
The HDAC Inhibitor Butyrate Impairs β Cell Function and Activates the Disallowed Gene Hexokinase I



Abstract
1. Introduction
2. Results
2.1. Butyrate Is Non-Toxic to BRIN-BD11 Cells
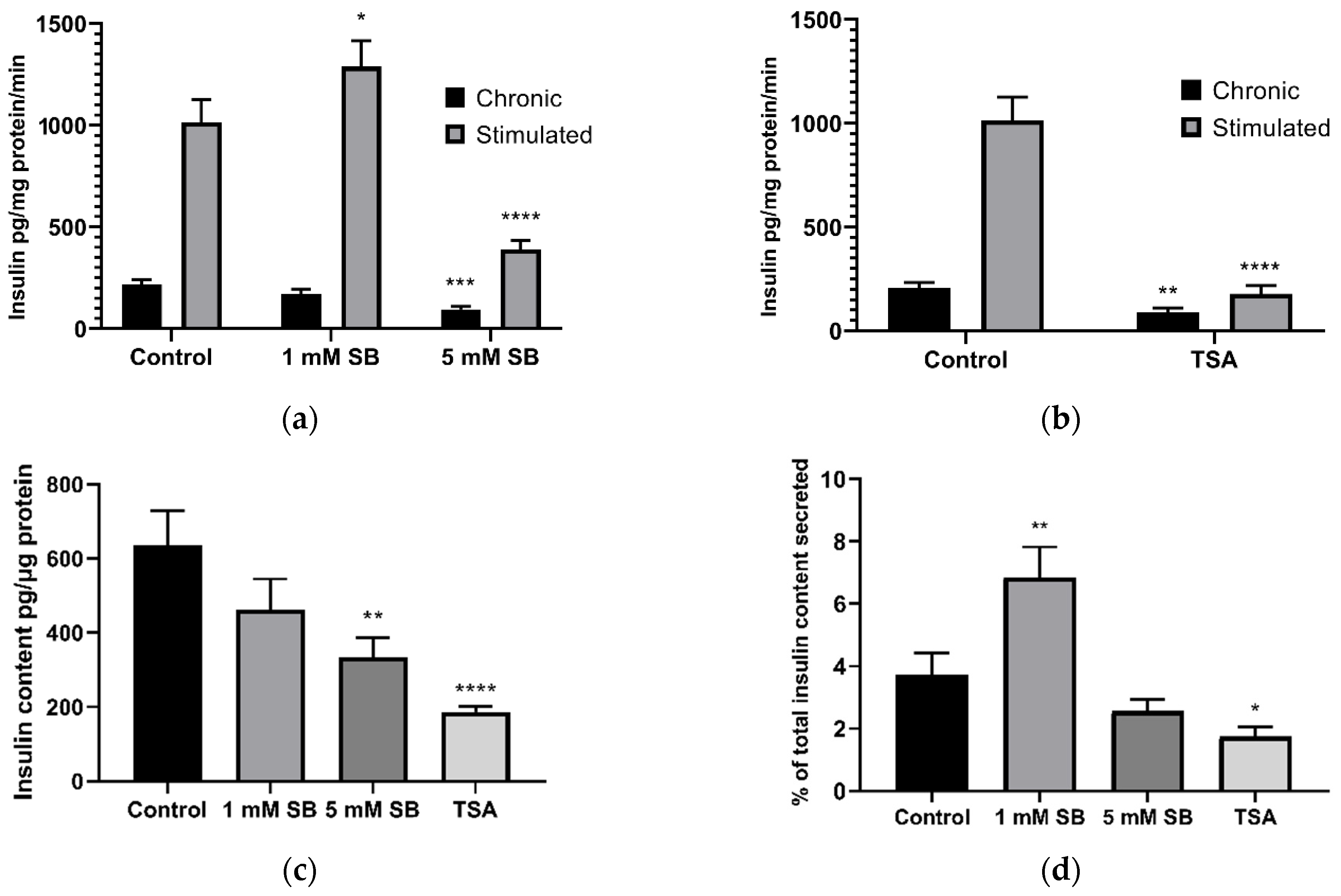
2.2. HDAC Inhibition Impairs Insulin Secretion and Reduces Insulin Content in BRIN-BD11 Cells
2.3. Acute Exposure to Butyrate Synergises Insulin Secretion by Secretagogues
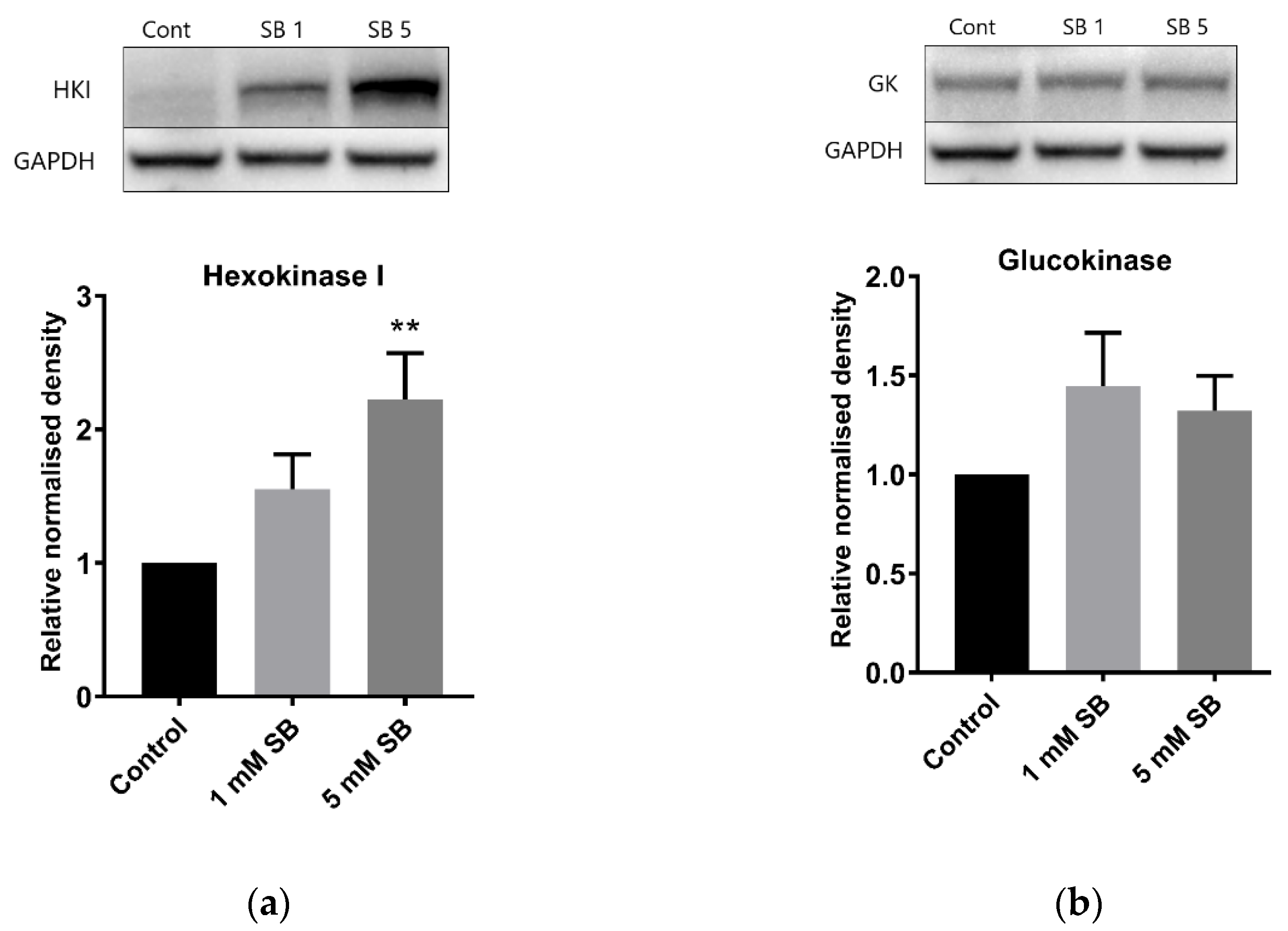
2.4. Butyrate Upregulates the Disallowed Gene Hexokinase I in BRIN-BD11 and HepG2 Cells
2.5. Butyrate Significantly Alters the Expression of Genes Implicated in β Cell Function and Identity
2.6. How Butyrate Activates HK1 Expression Remains Unclear
3. Discussion
4. Materials and Methods
4.1. Creation of Stock Solutions
4.2. Cell Culture and Treatments
4.3. Viability Assays
4.4. Protein Extraction
4.5. Immunoblotting
4.6. Insulin Secretion
4.6.1. Insulin Secretion Assay
4.6.2. Cellular Insulin Content
4.7. Gene Expression
4.7.1. RNA Extraction
4.7.2. Reverse Transcription
4.7.3. qPCR
4.8. Chromatin Immunoprecipitation
ChIP qPCR
4.9. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Bridgeman, S.C.; Northrop, W.; Melton, P.E.; Ellison, G.C.; Newsholme, P.; Mamotte, C.D.S. Butyrate, generated by gut microbiota, and its therapeutic role in metabolic syndrome. Pharmacol. Res. 2020, 160, 105174. [Google Scholar] [CrossRef]
- McNabney, S.M.; Henagan, T.M. Short chain fatty acids in the colon and peripheral tissues: A focus on butyrate, colon cancer, obesity and insulin resistance. Nutrients 2017, 9, 1348. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef]
- Khan, S.; Jena, G. Sodium butyrate reduces insulin-resistance, fat accumulation and dyslipidemia in type-2 diabetic rat: A comparative study with metformin. Chem. Biol. Interact. 2016, 254, 124–134. [Google Scholar] [CrossRef]
- Matheus, V.; Monteiro, L.; Oliveira, R.; Maschio, D.A.; Collares-Buzato, C.B. Butyrate reduces high-fat diet-induced metabolic alterations, hepatic steatosis and pancreatic beta cell and intestinal barrier dysfunctions in prediabetic mice. Exp. Biol. Med. 2017, 242, 1214–1226. [Google Scholar] [CrossRef] [PubMed]
