
Misfolded G Protein-Coupled Receptors and Endocrine Disease. Molecular Mechanisms and Therapeutic Prospects


Abstract
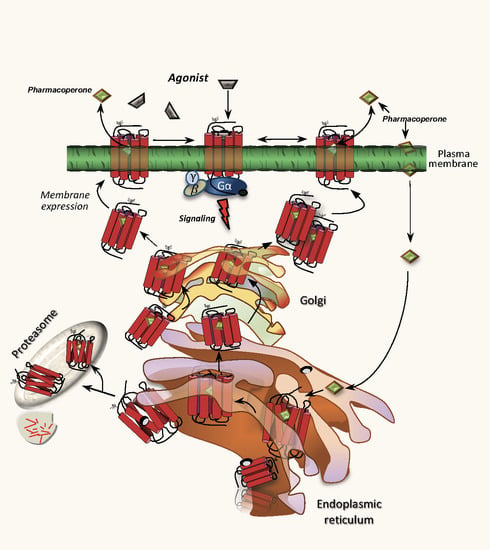
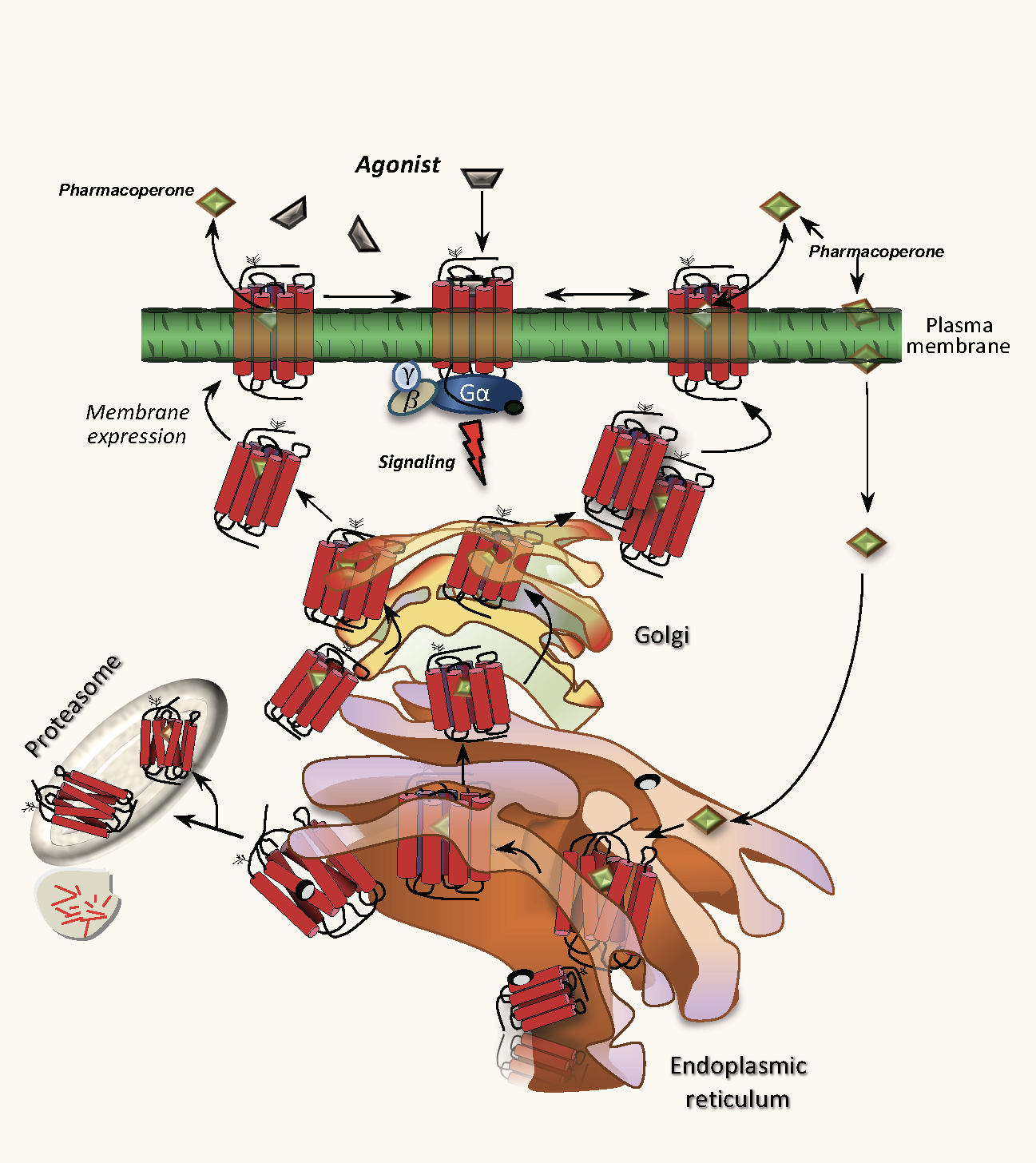
1. Introduction
2. Loss-of-Function Diseases Caused by Folding Defects in GPCRs Associated Endocrine Function
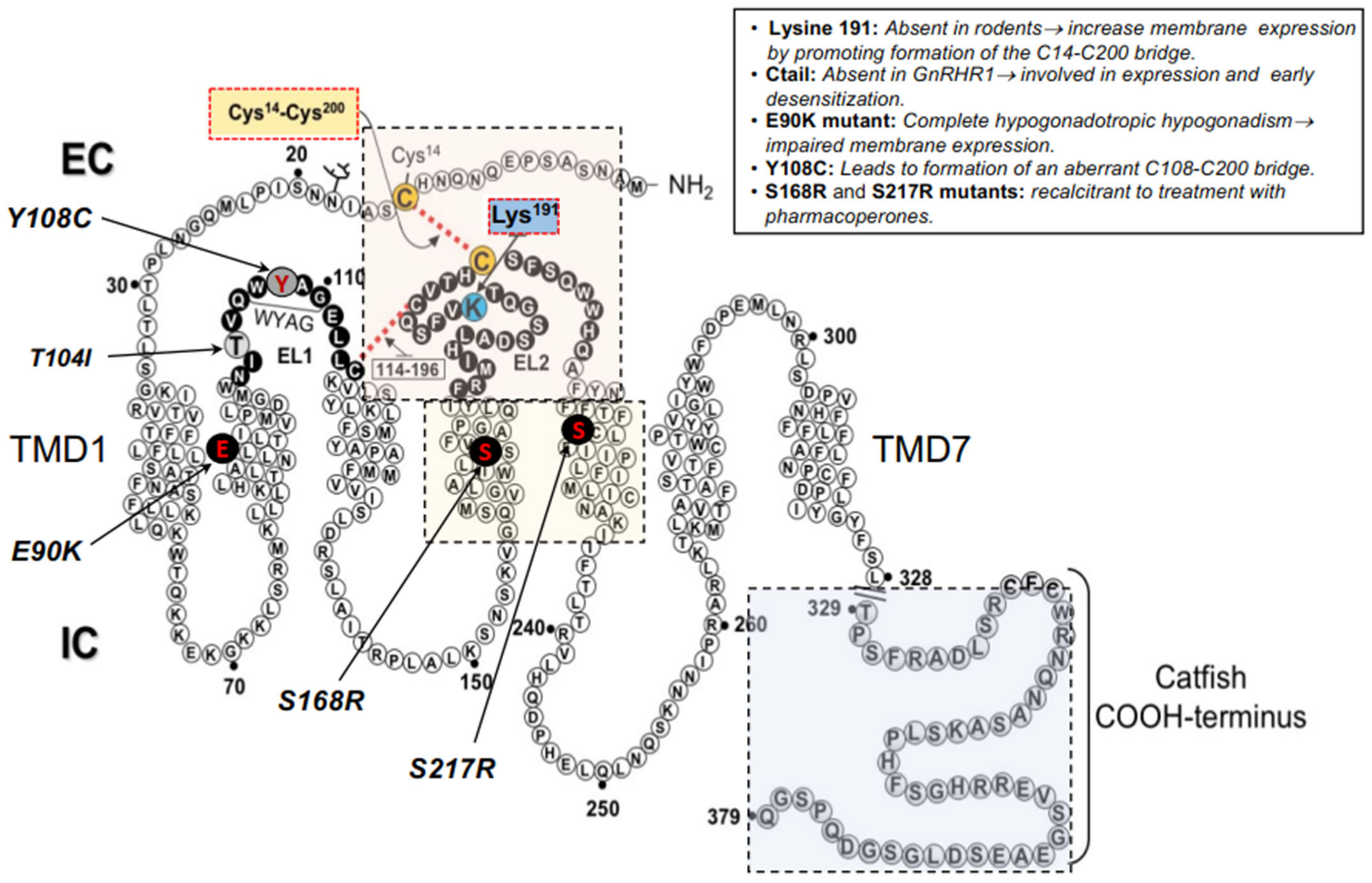
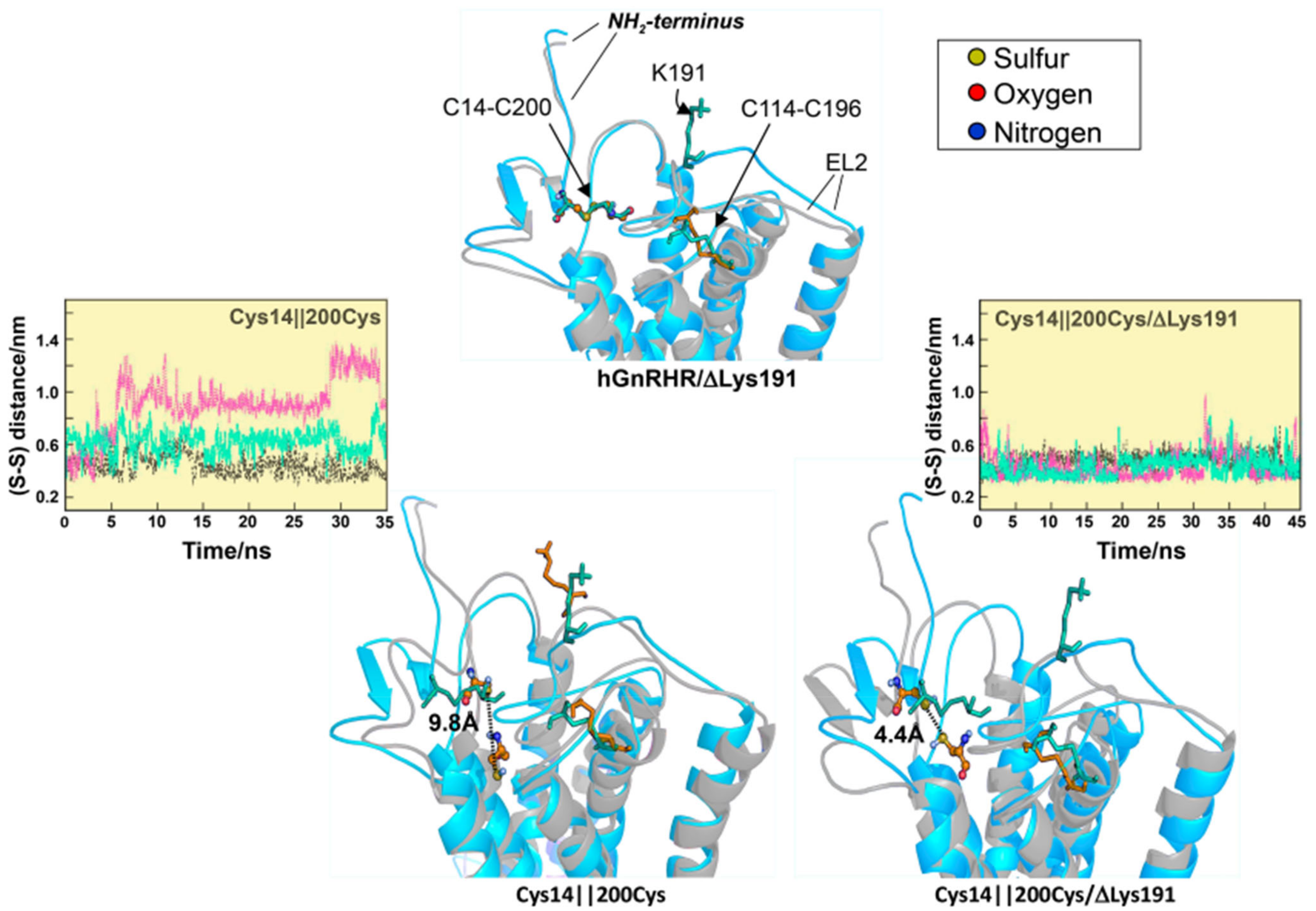
2.1. Misfolded GnRHRs and Hypogonadotropic Hypogonadism
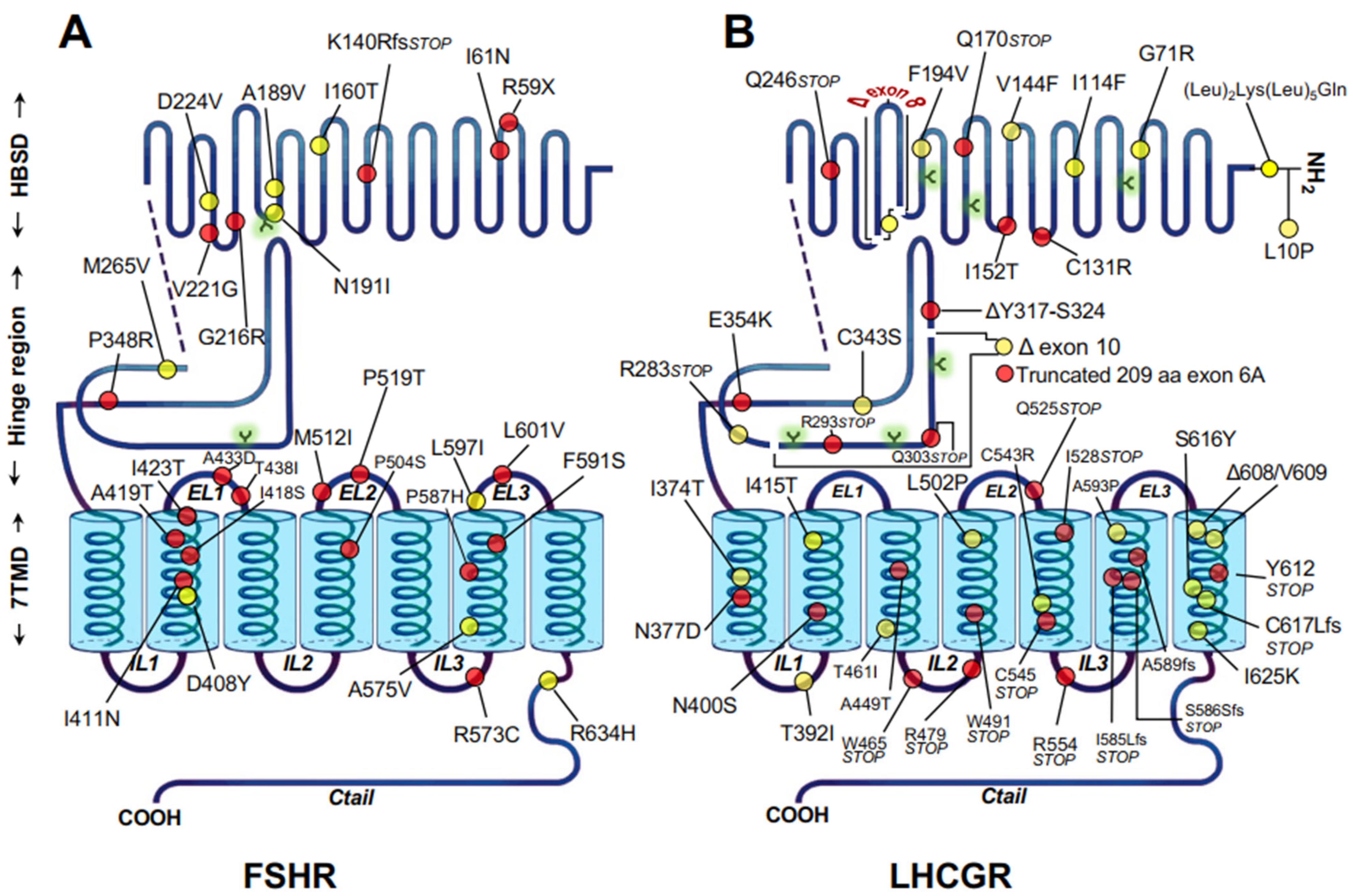
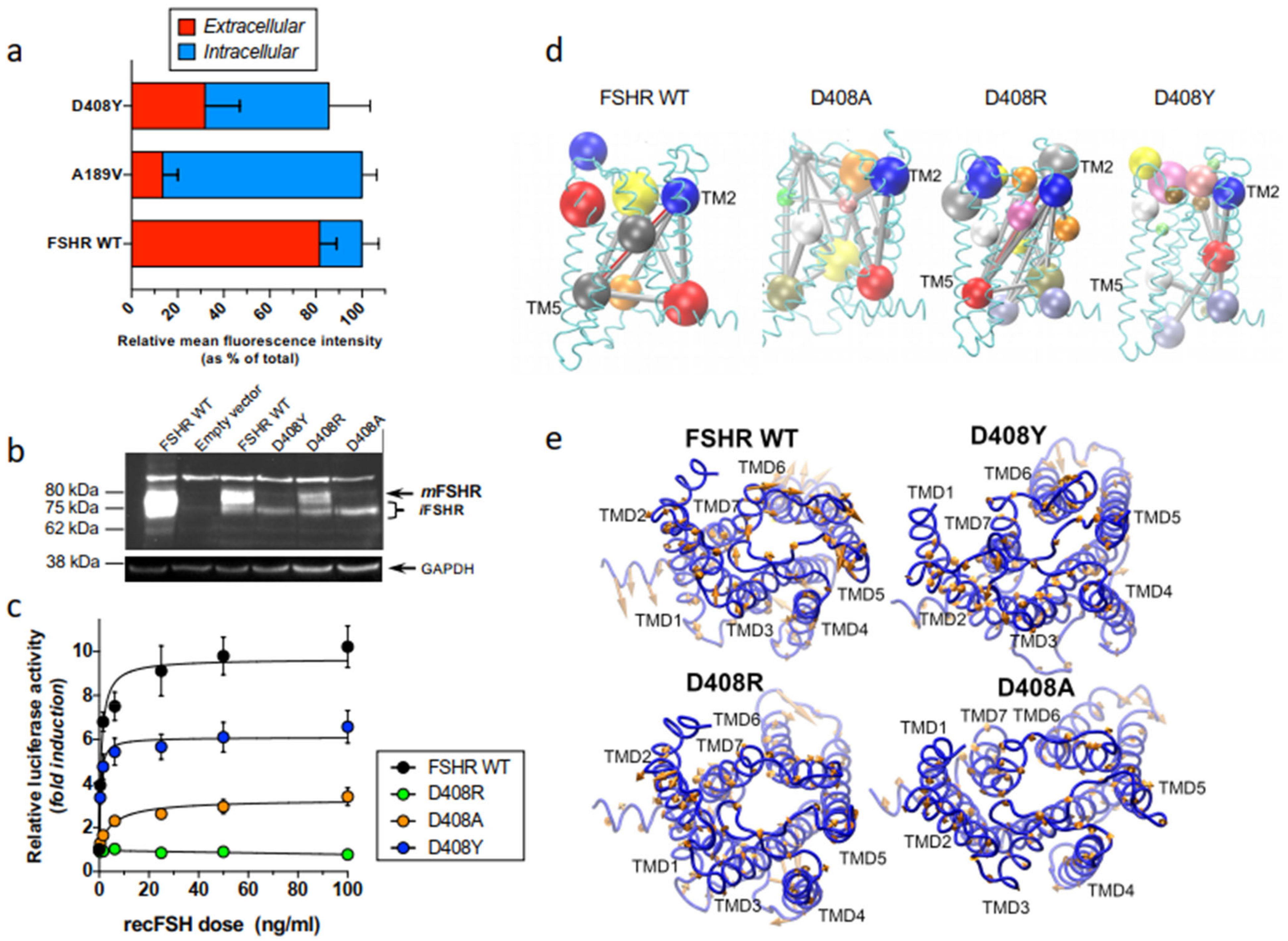
2.2. Misfolded Gonadotropin Receptors as a Cause of Hypergonadotropic Hypergonadism
3. Strategies to Rescue Misfolded GPCRs Leading to Endocrine Disorders
3.1. General Approaches to Rescue Function of Misfolded Proteins
3.2. Chemical Approaches
3.3. Pharmacological Approaches
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aridor, M. Visiting the ER: The endoplasmic reticulum as a target for therapeutics in traffic related diseases. Adv. Drug Deliv. Rev. 2007, 59, 759–781. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Principles of protein folding, misfolding and aggregation. Semin. Cell Dev. Biol. 2004, 15, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Hayer-Hartl, M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 2009, 16, 574–581. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Bernier, V.; Lagacé, M.; Bichet, D.-G.; Bouvier, M. Pharmacological chaperones: Potential treatment for conformational diseases. Trends Endocrinol. Metab. 2004, 15, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Ulloa-Aguirre, A.; Conn, P.M. Targeting of G protein-coupled receptors to the plasma membrane in health and disease. Front. Biosci. 2009, 14, 973–994. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ulloa-Aguirre, A.; Janovick, J.A.; Brothers, S.P.; Conn, P.M. Pharmacologic Rescue of Conformationally-Defective Proteins: Implications for the Treatment of Human Disease. Traffic 2004, 5, 821–837. [Google Scholar] [CrossRef]
- Tao, Y.-X.; Conn, P.M. Pharmacoperones as Novel Therapeutics for Diverse Protein Conformational Diseases. Physiol. Rev. 2018, 98, 697–725. [Google Scholar] [CrossRef]
- Conn, P.M.; Ulloa-Aguirre, A. Trafficking of G-protein-coupled receptors to the plasma membrane: Insights for pharmacoperone drugs. Trends Endocrinol. Metab. 2010, 21, 190–197. [Google Scholar] [CrossRef]
- Sharma, M.; Pampinella, F.; Nemes, C.; Benharouga, M.; So, J.; Du, K.; Bache, K.G.; Papsin, B.; Zerangue, N.; Stenmark, H.; et al. Misfolding diverts CFTR from recycling to degradation: Quality control at early endosomes. J. Cell Biol. 2004, 164, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Ulloa-Aguirre, A.; Janovick, J.A. Modulation of proteostasis and protein trafficking: A therapeutic avenue for misfolded G protein-coupled receptors causing disease in humans. Emerg. Top. Life Sci. 2019, 3, 39–52. [Google Scholar] [CrossRef]
- Hou, Z.-S.; Ulloa-Aguirre, A.; Tao, Y.-X. Pharmacoperone drugs: Targeting misfolded proteins causing lysosomal storage-, ion channels-, and G protein-coupled receptors-associated conformational disorders. Expert Rev. Clin. Pharmacol. 2018, 11, 611–624. [Google Scholar] [CrossRef]
- Shastry, B.S. Neurodegenerative disorders of protein aggregation. Neurochem. Int. 2003, 43, 1–7. [Google Scholar] [CrossRef]
- Horwich, A. Protein aggregation in disease: A role for folding intermediates forming specific multimeric interactions. J. Clin. Investig. 2002, 110, 1221–1232. [Google Scholar] [CrossRef]
- Gragg, M.; Park, P.S.-H. Misfolded rhodopsin mutants display variable aggregation properties. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2018, 1864, 2938–2948. [Google Scholar] [CrossRef]
- Park, S.H.O. Rhodopsin oligomerization and aggregation. J. Membr. Biol. 2019, 252, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Clarke, D.M. Chemical and pharmacological chaperones as new therapeutic agents. Expert Rev. Mol. Med. 2007, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Madoux, F.; Janovick, J.A.; Smithson, D.; Fargue, S.; Danpure, C.J.; Scampavia, L.; Chen, Y.-T.; Spicer, T.P.; Conn, P.M. Development of a Phenotypic High-Content Assay to Identify Pharmacoperone Drugs for the Treatment of Primary Hyperoxaluria Type 1 by High-Throughput Screening. ASSAY Drug Dev. Technol. 2015, 13, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Bernier, V.; Morello, J.-P.; Zarruk, A.; Debrand, N.; Salahpour, A.; Lonergan, M.; Arthus, M.-F.; Laperriere, A.; Brouard, R.; Bouvier, M.; et al. Pharmacologic Chaperones as a Potential Treatment for X-Linked Nephrogenic Diabetes Insipidus. J. Am. Soc. Nephrol. 2006, 17, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Bernier, V.; Bichet, D.-G.; Bouvier, M. Pharmacological chaperone action on G-protein-coupled receptors. Curr. Opin. Pharmacol. 2004, 4, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Conn, P.M.; Ulloa-Aguirre, A. Pharmacological Chaperones for Misfolded Gonadotropin-Releasing Hormone Receptors. Adv. Pharmacol. 2011, 62, 109–141. [Google Scholar] [CrossRef]
- Ulloa-Aguirre, A.; Conn, P.M. Pharmacoperones as a New Therapeutic Approach: In Vitro Identification and In vivo Validation of Bioactive Molecules. Curr. Drug Targets 2016, 17, 1471–1481. [Google Scholar] [CrossRef] [PubMed]
