
Recent Discoveries in Epigenetic Modifications of Polycystic Kidney Disease

 , , , and
, , , and
Abstract
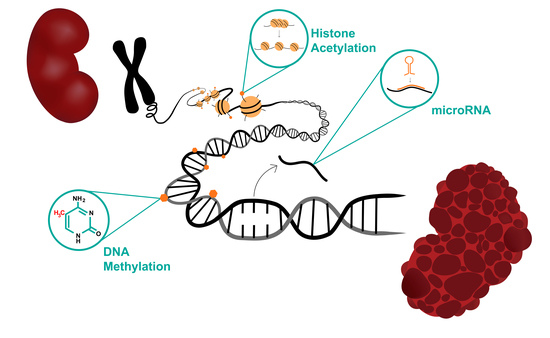
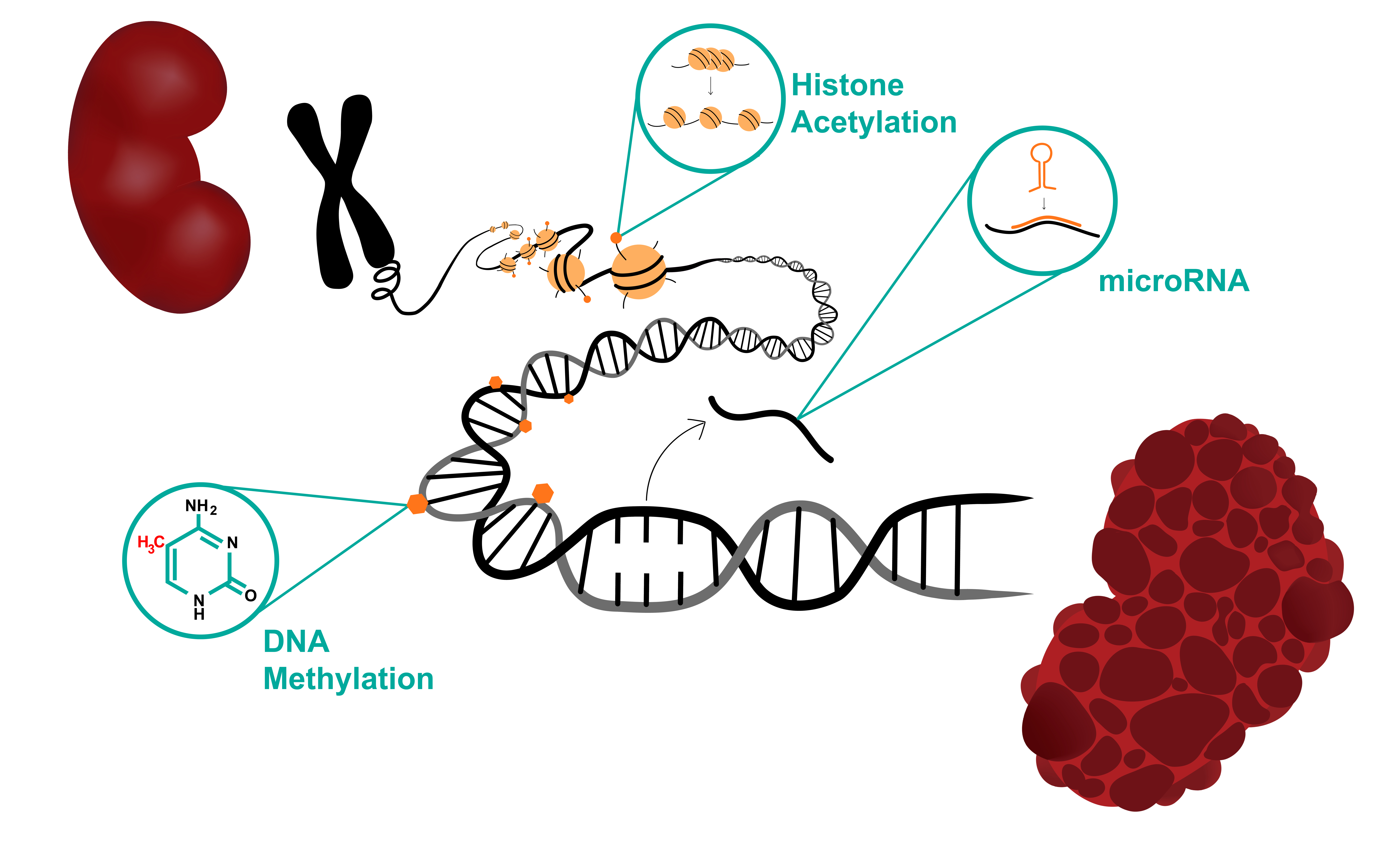
1. Introduction
1.1. PKD-Causing Genes
1.2. ADPKD as ‘Neoplasia in Disguise’
2. Epigenetic and Post-Translational Modification in ADPKD
2.1. Potential Epigenetic Changes Associated with ADPKD
2.2. MicroRNA
2.3. Histone Deacetylases (HDACs)
2.4. DNA Methylation
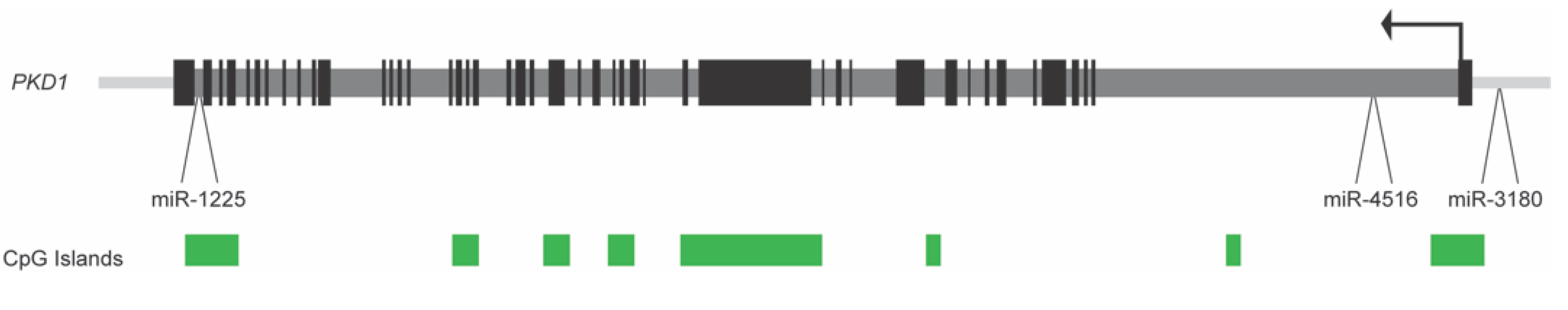
2.5. DNA Methylation in Human ADPKD
3. Implications for Future DNA Methylation Research in ADPKD
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Solazzo, A.; Testa, F.; Giovanella, S.; Busutti, M.; Furci, L.; Carrera, P.; Ferrari, M.; Ligabue, G.; Mori, G.; Leonelli, M.; et al. The prevalence of autosomal dominant polycystic kidney disease (adpkd): A meta-analysis of european literature and prevalence evaluation in the province of modena suggest that adpkd is a rare and underdiagnosed condition. PLoS ONE 2018, 13, e0190430. [Google Scholar] [CrossRef]
- Gabow, P.A.; Johnson, A.M.; Kaehny, W.D.; Kimberling, W.J.; Lezotte, D.C.; Duley, I.T.; Jones, R.H. Factors affecting the progression of renal-disease in autosomal-dominant polycystic kidney-disease. Kidney Int. 1992, 41, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Fick, G.M.; Johnson, A.M.; Strain, J.D.; Kimberling, W.J.; Kumar, S.; Mancojohnson, M.L.; Duley, I.T.; Gabow, P.A. Characteristics of very early-onset autosomal-dominant polycystic kidney-disease. J. Am. Soc. Nephrol. 1993, 3, 1863–1870. [Google Scholar] [CrossRef] [PubMed]
- Reed-Gitomer, B. Autosomal dominant polycystic kidney disease: Genetics, epidemiology, and treatment. Adv. Genom. Genet. 2014, 2014, 173–183. [Google Scholar] [CrossRef]
- Wilson, P.D. Mechanisms of disease: Polycystic kidney disease. N. Engl. J. Med. 2004, 350, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Segal, Y.; Peissel, B.; Deng, N.H.; Pei, Y.; Carone, F.; Rennke, H.G.; GlucksmannKuis, A.M.; Schneider, M.C.; Ericsson, M.; et al. Identification and localization of polycystin, the pkd1 gene product. J. Clin. Investig. 1996, 98, 2674–2682. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, C.M.D.; Souza-Menezes, J.; Amaral, A.G.; Onuchic, L.F.; Cebotaru, L.; Guggino, W.B.; Morales, M.M. Regulation of cftr expression and arginine vasopressin activity are dependent on polycystin-1 in kidney-derived cells. Cell. Physiol. Biochem. 2016, 38, 28–39. [Google Scholar] [CrossRef]
