
The Effect of Conjugation with Octaarginine, a Cell-Penetrating Peptide on Antifungal Activity of Imidazoacridinone Derivative

 ,
, 

Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.1.1. Synthesis of Triazoloacridinone and Imidazoacridinone Derivatives
2.1.2. Peptide Synthesis
2.2. Susceptibility Testing of Novel Compounds against Fungal Strains
2.3. Molecular Mechanism of Antifungal Activity
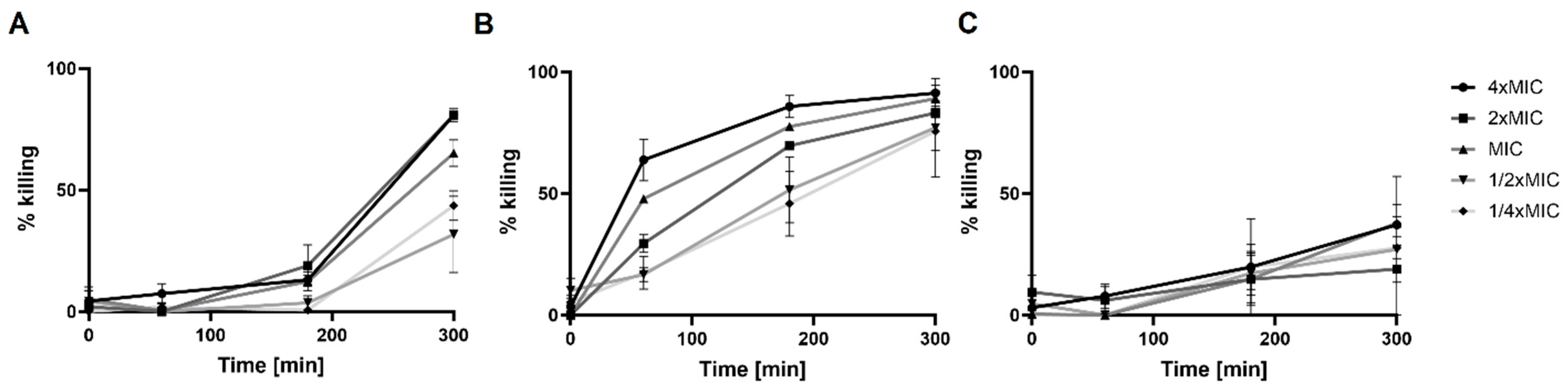
2.3.1. Killing Activity of Compound 1-R8
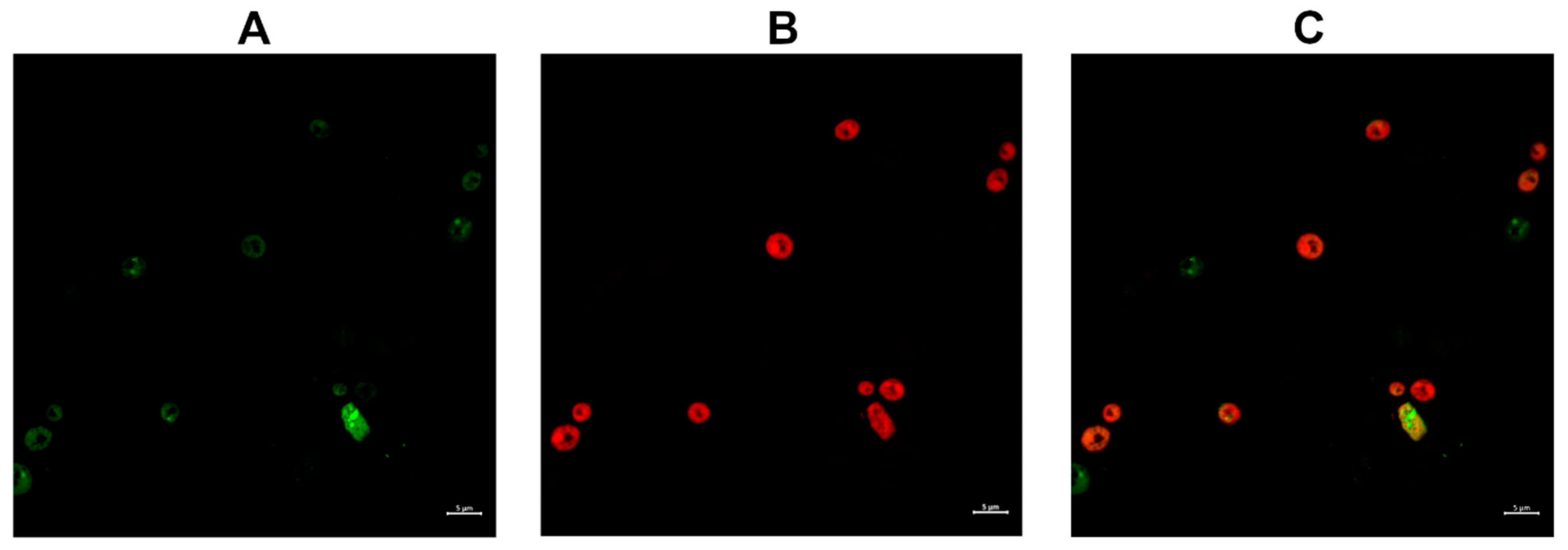
2.3.2. Compound 1 and Compound 1-R8 Accumulation in Fungal Cells
2.3.3. Inhibition of the Relaxation Activity of Yeast Topoisomerase II In Vitro
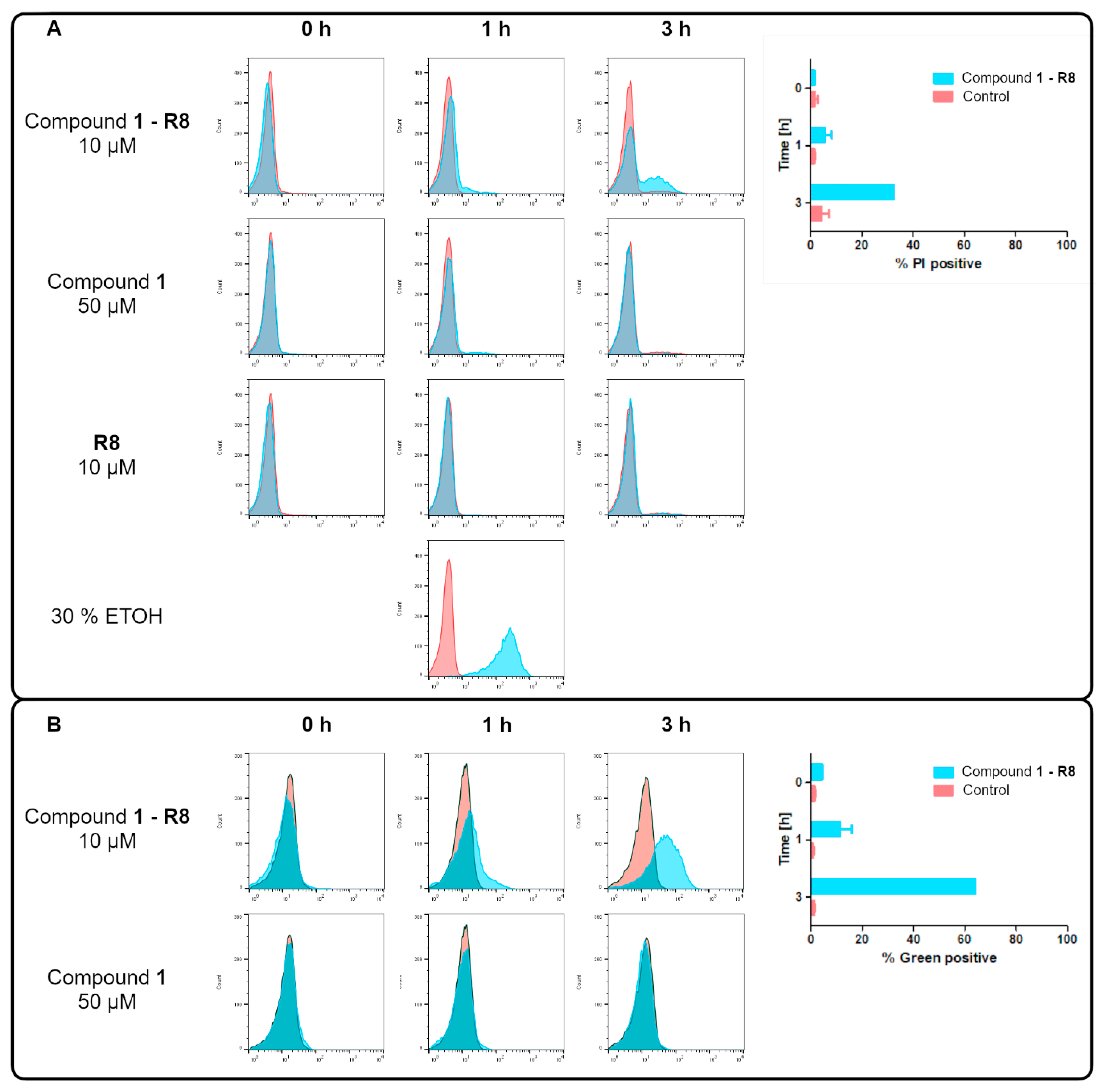
2.4. Selectivity in Relation to Mammalian Cells
3. Material and Methods
3.1. Chemical Synthesis
3.1.1. General Formula for Imidazo- and Triazoloacridinone Synthesis
Synthesis of 5-[-(3′-Dimethylamino)propylamino]-8-hydroxy-6H-[1,2,3]-triazolo[4,5,l-de]acridin-6-one (C-1305)
Synthesis of 5-[-(3′-Dimethylamino)propylamino]-8-methyl-6H-[1,2,3]-triazolo[4,5,l-de]acridin-6-one (C-1296)
Synthesis of 5-[2′-(Ethyl)-ethylenediamine]-8-hydroxy-6H-[1,2,3]-triazolo[4,5,l-de]acridin-6-one (C-1410)
Synthesis of 5-((2-(Diethylamino)ethyl)amino)-8-hydroxy-6H-imidazo[4,5,1-de]acridin-6-one (C-1311)
Synthesis of 5-((2-Aminoethyl)amino)-8-hydroxy-6H-imidazo[4,5,1-de]acridin-6-one (Compound 1)
3.1.2. R8-Conjugated Compound 1 Synthesis and Purification
3.2. Microorganisms Strains and Growth Conditions
3.3. Antimicrobial Activity Assay
3.4. Fungicidal Activity Analysis
3.5. Monitoring of the Cellular Uptake
3.6. Flow Cytometry Analysis
3.7. Yeast Topoisomerase II Relaxation Assay and Inhibition
3.8. Formation of Cleavable Complexes In Vitro
3.9. Antiproliferative Activity Determination
3.10. Hemolytic Activity Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Janočková, J.; Plšíková, J.; Kašpárková, J.; Brabec, V.; Jendželovský, R.; Mikeš, J.; Kovaľ, J.; Hamuľaková, S.; Fedoročko, P.; Kuča, K.; et al. Inhibition of DNA topoisomerases I and II and growth inhibition of HL-60 cells by novel acridine-based compounds. Eur. J. Pharm. Sci. 2015, 76, 192–202. [Google Scholar] [CrossRef]
- Zhang, B.; Li, X.; Li, B.; Gao, C.; Jiang, Y. Acridine and its derivatives: A patent review (2009–2013). Expert Opin. Ther. Pat. 2014, 24, 647–664. [Google Scholar] [CrossRef]