- Ulloa-Aguirre, A.; Conn, P.M. G protein-coupled receptors and the G protein family. In Handbook of Physiology-Endocrinology: Section 7, Cellular Endocrinology; Conn, P.M., Ed.; Oxford University Press: New York, NY, USA, 1998; pp. 87–141. [Google Scholar]
- Gershengorn, M.C.; Osman, R. Minireview: Insights into G Protein-Coupled Receptor Function Using Molecular Models. Endocrinology 2001, 142, 2–10. [Google Scholar] [CrossRef]
- Oldham, W.; Hamm, H.E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2008, 9, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Hanyaloglu, A.C.; von Zastrow, M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 537–568. [Google Scholar] [CrossRef]
- Ulloa-Aguirre, A.; Hanyaloglu, A.C.; Zarinan, T.; Janovick, J.A. Protein homeostasis and regulation of intreacellular trafficking of G protein-coupled receptors. In Protein Homeostasis Diseases; Pey, A.L., Ed.; Academic Press: London, UK, 2020; pp. 247–277. [Google Scholar]
- Fredriksson, R.; Lagerstrom, M.C.; Lundin, L.-G.; Schioth, H.B. The G-Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef]
- Insel, P.A.; Sriram, K.; Wiley, S.Z.; Wilderman, A.; Katakia, T.; McCann, T.; Yokouchi, H.; Zhang, L.; Corriden, R.; Liu, D.; et al. GPCRomics: GPCR Expression in Cancer Cells and Tumors Identifies New, Potential Biomarkers and Therapeutic Targets. Front. Pharmacol. 2018, 9, 431. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Conn, P.M.; Ulloa-Aguirre, A.; Janovick, J.A. “Pharmacoperone”: What’s in a word? Pharmacol. Res. 2013, 83, 1–2. [Google Scholar] [CrossRef]
- Bichet, D.G. Nephrogenic diabetes insipidus. Nephrol. Ther. 2006, 2, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Conn, P.M.; Ulloa-Aguirre, A.; Ito, J.; Janovick, J.A. G Protein-Coupled Receptor Trafficking in Health and Disease: Lessons Learned to Prepare for Therapeutic Mutant Rescue In Vivo. Pharmacol. Rev. 2007, 59, 225–250. [Google Scholar] [CrossRef] [PubMed]
- Calebiro, D.; De Filippis, T.; Lucchi, S.; Covino, C.; Panigone, S.; Beck-Peccoz, P.; Dunlap, D.; Persani, L. Intracellular entrapment of wild-type TSH receptor by oligomerization with mutants linked to dominant TSH resistance. Hum. Mol. Genet. 2005, 14, 2991–3002. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Cavanaugh, A.; Breitwieser, G.E. Regulation of Stability and Trafficking of Calcium-Sensing Receptors by Pharmacologic Chaperones. Adv. Pharmacol. 2011, 62, 143–173. [Google Scholar] [CrossRef]
- Tao, Y.-X. Molecular mechanisms of the neural melanocortin receptor dysfunction in severe early onset obesity. Mol. Cell. Endocrinol. 2005, 239, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.-X. The Melanocortin-4 Receptor: Physiology, Pharmacology, and Pathophysiology. Endocr. Rev. 2010, 31, 506–543. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.-X.; Conn, P.M. Chaperoning G Protein-Coupled Receptors: From Cell Biology to Therapeutics. Endocr. Rev. 2014, 35, 602–647. [Google Scholar] [CrossRef]
- Tao, Y.-X.; Segaloff, D.L. Functional Characterization of Melanocortin-4 Receptor Mutations Associated with Childhood Obesity. Endocrinology 2003, 144, 4544–4551. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Metherell, L.A.; Cheetham, M.E.; Huebner, A. Inherited ACTH insensitivity illuminates the mechanisms of ACTH action. Trends Endocrinol. Metab. 2005, 16, 451–457. [Google Scholar] [CrossRef]
- Ulloa-Aguirre, A.; Zarinan, T.; Dias, J.A.; Conn, P.M. Mutations in G protein-coupled receptors that impact receptor trafficking and reproductive function. Mol. Cell. Endocrinol. 2014, 382, 411–423. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ulloa-Aguirre, A.Z.T.; Lira-Albarrán, S. Mutations of gonadotropin-releasing hormone receptor gene. In Encyclopedia of Endocrine Diseases, 2nd ed.; Huhtaniemi, I.M.L., Ed.; Elsevier Inc.: Oxford, Academic Press, United Kingdom, 2019; pp. 704–712. [Google Scholar]
- Francou, B.; Bouligand, J.; Voican, A.; Amazit, L.; Trabado, S.; Fagart, J.; Meduri, G.; Brailly-Tabard, S.; Chanson, P.; LeComte, P.; et al. Normosmic Congenital Hypogonadotropic Hypogonadism Due to TAC3/TACR3 Mutations: Characterization of Neuroendocrine Phenotypes and Novel Mutations. PLoS ONE 2011, 6, e25614. [Google Scholar] [CrossRef]
- Monnier, C.; Dode, C.; Fabre, L.; Teixeira, L.; Labesse, G.; Pin, J.-P.; Hardelin, J.-P.; Rondard, P. PROKR2 missense mutations associated with Kallmann syndrome impair receptor signalling activity. Hum. Mol. Genet. 2009, 18, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Nimri, R.; Lebenthal, Y.; Lazar, L.; Chevrier, L.; Phillip, M.; Bar, M.; Hernandez-Mora, E.; De Roux, N.; Gat-Yablonski, G. A Novel Loss-of-Function Mutation in GPR54/KISS1R Leads to Hypogonadotropic Hypogonadism in a Highly Consanguineous Family. Endocrinology 2011, 152, 333. [Google Scholar] [CrossRef]
- Castro-Fernaández, C.; Maya-Nuúnñez, G.; Conn, P.M. Beyond the Signal Sequence: Protein Routing in Health and Disease. Endocr. Rev. 2004, 26, 479–503. [Google Scholar] [CrossRef][Green Version]
- Morello, J.-P.; Bouvier, M.; Petäjä-Repo, U.E.; Bichet, D.G. Pharmacological chaperones: A new twist on receptor folding. Trends Pharmacol. Sci. 2000, 21, 466–469. [Google Scholar] [CrossRef]
