
Honokiol Acts as a Potent Anti-Fibrotic Agent in the Liver through Inhibition of TGF-β1/SMAD Signaling and Autophagy in Hepatic Stellate Cells

 ,
,  , ,
, ,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
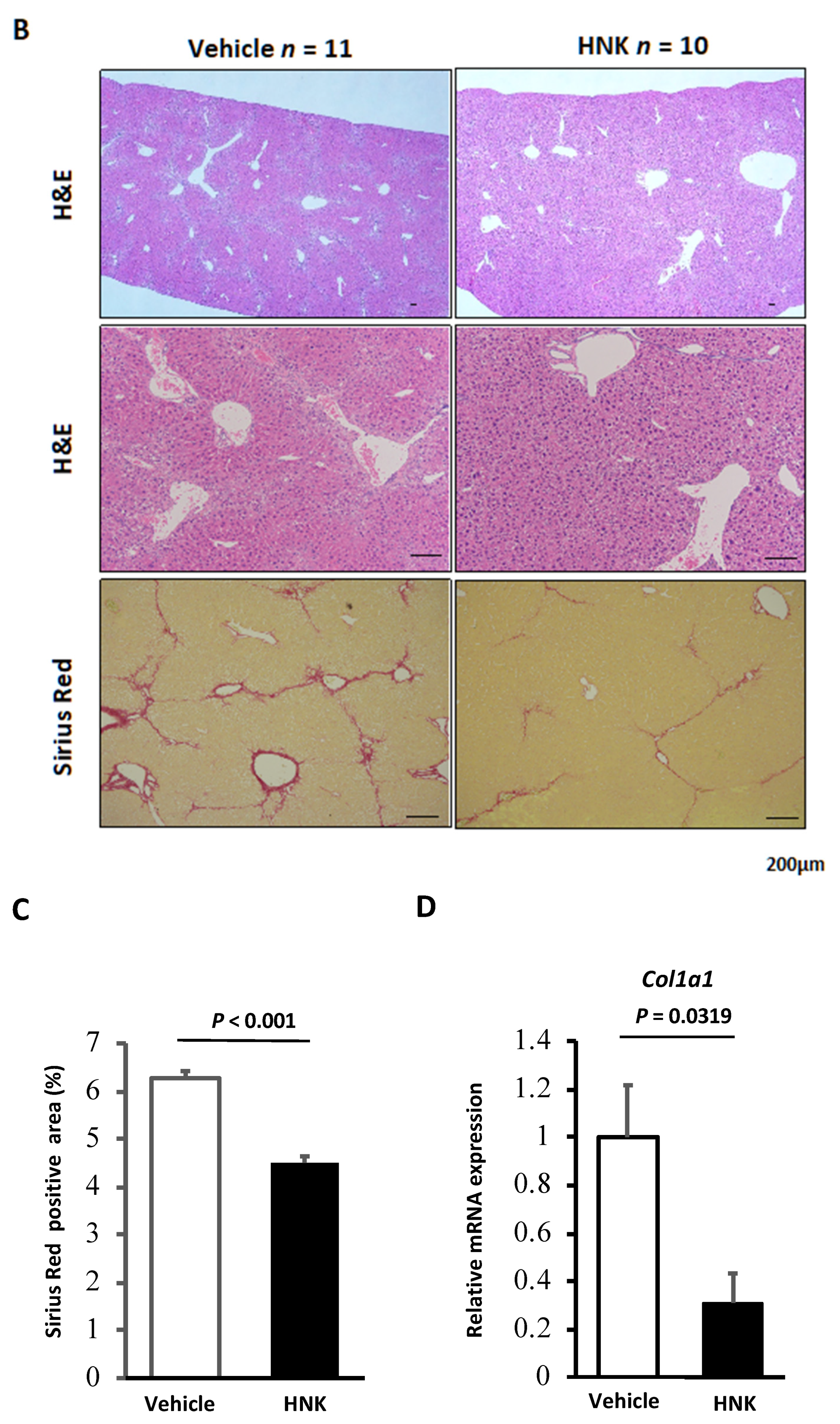
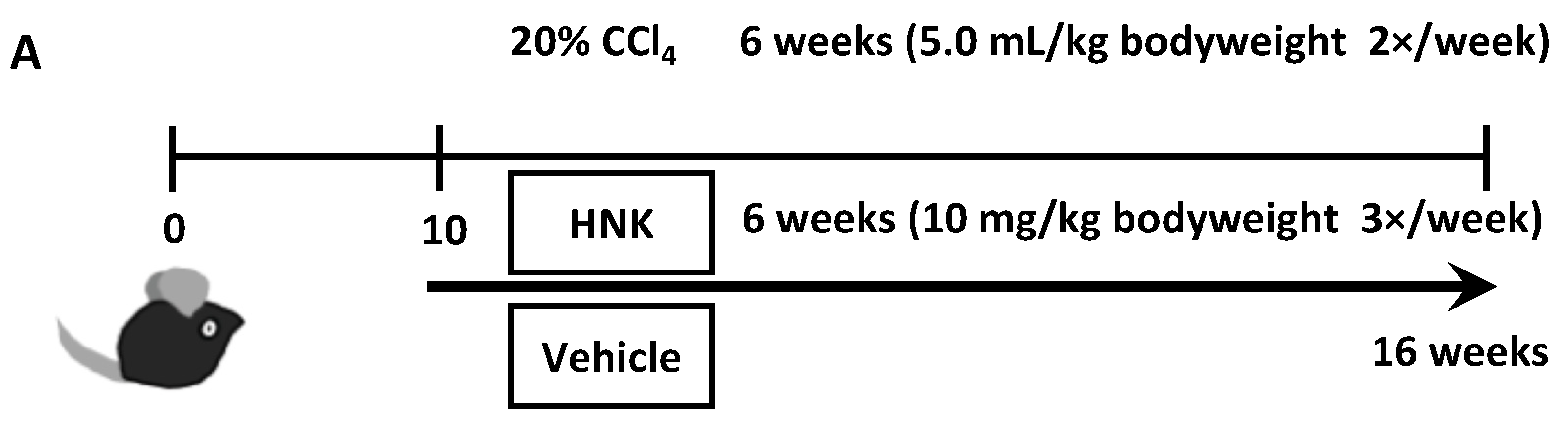
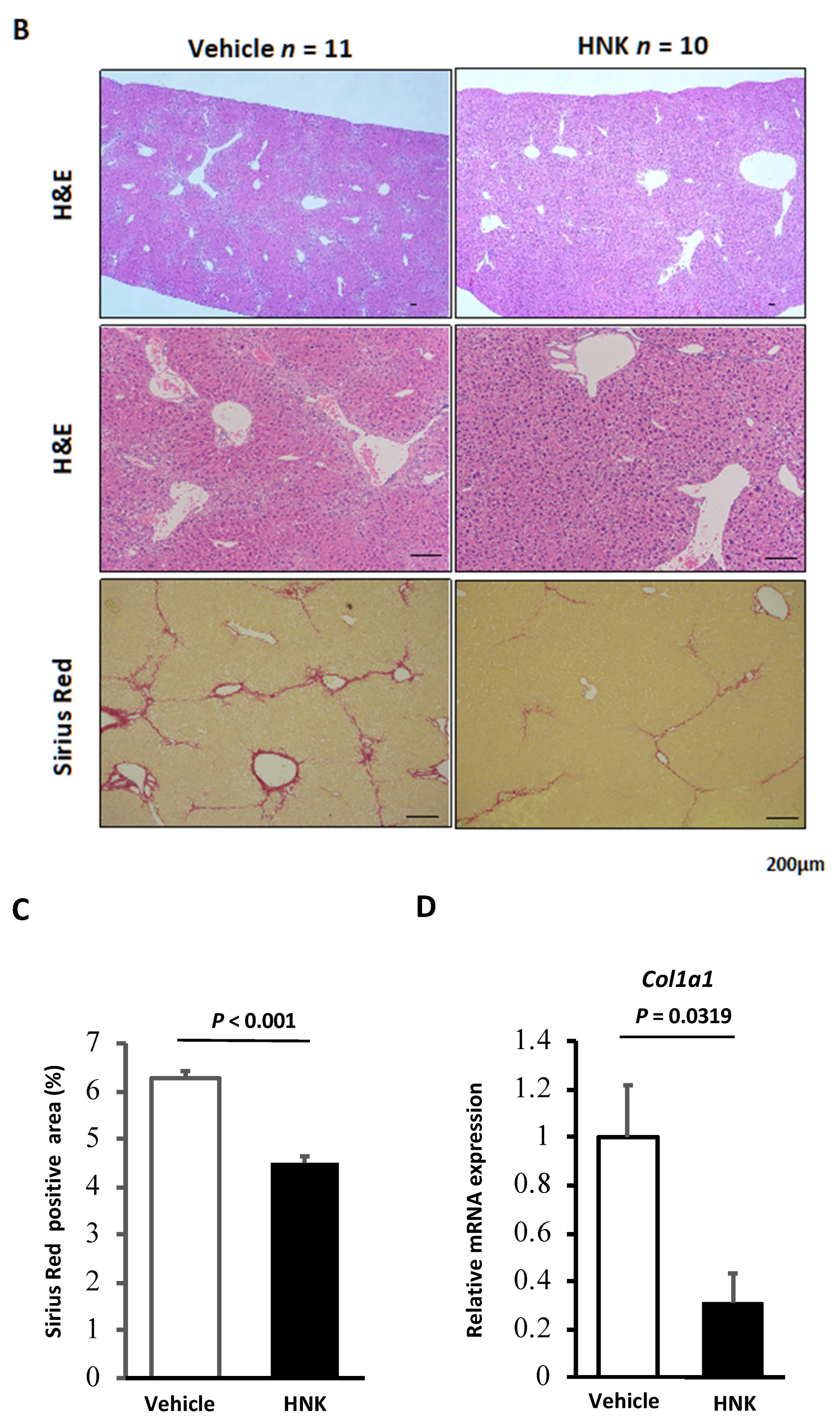
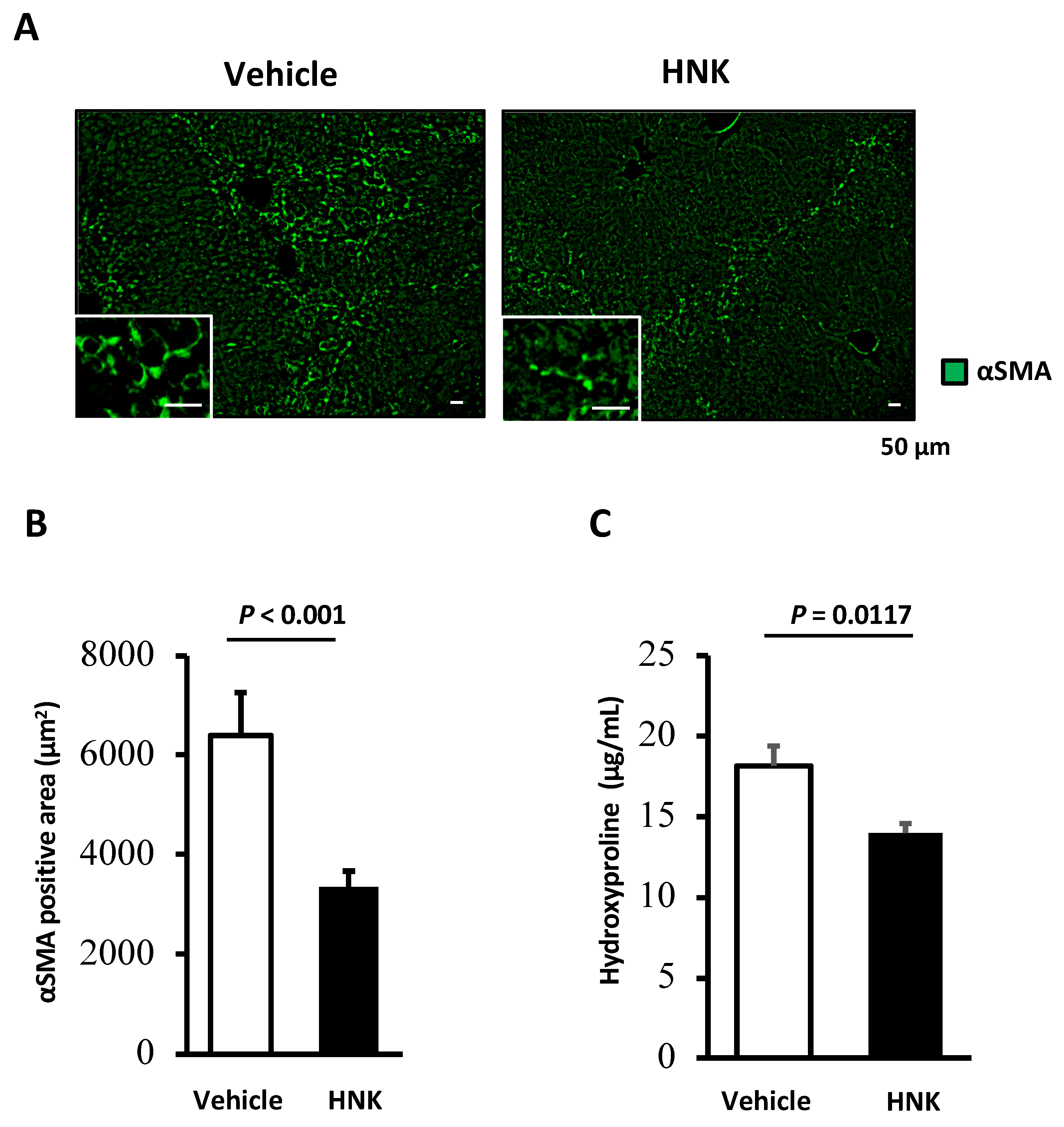
2.1. HNK Treatment Attenuates Fibrosis Development in Mice with CCl4-Induced Liver Injury
2.2. HNK Treatment Suppresses HSC Activation in the CCl4-Induced Fibrosis Mouse Model
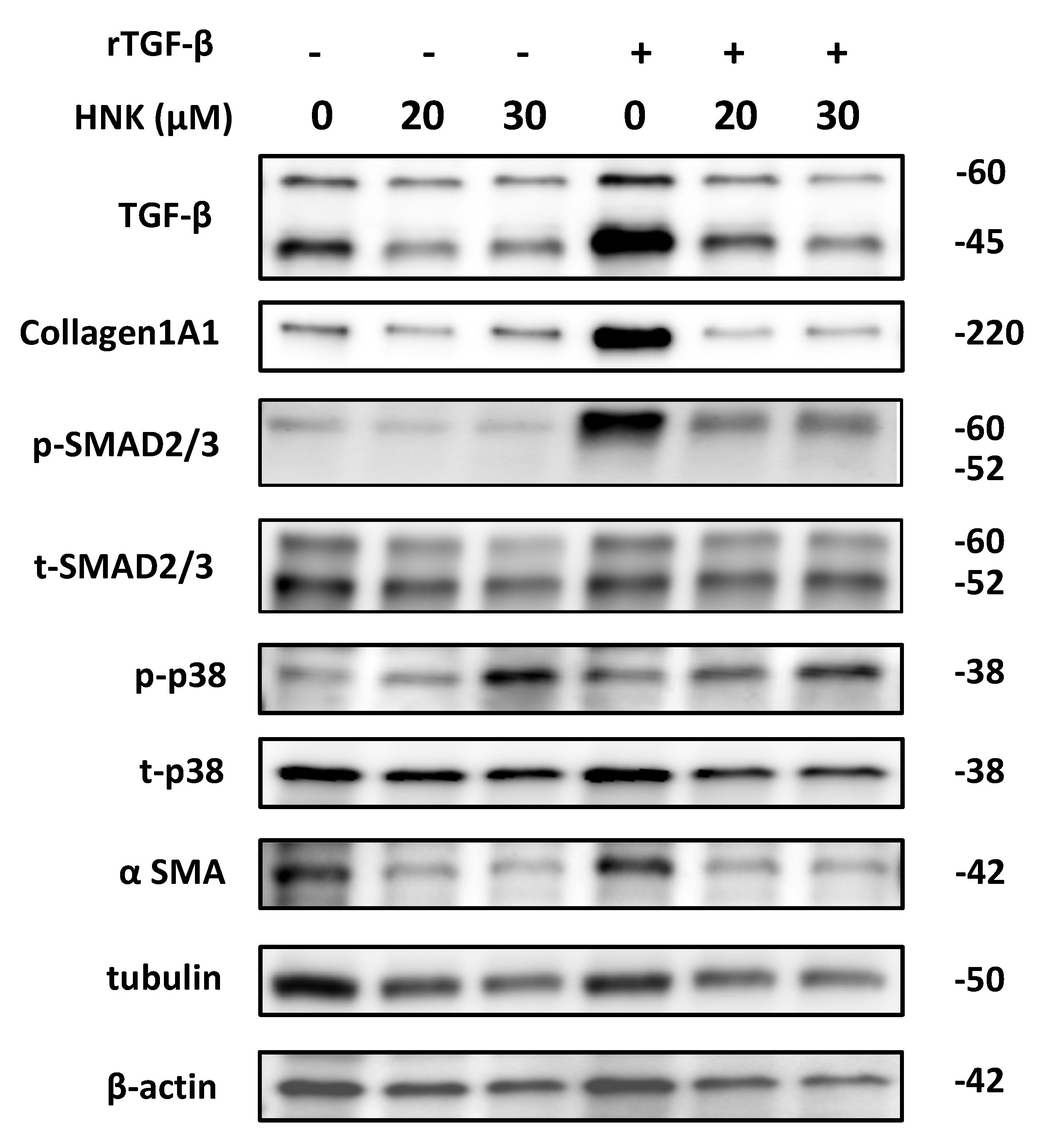
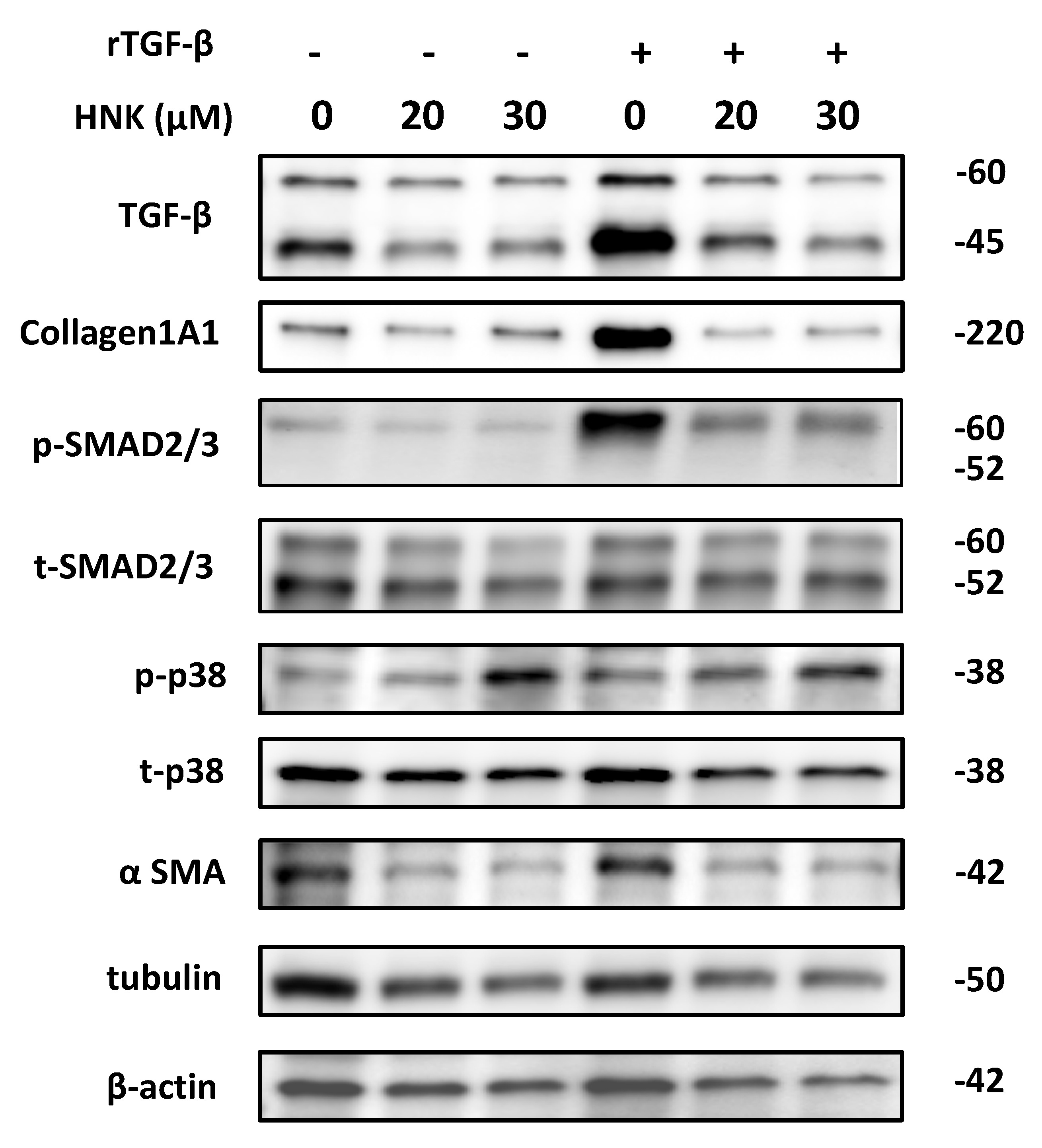
2.3. HNK Treatment Suppresses Proliferative and Fibrogenic Activities in Human Activated Hepatic Stellate Cells
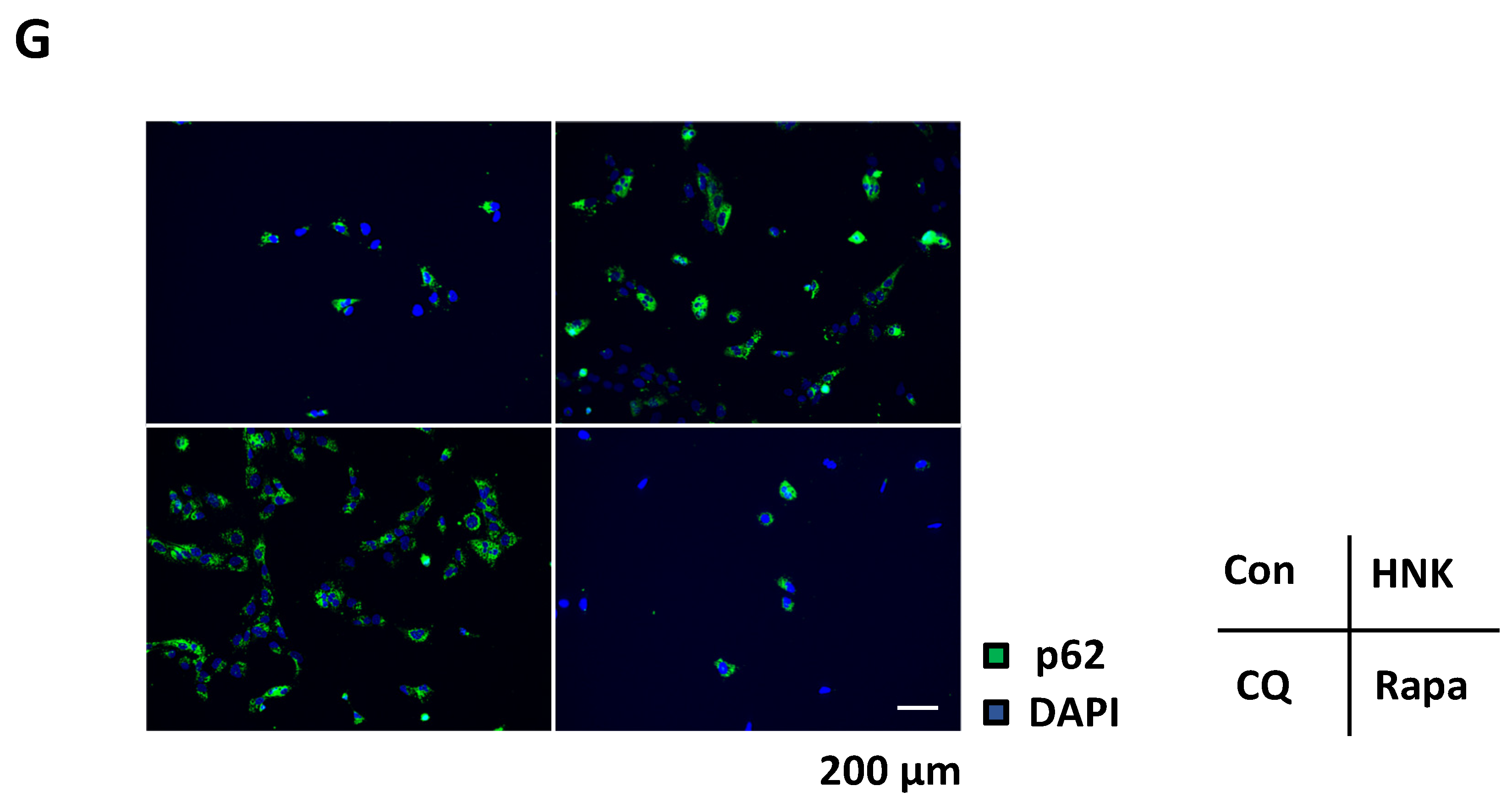
2.4. HNK Suppresses Autophagy in LX-2 Cells
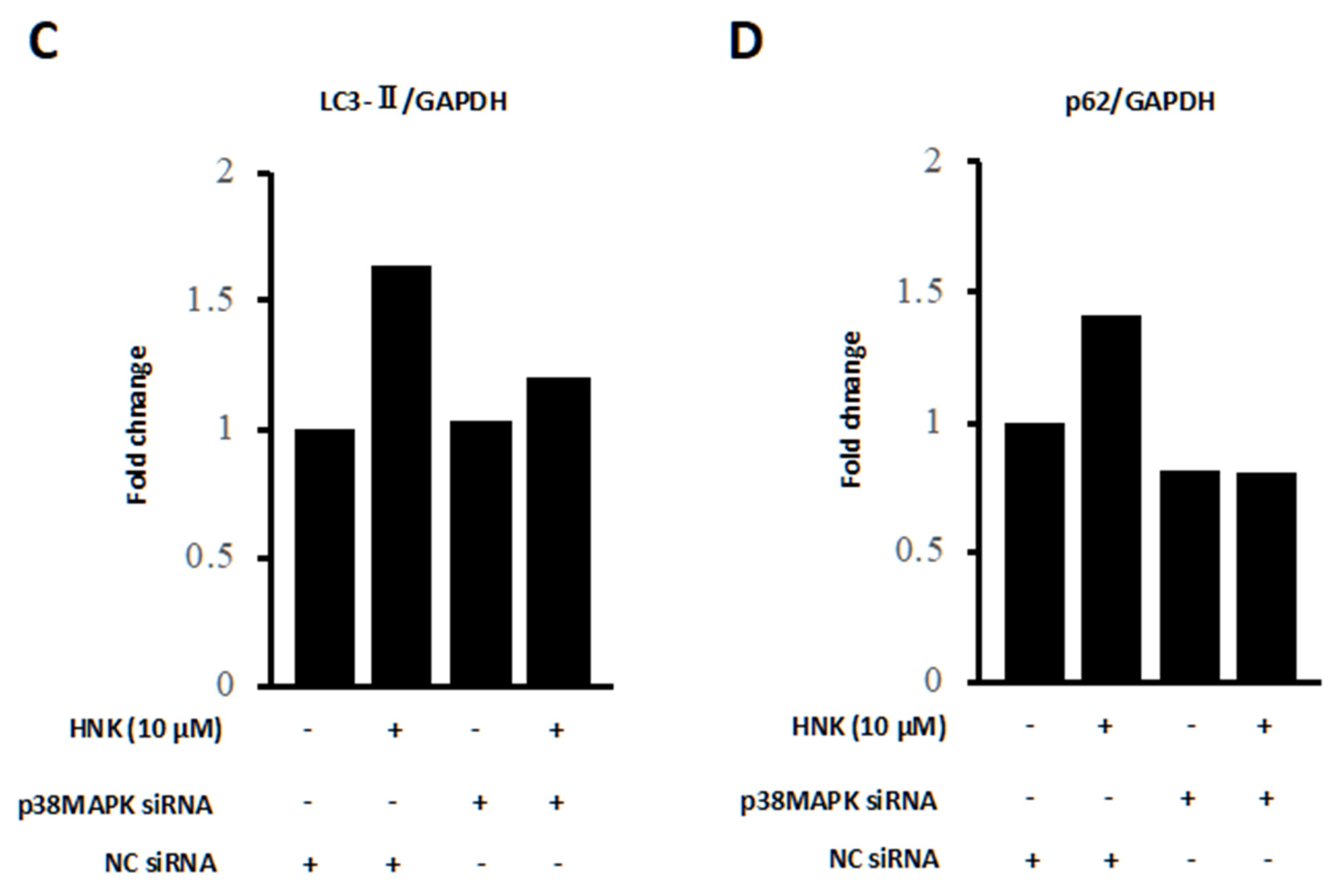
2.5. HNK Suppresses mTOR Signaling, but Activates p38
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Human HSC Cells
4.3. Immunofluorescence Analysis
4.4. RNA Isolation and Quantitative RT-PCR
4.5. Immunoblot Analysis
4.6. Autophagy Analysis
4.7. Apoptosis Detection Assay
4.8. Small Interfering RNA (siRNA) Transfection
