
Microglia-Derived Olfactomedin-like 3 Promotes Pro-Tumorigenic Microglial Function and Malignant Features of Glioma Cells

 , and
, and
Abstract
:1. Introduction
2. Results
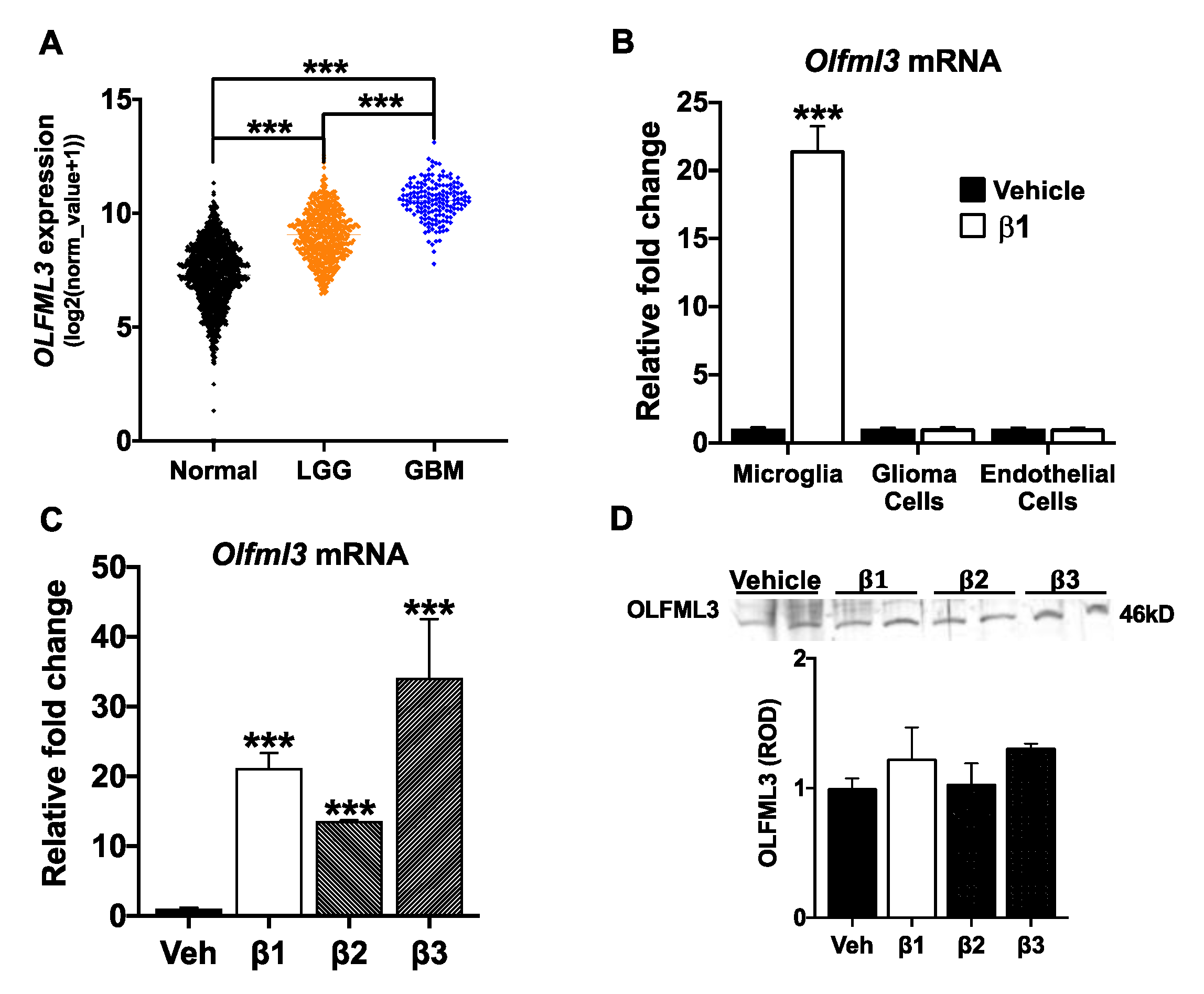
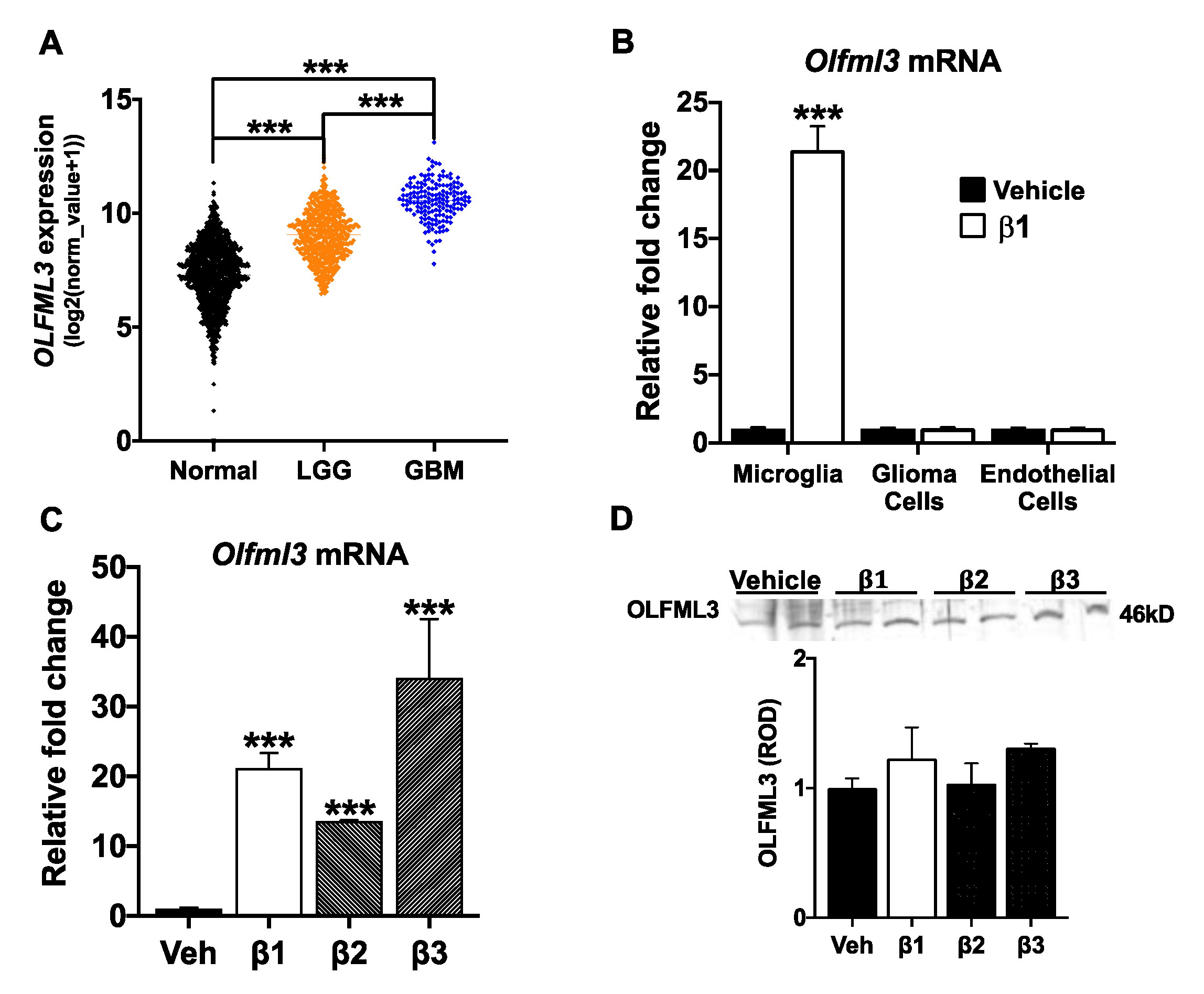
2.1. OLFML3 Is Up-Regulated in GBM and Is a TGFβ Target Gene in Microglia
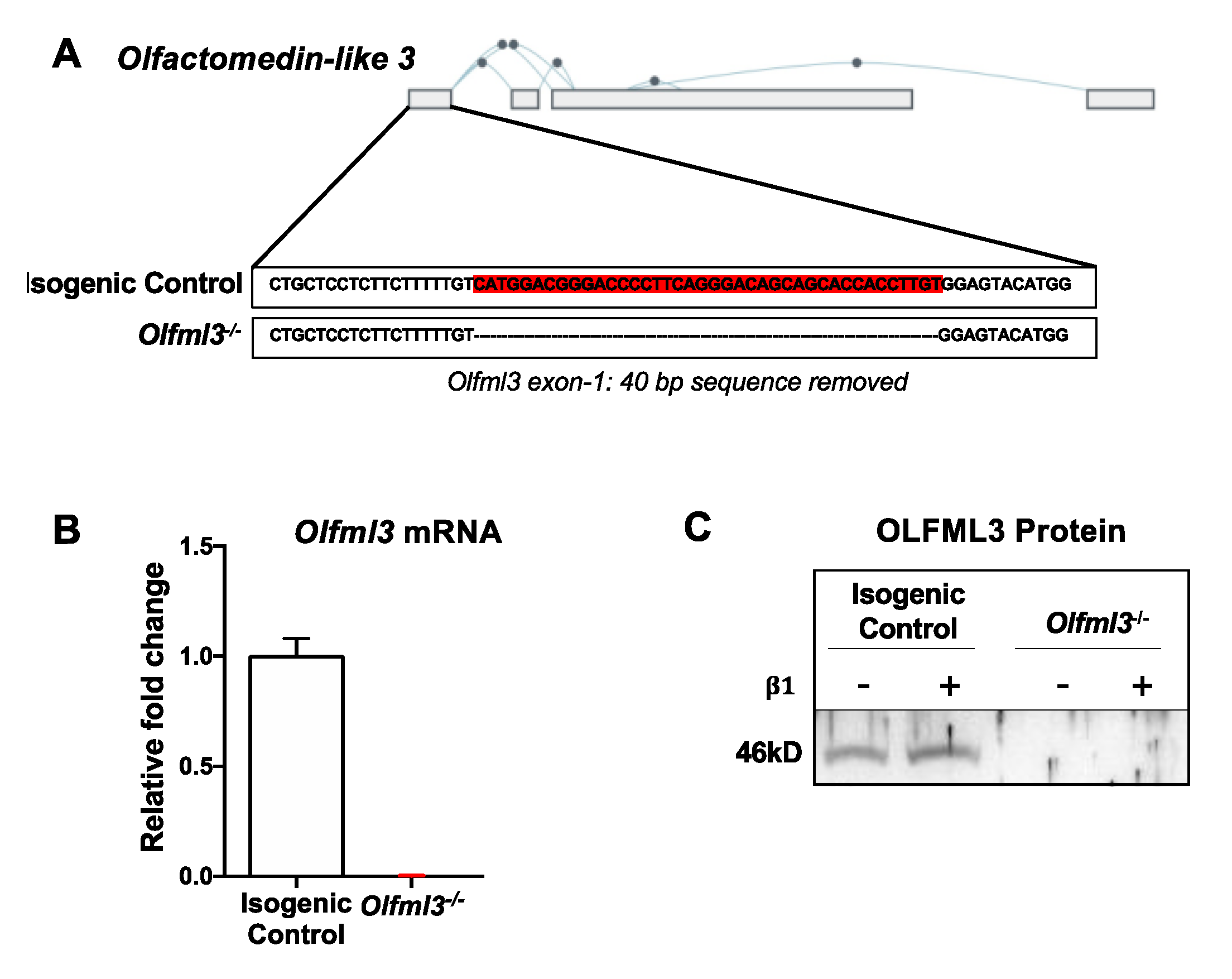
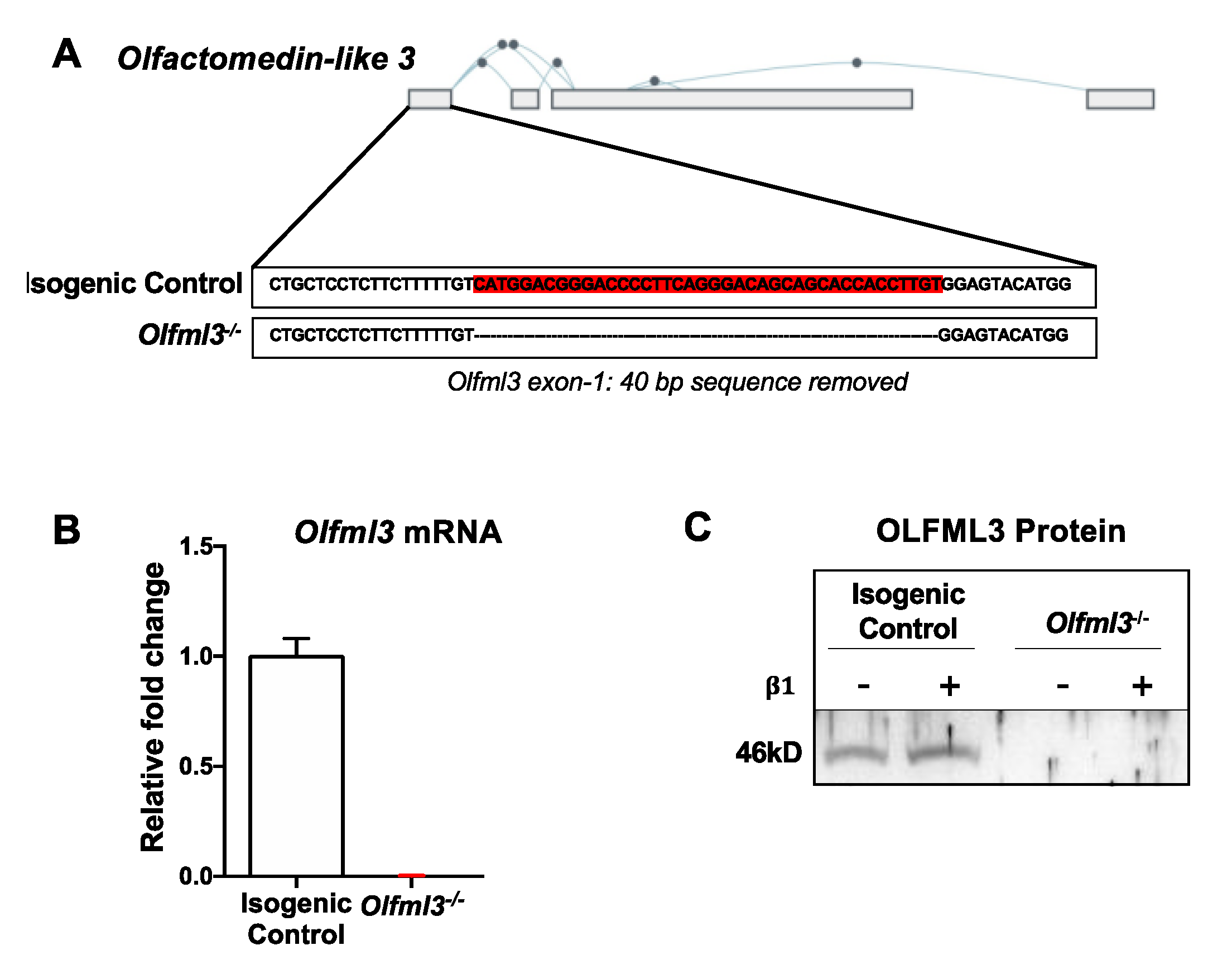
2.2. CRISPR/Cas9-Mediated Knockout of Olfml3 in Microglia
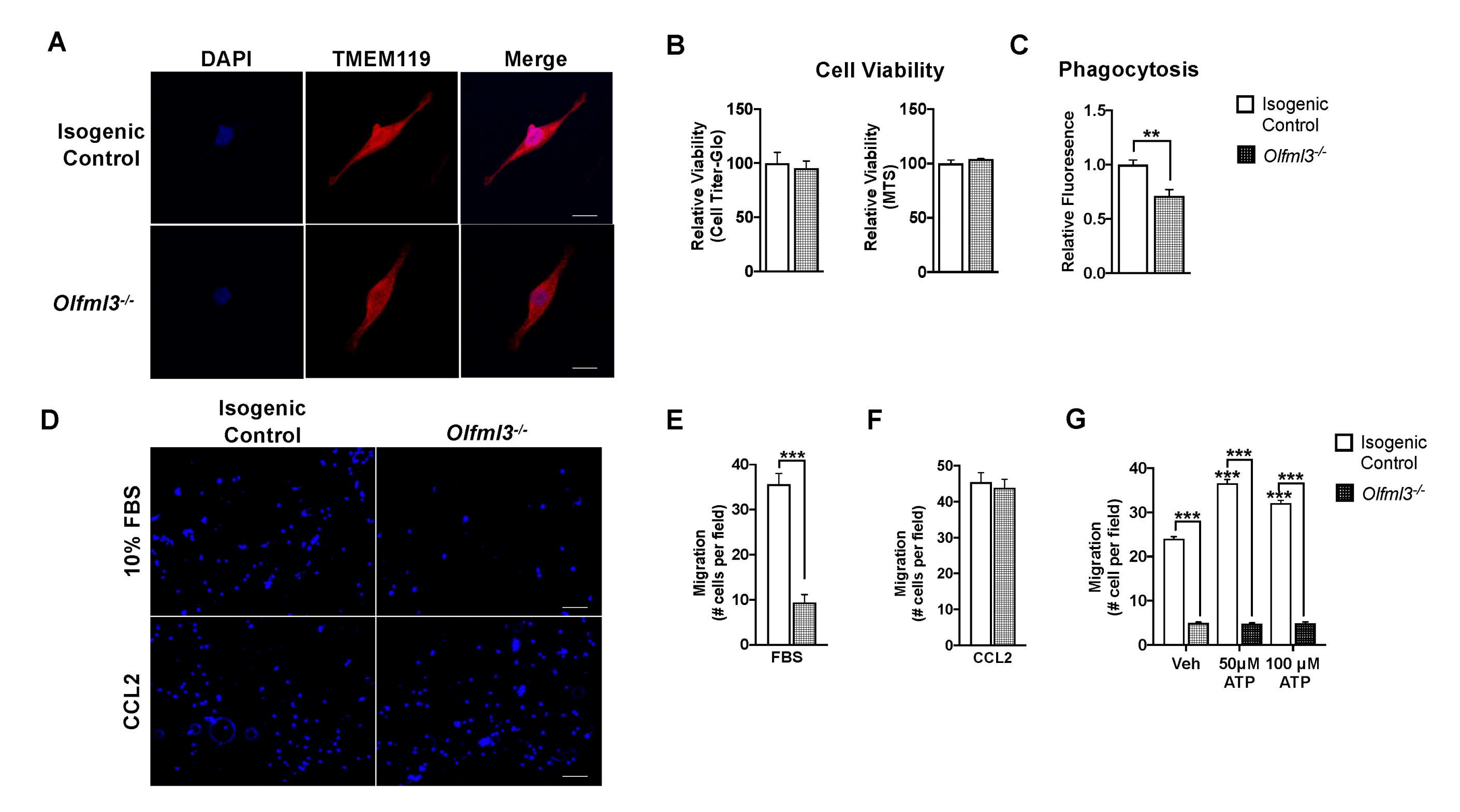
2.3. Loss of Olfml3 Impaired Microglial Phagocytosis and Chemotaxis
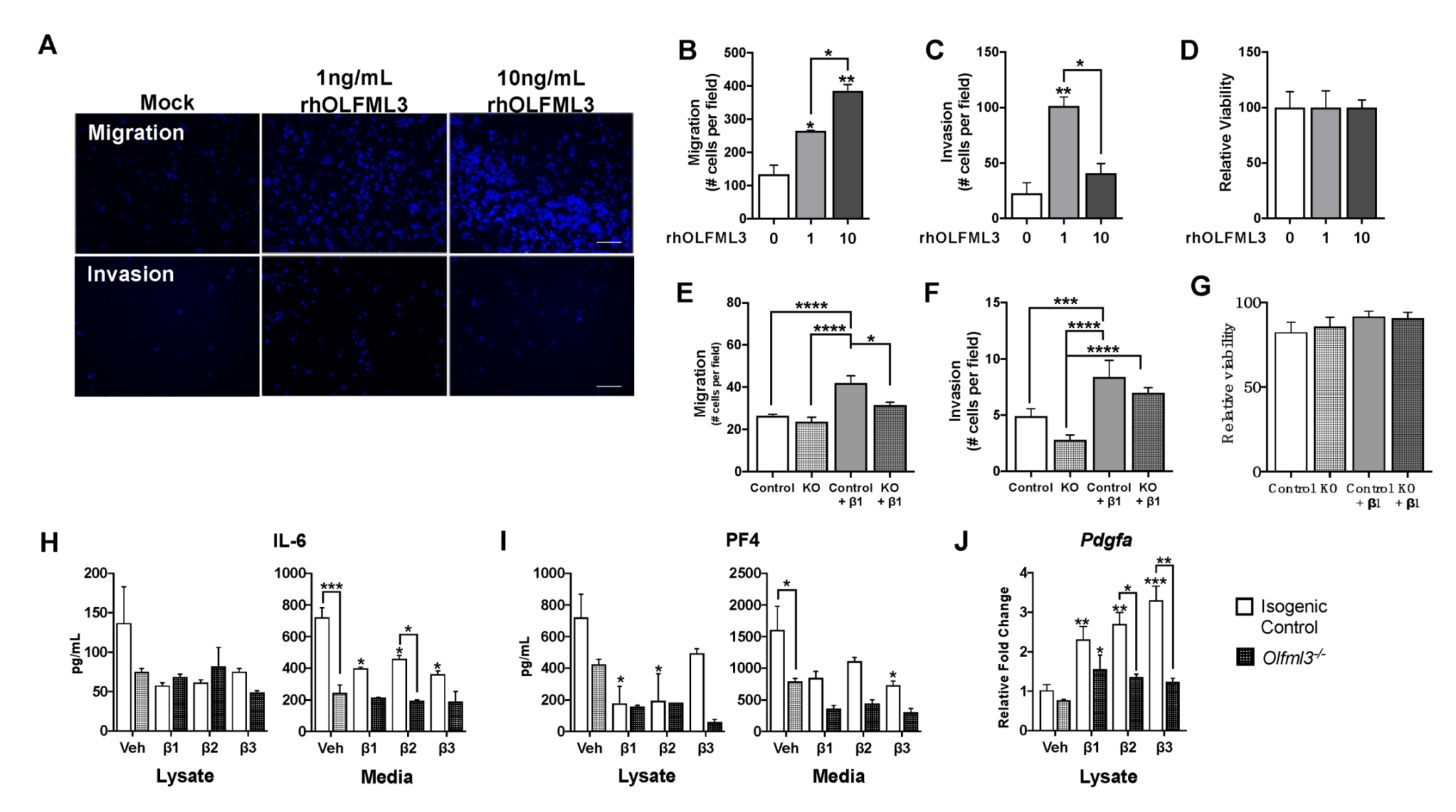
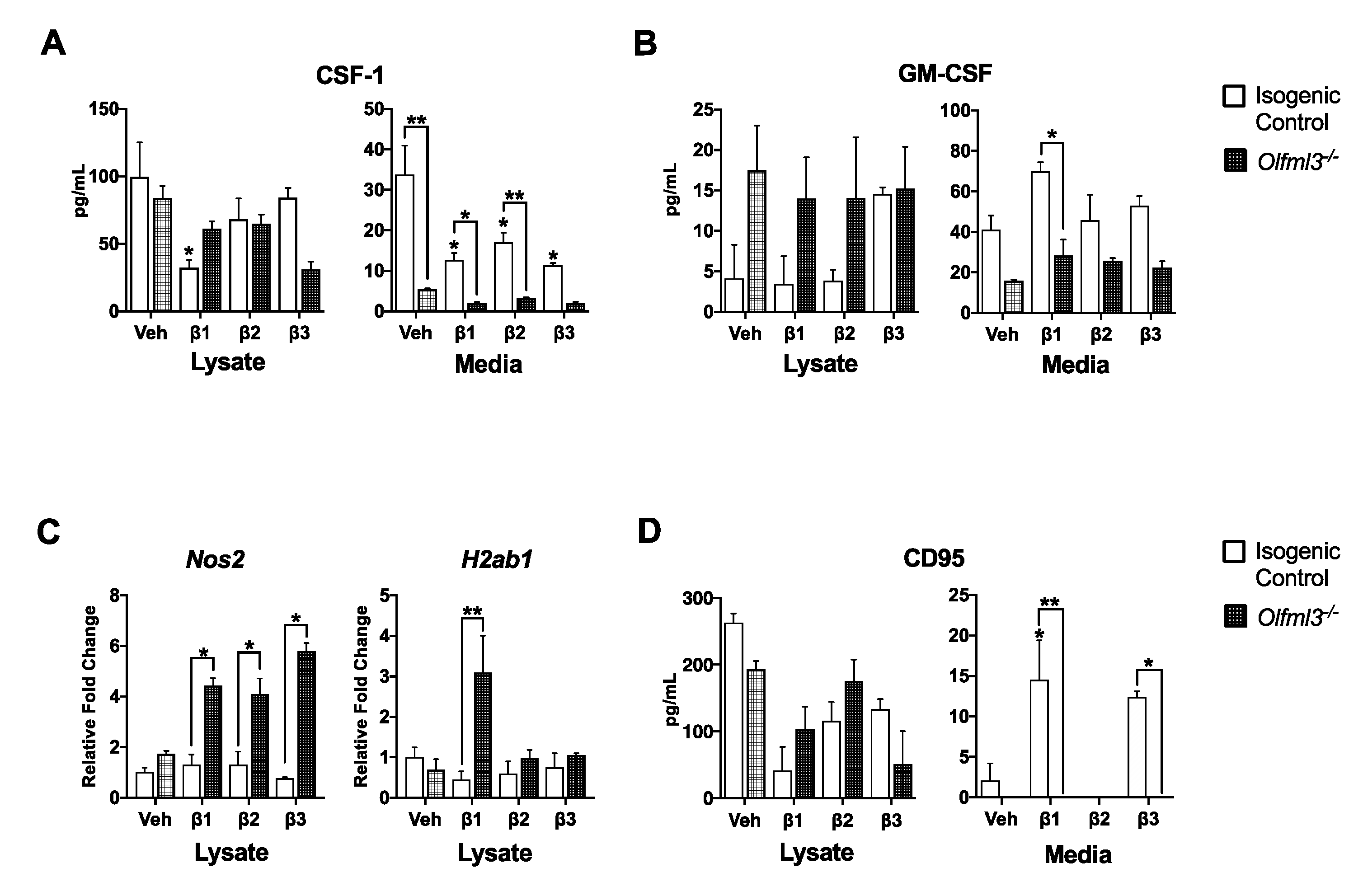
2.4. Microglial Olfml3 Is Necessary for the Pro-Tumorigenic GAM Phenotype
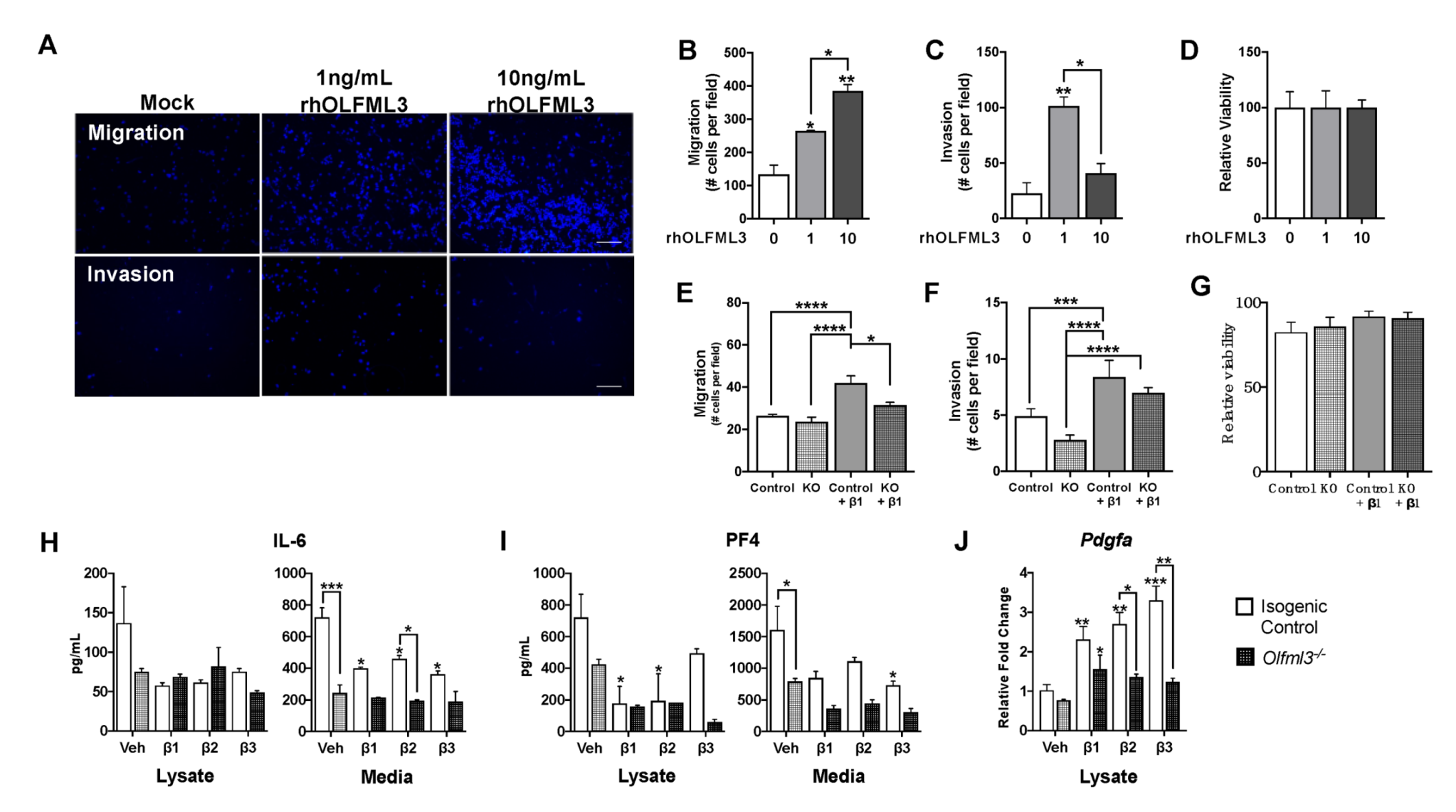
2.5. OLFML3 Promotes Glioma Cell Migration and Invasion
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. CRISPR/Cas9-Mediated Olfml3 Knockout
4.3. Human Recombinant OLFML3 Protein Generation
4.4. Generation of Anti-OLFML3 Antibody
4.5. Quantitative Real-Time PCR
4.6. Immunofluorescence and Confocal Microscopy
4.7. Western Blot Analysis
4.8. Murine Protein Arrays
4.9. Cell Viability
4.10. Transwell Migration and Invasion Assays
4.11. Phagocytosis Assay
4.12. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Gutmann, D.H.; McLellan, M.D.; Hussain, I.; Wallis, J.W.; Fulton, L.L.; Fulton, R.S.; Magrini, V.; Demeter, R.; Wylie, T.; Kandoth, C.; et al. Somatic neurofibromatosis type 1 (NF1) inactivation characterizes NF1-associated pilocytic astrocytoma. Genome Res. 2013, 23, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Kristensen, B.W. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol. Appl. Neurobiol. 2018, 44, 185–206. [Google Scholar] [CrossRef] [Green Version]