- Rangan, G.K.; Tchan, M.C.; Tong, A.; Wong, A.T.Y.; Nankivell, B.J. Recent advances in autosomal-dominant polycystic kidney disease. Intern. Med. J. 2016, 46, 883–892. [Google Scholar] [CrossRef]
- Gabow, P.A. Autosomal-dominant polycystic kidney-disease. N. Engl. J. Med. 1993, 329, 332–342. [Google Scholar] [CrossRef]
- Rinkel, G.J.E.; Djibuti, M.; Algra, A.; van Gijn, J. Prevalence and risk of rupture of intracranial aneurysms—A systematic review. Stroke 1998, 29, 251–256. [Google Scholar] [CrossRef]
- Shin, Y.B.; Park, J.H. Recent trends in adpkd research. Adv. Exp. Med. Biol. 2016, 933, 3–11. [Google Scholar]
- Grantham, J.J. Autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2008, 359, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.Y.; Ong, A.C.M. New treatments for autosomal dominant polycystic kidney disease. Br. J. Clin. Pharmacol. 2013, 76, 524–535. [Google Scholar] [CrossRef]
- Braun, W.E. Autosomal dominant polycystic kidney disease: Emerging concepts of pathogenesis and new treatments. Clevel. Clin. J. Med. 2009, 76, 97–104. [Google Scholar] [CrossRef]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Grantham, J.J.; Higashihara, E.; Perrone, R.D.; Krasa, H.B.; Ouyang, J.; Czerwiec, F.S.; et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2012, 367, 2407–2418. [Google Scholar] [CrossRef] [PubMed]
- Hateboer, N.; Dijk, M.A.V.; Bogdanova, N.; Coto, E.; Saggar-Malik, A.K.; San Millan, J.L.; Torra, R.; Breuning, M.; Ravine, D. Comparison of phenotypes of polycystic kidney disease types 1 and 2. Lancet 1999, 353, 103–107. [Google Scholar] [CrossRef]
- Porath, B.; Gainullin, V.G.; Cornec-Le Gall, E.; Dillinger, E.K.; Heyer, C.M.; Hopp, K.; Edwards, M.E.; Madsen, C.D.; Mauritz, S.R.; Banks, C.J.; et al. Mutations in ganab, encoding the glucosidase iia subunit, cause autosomal-dominant polycystic kidney and liver disease. Am. J. Hum. Genet. 2016, 98, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Cornec-Le Gall, E.; Olson, R.J.; Besse, W.; Heyer, C.M.; Gainullin, V.G.; Smith, J.M.; Audrezet, M.P.; Hopp, K.; Porath, B.; Shi, B.; et al. Monoallelic mutations to dnajb11 cause atypical autosomal-dominant polycystic kidney disease. Am. J. Hum. Genet. 2018, 102, 832–844. [Google Scholar] [CrossRef]
