
Genome-Wide Mutation Scoring for Machine-Learning-Based Antimicrobial Resistance Prediction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Overview of the Data
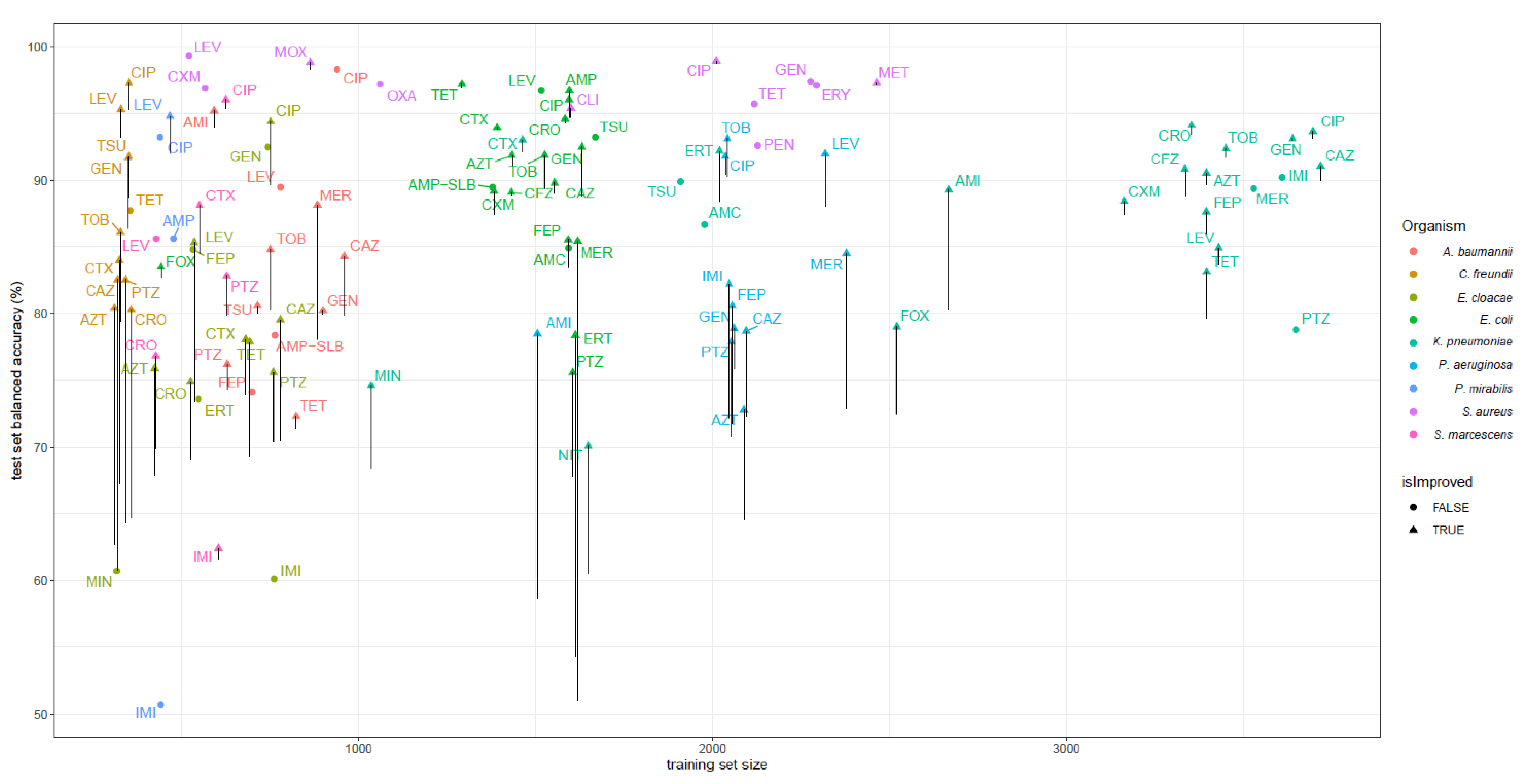
2.2. WGS-AST Predictive Models on DNA K-Mers
2.3. Feature Engineering
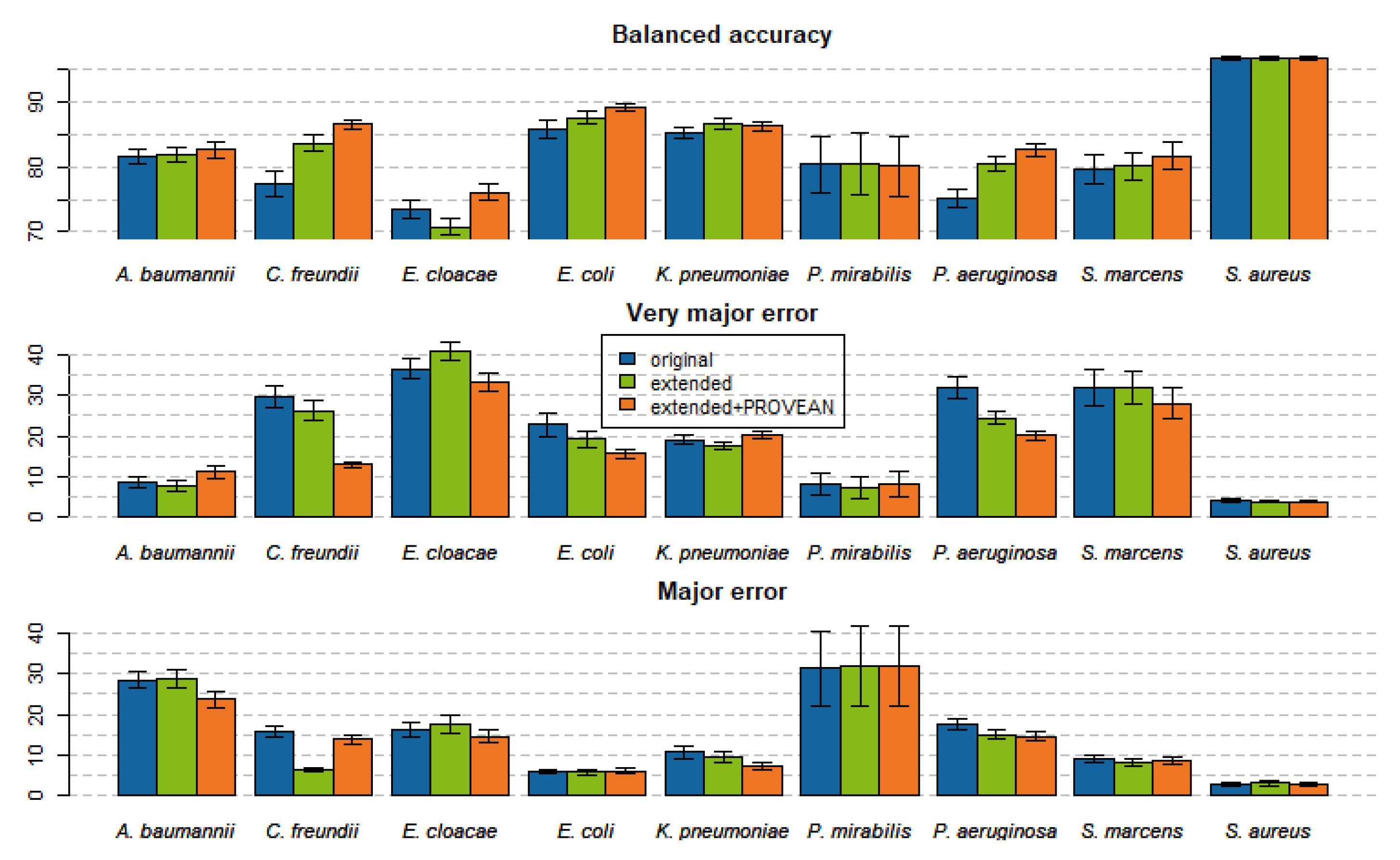
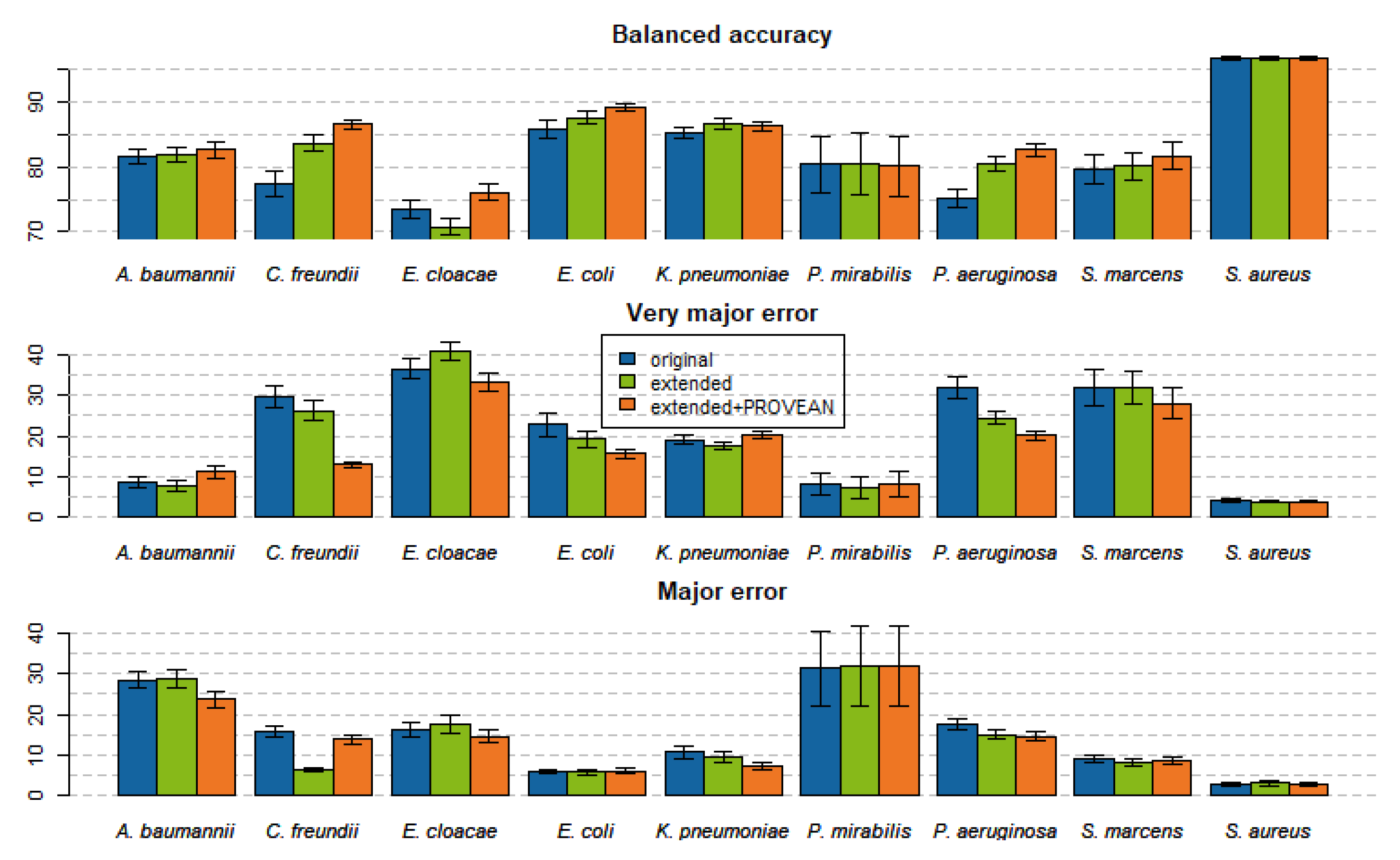