- Markovic, D.S.; Glass, R.; Synowitz, M.; van Rooijen, N.; Kettenmann, H. Microglia Stimulate the Invasiveness of Glioma Cells by Increasing the Activity of Metalloprotease-2. J. Neuropathol. Exp. Neurol. 2005, 64, 754–762. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Kowal, J.; Akkari, L.; Schuhmacher, A.J.; Huse, J.T.; West, B.; A J, S. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 2017, 36, 6049–6058. [Google Scholar] [CrossRef]
- Wang, W.; Cho, H.-Y.; Rosenstein-Sisson, R.; Ramos, N.I.M.; Price, R.; Hurth, K.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C. Intratumoral delivery of bortezomib: Impact on survival in an intracranial glioma tumor model. J. Neurosurg. 2018, 128, 695–700. [Google Scholar] [CrossRef] [Green Version]
- Brandenburg, S.; Müller, A.; Turkowski, K.; Radev, Y.T.; Rot, S.; Schmidt, C.; Bungert, A.D.; Acker, G.; Schorr, A.; Hippe, A.; et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2015, 131, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Wesolowska, A.; Kwiatkowska, A.J.; Slomnicki, L.; Dembinski, M.; Master, A.M.; Sliwa, M.; Franciszkiewicz, K.; Chouaib, S.; Kaminska, B. Microglia-derived TGF-β as an important regulator of glioblastoma invasion—An inhibition of TGF-β-dependent effects by shRNA against human TGF-β type II receptor. Oncogene 2007, 27, 918–930. [Google Scholar] [CrossRef] [Green Version]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial Stimulation of Glioblastoma Invasion Involves Epidermal Growth Factor Receptor (EGFR) and Colony Stimulating Factor 1 Receptor (CSF-1R) Signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Graeber, M.B. The molecular profile of microglia under the influence of glioma. Neuro-Oncology 2012, 14, 958–978. [Google Scholar] [CrossRef] [Green Version]
- Seystahl, K.; Papachristodoulou, A.; Burghardt, I.; Schneider, H.; Hasenbach, K.; Janicot, M.; Roth, P.; Weller, M. Biological Role and Therapeutic Targeting of TGF-β3 in Glioblastoma. Mol. Cancer Ther. 2017, 16, 1177–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.E.; Yu, J. TGF-beta signaling and its