- Mollica, M.P.; Mattace Raso, G.; Cavaliere, G.; Trinchese, G.; De Filippo, C.; Aceto, S.; Prisco, M.; Pirozzi, C.; Di Guida, F.; Lama, A.; et al. Butyrate regulates liver mitochondrial function, efficiency, and dynamics in insulin-resistant obese mice. Diabetes 2017, 66, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Liu, J.; Yuan, Y.; Chen, J.; Cheng, S.; Wang, H.; Xu, Y. Sodium butyrate mitigates type 2 diabetes by inhibiting PERK-CHOP pathway of endoplasmic reticulum stress. Environ. Toxicol. Pharmacol. 2018, 64, 112–121. [Google Scholar] [CrossRef]
- Liu, H.K.; Green, B.D.; Flatt, P.R.; McClenaghan, N.H.; McCluskey, J.T. Effects of long-term exposure to nicotinamide and sodium butyrate on growth, viability, and the function of clonal insulin secreting cells. Endocr. Res. 2004, 30, 61–68. [Google Scholar] [CrossRef]
- Fernandez-Mejia, C.; Davidson, M.B. Effect of sodium butyrate on glucose transport and glucose-phosphorylating enzymes in RIN-m5F cells. Pancreas 1993, 8, 589–596. [Google Scholar] [CrossRef]
- Tiedge, M.; Lenzen, S. Effects of sodium butyrate on glucose transporter and glucose-phosphorylating enzyme gene expression in RINm5F insulinoma cells. J. Mol. Endocrinol. 1996, 17, 19–26. [Google Scholar] [CrossRef]
- Yamato, E. High dose of histone deacetylase inhibitors affects insulin secretory mechanism of pancreatic beta cell line. Endocr. Regul. 2018, 52, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, S.; Northrop, W.; Ellison, G.; Sabapathy, T.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Statins do not directly inhabit the activity of major epigenetic modifying enzymes. Cancers 2019, 11, 516. [Google Scholar] [CrossRef] [PubMed]
- Carlessi, R.; Chen, Y.; Rowlands, J.; Cruzat, V.F.; Keane, K.N.; Egan, L.; Mamotte, C.; Stokes, R.; Gunton, J.E.; de Bittencourt, P.I.H.; et al. GLP-1 receptor signalling promotes β-cell glucose metabolism via mTOR-dependent HIF-1α activation. Sci. Rep. 2017, 7, 2661. [Google Scholar] [CrossRef]
- Liu, Z.; Jeppesen, P.B.; Gregersen, S.; Chen, X.; Hermansen, K. Dose- and glucose dependent effects of amino acids in insulin secretion from isolated mouse islets and clonal INS-1E Beta-Cells. Rev. Diabet. Stud. 2008, 5, 232–244. [Google Scholar] [CrossRef]
- Lemaire, K.; Thorrez, L.; Schuit, F. Disallowed and allowed gene expression: Two faces of mature islet beta cells. Annu. Rev. Nutr. 2016, 36, 45–71. [Google Scholar] [CrossRef] [PubMed]
- Green, A.D.; Vasu, S.; Flatt, P.R. Cellular models for beta-cell function and diabetes gene therapy. Acta Physiol. 2018, 222, e13012. [Google Scholar] [CrossRef]
