
Heat Shock Factor 1 Directly Regulates Postsynaptic Scaffolding PSD-95 in Aging and Huntington’s Disease and Influences Striatal Synaptic Density

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
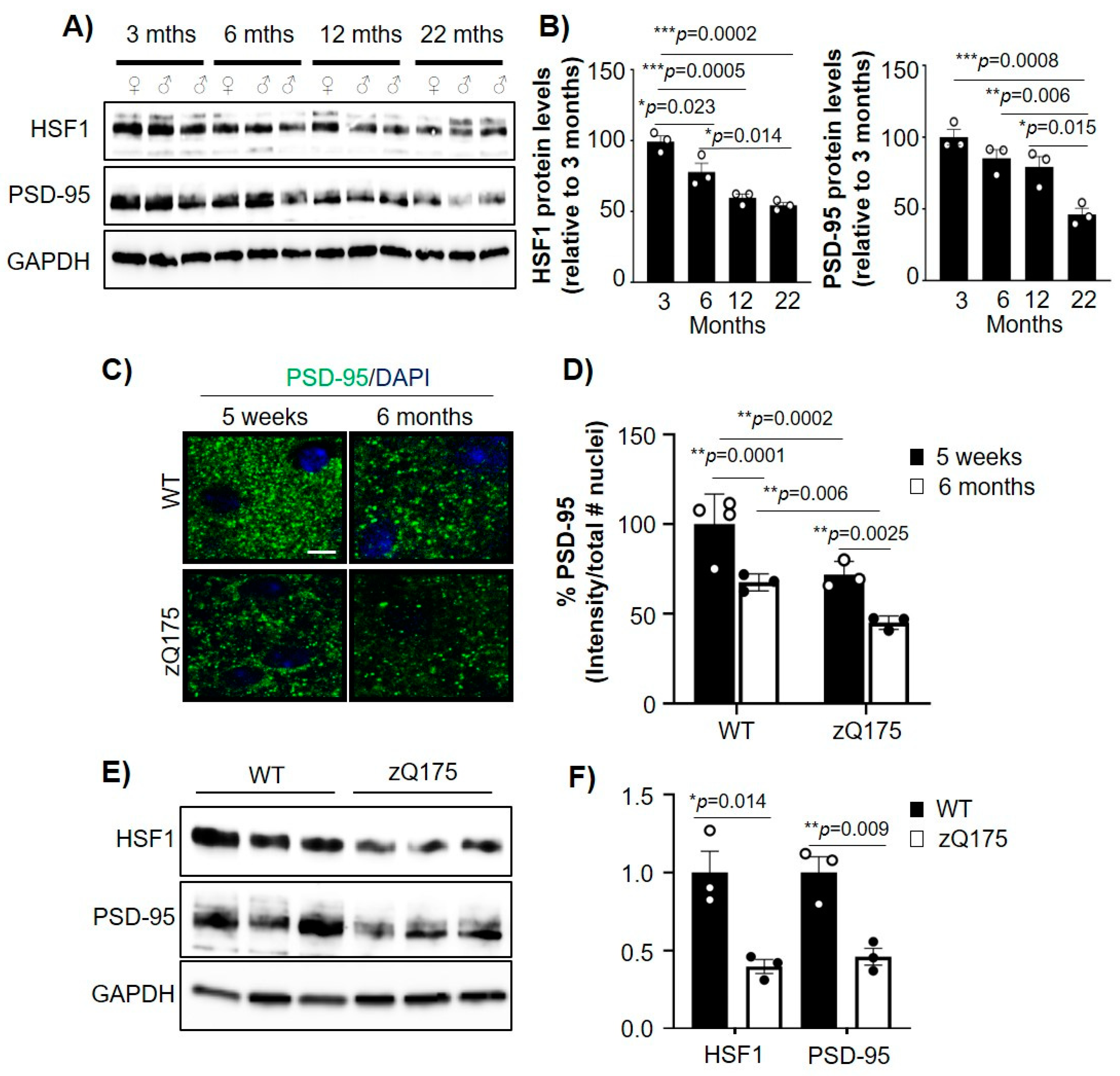
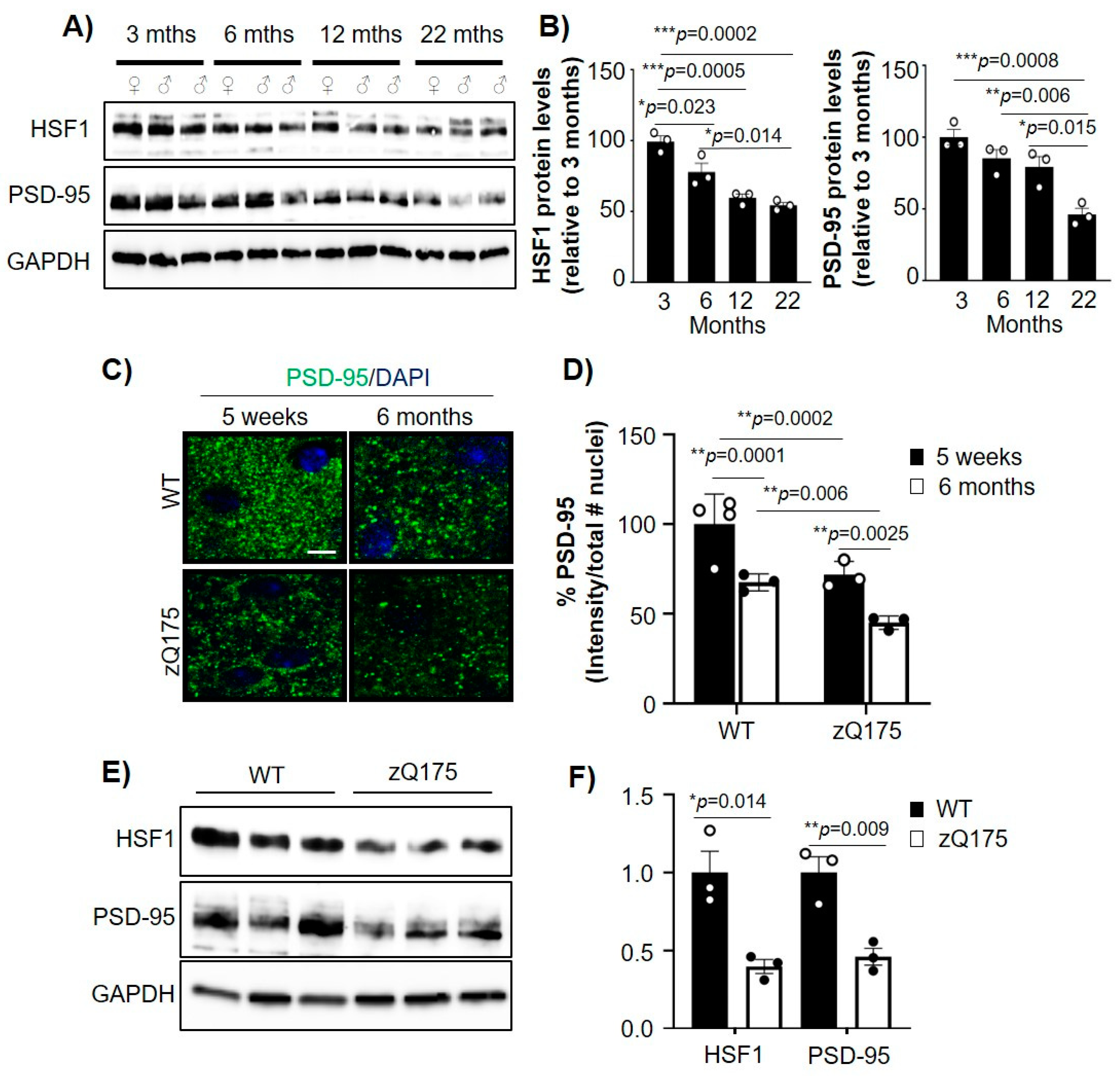
2.1. Aging-Related Reduction of PSD-95 and HSF1 Is Increased in HD
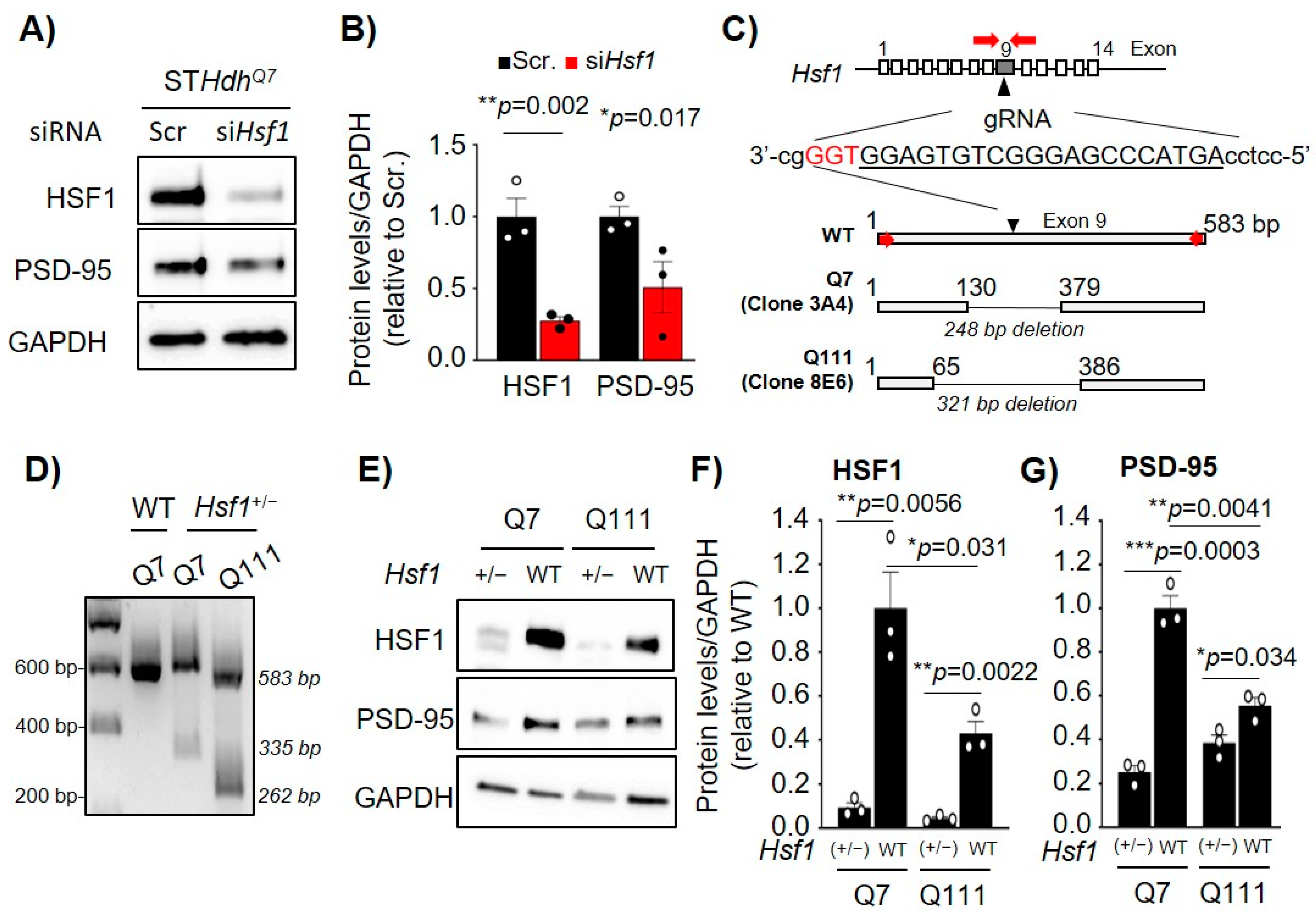
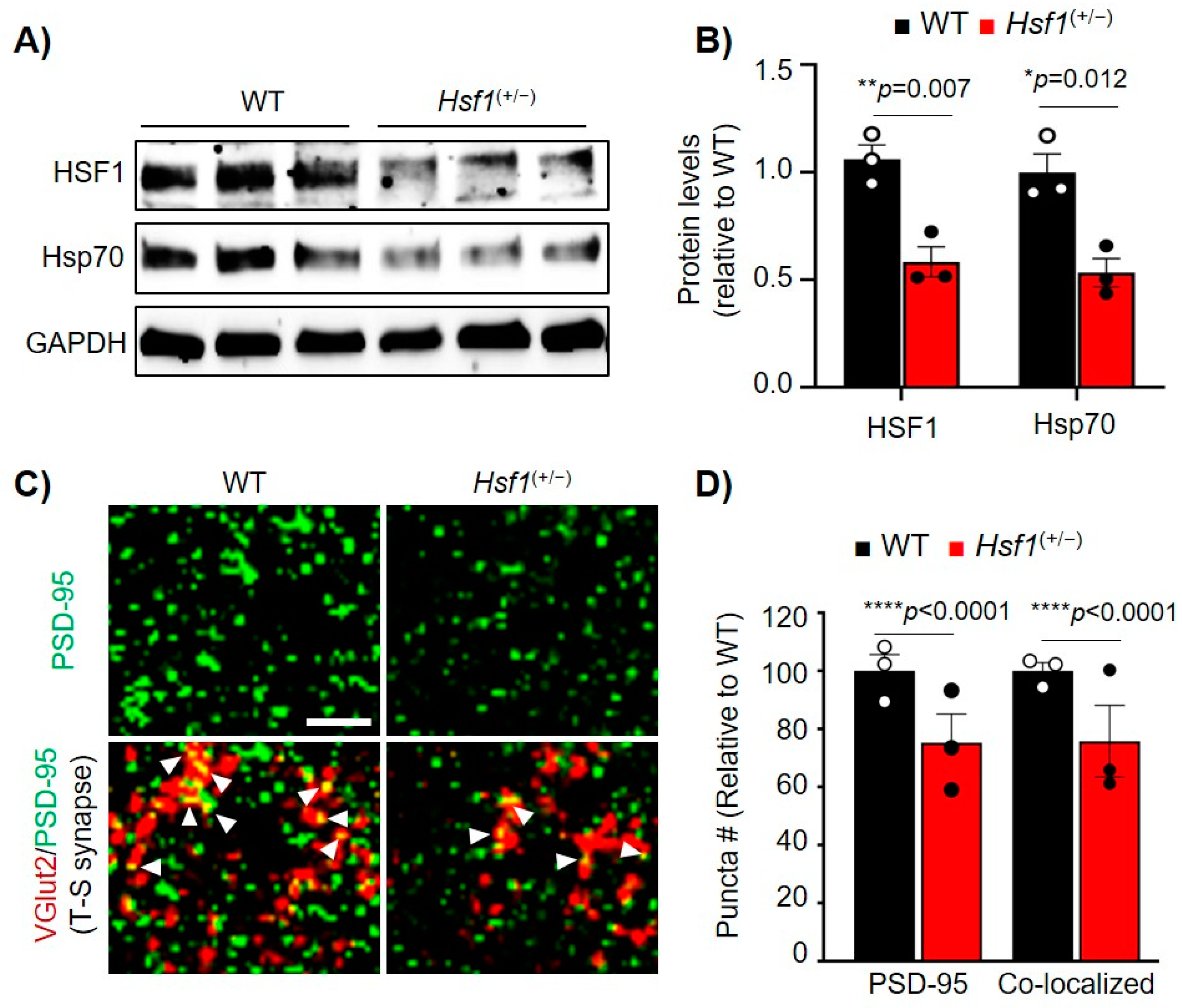
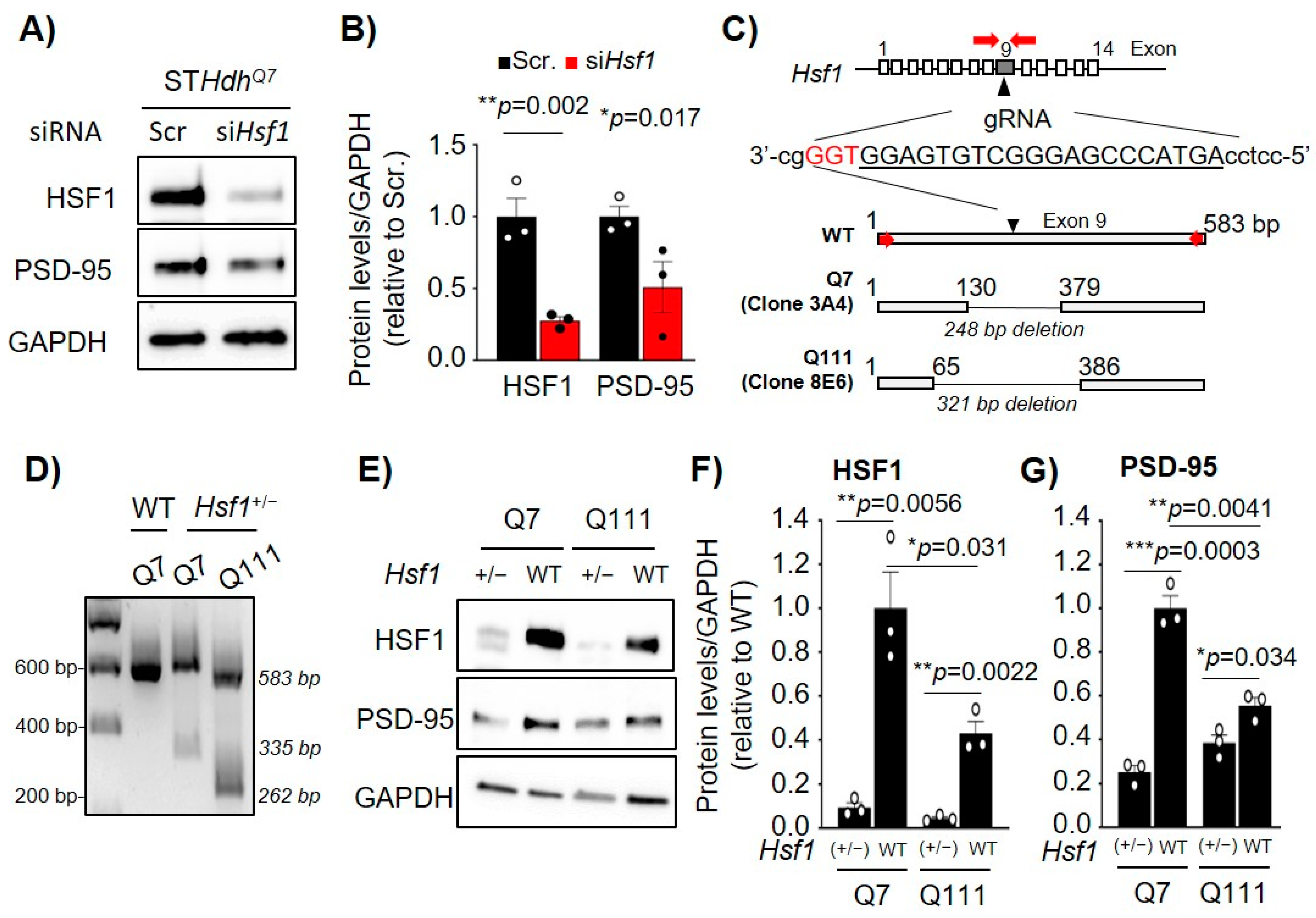
2.2. Acute or Chronic Reduction of HSF1 Results in Reduced PSD-95 Expression
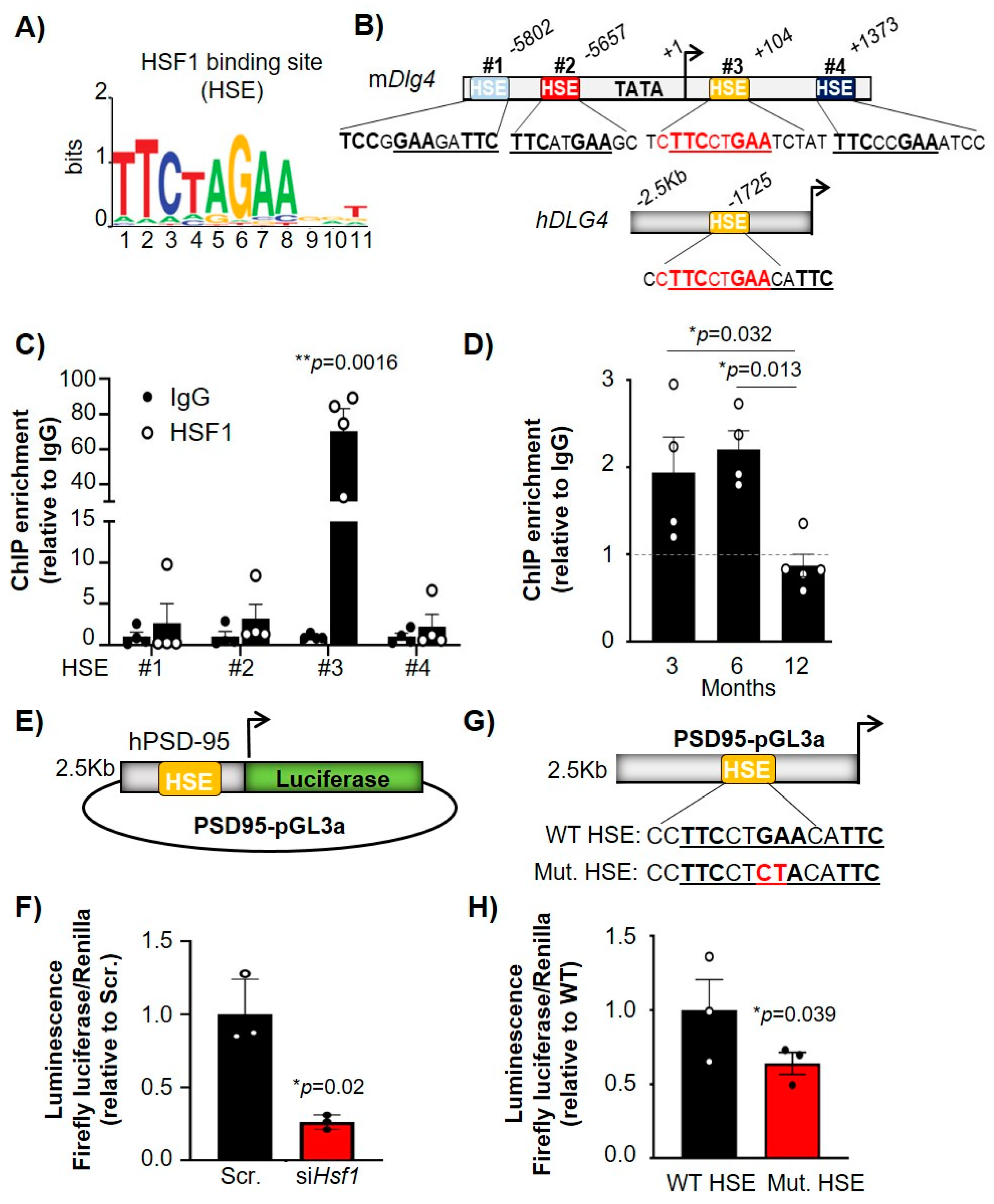
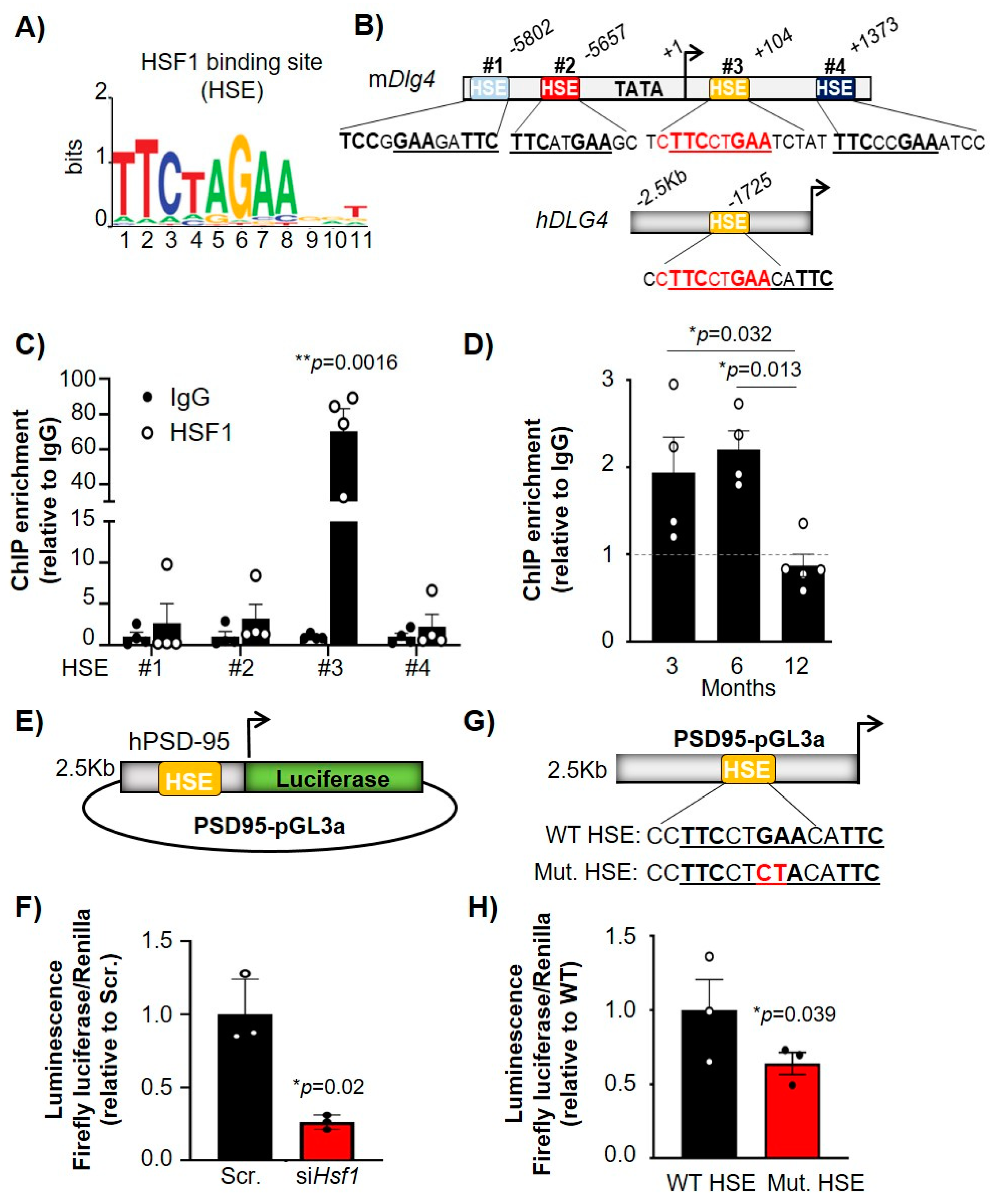
2.3. HSF1 Directly Binds to HSEs Present in the PSD-95 (Dlg4) Gene and Regulates Its Transcription
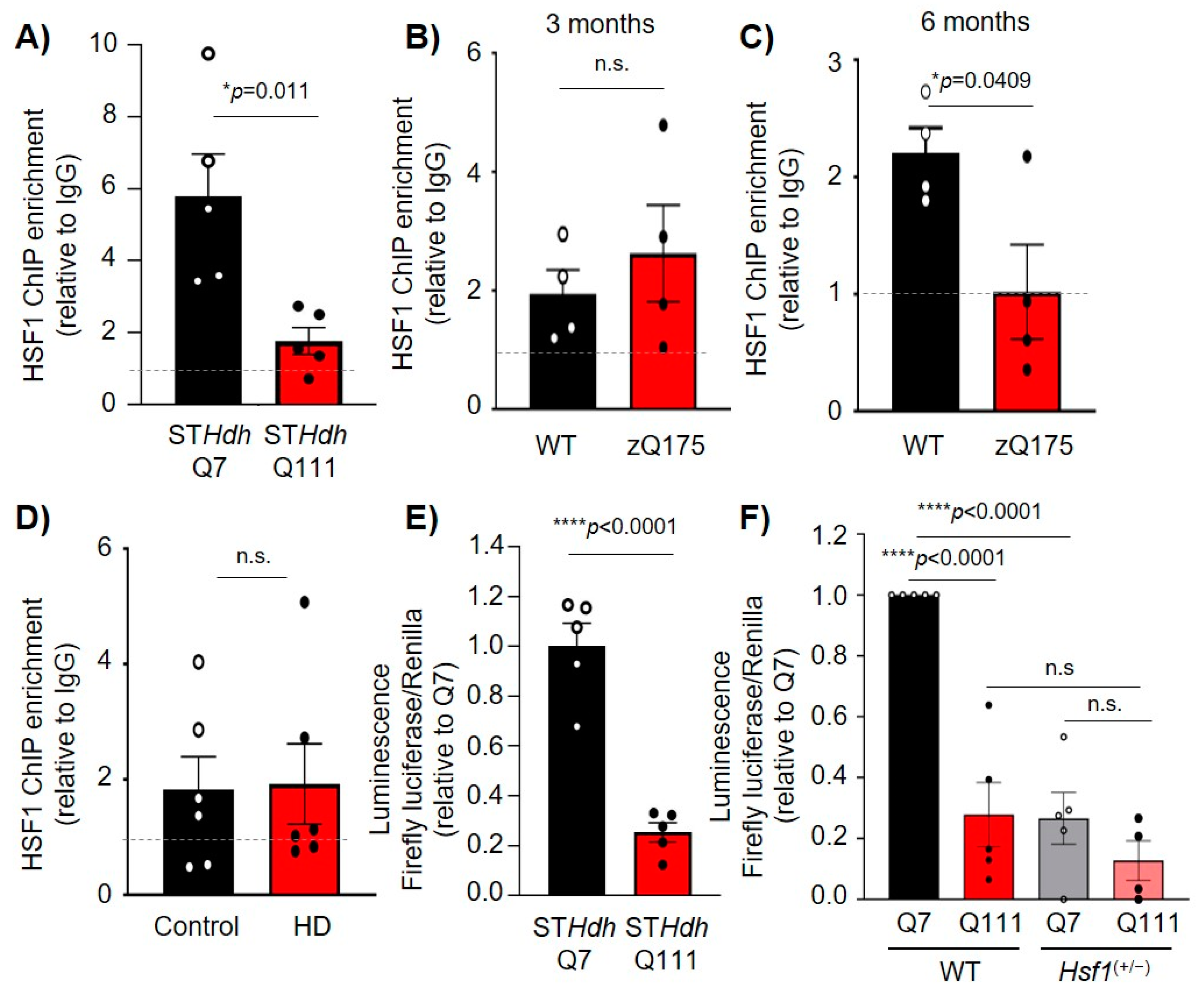
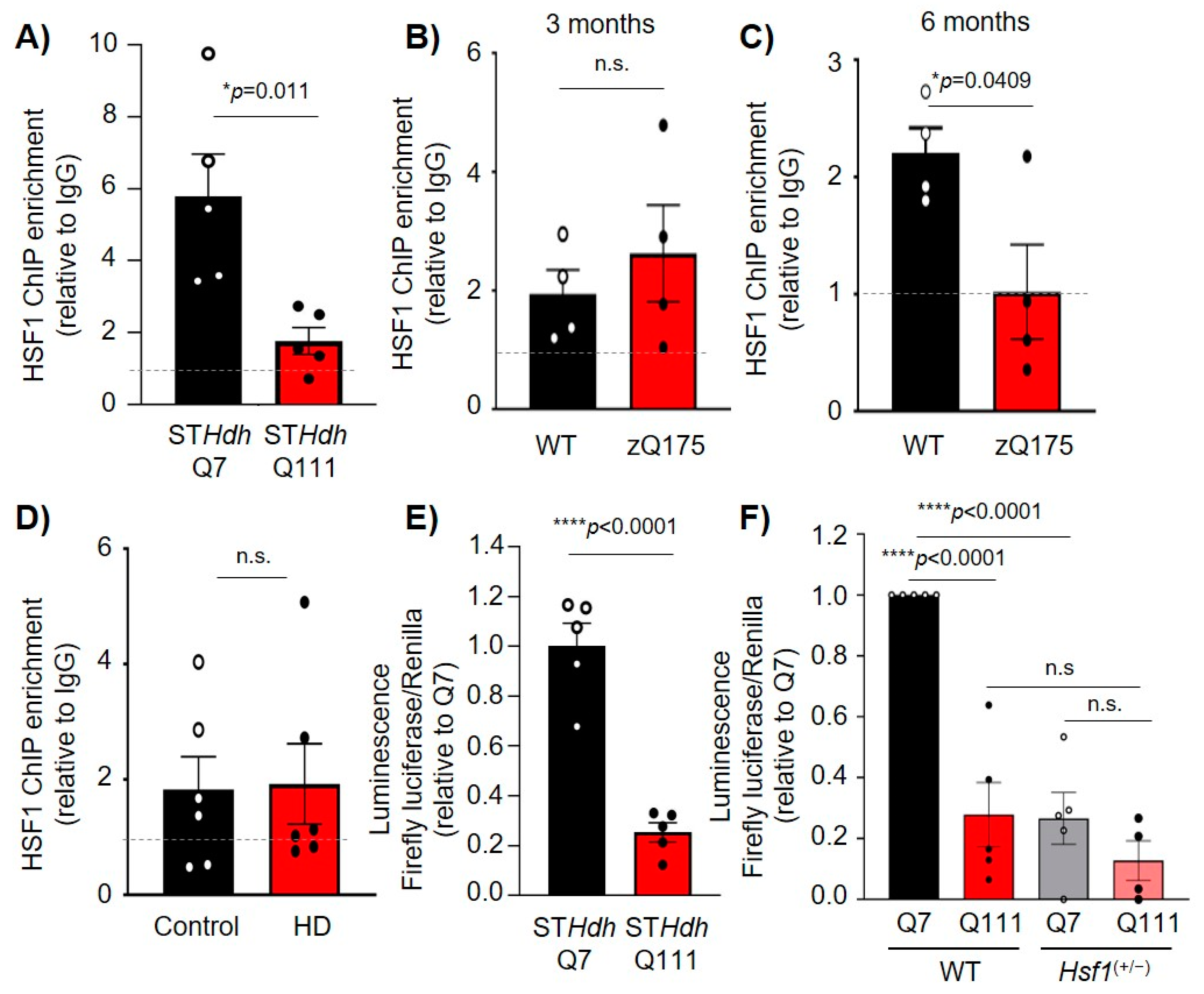
2.4. HSF1 Binding to Dlg4 Regulatory Elements Is Impaired in HD
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Mouse Strains
4.3. Human Samples
4.4. Immunoblot Analysis
4.5. Immunohistochemistry & Synapse Density Analyses
4.6. RNA Preparation and RT-qPCR
4.7. siRNA Transfection
4.8. Chromatin Immunoprecipitation
4.9. Luciferase Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- El-Husseini, A.; Schnell, E.; Chetkovich, D.; Nicoll, R.; Bredt, D. PSD-95 involvement in maturation of excitatory synapses. Science 2000, 290, 1364–1368. [Google Scholar] [CrossRef]
- Béïque, J.-C.; Lin, D.-T.; Kang, M.-G.; Aizawa, H.; Takamiya, K.; Huganir, R.L. Synapse-specific regulation of AMPA receptor function by PSD-95. Proc. Natl. Acad. Sci. USA 2006, 103, 19535–19540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Abdourahman, A.; Tamm, J.; Pehrson, A.; Sánchez, C.; Gulinello, M. Reversal of age-associated cognitive deficits is accompanied by increased plasticity-related gene expression after chronic antidepressant administration in middle-aged mice. Pharmacol. Biochem. Behav. 