- Robben, J.H.; Deen, P.M. Pharmacological chaperones in nephrogenic diabetes insipidus: Possibilities for clinical application. BioDrugs 2007, 21, 157–166. [Google Scholar] [CrossRef]
- Robben, J.H.; Sze, M.; Knoers, N.V.; Deen, P.M. Functional rescue of vasopressin V2 receptor mutants in MDCK cells by pharmacochaperones: Relevance to therapy of nephrogenic diabetes insipidus. Am. J. Physiol. Renal. Physiol. 2007, 292, F253–F260. [Google Scholar] [CrossRef]
- Newton, C.L.; Anderson, R.C.; Kreuchwig, A.; Krause, G.; Katz, A.A.; Millar, R.P. Rescue of Function of Mutant Luteinising Hormone Receptors with Deficiencies in Cell Surface Expression, Hormone Binding, and Hormone Signalling. Neuroendocrinology 2021, 111, 451–464. [Google Scholar] [CrossRef]
- Newton, C.L.; Whay, A.M.; McArdle, C.A.; Zhang, M.; van Koppen, C.J.; van de Lagemaat, R.; Segaloff, D.L.; Millar, R.P. Rescue of expression and signaling of human luteinizing hormone G protein-coupled receptor mutants with an allosterically binding small-molecule agonist. Proc. Natl. Acad. Sci. USA 2011, 108, 7172–7176. [Google Scholar] [CrossRef]
- Hanyroup, S.; Anderson, R.C.; Nataraja, S.; Yu, H.N.; Millar, R.P.; Newton, C.L. Rescue of cell surface expression and signalling of mutant follicle-stimulating hormone receptors. Endocrinology 2021, 162, bqab134. [Google Scholar] [CrossRef]
- Janovick, J.A.; Maya-Nunez, G.; Ulloa-Aguirre, A.; Huhtaniemi, I.T.; Dias, J.A.; Verbost, P.; Conn, P.M. Increased plasma membrane expression of human follicle-stimulating hormone receptor by a small molecule thienopyr(im)idine. Mol. Cell. Endocrinol. 2009, 298, 84–88. [Google Scholar] [CrossRef]
- Tao, Y.-X.; Huang, H. Ipsen 5i is a Novel Potent Pharmacoperone for Intracellularly Retained Melanocortin-4 Receptor Mutants. Front. Endocrinol. 2014, 5, 131. [Google Scholar] [CrossRef] [PubMed]
- Millar, R.P. GnRHs and GnRH receptors. Anim. Reprod. Sci. 2005, 88, 5–28. [Google Scholar] [CrossRef]
- Ulloa-Aguirre, A.; Timossi, C. Biochemical and functional aspects of gonadotrophin-releasing hormone and gonadotrophins. Reprod. Biomed. Online 2000, 1, 48–62. [Google Scholar] [CrossRef]
- Millar, R.P.; Lu, Z.L.; Pawson, A.J.; Flanagan, C.A.; Morgan, K.; Maudsley, S.R. Gonadotropin-releasing hormone receptors. Endocr. Rev. 2004, 25, 235–275. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Janovick, J.A.; Brothers, S.; Blomenrohr, M.; Bogerd, J.; Conn, P.M. Addition of catfish gonadotropin-releasing hormone (GnRH) receptor intracellular carboxyl-terminal tail to rat GnRH receptor alters receptor expression and regulation. Mol. Endocrinol. 1998, 12, 161–171. [Google Scholar] [CrossRef] [PubMed]
- McArdle, C.A.; Davidson, J.S.; Willars, G.B. The tail of the gonadotrophin-releasing hormone receptor: Desensitization at, and distal to, G protein-coupled receptors. Mol. Cell. Endocrinol. 1999, 151, 129–136. [Google Scholar] [CrossRef]
- Janovick, J.A.; Ulloa-Aguirre, A.; Conn, P.M. Evolved Regulation of Gonadotropin-Releasing Hormone Receptor Cell Surface Expression. Endocrine 2003, 22, 317–328. [Google Scholar] [CrossRef]
- Klco, J.M.; Nikiforovich, G.V.; Baranski, T.J. Genetic Analysis of the First and Third Extracellular Loops of the C5a Receptor Reveals an Essential WXFG Motif in the First Loop. J. Biol. Chem. 2006, 281, 12010–12019. [Google Scholar] [CrossRef]
- Maya-Nunez, G.; Janovick, J.A.; Aguilar-Rojas, A.; Jardon-Valadez, E.; Leanos-Miranda, A.; Zarinan, T.; Ulloa-Aguirre, A.; Conn, P.M. Biochemical mechanism of pathogenesis of human gonadotropin-releasing hormone receptor mutants Thr104Ile and Tyr108Cys associated with familial hypogonadotropic hypogonadism. Mol. Cell. Endocrinol. 2011, 337, 16–23. [Google Scholar] [CrossRef]
- Ulloa-Aguirre, A.; Janovick, J.A.; Miranda, A.L.; Conn, P.M. G-Protein-Coupled Receptor Trafficking: Understanding the Chemical Basis of Health and Disease. ACS Chem. Biol. 2006, 1, 631–638. [Google Scholar] [CrossRef]
- Arora, K.K.; Chung, H.O.; Catt, K.J. Influence of a species-specific extracellular amino acid on expression and function of the human gonadotropin-releasing hormone receptor. Mol. Endocrinol. 1999, 13, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Conn, P.M.; Knollman, P.E.; Brothers, S.P.; Janovick, J.A. Protein Folding as Posttranslational Regulation: Evolution of a Mechanism for Controlled Plasma Membrane Expression of a G Protein-Coupled Receptor. Mol. Endocrinol. 2006, 20, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Knollman, P.E.; Janovick, J.A.; Brothers, S.P.; Conn, P.M. Parallel Regulation of Membrane Trafficking and Dominant-negative Effects by Misrouted Gonadotropin-releasing Hormone Receptor Mutants. J. Biol. Chem. 2005, 280, 24506–24514. [Google Scholar] [CrossRef] [PubMed]
- Beranova, M.; Oliveira, L.M.; Bedecarrats, G.Y.; Schipani, E.; Vallejo, M.; Ammini, A.C.; Quintos, J.B.; Hall, J.E.; Martin, K.A.; Hayes, F.J.; et al. Prevalence, Phenotypic Spectrum, and Modes of Inheritance of Gonadotropin-Releasing Hormone Receptor Mutations in Idiopathic Hypogonadotropic Hypogonadism1. J. Clin. Endocrinol. Metab. 2001, 86, 1580–1588. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Janovick, J.A.; Knollman, P.E.; Brothers, S.P.; Ayala-Yanez, R.; Aziz, A.S.; Conn, P.M. Regulation of G protein-coupled receptor trafficking by inefficient plasma membrane expression: Molecular basis of an evolved strategy. J. Biol. Chem. 2006, 281, 8417–8425. [Google Scholar] [CrossRef]
