
A Brief Journey through Protein Misfolding in Transthyretin Amyloidosis (ATTR Amyloidosis)


Abstract
:1. Introduction
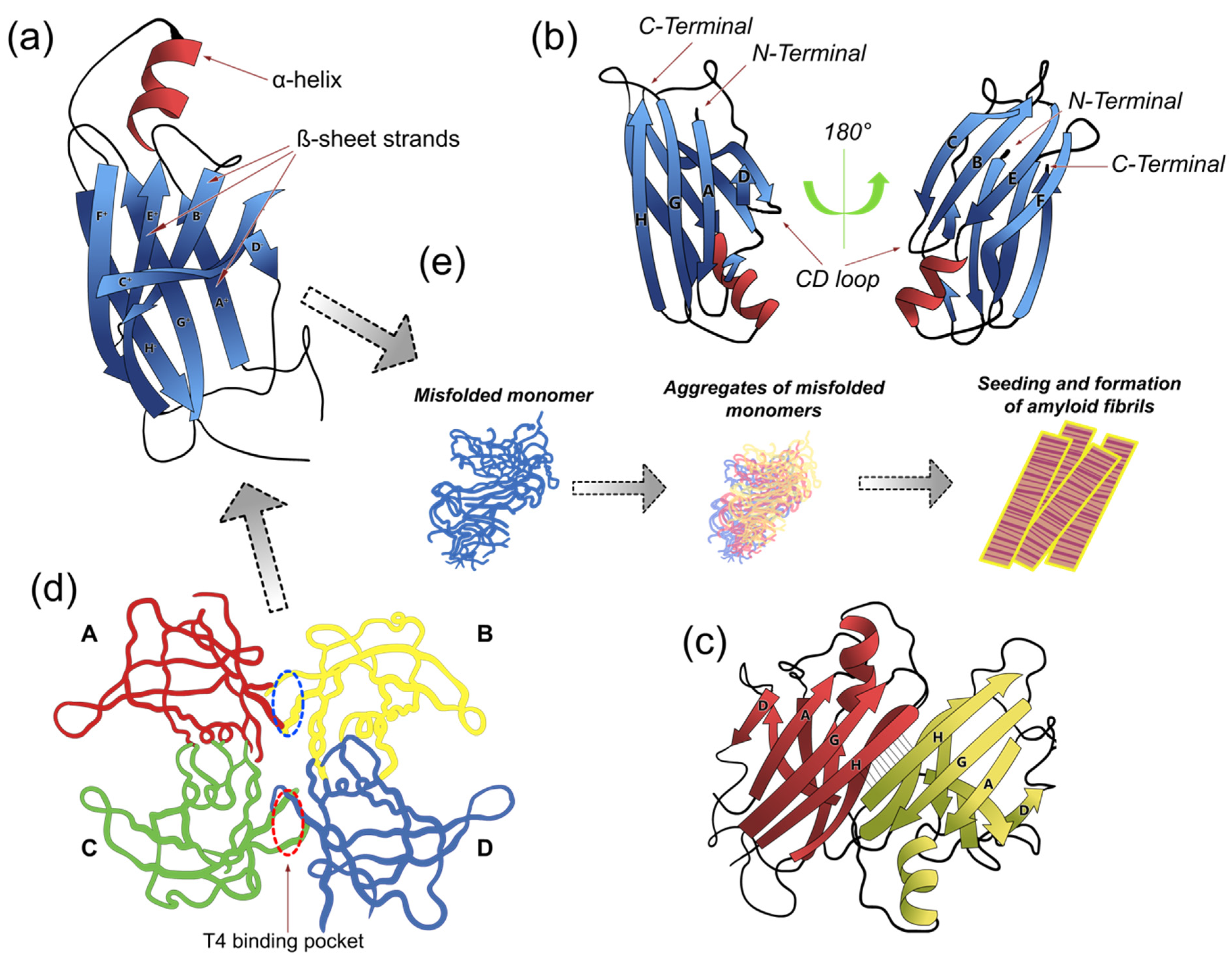
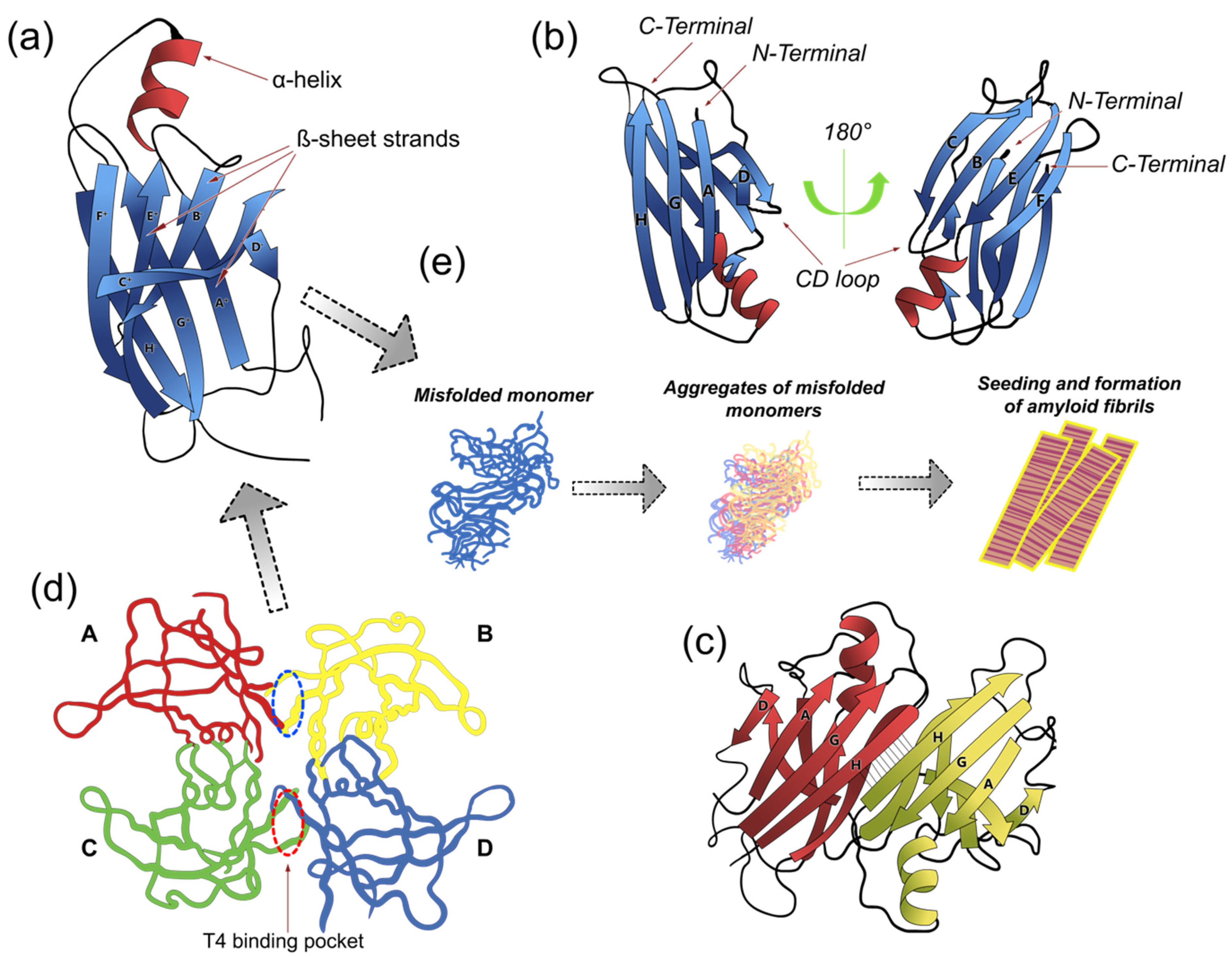
2. Structure of TTR
3. Protein Misfolding, Toxicity, and ATTR Amyloidosis
4. Endoplasmic Reticulum (ER) and Quality Control System (QCS) of the Cell and ATTR
5. Amyloid Formation
5.1. Dimers
5.2. Oligomers
5.3. Protofibrils and Fibrils
5.4. Seeding and Nucleation
5.5. Milieu Factors
5.6. Toxic Effects of Fibrils in Tissues
5.7. Dissociation, Misfolding, and Proteolysis
6. Diagnostic Methods
7. Novel Treatments for ATTR Amyloidosis
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Plante-Bordeneuve, V. Transthyretin familial amyloid polyneuropathy: An update. J. Neurol. 2018, 265, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Katsuno, M. Transthyretin amyloidosis: Update on the clinical spectrum, pathogenesis, and disease-modifying therapies. Neurol. Ther. 2020, 9, 317–333. [Google Scholar] [CrossRef]
- Suhr, O.B.; Lundgren, E.; Westermark, P. One mutation, two distinct disease variants: Unravelling the impact of transthyretin amyloid fibril composition. J. Intern. Med. 2017, 281, 337–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasperini, R.J.; Klaver, D.W.; Hou, X.; Aguilar, M.I.; Small, D.H. Mechanisms of transthyretin aggregation and toxicity. Subcell. Biochem. 2012, 65, 211–224. [Google Scholar] [PubMed]
- Kelly, J.W.; Colon, W.; Lai, Z.; Lashuel, H.A.; McCulloch, J.; McCutchen, S.L.; Miroy, G.J.; Peterson, S.A. Transthyretin quaternary and tertiary structural changes facilitate misassembly into amyloid. Adv. Protein. Chem. 1997, 50, 161–181. [Google Scholar]
- Artigas, F.; Perez, V.; Alvarez, E. Pindolol induces a rapid improvement of depressed patients treated with serotonin reuptake inhibitors. Arch. Gen. Psychiatry 1994, 51, 248–251. [Google Scholar] [CrossRef]
- Koike, H.; Katsuno, M. Ultrastructure in transthyretin amyloidosis: From pathophysiology to therapeutic insights. Biomedicines 2019, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Leach, B.I.; Zhang, X.; Kelly, J.W.; Dyson, H.J.; Wright, P.E. NMR Measurements reveal the structural basis of transthyretin destabilization by pathogenic Mutations. Biochemistry 2018, 57, 4421–4430. [Google Scholar] [CrossRef]