2015, 135, 70–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carretón, O.; Giralt, A.; Torres-Peraza, J.; Brito, V.; Lucas, J.; Ginés, S.; Canals, J.; Alberch, J. Age-dependent decline of motor neocortex but not hippocampal performance in heterozygous BDNF mice correlates with a decrease of cortical PSD-95 but an increase of hippocampal TrkB levels. Exp. Neurol. 2012, 237, 335–345. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, B.; Liu, D.; Li, J.J.; Xue, Y.; Sakata, K.; Zhu, L.Q.; Heldt, S.A.; Xu, H.; Liao, F.F. Hsp90 chaperone inhibitor 17-AAG Attenuates Aβ-induced synaptic toxicity and memory impairment. J. Neurosci. 2014, 34, 2464–2470. [Google Scholar] [CrossRef]
- Hodges, A.; Strand, A.; Aragaki, A.; Kuhn, A.; Sengstag, T.; Hughes, G.; Elliston, L.; Hartog, C.; Goldstein, D.; Thu, D.; et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 2006, 15, 965–977. [Google Scholar] [CrossRef]
- Qin, X.; Jiang, Y.; Tse, Y.; Wang, Y.; Wong, T.; Paudel, H. Early Growth Response 1 (Egr-1) Regulates N-Methyl-d-aspartate Receptor (NMDAR)-dependent Transcription of PSD-95 and α-Amino-3-hydroxy-5-methyl-4-isoxazole Propionic Acid Receptor (AMPAR) Trafficking in Hippocampal Primary Neurons. J. Biol. Chem. 2015, 290, 29603–29616. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Lin, H.; Ouyang, Y.; Lei, D.; Osman, A.; Kim, T.-W.; Mei, L.; Dai, P.; Ohlemiller, K.K.; Ambron, R.T. Activity-dependent transcription regulation of PSD-95 by neuregulin-1 and Eos. Nat. Neurosci. 2004, 7, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Gray, E.E.; Chawla, G.; Porse, B.T.; O’Dell, T.J.; Black, D.L. PSD-95 is post-transcriptionally repressed during early neural development by PTBP1 and PTBP2. Nat. Neurosci. 2012, 15, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Willett, C.E.; Kawasaki, H.; Amemiya, C.T.; Lin, S.; Steiner, L.A. Ikaros expression as a marker for lymphoid progenitors during zebrafish development. Dev. Dyn. 2001, 222, 694–698. [Google Scholar] [CrossRef]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell. Biol. 2017, 19, 4–19. [Google Scholar] [CrossRef]