- Leanos-Miranda, A.; Janovick, J.A.; Conn, P.M. Receptor-Misrouting: An Unexpectedly Prevalent and Rescuable Etiology in Gonadotropin-Releasing Hormone Receptor-Mediated Hypogonadotropic Hypogonadism. J. Clin. Endocrinol. Metab. 2002, 87, 4825–4828. [Google Scholar] [CrossRef] [PubMed]
- Janovick, J.A.; Goulet, M.; Bush, E.; Greer, J.; Wettlaufer, D.G.; Conn, P.M. Structure-Activity Relations of Successful Pharmacologic Chaperones for Rescue of Naturally Occurring and Manufactured Mutants of the Gonadotropin-Releasing Hormone Receptor. J. Pharmacol. Exp. Ther. 2003, 305, 608–614. [Google Scholar] [CrossRef]
- Maya-Nunez, G.; Janovick, J.A.; Ulloa-Aguirre, A.; Soderlund, D.; Conn, P.M.; Mendez, J.P. Molecular Basis of Hypogonadotropic Hypogonadism: Restoration of Mutant (E 90 K) GnRH Receptor Function by a Deletion at a Distant Site. J. Clin. Endocrinol. Metab. 2002, 87, 2144–2149. [Google Scholar] [CrossRef]
- Conn, P.M.; Smithson, D.C.; Hodder, P.S.; Stewart, M.D.; Behringer, R.R.; Smith, E.; Ulloa-Aguirre, A.; Janovick, J.A. Transitioning pharmacoperones to therapeutic use: In vivo proof-of-principle and design of high throughput screens. Pharmacol. Res. 2013, 83, 38–51. [Google Scholar] [CrossRef]
- Janovick, J.A.; Stewart, M.D.; Jacob, D.; Martin, L.D.; Deng, J.M.; Stewart, C.A.; Wang, Y.; Cornea, A.; Chavali, L.; Lopez, S.; et al. Restoration of testis function in hypogonadotropic hypogonadal mice harboring a misfolded GnRHR mutant by pharmacoperone drug therapy. Proc. Natl. Acad. Sci. USA 2013, 110, 21030–21035. [Google Scholar] [CrossRef]
- Janovick, J.A.; Conn, P.M. Salt bridge integrates GPCR activation with protein trafficking. Proc. Natl. Acad. Sci. USA 2010, 107, 4454–4458. [Google Scholar] [CrossRef]
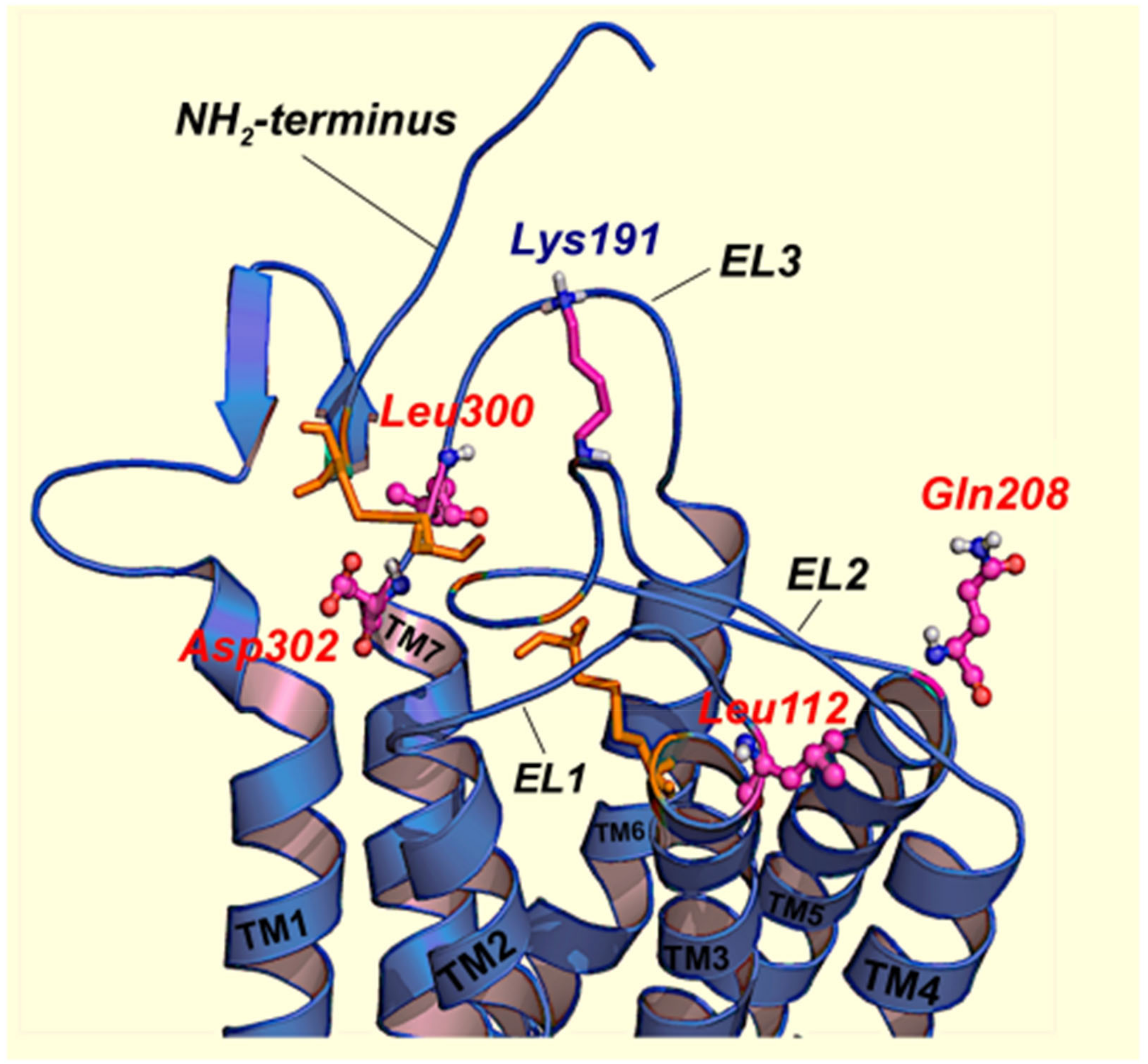
- Jardon-Valadez, E.; Aguilar-Rojas, A.; Maya-Nunez, G.; Leanos-Miranda, A.; Pineiro, A.; Conn, P.M.; Ulloa-Aguirre, A. Conformational effects of Lys191 in the human GnRH receptor: Mutagenesis and molecular dynamics simulations studies. J. Endocrinol. 2009, 201, 297–307. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Soderlund, D.; Canto, P.; De La Chesnaye, E.; Ulloa-Aguirre, A.; Mendez, J.P. A novel homozygous mutation in the second transmembrane domain of the gonadotrophin releasing hormone receptor gene. Clin. Endocrinol. 2001, 54, 493–498. [Google Scholar] [CrossRef]
- Brothers, S.P.; Cornea, A.; Janovick, J.A.; Conn, P.M. Human Loss-of-Function Gonadotropin-Releasing Hormone Receptor Mutants Retain Wild-Type Receptors in the Endoplasmic Reticulum: Molecular Basis of the Dominant-Negative Effect. Mol. Endocrinol. 2004, 18, 1787–1797. [Google Scholar] [CrossRef]
- Antelli, A.; Baldazzi, L.; Balsamo, A.; Pirazzoli, P.; Nicoletti, A.; Gennari, M.; Cicognani, A. Two novel GnRHR gene mutations in two siblings with hypogonadotropic hypogonadism. Eur. J. Endocrinol. 2006, 155, 201–205. [Google Scholar] [CrossRef]
- Cook, J.V.; Eidne, K.A. An Intramolecular Disulfide Bond between Conserved Extracellular Cysteines in the Gonadotropin-Releasing Hormone Receptor Is Essential for Binding and Activation1. Endocrinology 1997, 138, 2800–2806. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Conn, P.M.; Janovick, J.A. Drug development and the cellular quality control system. Trends Pharmacol. Sci. 2009, 30, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Leskela, T.T.; Markkanen, P.M.; Pietila, E.M.; Tuusa, J.T.; Petaja-Repo, U.E. Opioid receptor pharmacological chaperones act by binding and stabilizing newly synthesized receptors in the endoplasmic reticulum. J. Biol. Chem. 2007, 282, 23171–23183. [Google Scholar] [CrossRef] [PubMed]
- Petaja-Repo, U.E.; Hogue, M.; Laperriere, A.; Bhalla, S.; Walker, P.; Bouvier, M. Newly synthesized human delta opioid receptors retained in the endoplasmic reticulum are retrotranslocated to the cytosol, deglycosylated, ubiquitinated, and degraded by the proteasome. J. Biol. Chem. 2001, 276, 4416–4423. [Google Scholar] [CrossRef]
- Petaja-Repo, U.E.; Hogue, M.; Laperriere, A.; Walker, P.; Bouvier, M. Export from the endoplasmic reticulum represents the limiting step in the maturation and cell surface expression of the human delta opioid receptor. J. Biol. Chem. 2000, 275, 13727–13736. [Google Scholar] [CrossRef] [PubMed]
- Petaja-Repo, U.E.; Lackman, J.J. Targeting opioid receptors with pharmacological chaperones. Pharmacol. Res. 2014, 83, 52–62. [Google Scholar] [CrossRef]
- Pietila, E.M.; Tuusa, J.T.; Apaja, P.M.; Aatsinki, J.T.; Hakalahti, A.E.; Rajaniemi, H.J.; Petaja-Repo, U.E. Inefficient maturation of the rat luteinizing hormone receptor. A putative way to regulate receptor numbers at the cell surface. J. Biol. Chem. 2005, 280, 26622–26629. [Google Scholar] [CrossRef] [PubMed]
- Ascoli, M.; Fanelli, F.; Segaloff, D.L. The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocr. Rev. 2002, 23, 141–174. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.S.; Pangas, S.A. The ovary: Basic biology and clinical implications. J. Clin. Investig. 2010, 120, 963–972. [Google Scholar] [CrossRef]