targeting for glioma treatment. Am J Cancer Res. 2015, 5, 945–955. [Google Scholar]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncology 2015, 18, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Haage, V.; Semtner, M.; Vidal, R.O.; Hernandez, D.P.; Pong, W.W.; Chen, Z.; Hambardzumyan, D.; Magrini, V.; Ly, A.; Walker, J.; et al. Comprehensive gene expression meta-analysis identifies signature genes that distinguish microglia from peripheral monocytes/macrophages in health and glioma. Acta Neuropathol. Commun. 2019, 7, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomarev, S.I.; Nakaya, N. Olfactomedin Domain-Containing Proteins: Possible Mechanisms of Action and Functions in Normal Development and Pathology. Mol. Neurobiol. 2009, 40, 122–138. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Rodgers, G.P. Olfactomedin 4 expression and functions in innate immunity, inflammation, and cancer. Cancer Metastasis Rev. 2016, 35, 201–212. [Google Scholar] [CrossRef]
- Halleskog, C.; Dijksterhuis, J.P.; Kilander, M.B.C.; Becerril-Ortega, J.; Villaescusa, J.C.; Lindgren, E.; Arenas, E.; Schulte, G. Heterotrimeric G protein-dependent WNT-5A signaling to ERK1/2 mediates distinct aspects of microglia proinflammatory transformation. J. Neuroinflammation 2012, 9, 111. [Google Scholar] [CrossRef] [Green Version]
- Zuccarini, M.; Giuliani, P.; Ziberi, S.; Carluccio, M.; Di Iorio, P.; Caciagli, F.; Ciccarelli, R. The Role of Wnt Signal in Glioblastoma Development and Progression: A Possible New Pharmacological Target for the Therapy of This Tumor. Genes 2018, 9, 105. [Google Scholar] [CrossRef] [Green Version]
- Torres, S.; Bartolome, R.A.; Mendes, M.; Barderas, R.; Fernández-Aceñerp, M.J.; Peláez-García, A.; Peña, C.; Lopez-Lucendo, M.; Villar-Vázquez, R.; De Herreros, A.G.; et al. Proteome Profiling of Cancer-Associated Fibroblasts Identifies Novel Proinflammatory Signatures and Prognostic Markers for Colorectal Cancer. Clin. Cancer Res. 2013, 19, 6006–6019. [Google Scholar] [CrossRef] [Green Version]