- Zarate, N.; Gomez-Pastor, R. Excitatory synapse impairment and mitochondrial dysfunction in Huntington’s disease: Heat shock factor 1 (HSF1) converging mechanisms. Neural Regen. Res. 2020, 15, 69–70. [Google Scholar] [CrossRef]
- Hooper, P.L.; Durham, H.D.; Török, Z.; Hooper, P.L.; Crul, T.; Vígh, L. The central role of heat shock factor 1 in synaptic fidelity and memory consolidation. Cell Stress Chaperones 2016, 21, 745–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homma, S.; Jin, X.; Wang, G.; Tu, N.; Min, J.; Yanasak, N.; Mivechi, N. Demyelination, astrogliosis, and accumulation of ubiquitinated proteins, hallmarks of CNS disease in hsf1-deficient mice. J. Neurosci. 2007, 27, 7974–7986. [Google Scholar] [CrossRef] [Green Version]
- Gorenberg, E.; Chandra, S. The Role of Co-chaperones in Synaptic Proteostasis and Neurodegenerative Disease. Front. Neurosci. 2017, 11, 248. [Google Scholar] [CrossRef] [Green Version]
- Bechtold, D.A.; Rush, S.J.; Brown, I.R. Localization of the Heat-Shock Protein Hsp70 to the Synapse Following Hyperthermic Stress in the Brain. J. Neurochem. 2000, 74, 641–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, S.; Hara, K.; Kobayashi, A.; Fujimoto, M.; Otsuki, K.; Yamagata, H.; Hobara, T.; Abe, N.; Higuchi, F.; Shibata, T.; et al. Impaired hippocampal spinogenesis and neurogenesis and altered affective behavior in mice lacking heat shock factor 1. Proc. Natl. Acad. Sci. USA 2011, 108, 1681–1686. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Cheng, M.; Peng, M.; Xiao, X.; Yao, S.; Zhang, X. Basal behavioral characterization of hsf1 deficient mice and its cellular and behavioral abnormalities underlying chronic unpredictable stressors. Behav. Brain Res. 2008, 193, 225–229. [Google Scholar] [CrossRef]