- Simoni, M.; Gromoll, J.; Nieschlag, E. The Follicle-Stimulating Hormone Receptor: Biochemistry, Molecular Biology, Physiology, and Pathophysiology. Endocr. Rev. 1997, 18, 739–773. [Google Scholar] [CrossRef]
- Huhtaniemi, I. A short evolutionary history of FSH-stimulated spermatogenesis. Hormones 2015, 14, 468–478. [Google Scholar] [CrossRef]
- Saez, J.M. Leydig cells: Endocrine, paracrine, and autocrine regulation. Endocr. Rev. 1994, 15, 574–626. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, J. Ligand-selective determinants in gonadotropin receptors. Mol. Cell. Endocrinol. 2007, 260–262, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-R.; McLachlan, S.M.; Rapoport, B. A Monoclonal Antibody with Thyrotropin (TSH) Receptor Inverse Agonist and TSH Antagonist Activities Binds to the Receptor Hinge Region as Well as to the Leucine-Rich Domain. Endocrinology 2009, 150, 3401–3408. [Google Scholar] [CrossRef]
- Krause, G.; Kreuchwig, A.; Kleinau, G. Extended and Structurally Supported Insights into Extracellular Hormone Binding, Signal Transduction and Organization of the Thyrotropin Receptor. PLoS ONE 2012, 7, e52920. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, R.; Dighe, R.R. The Hinge Region of Human Thyroid-Stimulating Hormone (TSH) Receptor Operates as a Tunable Switch between Hormone Binding and Receptor Activation. PLoS ONE 2012, 7, e40291. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Liu, H.; Chen, X.; Chen, P.-H.; Fischer, D.; Sriraman, V.; Yu, H.N.; Arkinstall, S.; He, X. Structure of follicle-stimulating hormone in complex with the entire ectodomain of its receptor. Proc. Natl. Acad. Sci. USA 2012, 109, 12491–12496. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Dias, J.A.; He, X. Structural biology of glycoprotein hormones and their receptors: Insights to signaling. Mol. Cell. Endocrinol. 2014, 382, 424–451. [Google Scholar] [CrossRef] [PubMed]
- Kleinau, G.; Krause, G. Thyrotropin and Homologous Glycoprotein Hormone Receptors: Structural and Functional Aspects of Extracellular Signaling Mechanisms. Endocr. Rev. 2009, 30, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.A.; Cohen, B.D.; Lindau-Shepard, B.; Nechamen, C.A.; Peterson, A.J.; Schmidt, A. Molecular, structural, and cellular biology of follitropin and follitropin receptor. Vitam. Horm. 2002, 64, 249–322. [Google Scholar] [CrossRef] [PubMed]
- Duvernay, M.T.; Zhou, F.; Wu, G. A Conserved Motif for the Transport of G Protein-coupled Receptors from the Endoplasmic Reticulum to the Cell Surface. J. Biol. Chem. 2004, 279, 30741–30750. [Google Scholar] [CrossRef] [PubMed]
- Timossi, C.; Ortiz-Elizondo, C.; Pineda, D.B.; Dias, J.A.; Conn, P.M.; Ulloa-Aguirre, A. Functional significance of the BBXXB motif reversed present in the cytoplasmic domains of the human follicle-stimulating hormone receptor. Mol. Cell. Endocrinol. 2004, 223, 17–26. [Google Scholar] [CrossRef]
- Ulloa-Aguirre, A.; Zarinan, T.; Gutierrez-Sagal, R.; Dias, J.A. Intracellular Trafficking of Gonadotropin Receptors in Health and Disease. Handb. Exp. Pharmacol. 2018, 245, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Ulloa-Aguirre, A.; Reiter, E.; Crepieux, P. FSH Receptor Signaling: Complexity of Interactions and Signal Diversity. Endocrinology 2018, 159, 3020–3035. [Google Scholar] [CrossRef]
- Musnier, A.; Heitzler, D.; Boulo, T.; Tesseraud, S.; Durand, G.; Lecureuil, C.; Guillou, H.; Poupon, A.; Reiter, E.; Crepieux, P. Developmental regulation of p70 S6 kinase by a G protein-coupled receptor dynamically modelized in primary cells. Cell. Mol. Life Sci. 2009, 66, 3487–3503. [Google Scholar] [CrossRef]
- Gromoll, J.; Schulz, A.; Borta, H.; Gudermann, T.; Teerds, K.J.; Greschniok, A.; Nieschlag, E.; Seif, F.J. Homozygous mutation within the conserved Ala-Phe-Asn-Glu-Thr motif of exon 7 of the LH receptor causes male pseudohermaphroditism. Eur. J. Endocrinol. 2002, 147, 597–608. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aittomaki, K.; Herva, R.; Stenman, U.H.; Juntunen, K.; Ylostalo, P.; Hovatta, O.; De La Chapelle, A. Clinical features of primary ovarian failure caused by a point mutation in the follicle-stimulating hormone receptor gene. J. Clin. Endocrinol. Metab. 1996, 81, 3722–3726. [Google Scholar] [CrossRef][Green Version]
- Huhtaniemi, I.T.; Themmen, A.P. Mutations in Human Gonadotropin and Gonadotropin-Receptor Genes. Endocrine 2005, 26, 207–218. [Google Scholar] [CrossRef]
- Ulloa-Aguirre, A.; Zarinan, T. The Follitropin Receptor: Matching Structure and Function. Mol. Pharmacol. 2016, 90, 596–608. [Google Scholar] [CrossRef]
- Althumairy, D.; Zhang, X.; Baez, N.; Barisas, G.; Roess, D.A.; Bousfield, G.R.; Crans, D.C. Glycoprotein G-protein Coupled Receptors in Disease: Luteinizing Hormone Receptors and Follicle Stimulating Hormone Receptors. Diseases 2020, 8, 35. [Google Scholar] [CrossRef]
- Newton, C.L.; Anderson, R.C.; Katz, A.A.; Millar, R.P. Loss-of-Function Mutations in the Human Luteinizing Hormone Receptor Predominantly Cause Intracellular Retention. Endocrinology 2016, 157, 4364–4377. [Google Scholar] [CrossRef]
- Martens, J.W.; Lumbroso, S.; Verhoef-Post, M.; Georget, V.; Richter-Unruh, A.; Szarras-Czapnik, M.; Romer, T.E.; Brunner, H.G.; Themmen, A.P.; Sultan, C. Mutant luteinizing hormone receptors in a compound heterozygous patient with complete Leydig cell hypoplasia: Abnormal processing causes signaling deficiency. J. Clin. Endocrinol. Metab. 2002, 87, 2506–2513. [Google Scholar] [CrossRef] [PubMed]