- Reed, B.; McFann, K.; Kimberling, W.J.; Pei, Y.; Gabow, P.A.; Christopher, K.; Petersen, E.; Kelleher, C.; Fain, P.R.; Johnson, A.; et al. Presence of de novo mutations in autosomal dominant polycystic kidney disease patients without family history. Am. J. Kidney. Dis. 2008, 52, 1042–1050. [Google Scholar] [CrossRef]
- Gout, A.M.; Martin, N.C.; Brown, A.F.; Ravine, D. Pkdb: Polycystic kidney disease mutation database—A gene variant database for autosomal dominant polycystic kidney disease. Hum. Mutat. 2007, 28, 654–659. [Google Scholar] [CrossRef]
- Grantham, J.J.; Geiser, J.L.; Evan, A.P. Cyst formation and growth in autosomal dominant polycystic kidney-disease. Kidney Int. 1987, 31, 1145–1152. [Google Scholar] [CrossRef]
- Badenas, C.; Torra, R.; Perez-Oller, L.; Mallolas, J.; Talbot-Wright, R.; Torregrosa, V.; Darnell, A. Loss of heterozygosity in renal and hepatic epithelial cystic cells from adpkd1 patients. Eur. J. Hum. Genet. 2000, 8, 487–492. [Google Scholar] [CrossRef]
- Reeders, S.T. Multilocus polycystic disease. Nat. Genet. 1992, 1, 235–237. [Google Scholar] [CrossRef]
- Brasier, J.L.; Henske, E.P. Loss of the polycystic kidney disease (pkd1) region of chromosome 16p13 in renal cyst cells supports a loss-of-function model for cyst pathogenesis. J. Clin. Investig. 1997, 99, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Koptides, M.; Constantinides, R.; Kyriakides, G.; Hadjigavriel, M.; Patsalis, P.C.; Pierides, A.; Deltas, C.C. Loss of heterozygosity in polycystic kidney disease with a missense mutation in the repeated region of pkd1. Hum. Genet. 1998, 103, 709–717. [Google Scholar] [CrossRef]
- Knudson, A.G. Mutation and cancer—Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef]
- Watnick, T.; He, N.; Wang, K.R.; Liang, Y.; Parfrey, P.; Hefferton, D.; St George-Hyslop, P.; Germino, G.; Pei, Y. Mutations of pkd1 in adpkd2 cysts suggest a pathogenic effect of trans-heterozygous mutations. Nat. Genet. 2000, 25, 143–144. [Google Scholar] [CrossRef]