- Prasher, P.; Sharma, M. Medicinal chemistry of acridine and its analogues. MedChemComm 2018, 9, 1589–1618. [Google Scholar] [CrossRef]
- Markovich, Y.D.; Kudryavtseva, T.N.; Bogatyrev, K.V.; Sysoev, P.I.; Klimova, L.G.; Nazarov, G.V. Synthesis of 2-(4-methyl-1,3-thiazol-5-yl) ethyl esters of acridone carboxylic acids and evaluation of their antibacterial activity. Russ. Chem. Bull. 2014, 63, 1153–1158. [Google Scholar] [CrossRef]
- Kaya, M.; Yildirir, Y.; Celik, G.Y. Synthesis, characterization, and in vitro antimicrobial and antifungal activity of novel acridines. Pharm. Chem. J. 2015, 48, 722–726. [Google Scholar] [CrossRef]
- Gabriel, I. ‘Acridines’ as new horizons in antifungal treatment. Molecules 2020, 25, 1480. [Google Scholar] [CrossRef] [PubMed]
- Rząd, K.; Paluszkiewicz, E.; Gabriel, I. A new 1-nitro-9-aminoacridine derivative targeting yeast topoisomerase II able to overcome fluconazole-resistance. Bioorg. Med. Chem Lett. 2021, 35, 127815. [Google Scholar] [CrossRef]
- Taraszkiewicz, A.; Grinholc, M.; Bielawski, K.P.; Kawiak, A.; Nakonieczna, J. Imidazoacridinone derivatives as efficient sensitizers in photoantimicrobial chemotherapy. Appl. Environ. Microbiol. 2013, 79, 3692–3702. [Google Scholar] [CrossRef]
- Hyzy, M.; Bozko, P.; Konopa, J.; Skladanowski, A. Antitumour imidazoacridone C-1311 induces cell death by mitotic catastrophe in human colon carcinoma cells. Biochem. Pharmacol. 2005, 69, 801–809. [Google Scholar] [CrossRef]
- Skwarska, A.; Augustin, E.; Konopa, J. Sequential induction of mitotic catastrophe followed by apoptosis in human leukemia MOLT4 cells by imidazoacridinone C-1311. Apoptosis 2007, 12, 2245–2257. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C. Contemporary challenges in the design of topoisomerase II inhibitors for cancer chemotherapy. Chem. Rev. 2012, 112, 3611–3640. [Google Scholar] [CrossRef]
- Laskowski, T.; Czub, J.; Sowiński, P.; Mazerski, J. Intercalation complex of imidazoacridinone C-1311, a potential anticancer drug, with DNA helix d(CGATCG)2: Stereostructural studies by 2D NMR spectroscopy. J. Biomol. Struct. Dyn. 2016, 34, 653–663. [Google Scholar] [CrossRef]