- Azevedo, E.P.; Pereira, H.M.; Garratt, R.C.; Kelly, J.W.; Foguel, D.; Palhano, F.L. Dissecting the structure, thermodynamic stability, and aggregation properties of the A25T transthyretin (A25T-TTR) variant involved in leptomeningeal amyloidosis: Identifying protein partners that co-aggregate during A25T-TTR fibrillogenesis in cerebrospinal fluid. Biochemistry 2011, 50, 11070–11083. [Google Scholar] [PubMed]
- Hurshman Babbes, A.R.; Powers, E.T.; Kelly, J.W. Quantification of the thermodynamically linked quaternary and tertiary structural stabilities of transthyretin and its disease-associated variants: The relationship between stability and amyloidosis. Biochemistry 2008, 47, 6969–6984. [Google Scholar] [CrossRef] [Green Version]
- Esperante, S.A.; Varejao, N.; Pinheiro, F.; Sant’Anna, R.; Luque-Ortega, J.R.; Alfonso, C.; Sora, V.; Papaleo, E.; Rivas, G.; Reverter, D.; et al. Disease-associated mutations impacting BC-loop flexibility trigger long-range transthyretin tetramer destabilization and aggregation. J. Biol. Chem. 2021, 297, 101039. [Google Scholar] [CrossRef]
- Jiang, X.; Buxbaum, J.N.; Kelly, J.W. The V122I cardiomyopathy variant of transthyretin increases the velocity of rate-limiting tetramer dissociation, resulting in accelerated amyloidosis. Proc. Natl. Acad. Sci. USA 2001, 98, 14943–14948. [Google Scholar] [CrossRef] [Green Version]
- Ulloa-Aguirre, A.; Janovick, J.A.; Brothers, S.P.; Conn, P.M. Pharmacologic rescue of conformationally-defective proteins: Implications for the treatment of human disease. Traffic 2004, 5, 821–837. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevet, E.; Cameron, P.H.; Pelletier, M.F.; Thomas, D.Y.; Bergeron, J.J. The endoplasmic reticulum: Integration of protein folding, quality control, signaling and degradation. Curr. Opin. Struct. Biol. 2001, 11, 120–124. [Google Scholar] [CrossRef]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Hayer-Hartl, M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef] [Green Version]
- Hiller, M.M.; Finger, A.; Schweiger, M.; Wolf, D.H. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science 1996, 273, 1725–1728. [Google Scholar] [CrossRef]
- Kincaid, M.M.; Cooper, A.A. ERADicate ER stress or die trying. Antioxid. Redox Signal. 2007, 9, 2373–2387. [Google Scholar] [CrossRef]
- Nakatsukasa, K.; Brodsky, J.L. The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic 2008, 9, 861–870. [Google Scholar] [CrossRef]