- Miljkovic-Licina, M.; Hammel, P.; Garrido-Urbani, S.; Lee, B.P.-L.; Meguenani, M.; Chaabane, C.; Bochaton-Piallat, M.-L.; Imhof, B.A. Targeting Olfactomedin-like 3 Inhibits Tumor Growth by Impairing Angiogenesis and Pericyte Coverage. Mol. Cancer Ther. 2012, 11, 2588–2599. [Google Scholar] [CrossRef] [Green Version]
- Stalin, J.; Imhof, B.A.; Coquoz, O.; Jeitziner, R.; Hammel, P.; McKee, T.A.; Jemelin, S.; Poittevin, M.; Pocard, M.; Matthes, T.; et al. Targeting OLFML3 in Colorectal Cancer Suppresses Tumor Growth and Angiogenesis, and Increases the Efficacy of Anti-PD1 Based Immunotherapy. Cancers 2021, 13, 4625. [Google Scholar] [CrossRef]
- Qiu, R.; Shi, H.; Wang, S.; Leng, S.; Liu, R.; Zheng, Y.; Huang, W.; Zeng, Y.; Gao, J.; Zhang, K.; et al. BRMS1 coordinates with LSD1 and suppresses breast cancer cell metastasis. Am. J. Cancer Res. 2018, 8, 2030–2045. [Google Scholar]
- Chen, P.; Hsu, W.-H.; Chang, A.; Tan, Z.; Lan, Z.; Zhou, A.; Spring, D.J.; Lang, F.F.; Wang, Y.A.; Depinho, R.A. Circadian Regulator CLOCK Recruits Immune-Suppressive Microglia into the GBM Tumor Microenvironment. Cancer Discov. 2020, 10, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Righi, M.; Mori, L.; De Libero, G.; Sironi, M.; Biondi, A.; Mantovani, A.; Donini, S.D.; Ricciardi-Castagnoli, P. Monokine production by microglial cell clones. Eur. J. Immunol. 1989, 19, 1443–1448. [Google Scholar] [CrossRef]
- Ausman, J.I.; Shapiro, W.R.; Rall, D.P. Studies on the chemotherapy of experimental brain tumors: Development of an experimental model. Cancer Res. 1970, 30, 2394–2400. [Google Scholar] [PubMed]
- Neidert, N.; Von Ehr, A.; Zöller, T.; Spittau, B. Microglia-Specific Expression of Olfml3 Is Directly Regulated by Transforming Growth Factor β1-Induced Smad2 Signaling. Front. Immunol. 2018, 9, 1728. [Google Scholar] [CrossRef] [Green Version]