- Dziegielewski, J.; Slusarski, B.; Konitz, A.; Skladanowski, A.; Konopa, J. Intercalation of imidazoacridinones to DNA and its relevance to cytotoxic and antitumor activity. Biochem. Pharmacol. 2002, 63, 1653–1662. [Google Scholar] [CrossRef]
- Skladanowski, A.; Plisov, S.Y.; Konopa, J.; Larsen, A.K. Inhibition of DNA topoisomerase II by imidazoacridinones, new antineoplastic agents with strong activity against solid tumors. Mol. Pharmacol. 1996, 49, 772–780. [Google Scholar]
- Gabriel, I.; Rząd, K.; Paluszkiewicz, E.; Kozłowska-Tylingo, K. Antifungal activity of Capridine β as a consequence of its biotransformation into metabolite affecting yeast topoisomerase II activity. Pathogens 2021, 11, 189. [Google Scholar] [CrossRef]
- Wainwright, M.; Maisch, T.; Nonell, S.; Plaetzer, K.; Almeida, A.; Tegos, G.P.; Hamblin, M.R. Photoantimicrobials—Are we afraid of the light? Lancet. Infect. Dis. 2017, 17, e49–e55. [Google Scholar] [CrossRef]
- Buzun, K.; Bielawska, A.; Bielawski, K.; Gornowicz, A. DNA topoisomerases as molecular targets for anticancer drugs. J. Enzyme Inhib. Med. Chem. 2020, 35, 1781–1799. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Cushman, M. The indenoisoquinolines non-camptothecin topoisomerase I inhibitors: Update and perspectives. Mol. Cancer Ther. 2009, 8, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.F.; Srikannathasan, V.; Huang, J.; Cui, H.; Fosberry, A.P.; Gu, M.; Hann, M.M.; Hibbs, M.; Homes, P.; Ingraham, K.; et al. Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nat. Commun. 2015, 6, 10048. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, S.; Voelkel, K.; Sternglanz, R. DNA topoisomerase II mutant of Saccharomyces cerevisiae: Topoisomerase II is required for segregation of daughter molecules at the termination of DNA replication. Proc. Natl. Acad. Sci. USA 1984, 81, 2616–2620. [Google Scholar] [CrossRef]
- Holm, C.; Stearns, T.; Botstein, D. DNA topoisomerase II must act at mitosis to prevent nondisjunction and chromosome breakage. Mol. Cell Biol. 1989, 9, 159–168. [Google Scholar] [CrossRef]
- Kwok, S.C.; Schelenz, S.; Wang, X.; Steverding, D. In vitro effect of DNA topoisomerase inhibitors on Candida albicans. Med. Mycol. 2010, 48, 155–160. [Google Scholar] [CrossRef]
- Khan, S.I.; Nimrod, A.C.; Mehrpooya, M.; Nitiss, J.L.; Walker, L.A.; Clark, A.M. Antifungal activity of eupolauridine and its action on DNA topoisomerases. Antimicrob. Agents Chemother. 2002, 46, 1785–1792. [Google Scholar] [CrossRef]