- Pei, Y.; Watnick, T.; He, N.; Wang, K.R.; Liang, Y.; Parfrey, P.; Germino, G.; St George-Hyslop, P. Somatic pkd2 mutations in individual kidney and liver cysts support a "two-hit" model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1999, 10, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Bai, H.; Blumenfeld, J.; Ramnauth, A.B.; Barash, I.; Prince, M.; Tan, A.Y.; Michaeel, A.; Liu, G.; Chicos, I.; et al. Detection of PKD1 and PKD2 somatic variants in autosomal dominant polycystic kidney cyst epithelial cells by whole-genome sequencing. J. Am. Soc. Nephrol. 2021, 32, 3114–3129. [Google Scholar] [CrossRef]
- Eccles, M.R.; Stayner, C.A. Polycystic kidney disease—Where gene dosage counts. F1000 Prime Rep. 2014, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.C.; Torres, V.E. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J. Clin. Investig. 2014, 124, 2315–2324. [Google Scholar] [CrossRef]
- Grantham, J.J. Polycystic kidney-disease—Neoplasia in disguise. Am. J. Kidney Dis. 1990, 15, 110–116. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Seeger-Nukpezah, T.; Geynisman, D.M.; Nikonova, A.S.; Benzing, T.; Golemis, E.A. The hallmarks of cancer: Relevance to the pathogenesis of polycystic kidney disease. Nat. Rev. Nephrol. 2015, 11, 515–534. [Google Scholar] [CrossRef]
- Stayner, C.; Brooke, D.G.; Bates, M.; Eccles, M.R. Targeted therapies for autosomal dominant polycystic kidney disease. Curr. Med. Chem. 2019, 26, 3081–3102. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Li, X.G. Epigenetics and cell cycle regulation in cystogenesis. Cell Signal. 2020, 68, 109509. [Google Scholar] [CrossRef]
- Kerr, K.; McAneney, H.; Flanagan, C.; Maxwell, A.P.; McKnight, A.J. Differential methylation as a diagnostic biomarker of rare renal diseases: A systematic review. BMC Nephrol. 2019, 20, 320. [Google Scholar] [CrossRef]
- Li, X.G. Epigenetics and autosomal dominant polycystic kidney disease. Biochim. Biophys. Acta 2011, 1812, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Cannell, I.G.; Kong, Y.W.; Bushell, M. How do micrornas regulate gene expression? Biochem. Soc. Trans. 