- Platten, M.; Kretz, A.; Naumann, U.; Aulwurm, S.; Egashira, K.; Isenmann, S.; Weller, M. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Ann. Neurol. 2003, 54, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Flores-Toro, J.A.; Luo, D.; Gopinath, A.; Sarkisian, M.R.; Campbell, J.J.; Charo, I.F.; Singh, R.; Schall, T.J.; Datta, M.; Jain, R.K.; et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc. Natl. Acad. Sci. USA 2020, 117, 1129–1138. [Google Scholar] [CrossRef]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad—Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [Green Version]
- De, I.; Nikodemova, M.; Steffen, M.D.; Sokn, E.; Maklakova, V.I.; Watters, J.J.; Collier, L.S. CSF1 overexpression has pleiotropic effects on microgliain vivo. Glia 2014, 62, 1955–1967. [Google Scholar] [CrossRef] [Green Version]
- Dikmen, H.O.; Hemmerich, M.; Lewen, A.; Hollnagel, J.-O.; Chausse, B.; Kann, O. GM-CSF induces noninflammatory proliferation of microglia and disturbs electrical neuronal network rhythms in situ. J. Neuroinflammation 2020, 17, 1–13. [Google Scholar] [CrossRef]
- Revoltella, R.P.; Menicagli, M.; Campani, D. Granulocyte–macrophage colony-stimulating factor as an autocrine survival-growth factor in human gliomas. Cytokine 2012, 57, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298, 249–258. [Google Scholar] [CrossRef]
- Schartner, J.M.; Hagar, A.R.; Van Handel, M.; Zhang, L.; Nadkarni, N.; Badie, B. Impaired capacity for upregulation of MHC class II in tumor-associated microglia. Glia 2005, 51, 279–285. [Google Scholar] [CrossRef]
- Badie, B.; Schartner, J.; Prabakaran, S.; Paul, J.; Vorpahl, J. Expression of Fas ligand by microglia: Possible role in glioma immune evasion. J. Neuroimmunol. 2001, 120, 19–24. [Google Scholar] [CrossRef]