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mokdad, A.; Lopez, A.D.; Shahraz, S.; Lozano, R.; Stanaway, J.; Murray, C.J.L.; Naghavi, M. Liver cirrhosis mortality in 187 countries between 1980 and 2010: A systematic analysis. BMC Med. 2014, 12, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Bissell, D.M.; Wang, S.S.; Jarnagin, W.R.; Roll, F.J. Cell-specific expression of transforming growth factor-beta in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J. Clin. Investig. 1995, 96, 447–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehal, W.; Schuppan, D. Antifibrotic therapies in the liver. Semin. Liver Dis. 2015, 35, 184–198. [Google Scholar]
- Hsu, C.C.; Schwabe, R.F. Autophagy and hepatic stellate cell activation—Partners in crime? J. Hepatol. 2011, 55, 1176–1177. [Google Scholar] [CrossRef] [Green Version]
- Hernández–Gea, V.; Ghiassi–Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiskirchen, R.; Tacke, F. Relevance of Autophagy in Parenchymal and Non-Parenchymal Liver Cells for Health and Disease. Cells 2019, 8, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zbidah, M.; Lupescu, A.; Herrmann, T.; Yang, W.; Foller, M.; Jilani, K.; Lang, F. Effect of honokiol on erythrocytes. Toxicol In Vitro 2013, 27, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.-Y.; Chen, S.-J.; Wu, H.-N.; Ping, Y.-H.; Lin, C.-Y.; Shiuan, D.; Chen, C.-L.; Lee, Y.-R.; Huang, K.-J. Honokiol, a Lignan Biphenol Derived from the Magnolia Tree, Inhibits Dengue Virus Type 2 Infection. Viruses 2015, 7, 4894–4910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, C.P.; Lee, W.L.; Tang, Y.