- Kruit, J.K.; Wijesekara, N.; Westwell-Roper, C.; Vanmierlo, T.; de Haan, W.; Bhattacharjee, A.; Tang, R.; Wellington, C.L.; LütJohann, D.; Johnson, J.D.; et al. Loss of both ABCA1 and ABCG1 results in increased disturbances in islet sterol homeostasis, inflammation, and impaired β-Cell function. Diabetes 2012, 61, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Alhawiti, N.M.; Al Mahri, S.; Aziz, M.A.; Malik, S.S.; Mohammad, S. TXNIP in metabolic regulation: Physiological role and therapeutic outlook. Curr. Drug Targets 2017, 18, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H.; Amemiya-Kudo, M.; Takahashi, A.; Kato, T.; Ishikawa, M.; Yamada, N. Sterol regulatory element–binding protein-1c and pancreatic β-cell dysfunction. Diabetes Obes. Metab. 2007, 9, 133–139. [Google Scholar] [CrossRef]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic beta-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53. [Google Scholar] [CrossRef]
- Maiztegui, B.; Borelli, M.I.; Massa, M.L.; Del Zotto, H.; Gagliardino, J.J. Enhanced expression of hexokinase I in pancreatic islets induced by sucrose administration. J. Endocrinol. 2006, 189, 311–317. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cockburn, B.N.; Ostrega, D.M.; Sturis, J.; Kubstrup, C.; Polonsky, C.S.; Bell, G.I. Changes in pancreatic islet glucokinase and hexokinase activities with increasing age, obesity, and the onset of diabetes. Diabetes 1997, 46, 1434–1439. [Google Scholar] [CrossRef]
- Weir, G.C.; Aguayo-Mazzucato, C.; Bonner-Weir, S. β-cell dedifferentiation in diabetes is important, but what is it? Islets 2013, 5, 233–237. [Google Scholar] [CrossRef]
- Björklund, A.; Lansner, A.; Grill, V.E. Glucose-induced [Ca2+]i abnormalities in human pancreatic islets: Important role of overstimulation. Diabetes 2000, 49, 1840–1848. [Google Scholar] [CrossRef][Green Version]
- Bjorklund, A.; Grill, V. Enhancing effects of long-term elevated glucose and palmitate on stored and secreted proinsulin-to-insulin ratios in human pancreatic islets. Diabetes 1999, 48, 1409–1414. [Google Scholar] [CrossRef]
- Grill, V.; Björklund, A. Overstimulation and beta-cell function. Diabetes 2001, 50, S122. [Google Scholar] [CrossRef]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Tanaka, Y.; Takahashi, H. Glucose toxicity in β-Cells: Type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 2003, 52, 581–587. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, S.; Tschen, S.-I.; Zeng, C.; Guo, T.; Hebrok, M.; Matveyenko, A.; Bhushan, A. DNA methylation directs functional maturation of pancreatic β cells. J. Clin. Investig. 2015, 125, 2851–2860. [Google Scholar] [CrossRef]