- Gomez-Pastor, R.; Burchfiel, E.T.; Neef, D.W.; Jaeger, A.M.; Cabiscol, E.; Mckinstry, S.U.; Doss, A.; Aballay, A.; Lo, D.C.; Akimov, S.S.; et al. Abnormal degradation of the neuronal stress-protective transcription factor HSF1 in Huntington’s disease. Nat. Commun. 2017, 8, 14405. [Google Scholar] [CrossRef] [Green Version]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Carlson, O.D.; Mustapic, M.; Kapogiannis, D. Low neural exosomal levels of cellular survival factors in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 769. [Google Scholar] [CrossRef] [PubMed]
- Chafekar, S.M.; Duennwald, M.L. Impaired Heat Shock Response in Cells Expressing Full-Length Polyglutamine-Expanded Huntingtin. PLoS ONE 2012, 7, e37929. [Google Scholar] [CrossRef]
- Maheshwari, M.; Bhutani, S.; Das, A.; Mukherjee, R.; Sharma, A.; Kino, Y.; Nukina, N.; Ranjan Jana, N. Dexamethasone induces heat shock response and slows down disease progression in mouse and fly models of Huntington’s disease. Hum. Mol. Genet. 2014, 23, 2737–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.; Wang, B.; Sastry, N.; Masliah, E.; Nelson, P.; Cai, H.; Liao, F. NEDD4-mediated HSF1 degradation underlies α-synucleinopathy. Hum. Mol. Genet. 2016, 25, 211–222. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Peng, Q.; Wu, B.; Jiang, M.; Jin, J.; Hou, Z.; Zheng, J.; Zhang, J.; Duan, W. Characterization of Behavioral, Neuropathological, Brain Metabolic and Key Molecular Changes in zQ175 Knock-In Mouse Model of Huntington’s Disease. PLoS ONE 2016, 11, e0148839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mckinstry, S.U.; Karadeniz, Y.B.; Worthington, A.K.; Hayrapetyan, V.Y.; Ozlu, M.I.; Serafin-Molina, K.; Risher, X.W.C.; Ustunkaya, T.; Dragatsis, I.; Zeitlin, X.S.; et al. Huntingtin Is Required for Normal Excitatory Synapse Development in Cortical and Striatal Circuits. J. Neurosci. 2014, 34, 9455–9472. [Google Scholar] [CrossRef] [PubMed]