-Q.; Yap, W.H. Honokiol: A Review of Its Anticancer Potential and Mechanisms. Cancers 2019, 12, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, A.H.; Vo, L.T.; King, R.G. Honokiol protects against carbon tetrachloride induced liver damage in the rat. Phytother. Res. 2005, 19, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Elfeky, M.G.; Mantawy, E.M.; Gad, A.M.; Fawzy, H.M.; El-Demerdash, E. Mechanistic aspects of antifibrotic effects of honokiol in Con A-induced liver fibrosis in rats: Emphasis on TGF-β/SMAD/MAPK signaling pathways. Life Sci. 2020, 240, 117096. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Im, E.; Lee, H.; Sim, D.Y.; Lee, J.H.; Jung, J.H.; Park, J.E.; Shim, B.S.; Kim, S. Apoptotic and antihepatofibrotic effect of honokiol via activation ofGSK3βand suppression of Wnt/β-catenin pathway in hepatic stellate cells. Phytother. Res. 2020, 35, 452–462. [Google Scholar] [CrossRef]
- Fried, L.E.; Arbiser, J.L. Honokiol, a Multifunctional Antiangiogenic and Antitumor Agent. Antioxid. Redox Signal. 2009, 11, 1139–1148. [Google Scholar] [CrossRef]
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; van den Bossche, B.; De Oliveira, C.P.M.S.; Andraus, W.; Alves, V.A.F.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef] [Green Version]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, N. TGF-β in hepatic stellate cell activation and liver fibrogenesis—Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [Green Version]