- Thorrez, L.; Laudadio, I.; Van Deun, K.; Quintens, R.; Hendrickx, N.; Granvik, M.; Lemaire, K.; Schraenen, A.; Van Lommel, L.; Lehnert, S.; et al. Tissue-specific disallowance of housekeeping genes: The other face of cell differentiation. Genome Res. 2011, 21, 95–105. [Google Scholar] [CrossRef]
- Bompada, P.; Atac, D.; Luan, C.; Andersson, R.; Omella, J.D.; Laakso, E.O.; Wright, J.; Groop, L.; De Marinis, Y. Histone acetylation of glucose-induced thioredoxin-interacting protein gene expression in pancreatic islets. Int. J. Biochem. Cell Biol. 2016, 81, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Feingold, P.L.; Surman, D.R.; Brown, K.; Xu, Y.; McDuffie, L.A.; Shukla, V.; Reardon, E.S.; Crooks, D.R.; Trepel, J.B.; Lee, S.; et al. Induction of thioredoxin-interacting protein by a histone deacetylase inhibitor, entinostat, is associated with DNA damage and apoptosis in esophageal adenocarcinoma. Mol. Cancer Ther. 2018, 17, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.; Devi, T.S.; Hosoya, K.I.; Terasaki, T.; Singh, L.P. Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis. 2010, 1, e65. [Google Scholar] [CrossRef] [PubMed]
- Elgort, M.G.; O’Shea, J.M.; Jiang, Y.; Ayer, D.E. Transcriptional and translational downregulation of thioredoxin interacting protein is required for metabolic reprogramming during G(1). Genes Cancer 2010, 1, 893–907. [Google Scholar] [CrossRef]
- Manns, J.; Boda, J.; Willes, R. Probable role of propionate and butyrate in control of insulin secretion in sheep. Am. J. Physiol. 1967, 212, 756–764. [Google Scholar] [CrossRef]
- Lin, H.V.; Frassetto, A.; Kowalik, E.J., Jr.; Nawrocki, A.R.; Lu, M.M.; Kosinski, J.R.; Hubert, J.A.; Szeto, D.; Yao, X.; Forrest, G.; et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE 2012, 7, e35240. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–626. [Google Scholar] [CrossRef]
- Traisaeng, S.; Batsukh, A.; Chuang, T.-H.; Herr, D.R.; Huang, Y.-F.; Chimeddorj, B.; Huang, C.-M. Leuconostoc mesenteroides fermentation produces butyric acid and mediates Ffar2 to regulate blood glucose and insulin in type 1 diabetic mice. Sci. Rep. 2020, 10, 7928. [Google Scholar] [CrossRef]