- Mizrachi, D.; Segaloff, D.L. Intracellularly Located Misfolded Glycoprotein Hormone Receptors Associate with Different Chaperone Proteins than Their Cognate Wild-Type Receptors. Mol. Endocrinol. 2004, 18, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.S.; Roy, B.S.; Mahale, S.D. Mutations and polymorphisms in FSH receptor: Functional implications in human reproduction. Reproduction 2013, 146, R235–R248. [Google Scholar] [CrossRef]
- Huhtaniemi, I.; Alevizaki, M. Mutations along the hypothalamic–pituitary–gonadal axis affecting male reproduction. Reprod. Biomed. Online 2007, 15, 622–632. [Google Scholar] [CrossRef]
- Zarinan, T.; Mayorga, J.; Jardon-Valadez, E.; Gutierrez-Sagal, R.; Maravillas-Montero, J.L.; Mejia-Dominguez, N.R.; Martínez-Luis, I.; Yacini-Torres, O.G.; Cravioto, M.-D.; Reiter, E.; et al. A Novel Mutation in the FSH Receptor (I423T) Affecting Receptor Activation and Leading to Primary Ovarian Failure. J. Clin. Endocrinol. Metab. 2020, 106, e534–e550. [Google Scholar] [CrossRef]
- Aittomaki, K.; Tapanainen, J.; Huhtaniemi, I.; De La Chapelle, A. Inherited primary amenorrhea. The first gynecological disease of Finnish heritage. Duodecim 1996, 112, 9–11. [Google Scholar] [PubMed]
- Tapanainen, J.S.; Aittomaki, K.; Min, J.; Vaskivuo, T.; Huhtaniemi, I.T. Men homozygous for an inactivating mutation of the follicle-stimulating hormone (FSH) receptor gene present variable suppression of spermatogenesis and fertility. Nat. Genet. 1997, 15, 205–206. [Google Scholar] [CrossRef]
- Meduri, G.; Touraine, P.; Beau, I.; Lahuna, O.; Desroches, A.; Vacher-Lavenu, M.C.; Kuttenn, F.; Misrahi, M. Delayed Puberty and Primary Amenorrhea Associated with a Novel Mutation of the Human Follicle-Stimulating Hormone Receptor: Clinical, Histological, and Molecular Studies. J. Clin. Endocrinol. Metab. 2003, 88, 3491–3498. [Google Scholar] [CrossRef] [PubMed]
- Rannikko, A.; Pakarinen, P.; Manna, P.R.; Beau, I.; Misrahi, M.; Aittomaki, K.; Huhtaniemi, I. Functional characterization of the human FSH receptor with an inactivating Ala189Val mutation. Mol. Hum. Reprod. 2002, 8, 311–317. [Google Scholar] [CrossRef]
- Tranchant, T.; Durand, G.; Gauthier, C.; Crepieux, P.; Ulloa-Aguirre, A.; Royere, D.; Reiter, E. Preferential beta-arrestin signalling at low receptor density revealed by functional characterization of the human FSH receptor A189 V mutation. Mol. Cell. Endocrinol. 2011, 331, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Gromoll, J.; Simoni, M.; Nordhoff, V.; Behre, H.M.; De Geyter, C.; Nieschlag, E. Functional and clinical consequences of mutations in the FSH receptor. Mol. Cell. Endocrinol. 1996, 125, 177–182. [Google Scholar] [CrossRef]
- Jardon-Valadez, E.; Castillo-Guajardo, D.; Martinez-Luis, I.; Gutierrez-Sagal, R.; Zarinan, T.; Ulloa-Aguirre, A. Molecular dynamics simulation of the follicle-stimulating hormone receptor. Understanding the conformational dynamics of receptor variants at positions N680 and D408 from in silico analysis. PLoS ONE 2018, 13, e0207526. [Google Scholar] [CrossRef]
- Bramble, M.S.; Goldstein, E.H.; Lipson, A.; Ngun, T.; Eskin, A.; Gosschalk, J.E.; Roach, L.; Vashist, N.; Barseghyan, H.; Lee, E.; et al. A novel follicle-stimulating hormone receptor mutation causing primary ovarian failure: A fertility application of whole exome sequencing. Hum. Reprod. 2016, 31, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Hess, B. Convergence of sampling in protein simulations. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2002, 65, 031910. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, B.P.; Freedman, S.D. Cystic fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef]
- Ratjen, F.A. Cystic Fibrosis: Pathogenesis and Future Treatment Strategies. Respir. Care 2009, 54, 595–605. [Google Scholar] [CrossRef]
- Welsh, M.J.; Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254. [Google Scholar] [CrossRef]
- Brown, C.R.; Hong-Brown, L.Q.; Welch, W.J. Correcting temperature-sensitive protein folding defects. J. Clin. Investig. 1997, 99, 1432–1444. [Google Scholar] [CrossRef]
- Jaquette, J.; Segaloff, D.L. Temperature Sensitivity of Some Mutants of the Lutropin/Choriogonadotropin Receptor 1. Endocrinology 1997, 138, 85–91. [Google Scholar] [CrossRef][Green Version]
- Filipeanu, C.M.; de Vries, R.; Danser, A.H.; Kapusta, D.R. Modulation of α2C adrenergic receptor temperature-sensitive trafficking by HSP90. Biochim. Biophys. Acta (BBA)—Bioenerg. 2011, 1813, 346–357. [Google Scholar] [CrossRef]
- Maya-Nunez, G.; Ulloa-Aguirre, A.; Janovick, J.A.; Conn, P.M. Pharmacological Chaperones Correct Misfolded GPCRs and Rescue Function: Protein Trafficking as a Therapeutic Target. Subcell. Biochem. 2012, 63, 263–289. [Google Scholar] [CrossRef]
- Ulloa-Aguirre, A.; Janovick, J.A.; Leanos-Miranda, A.; Conn, P.M. Misrouted cell surface GnRH receptors as a disease aetiology for congenital isolated hypogonadotrophic hypogonadism. Hum. Reprod. Update 2004, 10, 177–192. [Google Scholar] [CrossRef]