- Lv, X.; Liu, F.; Shang, Y.; Chen, S.-Z. Honokiol exhibits enhanced antitumor effects with chloroquine by inducing cell death and inhibiting autophagy in human non-small cell lung cancer cells. Oncol. Rep. 2015, 34, 1289–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Xiao, D.; Ma, C.; Zhang, L.; Duan, Q.; Zheng, X.; Mao, M.; Zhu, D.; Li, Q. The effect of honokiol on pulmonary artery endothelium cell autophagy mediated by cyclo-philin A in hypoxic pulmonary arterial hypertension. J. Pharmacol. Sci. 2019, 139, 158–165. [Google Scholar] [CrossRef]
- Chen, M.-S.; Yeh, H.-T.; Li, Y.-Z.; Lin, W.-C.; Lee, Y.-R.; Tseng, Y.-S.; Sheu, S.-M. Flavopereirine Inhibits Autophagy via the AKT/p38 MAPK Signaling Pathway in MDA-MB-231 Cells. Int. J. Mol. Sci. 2020, 21, 5362. [Google Scholar] [CrossRef]
- He, Y.; She, H.; Zhang, T.; Xu, H.; Cheng, L.; Yepes, M.; Zhao, Y.; Mao, Z. p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1p38 promotes inflammation by phosphorylating ULK1. J. Cell Biol. 2018, 217, 315–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henson, S.M.; Lanna, A.; Riddell, N.E.; Franzese, O.; Macaulay, R.; Griffiths, S.J.; Puleston, D.J.; Watson, A.S.; Simon, A.K.; Tooze, S.A.; et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8+ T cells. J. Clin. Investig. 