- Pingitore, A.; Gonzalez-Abuin, N.; Ruz-Maldonado, I.; Huang, G.C.; Frost, G.; Persaud, S.J. Short chain fatty acids stimulate insulin secretion and reduce apoptosis in mouse and human islets in vitro: Role of free fatty acid receptor 2. Diabetes Obes. Metab. 2019, 21, 330–339. [Google Scholar] [CrossRef]
- Priyadarshini, M.; Villa, S.R.; Fuller, M.; Wicksteed, B.; Mackay, C.R.; Alquier, T.; Poitout, V.; Mancebo, H.; Mirmira, R.G.; Gilchrist, A.; et al. An acetate-specific GPCR, FFAR2, regulates insulin secretion. Mol. Endocrinol. 2015, 29, 1055–1066. [Google Scholar] [CrossRef]
- Priyadarshini, M.; Layden, B.T. FFAR3 modulates insulin secretion and global gene expression in mouse islets. Islets 2015, 7, e1045182. [Google Scholar] [CrossRef]
- Tang, C.; Ahmed, K.; Gille, A.; Lu, S.; Gröne, H.-J.; Tunaru, S.; Offermanns, S. Loss of FFA2 and FFA3 increases insulin secretion and improves glucose tolerance in type 2 diabetes. Nat. Med. 2015, 21, 173–177. [Google Scholar] [CrossRef]
- Damaskos, C.; Garmpis, N.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; et al. Histone deacetylase inhibitors: An attractive therapeutic strategy against breast cancer. Anticancer Res. 2017, 37, 35–46. [Google Scholar] [CrossRef]
- van der Beek, C.M.; Bloemen, J.G.; van den Broek, M.A.; Lenaerts, K.; Venema, K.; Buurman, W.A.; Dejong, C.H. Hepatic uptake of rectally administered butyrate prevents an increase in systemic butyrate concentrations in humans. J. Nutr. 2015, 145, 2019–2024. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.A.; Kurschel, E.; Osieka, R.; Schmidt, C.G. Clinical pharmacology of sodium butyrate in patients with acute leukemia. Eur. J. Cancer Clin. Oncol. 1987, 23, 1283–1287. [Google Scholar] [CrossRef]
- Mattace Raso, G.; Simeoli, R.; Russo, R.; Iacono, A.; Santoro, A.; Paciello, O.; Ferrante, M.C.; Canani, R.B.; Calignano, A.; Meli, R. Effects of sodium butyrate and its synthetic amide derivative on liver inflammation and glucose tolerance in an animal model of steatosis induced by high fat diet. PLoS ONE 2013, 8, e68626. [Google Scholar] [CrossRef]
- Li, H.-P.; Chen, X.; Li, M.-Q. Butyrate alleviates metabolic impairments and protects pancreatic β cell function in pregnant mice with obesity. Int. J. Clin. Exp. Pathol. 2013, 6, 1574–1584. [Google Scholar]