- Rieckmann, A.; Johnson, K.A.; Sperling, R.A.; Buckner, R.L.; Hedden, T. Dedifferentiation of caudate functional connectivity and striatal dopamine transporter density predict memory change in normal aging. Proc. Natl. Acad. Sci. USA 2018, 115, 10160–10165. [Google Scholar] [CrossRef] [Green Version]
- Lighthall, N.R.; Pearson, J.M.; Huettel, S.A.; Cabeza, R. Feedback-Based Learning in Aging: Contributions and Trajectories of Change in Striatal and Hippocampal Systems. J. Neurosci. 2018, 38, 8453–8462. [Google Scholar] [CrossRef] [Green Version]
- Ou, X.; Buckwalter, G.; McNeill, T.H.; Walsh, J.P. Age-related change in short-term synaptic plasticity intrinsic to excitatory striatal synapses. Synapse 1997, 27, 57–68. [Google Scholar] [CrossRef]
- Menalled, L.B.; Kudwa, A.E.; Miller, S.; Fitzpatrick, J.; Watson-Johnson, J.; Keating, N.; Ruiz, M.; Mushlin, R.; Alosio, W.; McConnell, K.; et al. Comprehensive Behavioral and Molecular Characterization of a New Knock-In Mouse Model of Huntington’s Disease: zQ175. PLoS ONE 2012, 7, e49838. [Google Scholar] [CrossRef] [PubMed]
- Vonsattel, J.; Myers, R.; Stevens, T.; Ferrante, R.; Bird, E.; Richardson, E. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Georgiou-Karistianis, N.; Stout, J.C.; Domínguez D, J.F.; Carron, S.P.; Ando, A.; Churchyard, A.; Chua, P.; Bohanna, I.; Dymowski, A.R.; Poudel, G.; et al. Functional magnetic resonance imaging of working memory in Huntington’s disease: Cross-sectional data from the IMAGE-HD study. Hum. Brain Mapp. 2014, 35, 1847–1864. [Google Scholar] [CrossRef] [PubMed]
- Henley, J.; Wilkinson, K. AMPA receptor trafficking and the mechanisms underlying synaptic plasticity and cognitive aging. Dialogues Clin. Neurosci. 2013, 15, 11–27. [Google Scholar] [CrossRef]