2014, 124, 4004–4016. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Weiskirchen, R. An update on the recent advances in antifibrotic therapy. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 1143–1152. [Google Scholar] [CrossRef]
- Wang, X.-D.; Wang, Y.-L.; Gao, W.-F. Honokiol possesses potential anti-inflammatory effects on rheumatoid arthritis and GM-CSF can be a target for its treatment. Int. J. Clin. Exp. Pathol. 2015, 8, 7929–7936. [Google Scholar] [PubMed]
- Chiang, C.-K.; Sheu, M.-L.; Lin, Y.-W.; Wu, C.-T.; Yang, C.-C.; Chen, M.-W.; Hung, K.-Y.; Wu, K.-D.; Liu, S.-H. Honokiol ameliorates renal fibrosis by inhibiting extracellular matrix and pro-inflammatory factors in vivo and in vitro. Br. J. Pharmacol. 2011, 163, 586–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulivendala, G.; Bale, S.; Godugu, C. Honokiol: A polyphenol neolignan ameliorates pulmonary fibrosis by inhibiting TGF-β/Smad signaling, matrix proteins and IL-6/CD44/STAT3 axis both in vitro and in vivo. Toxicol. Appl. Pharm. 2020, 391, 114913. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.C.; Issa, R.; Smart, D.E.; Trim, N.; Murray, G.I.; Primrose, J.N.; Arthur, M.J.P.; Iredale, J.P.; Mann, D.A. Gliotoxin stimulates the apoptosis of human and rat hepatic stellate cells and enhances the resolution of liver fibrosis