- Christiansen, C.B.; Gabe, M.B.N.; Svendsen, B.; Dragsted, L.O.; Rosenkilde, M.M.; Holst, J.J. The impact of short-chain fatty acids on GLP-1 and PYY secretion from the isolated perfused rat colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G53–G65. [Google Scholar] [CrossRef]
- Yadav, H.; Lee, J.H.; Lloyd, J.; Walter, P.; Rane, S.G. Beneficial metabolic effects of a probiotic via butyrate-induced GLP-1 hormone secretion. J. Biol. Chem. 2013, 288, 25088–25097. [Google Scholar] [CrossRef]
- Roshanravan, N.; Mahdavi, R.; Alizadeh, E.; Jafarabadi, M.A.; Hedayati, M.; Ghavami, A.; Alipour, S.; Alamdari, N.M.; Barati, M.; Ostadrahimi, A. Effect of butyrate and inulin supplementation on glycemic status, lipid profile and glucagon-like peptide 1 level in patients with type 2 diabetes: A randomized double-blind, placebo-controlled trial. Horm. Metab. Res. 2017, 49, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, J.; Heng, J.; Newsholme, P.; Carlessi, R. Pleiotropic effects of GLP-1 and analogs on cell signaling, metabolism, and function. Front. Endocrinol. 2018, 9, 672. [Google Scholar] [CrossRef]
- Brennan, L.; Shine, A.; Hewage, C.; Malthouse, J.P.G.; Brindle, K.M.; McClenaghan, N.; Flatt, P.R.; Newsholme, P. A nuclear magnetic resonance-based demonstration of substantial oxidative L-alanine metabolism and L-alanine-enhanced glucose metabolism in a clonal pancreatic beta-cell line: Metabolism of L-alanine is important to the regulation of insulin secretion. Diabetes 2002, 51, 1714–1721. [Google Scholar] [CrossRef] [PubMed]
- Rio, D.C.; Ares, M., Jr.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5439. [Google Scholar] [CrossRef]
- Chomczynski, P.; Mackey, K. Substitution of chloroform by bromo-chloropropane in the single-step method of RNA isolation. Anal. Biochem. 1995, 225, 163–164. [Google Scholar] [CrossRef]
- Ponchel, F.; Toomes, C.; Bransfield, K.; Leong, F.T.; Douglas, S.H.; Field, S.L.; Bell, S.M.; Combaret, V.; Puisieux, A.; Mighell, A.J.; et al. Real-time PCR based on SYBR-Green I fluorescence: An alternative to the TaqMan assay for a relative quantification of gene rearrangements, gene amplifications and micro gene deletions. BMC Biotechnol. 2003, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- RT-PCR Primer Design for ChIP Michigan: The University of Michigan. 2016. Available online: http://bridgeslab.sph.umich.edu/protocols/index.php/RT-PCR_primer_design_for_ChIP (accessed on 30 July 2019).