- Liu, C.; Luo, D.; Reynolds, B.A.; Meher, G.; Katritzky, A.R.; Lu, B.; Gerard, C.J.; Bhadha, C.P.; Harrison, J.K. Chemokine receptor CXCR3 promotes growth of glioma. Carcinog. 2010, 32, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Cantanhede, I.G.; De Oliveira, J.R.M. PDGF Family Expression in Glioblastoma Multiforme: Data Compilation from Ivy Glioblastoma Atlas Project Database. Sci. Rep. 2017, 7, 15271. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Yan, M.; Liu, Y.; McLeish, K.R.; Coleman, W.G.; Rodgers, G.P. Olfactomedin 4 Inhibits Cathepsin C-Mediated Protease Activities, Thereby Modulating Neutrophil Killing ofStaphylococcus aureusandEscherichia coliin Mice. J. Immunol. 2012, 189, 2460–2467. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Yan, M.; Liu, Y.; Wang, R.; Li, C.; Deng, C.; Singh, A.; Coleman, W.G.; Rodgers, G.P. Olfactomedin 4 down-regulates innate immunity against Helicobacter pylori infection. Proc. Natl. Acad. Sci. USA 2010, 107, 11056–11061. [Google Scholar] [CrossRef] [Green Version]
- Amirbeagi, F.; Thulin, P.; Pullerits, R.; Pedersen, B.; Andersson, B.A.; Dahlgren, C.; Welin, A.; Bylund, J. Olfactomedin-4 autoantibodies give unusual c-ANCA staining patterns with reactivity to a subpopulation of neutrophils. J. Leukoc. Biol. 2014, 97, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Gottfried-Blackmore, A.; Kaunzner, U.W.; Idoyaga, J.; Felger, J.C.; McEwen, B.S.; Bulloch, K. Acute in vivo exposure to interferon- enables resident brain dendritic cells to become effective antigen presenting cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20918–20923. [Google Scholar] [CrossRef] [Green Version]
- Facoetti, A.; Nano, R.; Zelini, P.; Morbini, P.; Benericetti, E.; Ceroni, M.; Campoli, M.; Ferrone, S. Human Leukocyte Antigen and Antigen Processing Machinery Component Defects in Astrocytic Tumors. Clin. Cancer Res. 2005, 11, 8304–8311. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Petit, P.-F.; Van den Eynde, B.J. Apoptosis of tumor-infiltrating T lymphocytes: A new immune checkpoint mechanism. Cancer Immunol. Immunother. 2019, 68, 835–847. [Google Scholar] [CrossRef]