in rats. Gastroenterology 2001, 121, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; Delvoux, B.; Streckert, M.; Bonzel, L.; Stopa, M.; ten Dijke, P.; Gressner, A.M. Transforming growth factor β signal transduction in hepatic stellate cells via Smad2/3 phosphorylation, a pathway that is abrogated during in vitro progression to myofibroblasts. FEBS Lett. 2001, 502, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-J.; Chen, T.-L.; Tseng, Y.-Y.; Wu, G.-J.; Hsieh, M.-H.; Lin, Y.W.; Chen, R.M. Honokiol induces autophagic cell death in malignant glioma through reactive oxygen species-mediated regulation of the p53/PI3K/Akt/mTOR signaling pathway. Toxicol. Appl. Pharm. 2016, 304, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Yeh, P.-S.; Wang, W.; Chang, Y.-A.; Lin, C.-J.; Wang, J.-J.; Chen, R.-M. Honokiol induces autophagy of neuroblastoma cells through activating the PI3K/Akt/mTOR and endoplasmic reticular stress/ERK1/2 signaling pathways and suppressing cell migration. Cancer Lett. 2016, 370, 66–77. [Google Scholar] [CrossRef]
- Huang, K.-J.; Kuo, C.-H.; Chen, S.-H.; Lin, C.-Y.; Lee, Y.-R. Honokiol inhibits in vitro and in vivo growth of oral squamous cell carcinoma through induction of apoptosis, cell cycle arrest and autophagy. J. Cell. Mol. Med. 2018, 22, 1894–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.-X.; Li, Y.; Liu, Z.-Q.; Fan, X.