2008, 36, 1224–1231. [Google Scholar] [CrossRef]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microrna targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—Micrornas with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Trionfini, P.; Benigni, A. Micrornas as master regulators of glomerular function in health and disease. J. Am. Soc. Nephrol. 2017, 28, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.M.; Kim, D.Y.; Koo, N.J.; Kim, Y.M.; Lee, S.; Ko, J.Y.; Shin, Y.; Kim, B.H.; Mun, H.; Choi, S.; et al. Profiling of mirnas and target genes related to cystogenesis in adpkd mouse models. Sci. Rep. 2017, 7, 14151. [Google Scholar] [CrossRef]
- Sun, L.J.; Zhu, J.Q.; Wu, M.; Sun, H.P.; Zhou, C.C.; Fu, L.L.; Xu, C.G.; Mei, C.L. Inhibition of mir-199a-5p reduced cell proliferation in autosomal dominant polycystic kidney disease through targeting cdkn1c. Med. Sci. Monit. 2015, 21, 195–200. [Google Scholar] [PubMed]
- Kim, D.Y.; Woo, Y.M.; Lee, S.; Oh, S.; Shin, Y.; Shin, J.O.; Park, E.Y.; Ko, J.Y.; Lee, E.J.; Bok, J.; et al. Impact of mir-192 and mir-194 on cyst enlargement through emt in autosomal dominant polycystic kidney disease. FASEB J. 2019, 33, 2870–2884. [Google Scholar] [CrossRef] [PubMed]
- de Stephanis, L.; Mangolini, A.; Servello, M.; Harris, P.C.; Dell’Atti, L.; Pinton, P.; Aguiari, G. Microrna501-5p induces p53 proteasome degradation through the activation of the mtor/mdm2 pathway in adpkd cells. J. Cell. Physiol. 2018, 233, 6911–6924. [Google Scholar] [CrossRef]
- Patel, V.; Williams, D.; Hajarnis, S.; Hunter, R.; Pontoglio, M.; Somlo, S.; Igarashi, P. Mir-17 similar to 92 mirna cluster promotes kidney cyst growth in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2013, 110, 10765–10770. [Google Scholar] [CrossRef]
- Yheskel, M.; Lakhia, R.; Cobo-Stark, P.; Flaten, A.; Patel, V. Anti-microrna screen uncovers mir-17 family within mir-17 similar to 92 cluster as the primary driver of kidney cyst growth. Sci. Rep. 2019, 9, 1920. [Google Scholar] [CrossRef]
- Perico, N.; Remuzzi, G. Do mtor inhibitors still have a future in adpkd? Nat. Rev. Nephrol. 2010, 6, 696–698. [Google Scholar] [CrossRef]
- Bowden, S. Genome-Wide DNA Methylation in Polycystic Kidney Disease. Master’s Thesis, University of Otago, Dunedin, New Zealand, 2019. Available online: http://hdl.handle.net/10523/9536 (accessed on 3 November 2019).