- Hao, C.; Chen, G.; Zhao, H.; Li, Y.; Chen, J.; Zhang, H.; Li, S.; Zhao, Y.; Chen, F.; Li, W.; et al. PD-L1 Expression in Glioblastoma, the Clinical and Prognostic Significance: A Systematic Literature Review and Meta-Analysis. Front. Oncol. 2020, 10, 1015. [Google Scholar] [CrossRef]
- Reardon, D.A.; Omuro, A.; Brandes, A.A.; Rieger, J.; Wick, A.; Sepulveda, J.; Phuphanich, S.; De Souza, P.; Ahluwalia, M.S.; Lim, M.; et al. OS10.3 Randomized Phase 3 Study Evaluating the Efficacy and Safety of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: CheckMate 143. Neuro-Oncology 2017, 19, iii21. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Chen, A.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Lamano, J.B.; Lamano, J.B.; Li, Y.D.; DiDomenico, J.D.; Choy, W.; Veliceasa, D.; Oyon, D.E.; Fakurnejad, S.; Ampie, L.; Kesavabhotla, K.; et al. Glioblastoma-Derived IL6 Induces Immunosuppressive Peripheral Myeloid Cell PD-L1 and Promotes Tumor Growth. Clin. Cancer Res. 2019, 25, 3643–3657. [Google Scholar] [CrossRef] [Green Version]
- Sloin, H.E.; Ruggiero, G.; Rubinstein, A.; Storz, S.S.; Foulkes, N.S.; Gothilf, Y. Intera ctions between the circadian clock and TGF-β signaling pathway in zebrafish. PLoS ONE 2018, 13, e0199777. [Google Scholar] [CrossRef] [PubMed]
- Kon, N.; Hirota, T.; Kawamoto, T.; Kato, Y.; Tsubota, T.; Fukada, Y. Activation of TGF-β/activin signalling resets the circadian clock through rapid induction of Dec1 transcripts. Nat. Cell Biol. 2008, 10, 1463–1469. [Google Scholar] [CrossRef]
- Toedebusch, R.; Grodzki, A.C.; Dickinson, P.J.; Woolard, K.; Vinson, N.; Sturges, B.; Snyder, J.; Li, C.-F.; Nagasaka, O.; Consales, B.; et al. Glioma-associated microglia/macrophages augment tumorigenicity in canine astrocytoma, a naturally occurring model of human glioma. Neuro-Oncol. Adv. 2021, 3, vdab062. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene. | Sequence-F (5′ to 3′) | Sequence-R (5′ to 3′) | Use |