-X.; Duan, F.-G.; Li, R.-Z.; Yao, X.-J.; Leung, E.L.-H.; Liu, L. Honokiol Induces Apoptosis, G1 Arrest, and Autophagy in KRAS Mutant Lung Cancer Cells. Front. Pharmacol. 2017, 8, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Qian, Y.; Geng, L.; Chen, J.; Wang, X.; Xie, H.; Yan, S.; Jiang, G.; Zhou, L.; Zheng, S. Involvement of p38 mitogen-activated protein kinase pathway in honokiol-induced apoptosis in a human hepatoma cell line (hepG2). Liver Int. 2008, 28, 1458–1464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ren, X.; Shi, M.; Jiang, Z.; Wang, H.; Su, Q.; Liu, Q.; Li, G.; Jiang, G. Downregulation of STAT3 and activation of MAPK are involved in the induction of apoptosis by HNK in glioblastoma cell line U87. Oncol. Rep. 2014, 32, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleiner, D.E.; Chalasani, N.P.; Lee, W.M.; Fontana, R.J.; Bonkovsky, H.L.; Watkins, P.B.; Hayashi, P.H.; Davern, T.J.; Navarro, V.; Reddy, R.; et al. Hepatic histological findings suspected drug-induced liver injury: Systematic evaluation and clinical associations. Hepatology 2014, 59, 661–670. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kataoka, S.; Umemura, A.; Okuda, K.; Taketani, H.; Seko, Y.; Nishikawa, T.; Yamaguchi, K.; Moriguchi, M.; Kanbara, Y.; Arbiser, J.L.; et al. Honokiol Acts as a Potent Anti-Fibrotic Agent in the Liver through Inhibition of TGF-β1/SMAD Signaling and Autophagy in Hepatic Stellate Cells. Int. J. Mol. Sci. 2021, 22, 13354. https://doi.org/10.3390/ijms222413354
