. 2021, 22, 8416. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E.R. The next-generation sequencing revolution and its impact on genomics. Cell 2013, 155, 27–38. [Google Scholar] [CrossRef]
- Woerner, A.C.; Gallagher, R.C.; Vockley, J.; Adhikari, A.N. The Use of Whole Genome and Exome Sequencing for Newborn Screening: Challenges and Opportunities for Population Health. Front. Pediatr. 2021, 9, 663752. [Google Scholar] [CrossRef]
- Pickett, S.B.; Raible, D.W. Water Waves to Sound Waves: Using Zebrafish to Explore Hair Cell Biology. J. Assoc. Res. Otolaryngol. JARO 2019, 20, 1–19. [Google Scholar] [CrossRef]
- Van Wijk, R.C.; Krekels, E.H.J.; Kantae, V.; Ordas, A.; Kreling, T.; Harms, A.C.; Hankemeier, T.; Spaink, H.P.; van der Graaf, P.H. Mechanistic and Quantitative Understanding of Pharmacokinetics in Zebrafish Larvae through Nanoscale Blood Sampling and Metabolite Modeling of Paracetamol. J. Pharmacol. Exp. Ther. 2019, 371, 15–24. [Google Scholar] [CrossRef]
- Guarin, M.; Ny, A.; De Croze, N.; Maes, J.; Léonard, M.; Annaert, P.; de Witte, P.A.M. Pharmacokinetics in Zebrafish Embryos (ZFE) following Immersion and Intrayolk Administration: A Fluorescence-Based Analysis. Pharmaceuticals 2021, 14, 576. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| LFP | iZAP | Multi-Electrode Array | Bioluminescence | |
|---|---|---|---|---|
| Number of zebrafish larvae | 1 | Multiple | 1 | Multiple |
| Possible recording time | Hours | Hours | Days | Days |
| Areas of neuronal activity | 1 | 4 (brain, muscle, eye and ear) | 61 | Multiple |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crouzier, L.; Richard, E.M.; Sourbron, J.; Lagae, L.; Maurice, T.; Delprat, B. Use of Zebrafish Models to Boost Research in Rare Genetic Diseases. Int. J. Mol. Sci. 2021, 22, 13356. https://doi.org/10.3390/ijms222413356
Crouzier L, Richard EM, Sourbron J, Lagae L, Maurice T, Delprat B. Use of Zebrafish Models to Boost Research in Rare Genetic Diseases. International Journal of Molecular Sciences. 2021; 22(24):13356. https://doi.org/10.3390/ijms222413356
Chicago/Turabian StyleCrouzier, Lucie, Elodie M. Richard, Jo Sourbron, Lieven Lagae, Tangui Maurice, and Benjamin Delprat. 2021. "Use of Zebrafish Models to Boost Research in Rare Genetic Diseases" International Journal of Molecular Sciences 22, no. 24: 13356. https://doi.org/10.3390/ijms222413356
APA StyleCrouzier, L., Richard, E. M., Sourbron, J., Lagae, L., Maurice, T., & Delprat, B. (2021). Use of Zebrafish Models to Boost Research in Rare Genetic Diseases. International Journal of Molecular Sciences, 22(24), 13356. https://doi.org/10.3390/ijms222413356