- Kukowska, M. Amino acid or peptide conjugates of acridine/acridone and quinoline/quinolone-containing drugs. A critical examination of their clinical effectiveness within a twenty-year timeframe in antitumor chemotherapy and treatment of infectious diseases. Eur. J. Pharm. Sci. 2017, 109, 587–615. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, X.; Sofang, J.; Zheng, X.; Chen, J.; Ma, P.; Zhang, B.; Wang, R. Conjugation with acridines turn nuclear localization sequence into highly active antimicrobial peptide. Engineering 2015, 1, 500–505. [Google Scholar] [CrossRef]
- Carlson, C.B.; Vuyisich, M.; Gooch, B.D.; Beal, P.A. Preferred RNA binding sites for a threading intercalator revealed by in vitro evolution. Chem. Biol. 2003, 10, 663–672. [Google Scholar] [CrossRef]
- Wynn, J.E.; Zhang, W.; Falkinham, J.O.; Santos, W.L. Branched peptides: Acridine and boronic acid derivatives as antimicrobial agents. ACS Med. Chem. Lett. 2017, 8, 820–823. [Google Scholar] [CrossRef]
- Ashok, B.T.; Tadi, K.; Banerjee, D.; Konopa, J.; Iatropoulos, M.; Tiwari, R.K. Pre-clinical toxicology and pathology of 9-(2′-hydroxyethylamino)-4-methyl-1-nitroacridine (C-1748), a novel anti-cancer agent in male Beagle dogs. Life Sci. 2006, 79, 1334–1342. [Google Scholar] [CrossRef]
- Evison, B.J.; Sleebs, B.E.; Watson, K.G.; Phillips, D.R.; Cutts, S.M. Mitoxantrone, more than just another Topoisomerase II poison. Med. Res. Rev. 2016, 36, 248–299. [Google Scholar] [CrossRef]
- Lemke, K.; Poindessous, V.; Skladanowski, A.; Larsen, A.K. The antitumor triazoloacridone C-1305 is a topoisomerase II poison with unusual properties. Mol. Pharmacol. 2004, 66, 1035–1042. [Google Scholar] [CrossRef]
- Ferguson, D.M.; Jacobson, B.A.; Jay-Dixon, J.; Patel, M.R.; Kratzke, R.A.; Raza, A. Targeting topoisomerase II activity in NSCLC with 9-aminoacridine derivatives. Anticancer Res. 2015, 35, 5211–5217. [Google Scholar]
- Cholody, W.M.; Martelli, S.; Konopa, J. 8-Substituted 5[(aminoalkyl-amino-6H-[1,2,3]-triazolo [4,5,l-de] acridin-6-ones as potential antineoplastic agents. Synthesis and biological activity. J. Med. Chem. 1990, 33, 2852–2856. [Google Scholar] [CrossRef]