- Morigaki, R.; Goto, S. Postsynaptic Density Protein 95 in the Striosome and Matrix Compartments of the Human Neostriatum. Front. Neuroanat. 2015, 9, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurivich, D.; Manocha, G.; Trivedi, R.; Lizakowski, M.; Rakoczy, S.; Brown-Borg, H. Multifactorial Attenuation of the Murine Heat Shock Response With Age. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1846–1852. [Google Scholar] [CrossRef]
- Nithianantharajah, J.; Barkus, C.; Murphy, M.; Hannan, A.J. Gene–environment interactions modulating cognitive function and molecular correlates of synaptic plasticity in Huntington’s disease transgenic mice. Neurobiol. Dis. 2008, 29, 490–504. [Google Scholar] [CrossRef]
- Smith, G.A.; Rocha, E.M.; McLean, J.R.; Hayes, M.A.; Izen, S.C.; Isacson, O.; Hallett, P.J. Progressive axonal transport and synaptic protein changes correlate with behavioral and neuropathological abnormalities in the heterozygous Q175 KI mouse model of Huntington’s disease. Hum. Mol. Genet. 2014, 23, 4510–4527. [Google Scholar] [CrossRef] [Green Version]
- Fourie, C.; Kim, E.; Waldvogel, H.; Wong, J.M.; McGregor, A.; Faull, R.L.M.; Montgomery, J.M. Differential Changes in Postsynaptic Density Proteins in Postmortem Huntington’s Disease and Parkinson’s Disease Human Brains. J. Neurodegener. Dis. 2014, 2014, 938530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carty, N.; Berson, N.; Tillack, K.; Thiede, C.; Scholz, D.; Kottig, K.; Sedaghat, Y.; Gabrysiak, C.; Yohrling, G.; Kammer, H.v.d.; et al. Characterization of HTT Inclusion Size, Location, and Timing in the zQ175 Mouse Model of Huntington’s Disease: An In Vivo High-Content Imaging Study. PLoS ONE 2015, 10, e0123527. [Google Scholar] [CrossRef] [Green Version]
- Smith, Y.; Raju, D.; Nanda, B.; Pare, J.-F.; Galvan, A.; Wichmann, T. The Thalamostriatal Systems: Anatomical and Functional Organization in Normal and Parkinsonian States. Brain Res. Bull. 2009, 78, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Ocampo, I.; Mena-Segovia, J.; Bolam, J.P. Convergence of cortical and thalamic input to direct and indirect pathway medium spiny neurons in the striatum. Brain Struct. Funct. 