- Maya-Nunez, G.; Janovick, J.A.; Conn, P.M. Combined Modification of Intracellular and Extracellular Loci on Human Gonadotropin-Releasing Hormone Receptor Provides a Mechanism for Enhanced Expression. Endocrine 2000, 13, 401–407. [Google Scholar] [CrossRef]
- Brothers, S.P.; Janovick, J.A.; Conn, P.M. Calnexin regulated gonadotropin-releasing hormone receptor plasma membrane expression. J. Mol. Endocrinol. 2006, 37, 479–488. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gorbatyuk, M.S.; Knox, T.; LaVail, M.M.; Gorbatyuk, O.S.; Noorwez, S.M.; Hauswirth, W.W.; Lin, J.H.; Muzyczka, N.; Lewin, A.S. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc. Natl. Acad. Sci. USA 2010, 107, 5961–5966. [Google Scholar] [CrossRef]
- Oueslati, M.; Hermosilla, R.; Schonenberger, E.; Oorschot, V.; Beyermann, M.; Wiesner, B.; Schmidt, A.; Klumperman, J.; Rosenthal, W.; Schulein, R. Rescue of a Nephrogenic Diabetes Insipidus-causing Vasopressin V2 Receptor Mutant by Cell-penetrating Peptides. J. Biol. Chem. 2007, 282, 20676–20685. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, T.; Ejima, D.; Kita, Y.; Tsumoto, K. Small molecule pharmacological chaperones: From thermodynamic stabilization to pharmaceutical drugs. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2006, 1764, 1677–1687. [Google Scholar] [CrossRef]
- Perlmutter, D.H. Chemical chaperones: A pharmacological strategy for disorders of protein folding and trafficking. Pediatr. Res. 2002, 52, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Janovick, J.A.; Park, B.S.; Conn, P.M. Therapeutic Rescue of Misfolded Mutants: Validation of Primary High Throughput Screens for Identification of Pharmacoperone Drugs. PLoS ONE 2011, 6, e22784. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leanños-Miranda, A.; Ulloa-Aguirre, A.; Janovick, J.A.; Conn, P.M. In Vitro Coexpression and Pharmacological Rescue of Mutant Gonadotropin-Releasing Hormone Receptors Causing Hypogonadotropic Hypogonadism in Humans Expressing Compound Heterozygous Alleles. J. Clin. Endocrinol. Metab. 2005, 90, 3001–3008. [Google Scholar] [CrossRef]
- Leanos-Miranda, A.; Ulloa-Aguirre, A.; Ji, T.H.; Janovick, J.A.; Conn, P.M. Dominant-Negative Action of Disease-Causing Gonadotropin-Releasing Hormone Receptor (GnRHR) Mutants: A Trait That Potentially Coevolved with Decreased Plasma Membrane Expression of GnRHR in Humans. J. Clin. Endocrinol. Metab. 2003, 88, 3360–3367. [Google Scholar] [CrossRef]
- Morello, J.P.; Salahpour, A.; Laperriere, A.; Bernier, V.; Arthus, M.-F.; Lonergan, M.; Petaja-Repo, U.; Angers, S.; Morin, D.; Bichet, D.G.; et al. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J. Clin. Investig. 2000, 105, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Noorwez, S.M.; Kuksa, V.; Imanishi, Y.; Zhu, L.; Filipek, S.; Palczewski, K.; Kaushal, S. Pharmacological Chaperone-mediated In Vivo Folding and Stabilization of the P23H-opsin Mutant Associated with Autosomal Dominant Retinitis Pigmentosa. J. Biol. Chem. 2003, 278, 14442–14450. [Google Scholar] [CrossRef]
- Rubenstein, R.C.; Egan, M.E.; Zeitlin, P.L. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J. Clin. Investig. 1997, 100, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Wuller, S.; Wiesner, B.; Loffler, A.; Furkert, J.; Krause, G.; Hermosilla, R.; Schaefer, M.; Schulein, R.; Rosenthal, W.; Oksche, A. Pharmacochaperones Post-translationally Enhance Cell Surface Expression by Increasing Conformational Stability of Wild-Type and Mutant Vasopressin V2 Receptors. J. Biol. Chem. 2004, 279, 47254–47263. [Google Scholar] [CrossRef] [PubMed]
- Sangkuhl, K.; Schulz, A.; Rompler, H.; Yun, J.; Wess, J.; Schoneberg, T. Aminoglycoside-mediated rescue of a disease-causing nonsense mutation in the V2 vasopressin receptor gene in vitro and in vivo. Hum. Mol. Genet. 2004, 13, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Barton-Davis, E.R.; Cordier, L.; Shoturma, D.I.; Leland, S.E.; Sweeney, H.L. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Investig. 1999, 104, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Birnbaumer, M. V2R structure and diabetes insipidus. Recept. Channels 2002, 8, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.R.; Kohler, G.; Schaerer, F.; Senn, C.; Weyermann, P.; Hofbauer, K.G. Peripheral Administration of a Melanocortin 4-Receptor Inverse Agonist Prevents Loss of Lean Body Mass in Tumor-Bearing Mice. J. Pharmacol. Exp. Ther. 2006, 317, 771–777. [Google Scholar] [CrossRef]
- Vos, T.J.; Caracoti, A.; Che, J.L.; Dai, M.; Farrer, C.A.; Forsyth, N.E.; Drabic, S.V.; Horlick, R.A.; Lamppu, D.; Yowe, D.L.; et al. Identification of 2-{2-[2-(5-Bromo-2- methoxyphenyl)-ethyl]-3-fluorophenyl}-4,5-dihydro-1H-imidazole (ML00253764), a Small Molecule Melanocortin 4 Receptor Antagonist That Effectively Reduces Tumor-Induced Weight Loss in a Mouse Model. J. Med. Chem. 2004, 47, 1602–1604. [Google Scholar] [CrossRef]
- Fan, Z.-C.; Tao, Y.-X. Functional characterization and pharmacological rescue of melanocortin-4 receptor mutations identified from obese patients. J. Cell. Mol. Med. 2009, 13, 3268–3282. [Google Scholar] [CrossRef]
- Huang, H.; Tao, Y.X. A Small Molecule Agonist THIQ as a Novel Pharmacoperone for Intracellularly Retained Melanocortin-4 Receptor Mutants. Int. J. Biol. Sci. 2014, 10, 817–824. [Google Scholar] [CrossRef]