- Cholody, W.M.; Martelli, S.; Pardziej-Lukowicz, J.; Konopa, J. 5-[(Aminoalkyl)amino]imidazo[4,5,1-de]acridin-6-ones as a novel class of antineoplastic agents. Synthesis and biological activity. J. Med. Chem. 1990, 33, 49–52. [Google Scholar] [CrossRef]
- Konieczny, M.T.; Konopa, J.K. Acridone Derivatives and Preparation of 8-Hydroxy-Imidazoacridinone Derivatives 1998. U.S. Patent GB 2317888, 8 April 1998. [Google Scholar]
- Franz, R.; Kelly, S.L.; Lamb, D.C.; Kelly, D.E.; Ruhnke, M.; Morschhäuser, J. Multiple molecular mechanisms contribute to a stepwise development of fluconazole resistance in clinical Candida albicans strains. Antimicrob. Agents Chemother. 1998, 42, 3065–3072. [Google Scholar] [CrossRef]
- Franz, R.; Ruhnke, M.; Morschhäuser, J. Molecular aspects of fluconazole resistance development in Candida albicans. Mycoses 1999, 42, 453–458. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Banerjee, R.; Deb, R.; Roy, K.; Chatterjee, S.; Nagotu, S. Uptake and intracellular fate of nona-arginine peptide in yeast. Pept. Sci. 2019, 111, e24101. [Google Scholar] [CrossRef]
- Achilles, J.; Harms, H.; Müller, S. Analysis of living S. cerevisiae cell states—A three color approach. Cytometry Part A 2006, 69, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Davey, H.M.; Hexley, P. Red but not dead? Membranes of stressed Saccharomyces cerevisiae are permeable to propidium iodide. Environ. Microbiol. 2011, 13, 163–171. [Google Scholar] [CrossRef]
- Arakawa, T.; Hirano, A.; Shiraki, K.; Kita, Y.; Koyama, A.H. Stabilizing and destabilizing effects of arginine on deoxyribonucleic acid. Int. J. Biol. Macromol. 2010, 46, 217–222. [Google Scholar] [CrossRef]
- Visone, V.; Szabó, I.; Perugino, G.; Hudecz, F.; Bánóczi, Z.; Valenti, A. Topoisomerases inhibition and DNA binding mode of daunomycin-oligoarginine conjugate. J. Enzyme Inhib. Med. Chem. 2020, 35, 1363–1371. [Google Scholar] [CrossRef]
- Upadhya, A.; Sangave, P.C. Hydrophobic and electrostatic interactions between cell penetrating peptides and plasmid DNA are important for stable non-covalent complexation and intracellular delivery. J. Pept. Sci. 2016, 22, 647–659. [Google Scholar] [CrossRef]
- Suzuki, T.; Futaki, S.; Niwa, M.; Tanaka, S.; Ueda, K.; Sugiura, Y. Possible existence of common internalization mechanisms among arginine-rich peptides. J. Biol. Chem. 2002, 277, 2437–2443. [Google Scholar] [CrossRef]