- Kocyigit, I.; Taheri, S.; Sener, E.F.; Eroglu, E.; Ozturk, F.; Unal, A.; Korkmaz, K.; Zararsiz, G.; Sipahioglu, M.H.; Ozkul, Y.; et al. Serum micro-rna profiles in patients with autosomal dominant polycystic kidney disease according to hypertension and renal function. BMC Nephrol. 2017, 18, 179. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncog. 2015, 20, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Van Bodegom, D.; Saifudeen, Z.; Dipp, S.; Puri, S.; Magenheimer, B.S.; Calvet, J.P.; El-Dahr, S.S. The polycystic kidney disease-1 gene is a target for p53-mediated transcriptional repression. J. Biol. Chem. 2006, 281, 31234–31244. [Google Scholar] [CrossRef]
- Xia, S.; Li, X.G.; Johnson, T.; Seidel, C.; Wallace, D.P.; Li, R. Polycystin-dependent fluid flow sensing targets histone deacetylase 5 to prevent the development of renal cysts. Development 2010, 137, 1075–1084. [Google Scholar] [CrossRef]
- Yanda, M.K.; Liu, Q.N.; Cebotaru, V.; Guggino, W.B.; Cebotaru, L. Histone deacetylase 6 inhibition reduces cysts by decreasing camp and Ca2+ in knock-out mouse models of polycystic kidney disease. J. Biol. Chem. 2017, 292, 17897–17908. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Fan, L.X.; Sweeney, W.E.; Denu, J.M.; Avner, E.D.; Li, X.G. Sirtuin 1 inhibition delays cyst formation in autosomal-dominant polycystic kidney disease. J. Clin. Investig. 2013, 123, 3084–3098. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging hdac inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Ozdag, H.; Teschendorff, A.E.; Ahmed, A.A.; Hyland, S.J.; Blenkiron, C.; Bobrow, L.; Veerakumarasivam, A.; Burtt, G.; Subkhankulova, T.; Arends, M.J.; et al. Differential expression of selected histone modifier genes in human solid cancers. BMC Genom. 2006, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.F.; Wang, D.D.; Wang, D.Y.; Jin, T.C.; Yang, L.P.; Wu, H.; Li, Y.Y.; Zhao, J.; Du, F.P.; Song, M.X.; et al. Hedd: The human epigenetic drug database. Database 2016, 2016, baw159. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.X.; Li, X.J.; Magenheimer, B.; Calvet, J.P.; Li, X.G. Inhibition of histone deacetylases targets the transcription regulator id2 to attenuate cystic epithelial cell proliferation. Kidney Int. 2012, 81, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Semanchik, N.; Lee, S.H.; Somlo, S.; Barbano, P.E.; Coifman, R.; Sun, Z.X. Chemical modifier screen identifies hdac inhibitors as suppressors of pkd models. Proc. Natl. Acad. Sci. USA 2009, 106, 21819–21824. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.P.; Hu, C.F.; Zhang, X.Z. Histone deacetylase inhibitors reduce cysts by activating autophagy in polycystic kidney disease. Kidney Dis. 2019, 5, 163–172. [Google Scholar] [CrossRef]
- Asawa, R.R.; Danchik, C.; Zahkarov, A.; Chen, Y.C.; Voss, T.; Jadhav, A.; Wallace, D.P.; Trott, J.F.; Weiss, R.H.; Simeonov, A.; et al. A high-throughput screening platform for polycystic kidney disease (pkd) drug repurposing utilizing murine and human adpkd cells. Sci. Rep. 2020, 10, 4203. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G.P. DNA methylation and its basic function. Neuropsychopharmacol 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Eccles, M.R. DNA methylation and epigenomics: New technologies and emerging concepts. Genome Biol. 2015, 16, 103. [Google Scholar] [CrossRef]