|---|---|---|---|
| Olfml3 s1 | GCTAACGGGCTGGAGGGAAA | AGTGGTACCATCCCATCCGA | PCR |
| Olfml3 s2 | AGCTGCCTTAGAGGAACGG | CCTCCCTTTCAAGACGGTCC | qPCR |
| H2-Ab1 | AGCCCCATCACTGTGGAGT | GATGCCGCTCAACATCTTGC | qPCR |
| Nos2 | TTCTCAGCCACCTTGGTGAAG | AAGTGAAATCCGATGTGGCC | qPCR |
| Pdgfa | GAGGAAGCCGAGATACCCC | TGCTGTGGATCTGACTTCGAG | qPCR |
| Rpl22 | AGCAGGTTTTGAAGTTCACCC | CAGCTTTCCCATTCACCTTGA | qPCR |
| Olfml3-gRNA | TCATGGACGGGACCCCTTCA | CRISPR | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toedebusch, R.G.; Lucchesi, C.A.; Debebe, E.T.; Wittenburg, L.A.; Chen, X.; Toedebusch, C.M. Microglia-Derived Olfactomedin-like 3 Promotes Pro-Tumorigenic Microglial Function and Malignant Features of Glioma Cells. Int. J. Mol. Sci. 2021, 22, 13052. https://doi.org/10.3390/ijms222313052
Toedebusch RG, Lucchesi CA, Debebe ET, Wittenburg LA, Chen X, Toedebusch CM. Microglia-Derived Olfactomedin-like 3 Promotes Pro-Tumorigenic Microglial Function and Malignant Features of Glioma Cells. International Journal of Molecular Sciences. 2021; 22(23):13052. https://doi.org/10.3390/ijms222313052
Chicago/Turabian StyleToedebusch, Ryan G., Christopher A. Lucchesi, Eshetu T. Debebe, Luke A. Wittenburg, Xinbin Chen, and Christine M. Toedebusch. 2021. "Microglia-Derived Olfactomedin-like 3 Promotes Pro-Tumorigenic Microglial Function and Malignant Features of Glioma Cells" International Journal of Molecular Sciences 22, no. 23: 13052. https://doi.org/10.3390/ijms222313052
APA StyleToedebusch, R. G., Lucchesi, C. A., Debebe, E. T., Wittenburg, L. A., Chen, X., & Toedebusch, C. M. (2021). Microglia-Derived Olfactomedin-like 3 Promotes Pro-Tumorigenic Microglial Function and Malignant Features of Glioma Cells. International Journal of Molecular Sciences, 22(23), 13052. https://doi.org/10.3390/ijms222313052