- Hammarstrom, P.; Sekijima, Y.; White, J.T.; Wiseman, R.L.; Lim, A.; Costello, C.E.; Altland, K.; Garzuly, F.; Budka, H.; Kelly, J.W. D18G transthyretin is monomeric, aggregation prone, and not detectable in plasma and cerebrospinal fluid: A prescription for central nervous system amyloidosis? Biochemistry 2003, 42, 6656–6663. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y.; Hammarstrom, P.; Matsumura, M.; Shimizu, Y.; Iwata, M.; Tokuda, T.; Ikeda, S.; Kelly, J.W. Energetic characteristics of the new transthyretin variant A25T may explain its atypical central nervous system pathology. Lab. Investig. 2003, 83, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Hammarstrom, P.; Jiang, X.; Hurshman, A.R.; Powers, E.T.; Kelly, J.W. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc. Natl. Acad. Sci. USA 2002, 99 (Suppl. 4), 16427–16432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, F.E.; Kelly, J.W. Therapeutic approaches to protein-misfolding diseases. Nature 2003, 426, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Susuki, S.; Suico, M.A.; Miyata, M.; Ando, Y.; Mizuguchi, M.; Takeuchi, M.; Dobashi, M.; Shuto, T.; Kai, H. Endoplasmic reticulum quality control regulates the fate of transthyretin variants in the cell. EMBO J. 2007, 26, 2501–2512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm, D.; Korhonen, L.; Eriksson, O.; Koks, S. Recent insights into the role of unfolded protein response in ER Stress in health and disease. Front. Cell Dev. Biol. 2017, 5, 48. [Google Scholar] [CrossRef] [Green Version]
- Reid, D.W.; Chen, Q.; Tay, A.S.; Shenolikar, S.; Nicchitta, C.V. The unfolded protein response triggers selective mRNA release from the endoplasmic reticulum. Cell 2014, 158, 1362–1374. [Google Scholar] [CrossRef] [Green Version]
- Buxbaum, J.N. The systemic amyloidoses. Curr. Opin. Rheumatol. 2004, 16, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Norgren, N.; Olsson, M.; Nystrom, H.; Ericzon, B.G.; de Tayrac, M.; Genin, E.; Plante-Bordeneuve, V.; Suhr, O.B. Gene expression profile in hereditary transthyretin amyloidosis: Differences in targeted and source organs. Amyloid 2014, 21, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Forman, M.S.; Lee, V.M.; Trojanowski, J.Q. ‘Unfolding’ pathways in neurodegenerative disease. Trends Neurosci. 2003, 26, 407–410. [Google Scholar] [CrossRef]
- Yang, Y.; Turner, R.S.; Gaut, J.R. The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J. Biol. Chem. 1998, 273, 25552–25555. [Google Scholar] [CrossRef] [Green Version]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 2013, 5, a013169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, J.B.; Kim, B.; Kim, J.H. Transthyretin misfolding, a fatal structural pathogenesis mechanism. Int. J. Mol. Sci. 2021, 22, 4429. [Google Scholar] [CrossRef] [PubMed]