- Chen, M.; Cai, M.; Aprahamian, C.J.; Georgeson, K.E.; Hruby, V.; Harmon, C.M.; Yang, Y. Contribution of the conserved amino acids of the melanocortin-4 receptor in [corrected] [Nle4,D-Phe7]-alpha-melanocyte-stimulating [corrected] hormone binding and signaling. J. Biol. Chem. 2007, 282, 21712–21719. [Google Scholar] [CrossRef]
- Cavanaugh, A.; McKenna, J.; Stepanchick, A.; Breitwieser, G.E. Calcium-sensing Receptor Biosynthesis Includes a Cotranslational Conformational Checkpoint and Endoplasmic Reticulum Retention. J. Biol. Chem. 2010, 285, 19854–19864. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Breitwieser, G.E. Rescue of Calcium-sensing Receptor Mutants by Allosteric Modulators Reveals a Conformational Checkpoint in Receptor Biogenesis. J. Biol. Chem. 2007, 282, 9517–9525. [Google Scholar] [CrossRef] [PubMed]
- Leidenheimer, N.J.; Ryder, K.G. Pharmacological chaperoning: A primer on mechanism and pharmacology. Pharmacol. Res. 2014, 83, 10–19. [Google Scholar] [CrossRef]
- White, E.; McKenna, J.; Cavanaugh, A.; Breitwieser, G.E. Pharmacochaperone-Mediated Rescue of Calcium-Sensing Receptor Loss-of-Function Mutants. Mol. Endocrinol. 2009, 23, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-N.; Ma, Y.-T.; Liu, H.; Zhou, Q.-Y.; Li, J.-D. Functional Rescue of Kallmann Syndrome-associated Prokineticin Receptor 2 (PKR2) Mutants Deficient in Trafficking. J. Biol. Chem. 2014, 289, 15518–15526. [Google Scholar] [CrossRef] [PubMed]
- Conn, P.M.; Janovick, J.A. Pharmacoperone identification for therapeutic rescue of misfolded mutant proteins. Front. Endocrinol. 2011, 2, 6. [Google Scholar] [CrossRef]
- Janovick, J.A.; Patny, A.; Mosley, R.; Goulet, M.T.; Altman, M.D.; Rush, T.S., 3rd; Cornea, A.; Conn, P.M. Molecular mechanism of action of pharmacoperone rescue of misrouted GPCR mutants: The GnRH receptor. Mol. Endocrinol. 2009, 23, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.D.; Deng, J.M.; Stewart, C.A.; Mullen, R.D.; Wang, Y.; Lopez, S.; Serna, M.K.; Huang, C.-C.; Janovick, J.A.; Pask, A.J.; et al. Mice harboring Gnrhr E90K, a mutation that causes protein misfolding and hypogonadotropic hypogonadism in humans, exhibit testis size reduction and ovulation failure. Mol. Endocrinol. 2012, 26, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- Conn, P.M.; Smith, E.; Hodder, P.; Janovick, J.A.; Smithson, D.C. High-Throughput Screen for Pharmacoperones of the Vasopressin Type 2 Receptor. J. Biomol. Screen. 2013, 18, 930–937. [Google Scholar] [CrossRef]
- Janovick, J.A.; Ulloa-Aguirre, A. Cellular high-throughput screening. In Protein Homeostasis Deseases; Pey, A.L., Ed.; Academic Press: London, UK, 2020; pp. 343–358. [Google Scholar]
- Krebs, M.P.; Holden, D.C.; Joshi, P.; Clark, C.L., 3rd; Lee, A.H.; Kaushal, S. Molecular mechanisms of rhodopsin retinitis pigmentosa and the efficacy of pharmacological rescue. J. Mol. Biol. 2010, 395, 1063–1078. [Google Scholar] [CrossRef]
- Noorwez, S.M.; Ostrov, D.A.; McDowell, J.H.; Krebs, M.P.; Kaushal, S. A High-Throughput Screening Method for Small-Molecule Pharmacologic Chaperones of Misfolded Rhodopsin. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3224–3230. [Google Scholar] [CrossRef] [PubMed]
- Pey, A.L.; Ying, M.; Cremades, N.; Velazquez-Campoy, A.; Scherer, T.; Thony, B.; Sancho, J.; Martinez, A. Identification of pharmacological chaperones as potential therapeutic agents to treat phenylketonuria. J. Clin. Investig. 2008, 118, 2858–2867. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | GPCR | Pharmacoperone |
|---|---|---|
| Nephrogenic diabetes insípidus | V2R | Satavaptan, relcovaptan, VPA985, YM087, tolvaptan, OPC31260 [20,48,49,50] |
| Isolated hypogonadotropic hypogonadism | GnRHR | IN3, IN30, Q89, A177775, TAK-013 (reviewed in Ref. [22]) |
| Familial hypocalciuric hypercalcemia | CaR | NPS R-568 (reviewed in Ref. [36]) |
| Male pseudohermaphroditism and hypergonadotropic hypogonadism | LHR | Org 42,599 [51,52] |
| Primary ovarian failure | FSHR | Org41841, CAN1404 [53,54] |
| Congenital hypothyroidism | TSHR | - |
| Familial glucocorticoid resistance | MC2R (ACTHR) | - |
| Obesity | MCR3, MCR4 | ML00253764, Ipsen 5i, THIQ [9,13,38,39,55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ulloa-Aguirre, A.; Zariñán, T.; Jardón-Valadez, E. Misfolded G Protein-Coupled Receptors and Endocrine Disease. Molecular Mechanisms and Therapeutic Prospects. Int. J. Mol. Sci. 2021, 22, 12329. https://doi.org/10.3390/ijms222212329
Ulloa-Aguirre A, Zariñán T, Jardón-Valadez E. Misfolded G Protein-Coupled Receptors and Endocrine Disease. Molecular Mechanisms and Therapeutic Prospects. International Journal of Molecular Sciences. 2021; 22(22):12329. https://doi.org/10.3390/ijms222212329
Chicago/Turabian StyleUlloa-Aguirre, Alfredo, Teresa Zariñán, and Eduardo Jardón-Valadez. 2021. "Misfolded G Protein-Coupled Receptors and Endocrine Disease. Molecular Mechanisms and Therapeutic Prospects" International Journal of Molecular Sciences 22, no. 22: 12329. https://doi.org/10.3390/ijms222212329
APA StyleUlloa-Aguirre, A., Zariñán, T., & Jardón-Valadez, E. (2021). Misfolded G Protein-Coupled Receptors and Endocrine Disease. Molecular Mechanisms and Therapeutic Prospects. International Journal of Molecular Sciences, 22(22), 12329. https://doi.org/10.3390/ijms222212329