2014, 219, 1787–1800. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, A.; Makley, L.; Gestwicki, J.; Thiele, D. Genomic heat shock element sequences drive cooperative human heat shock factor 1 DNA binding and selectivity. J. Biol. Chem. 2014, 289, 30459–30469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gylys, K.; Fein, J.; Yang, F.; Wiley, D.; Miller, C.; Cole, G. Synaptic changes in Alzheimer’s disease: Increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am. J. Pathol. 2004, 165, 1809–1817. [Google Scholar] [CrossRef]
- Morimoto, R. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008, 22, 1427–1438. [Google Scholar] [CrossRef] [Green Version]
- Ting, Y.K.; Morikawa, K.; Kurata, Y.; Li, P.; Bahrudin, U.; Mizuta, E.; Kato, M.; Miake, J.; Yamamoto, Y.; Yoshida, A.; et al. Transcriptional activation of the anchoring protein SAP97 by heat shock factor (HSF)-1 stabilizes K v 1.5 channels in HL-1 cells. Br. J. Pharmacol. 2011, 162, 1832–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riva, L.; Koeva, M.; Yildirim, F.; Pirhaji, L.; Dinesh, D.; Mazor, T.; Duennwald, M.L.; Fraenkel, E. Poly-glutamine expanded huntingtin dramatically alters the genome wide binding of HSF1. J. Huntingtons. Dis. 2012, 1, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Zarate, N.; Cuccu, F.; Yue, J.S.; Brown, T.G.; Tsai, A.; Mansky, R.; Jiang, K.; Kim, H.; Nanclares, C.; et al. Protein kinase CK2 alpha prime and alpha-synuclein constitute a key regulatory pathway in Huntington’s disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Nithianantharajah, J.; Hannan, A. Dysregulation of synaptic proteins, dendritic spine abnormalities and pathological plasticity of synapses as experience-dependent mediators of cognitive and psychiatric symptoms in Huntington’s disease. Neuroscience 2013, 251, 66–74. [Google Scholar] [CrossRef]