- Chatani, E.; Yamamoto, N. Recent progress on understanding the mechanisms of amyloid nucleation. Biophys. Rev. 2018, 10, 527–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saelices, L.; Chung, K.; Lee, J.H.; Cohn, W.; Whitelegge, J.P.; Benson, M.D.; Eisenberg, D.S. Amyloid seeding of transthyretin by ex vivo cardiac fibrils and its inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E6741–E6750. [Google Scholar] [CrossRef] [Green Version]
- Jung, D.K.; Lee, Y.; Park, S.G.; Park, B.C.; Kim, G.H.; Rhee, S. Structural and functional analysis of PucM, a hydrolase in the ureide pathway and a member of the transthyretin-related protein family. Proc. Natl. Acad. Sci. USA 2006, 103, 9790–9795. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.M.; Schmuck, B.; Hansson, H.; Hard, T.; Westermark, G.T.; Sandgren, M. Enhanced detection of ATTR amyloid using a nanofibril-based assay. Amyloid 2021, 28, 158–167. [Google Scholar] [CrossRef]
- Lee, J.; Kim, K.; Choi, J.O.; Kim, S.J.; Jeon, E.S.; Choi, J.Y. 99mTc-DPD scintigraphy and SPECT/CT in patients with AL and ATTR type amyloidosis: Potential clinical implications. Medicine 2020, 99, e18905. [Google Scholar] [CrossRef] [PubMed]
- Bokhari, S.; Castano, A.; Pozniakoff, T.; Deslisle, S.; Latif, F.; Maurer, M.S. (99 m)Tc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ. Cardiovasc. Imaging 2013, 6, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Judge, D.P.; Heitner, S.B.; Falk, R.H.; Maurer, M.S.; Shah, S.J.; Witteles, R.M.; Grogan, M.; Selby, V.N.; Jacoby, D.; Hanna, M.; et al. Transthyretin stabilization by AG10 in symptomatic transthyretin amyloid cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.M.; Tu, M.J. Deliver the promise: RNAs as a new class of molecular entities for therapy and vaccination. Pharmacol. Ther. 2021, 107967. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Plante-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Kristen, A.V.; Lehrke, S.; Buss, S.; Mereles, D.; Steen, H.; Ehlermann, P.; Hardt, S.; Giannitsis, E.; Schreiner, R.; Haberkorn, U.; et al. Green tea halts progression of cardiac transthyretin amyloidosis: An observational report. Clin. Res. Cardiol. 2012, 101, 805–813. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, N.; Saraiva, M.J.; Almeida, M.R. Epigallocatechin-3-gallate as a potential therapeutic drug for TTR-related amyloidosis: “in vivo” evidence from FAP mice models. PLoS ONE 2012, 7, e29933. [Google Scholar] [CrossRef] [PubMed]