- Ehrlich, M.; Gamasosa, M.A.; Huang, L.H.; Midgett, R.M.; Kuo, K.C.; Mccune, R.A.; Gehrke, C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues or cells. Nucleic Acids Res. 1982, 10, 2709–2721. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.; Molloy, P.L. Cytosine methylation prevents binding to DNA of a hela-cell transcription factor required for optimal expression of the adenovirus major late promoter. Gene Dev. 1988, 2, 1136–1143. [Google Scholar] [CrossRef]
- Nan, X.S.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-cpg-binding protein mecp2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef]
- Flanagan, J.M.; Wild, L. An epigenetic role for noncoding rnas and intragenic DNA methylation. Genome Biol. 2007, 8, 307. [Google Scholar] [CrossRef]
- Rauch, T.A.; Wu, X.W.; Zhong, X.; Riggs, A.D.; Pfeifer, G.P. A human b cell methylome at 100-base pair resolution. Proc. Natl. Acad. Sci. USA 2009, 106, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Issa, J.P.J.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.; Bae, J.B.; Oh, Y.H.; Lee, Y.G.; Lee, M.; Park, E.; Choi, J.K.; Lee, S.; Shin, Y.; Lyu, J.; et al. Genome-wide methylation profiling of adpkd identified epigenetically regulated genes associated with renal cyst development. Hum. Genet. 2014, 133, 281–297. [Google Scholar] [CrossRef]
- Woo, Y.M.; Shin, Y.B.; Hwang, J.A.; Hwang, Y.H.; Lee, S.; Park, E.Y.; Kong, H.K.; Park, H.C.; Lee, Y.S.; Park, J.H. Epigenetic silencing of the mupcdh gene as a possible prognostic biomarker for cyst growth in ADPKD. Sci. Rep. 2015, 5, 15238. [Google Scholar] [CrossRef]
- Bowden, S.A.; Rodger, E.J.; Bates, M.; Chatterjee, A.; Eccles, M.R.; Stayner, C. Genome-scale single nucleotide resolution analysis of DNA methylation in human autosomal dominant polycystic kidney disease. Am. J. Nephrol. 2018, 48, 415–424. [Google Scholar] [CrossRef]
- Hajirezaei, F.; Ghaderian, S.M.H.; Hasanzad, M.; Nafar, M.; Ghadiani, M.H.; Biglari, S.; Sohrabifar, N.; Jafari, H. Methylation of the pkd1 promoter inversely correlates with its expression in autosomal dominant polycystic kidney disease. Rep. Biochem. Mol. Biol. 2020, 9, 193–198. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Eccles, M.R. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin. Cancer Biol. 2018, 51, 149–159. [Google Scholar] [CrossRef]
- Bowden, S.A.; Stockwell, P.A.; Rodger, E.J.; Parry, M.F.; Eccles, M.R.; Stayner, C.; Chatterjee, A. Extensive inter-cyst DNA methylation variation in autosomal dominant polycystic kidney disease revealed by genome scale sequencing. Front. Genet. 2020, 11, 348. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Morison, I.M.; Eccles, M.R.; Stockwell, P.A. Tools and strategies for analysis of genome-wide and gene-specific DNA methylation patterns. Methods Mol. Biol. 2017, 1537, 249–277. [Google Scholar] [PubMed]
- Kenter, A.T.; Rentmeester, E.; van Riet, J.; Boers, R.; Boers, J.; Ghazvini, M.; Xavier, V.J.; van Leenders, G.J.L.H.; Verhagen, P.C.M.S.; van Til, M.E.; et al. Cystic renal-epithelial derived induced pluripotent stem cells from polycystic kidney disease patients. Stem Cell Transl. Med. 2020, 9, 478–490. [Google Scholar] [CrossRef]