Kataoka S, Umemura A, Okuda K, Taketani H, Seko Y, Nishikawa T, Yamaguchi K, Moriguchi M, Kanbara Y, Arbiser JL, et al. Honokiol Acts as a Potent Anti-Fibrotic Agent in the Liver through Inhibition of TGF-β1/SMAD Signaling and Autophagy in Hepatic Stellate Cells. International Journal of Molecular Sciences. 2021; 22(24):13354. https://doi.org/10.3390/ijms222413354
Chicago/Turabian StyleKataoka, Seita, Atsushi Umemura, Keiichiro Okuda, Hiroyoshi Taketani, Yuya Seko, Taichiro Nishikawa, Kanji Yamaguchi, Michihisa Moriguchi, Yoshihiro Kanbara, Jack L. Arbiser, and et al. 2021. "Honokiol Acts as a Potent Anti-Fibrotic Agent in the Liver through Inhibition of TGF-β1/SMAD Signaling and Autophagy in Hepatic Stellate Cells" International Journal of Molecular Sciences 22, no. 24: 13354. https://doi.org/10.3390/ijms222413354
APA StyleKataoka, S., Umemura, A., Okuda, K., Taketani, H., Seko, Y., Nishikawa, T., Yamaguchi, K., Moriguchi, M., Kanbara, Y., Arbiser, J. L., Shima, T., Okanoue, T., & Itoh, Y. (2021). Honokiol Acts as a Potent Anti-Fibrotic Agent in the Liver through Inhibition of TGF-β1/SMAD Signaling and Autophagy in Hepatic Stellate Cells. International Journal of Molecular Sciences, 22(24), 13354. https://doi.org/10.3390/ijms222413354