- Milnerwood, A.J.; Raymond, L.A. Early synaptic pathophysiology in neurodegeneration: Insights from Huntington’s disease. Trends Neurosci. 2010, 33, 513–523. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Heikkinen, T.; Lehtimäki, K.; Vartiainen, N.; Puoliväli, J.; Hendricks, S.J.; Glaser, J.R.; Bradaia, A.; Wadel, K.; Touller, C.; Kontkanen, O.; et al. Characterization of Neurophysiological and Behavioral Changes, MRI Brain Volumetry and 1H MRS in zQ175 Knock-In Mouse Model of Huntington’s Disease. PLoS ONE 2012, 7, e50717. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zarate, N.; Intihar, T.A.; Yu, D.; Sawyer, J.; Tsai, W.; Syed, M.; Carlson, L.; Gomez-Pastor, R. Heat Shock Factor 1 Directly Regulates Postsynaptic Scaffolding PSD-95 in Aging and Huntington’s Disease and Influences Striatal Synaptic Density. Int. J. Mol. Sci. 2021, 22, 13113. https://doi.org/10.3390/ijms222313113
Zarate N, Intihar TA, Yu D, Sawyer J, Tsai W, Syed M, Carlson L, Gomez-Pastor R. Heat Shock Factor 1 Directly Regulates Postsynaptic Scaffolding PSD-95 in Aging and Huntington’s Disease and Influences Striatal Synaptic Density. International Journal of Molecular Sciences. 2021; 22(23):13113. https://doi.org/10.3390/ijms222313113
Chicago/Turabian StyleZarate, Nicole, Taylor A. Intihar, Dahyun Yu, Jacob Sawyer, Wei Tsai, Maha Syed, Luke Carlson, and Rocio Gomez-Pastor. 2021. "Heat Shock Factor 1 Directly Regulates Postsynaptic Scaffolding PSD-95 in Aging and Huntington’s Disease and Influences Striatal Synaptic Density" International Journal of Molecular Sciences 22, no. 23: 13113. https://doi.org/10.3390/ijms222313113
APA StyleZarate, N., Intihar, T. A., Yu, D., Sawyer, J., Tsai, W., Syed, M., Carlson, L., & Gomez-Pastor, R. (2021). Heat Shock Factor 1 Directly Regulates Postsynaptic Scaffolding PSD-95 in Aging and Huntington’s Disease and Influences Striatal Synaptic Density. International Journal of Molecular Sciences, 22(23), 13113. https://doi.org/10.3390/ijms222313113