- Clinical and laboratory Standards Institute (CLSI). Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts, Approved Standard, 4th ed.; CLSI document M27-A3; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]
- ZEN 2 (blue edition), Version 2.6; Carl Zeiss Microscopy GmbH: Jena, Germany, 2014.
- FlowJo™ Software (for Windows), Version v10.8.0; Dickinson and Becton Company: Ashland, OR, USA, 2021.
- Slisz, M.; Cybulska, B.; Mazerski, J.; Grzybowska, J.; Borowski, E. Studies of the effects of antifungal cationic derivatives of amphotericin B on human erythrocytes. J. Antibiot. 2004, 57, 669–678. [Google Scholar] [CrossRef][Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | * MIC90 (μg mL−1) | ||||
|---|---|---|---|---|---|
| Candida albicans ATCC 10231 | Candida glabrata ATCC 90030 | Candida krusei ATCC 6258 | Candida parapsilosis ATCC 22019 | Saccharomyces cerevisiae ATCC 9763 | |
| C-1305 | >64 | >64 | >64 | >64 | >64 |
| C-1311 | >64 | >64 | >64 | >64 | >64 |
| C-1296 | >64 | >64 | >64 | >64 | 32 |
| C-1410 | >64 | >64 | >64 | >64 | >64 |
| Compound 1 | >64 | >64 | >64 | >64 | >64 |
| Compound 1-R8 | 1 | 4 | 0.5 | 0.25 | 1 |
| R8 | >64 | >64 | >64 | 64 | 64 |
| Amphotericin B | 0.5 | 1 | 1 | 1 | 0.5 |
| Compound | * MIC90 (μg mL−1) | ||||||
|---|---|---|---|---|---|---|---|
| Candida albicans ATCC 10231 | Candida albicans B3 | Candida albicans B4 | Candida albicans Gu4 | Candida albicans Gu5 | Candida albicans F2 | Candida albicans F5 | |
| Compound 1 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| Compound 1-R8 | 0.5 | 2 | 2 | 2 | 2 | 1 | 1 |
| R8 | >64 | >64 | >64 | >64 | >64 | >64 | >64 |
| Fluconazole | 8 | 1 | 16 | 4 | >64 | 8 | >64 |
| Compound | Consensus * LogP o/w | Antifungal Activity ** |
|---|---|---|
| Compound 1 | 0.52 | − |
| C-1410 | 1.59 | − |
| C-1305 | 1.72 | − |
| C-1311 | 1.84 | − |
| C-1296 | 2.47 | ± |
| C-1330 *** | 3.04 | + |
| C-1415 *** | 3.06 | + |
| C-1558 *** | 4.25 | + |
| Compound 1-R8 | −5.96 | ++ |
| Compound | * MFC (μg mL−1) | ||||
|---|---|---|---|---|---|
| C. albicans ATCC 10231 | C. glabrata DSM 11226 | C. krusei DSM 6128 | C. parapsilosis DSM 5784 | S. cerevisiae ATCC 9763 | |
| Compound 1-R8 | 32 | 128 | 16 | 8 | 2 |
| Amphotericin B | 2 | 2 | 2 | 2 | 2 |