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Type | Product | Company |
|---|---|---|---|
| GAPDH | Mouse monoclonal | ab8245 | abcam |
| Glucokinase | Rabbit polyclonal | ab37796 | abcam |
| Hexokinase 1 | Rabbit monoclonal | 2024 | Cell Signalling |
| Goat anti-mouse | IgG H&L (HRP) | ab6789 | abcam |
| Gene | Forward Primer | Reverse Primer | Annealing Temperature |
|---|---|---|---|
| Actb | AAGACCTCTATGCCAACAC | TGATCTTCATGGTGCTAGG | 57–61° 1 |
| Rpl13a | GCACAAGACCAAAAGAGG | CGCTTTTTCTTGTCATAGGG | 57–61° 1 |
| Hk1 | AAACTCTGGGAAACAAAGG | AAACTTGGTCTCAAAGATGC | 57° |
| Hnf4a | TGTGTGAGTCTATGAAGGAG | ATGTAGTCATTGCCTAGGAG | 61° |
| Pdx1 | AAAGGTTACAAACTTGAGCG | AAACAGCTCCCTTTATTCTC | 59° |
| Srebf1 | AAACCTGAAGTGGTAGAAAC | TTATCCTCAAAGGCTGGG | 59° |
| Ins1 | AACGTGGTTTCTTCTACAC | TCTCCAGTTGGTAGAGGG | 61° |
| Abcg1 | GTTATGTTCTTTGATGAGCCC | CCTTGACTTAGGACATAAAGC | 57° |
| Abca1 | CATCTGAAAAACAGGTTTGG | GGGAGAGAATGCTGAATATC | 57° |
| Txnip | CGTCAATACTCCTGACTTAATG | AAATGTCATCACCTTCACAG | 61° |
| Target | Type | Product | Company |
|---|---|---|---|
| H3 | Rabbit polyclonal | ab1791 | abcam |
| H3K9ac | Rabbit polyclonal | ab10812 | abcam |
| H4K8ac | Rabbit polyclonal | ab15823 | abcam |
| H3K9me3 | Rabbit polyclonal | ab8898 | abcam |
| Genomic Region | Forward Primer | Reverse Primer |
|---|---|---|
| GAPDH exon 2 | CCACATCGCTCAGACACCAT | CATACGACTGCAAAGACCCG |
| HK1 intron 1 | AGGTCAAAAAGGTGAGCCCC | CTGCAGTCCAACTCGATGCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bridgeman, S.; Ellison, G.; Newsholme, P.; Mamotte, C. The HDAC Inhibitor Butyrate Impairs β Cell Function and Activates the Disallowed Gene Hexokinase I. Int. J. Mol. Sci. 2021, 22, 13330. https://doi.org/10.3390/ijms222413330
Bridgeman S, Ellison G, Newsholme P, Mamotte C. The HDAC Inhibitor Butyrate Impairs β Cell Function and Activates the Disallowed Gene Hexokinase I. International Journal of Molecular Sciences. 2021; 22(24):13330. https://doi.org/10.3390/ijms222413330
Chicago/Turabian StyleBridgeman, Stephanie, Gaewyn Ellison, Philip Newsholme, and Cyril Mamotte. 2021. "The HDAC Inhibitor Butyrate Impairs β Cell Function and Activates the Disallowed Gene Hexokinase I" International Journal of Molecular Sciences 22, no. 24: 13330. https://doi.org/10.3390/ijms222413330
APA StyleBridgeman, S., Ellison, G., Newsholme, P., & Mamotte, C. (2021). The HDAC Inhibitor Butyrate Impairs β Cell Function and Activates the Disallowed Gene Hexokinase I. International Journal of Molecular Sciences, 22(24), 13330. https://doi.org/10.3390/ijms222413330