- aus dem Siepen, F.; Bauer, R.; Aurich, M.; Buss, S.J.; Steen, H.; Altland, K.; Katus, H.A.; Kristen, A.V. Green tea extract as a treatment for patients with wild-type transthyretin amyloidosis: An observational study. Drug Des. Devel. Ther. 2015, 9, 6319–6325. [Google Scholar] [CrossRef] [Green Version]
- Miyata, M.; Sato, T.; Kugimiya, M.; Sho, M.; Nakamura, T.; Ikemizu, S.; Chirifu, M.; Mizuguchi, M.; Nabeshima, Y.; Suwa, Y.; et al. The crystal structure of the green tea polyphenol (-)-epigallocatechin gallate-transthyretin complex reveals a novel binding site distinct from the thyroxine binding site. Biochemistry 2010, 49, 6104–6114. [Google Scholar] [CrossRef]
- Ferreira, N.; Cardoso, I.; Domingues, M.R.; Vitorino, R.; Bastos, M.; Bai, G.; Saraiva, M.J.; Almeida, M.R. Binding of epigallocatechin-3-gallate to transthyretin modulates its amyloidogenicity. FEBS Lett. 2009, 583, 3569–3576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, N.; Santos, S.A.; Domingues, M.R.; Saraiva, M.J.; Almeida, M.R. Dietary curcumin counteracts extracellular transthyretin deposition: Insights on the mechanism of amyloid inhibition. Biochim. Biophys. Acta 2013, 1832, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, M.; Rossor, A.M.; Laura, M.; Reilly, M.M. Clinical Presentation, Diagnosis and Treatment of TTR Amyloidosis. J. Neuromuscul. Dis. 2019, 6, 189–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Amyloid Fibril Formation |
|
| Tetramer Stabilization Mechanisms |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez-Duarte, A.; Ulloa-Aguirre, A. A Brief Journey through Protein Misfolding in Transthyretin Amyloidosis (ATTR Amyloidosis). Int. J. Mol. Sci. 2021, 22, 13158. https://doi.org/10.3390/ijms222313158
Gonzalez-Duarte A, Ulloa-Aguirre A. A Brief Journey through Protein Misfolding in Transthyretin Amyloidosis (ATTR Amyloidosis). International Journal of Molecular Sciences. 2021; 22(23):13158. https://doi.org/10.3390/ijms222313158
Chicago/Turabian StyleGonzalez-Duarte, Alejandra, and Alfredo Ulloa-Aguirre. 2021. "A Brief Journey through Protein Misfolding in Transthyretin Amyloidosis (ATTR Amyloidosis)" International Journal of Molecular Sciences 22, no. 23: 13158. https://doi.org/10.3390/ijms222313158
APA StyleGonzalez-Duarte, A., & Ulloa-Aguirre, A. (2021). A Brief Journey through Protein Misfolding in Transthyretin Amyloidosis (ATTR Amyloidosis). International Journal of Molecular Sciences, 22(23), 13158. https://doi.org/10.3390/ijms222313158