| Compound | * IC50 μM | |
|---|---|---|
| HEK293 | HEPG2 | |
| Compound 1 | 0.550 ± 0.125 | 0.364 ± 0.013 |
| Compound 1-R8 | 1.390 ± 0.099 | 0.991 ± 0.097 |
| R8 | >50 | >50 |
| C-1311 | 0.008 ± 0.007 | 0.129 ± 0.037 |
| Compound | IC50 HEK293/MIC90 | ||||
|---|---|---|---|---|---|
| Candida albicans ATCC 10231 | Candida glabrata ATCC 90030 | Candida krusei ATCC 6258 | Candida parapsilosis ATCC 22019 | Saccharomyces cerevisiae ATCC 9763 | |
| R8 | - | - | - | - | - |
| C-1311 | <0.00005 | <0.00005 | <0.00005 | <0.00005 | <0.00005 |
| Compound 1 | <0.0058 | <0.0058 | <0.0058 | <0.0058 | <0.0058 |
| Compound 1-R8 | 2.22 | 0.56 | 4.44 | 8.88 | 2.22 |
| IC50 HEPG2/MIC90 | |||||
| R8 | - | - | - | - | - |
| C-1311 | <0.00078 | <0.00078 | <0.00078 | <0.00078 | <0.00078 |
| Compound 1 | <0.0038 | <0.0038 | <0.0038 | <0.0038 | <0.0038 |
| Compound 1-R8 | 1.58 | 0.40 | 3.17 | 6.33 | 1.58 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rząd, K.; Paluszkiewicz, E.; Neubauer, D.; Olszewski, M.; Kozłowska-Tylingo, K.; Kamysz, W.; Gabriel, I. The Effect of Conjugation with Octaarginine, a Cell-Penetrating Peptide on Antifungal Activity of Imidazoacridinone Derivative. Int. J. Mol. Sci. 2021, 22, 13190. https://doi.org/10.3390/ijms222413190
Rząd K, Paluszkiewicz E, Neubauer D, Olszewski M, Kozłowska-Tylingo K, Kamysz W, Gabriel I. The Effect of Conjugation with Octaarginine, a Cell-Penetrating Peptide on Antifungal Activity of Imidazoacridinone Derivative. International Journal of Molecular Sciences. 2021; 22(24):13190. https://doi.org/10.3390/ijms222413190
Chicago/Turabian StyleRząd, Kamila, Ewa Paluszkiewicz, Damian Neubauer, Mateusz Olszewski, Katarzyna Kozłowska-Tylingo, Wojciech Kamysz, and Iwona Gabriel. 2021. "The Effect of Conjugation with Octaarginine, a Cell-Penetrating Peptide on Antifungal Activity of Imidazoacridinone Derivative" International Journal of Molecular Sciences 22, no. 24: 13190. https://doi.org/10.3390/ijms222413190
APA StyleRząd, K., Paluszkiewicz, E., Neubauer, D., Olszewski, M., Kozłowska-Tylingo, K., Kamysz, W., & Gabriel, I. (2021). The Effect of Conjugation with Octaarginine, a Cell-Penetrating Peptide on Antifungal Activity of Imidazoacridinone Derivative. International Journal of Molecular Sciences, 22(24), 13190. https://doi.org/10.3390/ijms222413190