- Huang, N.; Tan, L.; Xue, Z.G.; Cang, J.; Wang, H. Reduction of DNA hydroxymethylation in the mouse kidney insulted by ischemia reperfusion. Biochem. Biophys. Res. Commun. 2012, 422, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.L.; Ding, C.G.; Xue, W.J.; Ding, X.M.; Zheng, J.; Gao, Y.; Xia, X.X.; Li, S.T.; Liu, J.; Han, F.; et al. Genome-wide DNA methylation analysis in renal ischemia reperfusion injury. Gene 2017, 610, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Chu, A.Y.; Tin, A.; Schlosser, P.; Ko, Y.A.; Qiu, C.X.; Yao, C.; Joehanes, R.; Grams, M.E.; Liang, L.M.; Gluck, C.A.; et al. Epigenome-wide association studies identify DNA methylation associated with kidney function. Nat. Commun. 2017, 8, 1286. [Google Scholar] [CrossRef] [PubMed]
- Shema, E.; Bernstein, B.E.; Buenrostro, J.D. Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nat. Genet. 2019, 51, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, I.; Torres-Padilla, M.E.; Almouzni, G. Epigenomics in the single cell era, an important read out for genome function and cell identity. Epigenomics 2021, 13, 981–984. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Study | Sample Type | Key Findings | PKD1 Methylation | Technical Platform |
|---|---|---|---|---|
| Woo et al. (2014) [75] | Patient renal tissue | Gene body hypermethylation of cystogenesis-related genes, which were also downregulated in ADPKD. | Hypermethylation of the PKD1 gene body associated with a reduction in expression. | MIRA-Seq |
| Woo et al. (2015) [76] | Patient renal tissue | Hypermethylation of the MUPCDH promoter associated with transcriptional repression. Potential novel biomarker. | Not examined | MIRA-Seq |
| Bowden et al. (2018) [77] | Patient renal tissue | Global hypomethylation of the ADPKD genome. Differentially methylated loci are associated with ADPKD. | Hypermethylation within PKD1 gene body not associated with a reduction in transcription. | RRBS |
| Bowden et al. (2020) [80] | Patient renal tissue (individual cysts) | Methylation values in cysts reflect whole tissue RRBS data; highly variable methylation patterns in specific loci between cysts in a single patient. | Too little coverage to analyse. | RRBS |
| Kenter et al. (2020) [82] | iPSCs | Cells derived from patients display a methylation pattern indicative of disease-specific epigenetic memory. | No epigenetic changes to PKD-causing genes were found in iPSCs. | MeDIP-Seq |
| Hajirezaei et al. (2021) [78] | Patient blood | Methylation of the PKD1 promoter inversely correlates with gene expression. | Lower PKD1 promoter DNA methylation correlated with greater PKD1 gene expression in patients. | MS-HRM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bowden, S.A.; Rodger, E.J.; Chatterjee, A.; Eccles, M.R.; Stayner, C. Recent Discoveries in Epigenetic Modifications of Polycystic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 13327. https://doi.org/10.3390/ijms222413327
Bowden SA, Rodger EJ, Chatterjee A, Eccles MR, Stayner C. Recent Discoveries in Epigenetic Modifications of Polycystic Kidney Disease. International Journal of Molecular Sciences. 2021; 22(24):13327. https://doi.org/10.3390/ijms222413327
Chicago/Turabian StyleBowden, Sarah A., Euan J. Rodger, Aniruddha Chatterjee, Michael R. Eccles, and Cherie Stayner. 2021. "Recent Discoveries in Epigenetic Modifications of Polycystic Kidney Disease" International Journal of Molecular Sciences 22, no. 24: 13327. https://doi.org/10.3390/ijms222413327
APA StyleBowden, S. A., Rodger, E. J., Chatterjee, A., Eccles, M. R., & Stayner, C. (2021). Recent Discoveries in Epigenetic Modifications of Polycystic Kidney Disease. International Journal of Molecular Sciences, 22(24), 13327. https://doi.org/10.3390/ijms222413327