2.4. Models Trained with PROVEAN Features
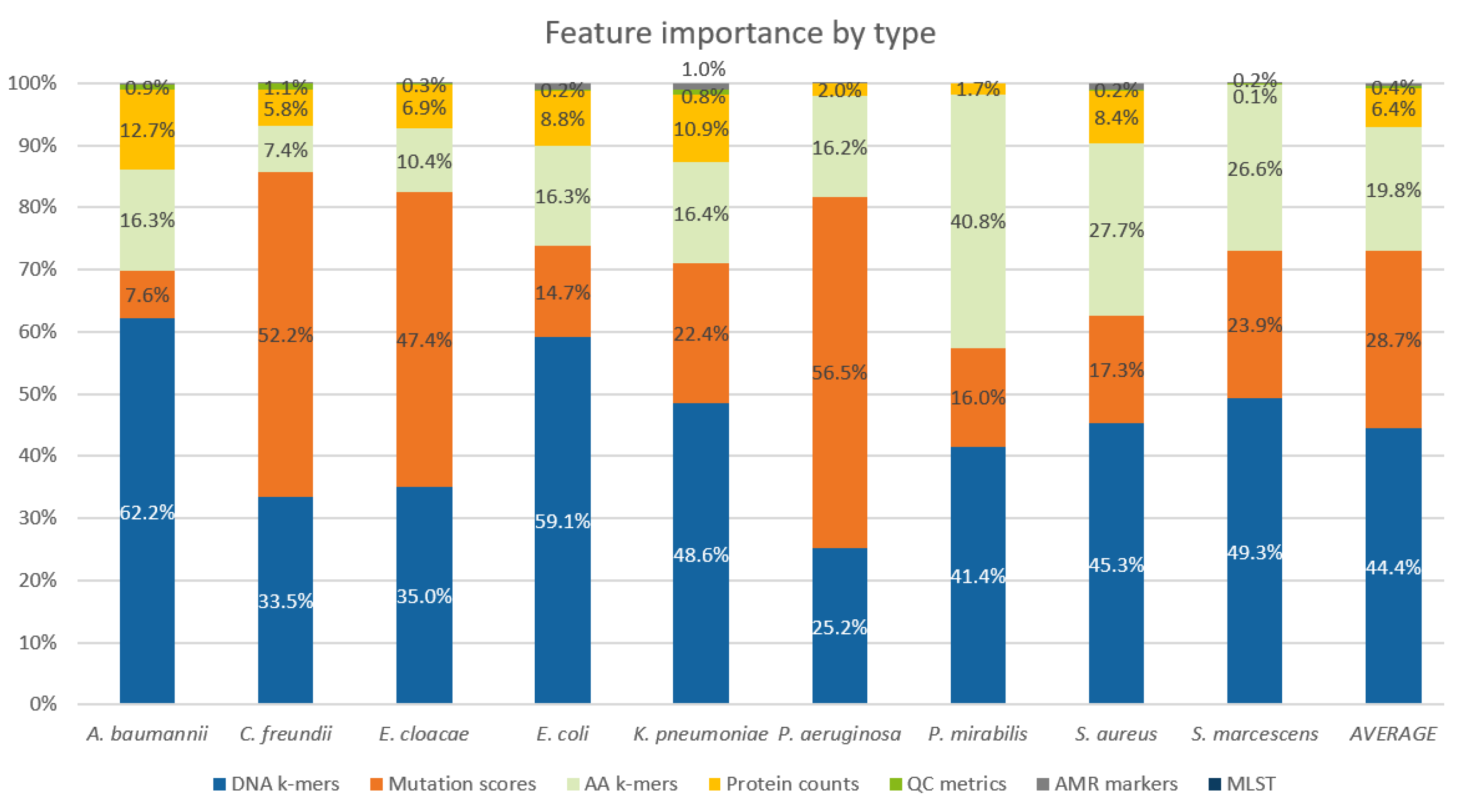
2.5. Feature Importance
3. Discussion
4. Materials and Methods
4.1. Data Retrieval
4.2. Antimicrobial Compounds
4.3. Machine Learning Feature Generation
4.4. Proteome-Wide Scoring of Functional Alterations
4.5. Feature Filtering
4.6. Model Training
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- O’Neill, J. The Review on Antimicrobial Resistance (Chaired by Jim O’Neill). Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. 2016. Available online: https://amr-review.org/sites/default/files/160525_Final%20paper_with%20cover.pdf (accessed on 1 December 2021).
- Břinda, K.; Callendrello, A.; Cowley, L.; Charalampous, T.; Lee, R.S.; MacFadden, D.R.; Kucherov, G.; O’Grady, J.; Baym, M.; Hanage, W.P. Lineage calling can identify antibiotic resistant clones within minutes. bioRxiv 2018, 40, 3204. [Google Scholar] [CrossRef] [Green Version]
- Bradley, P.; Gordon, N.C.; Walker, T.M.; Dunn, L.; Heys, S.; Huang, B.; Earle, S.; Pankhurst, L.J.; Anson, L.; De Cesare, M.; et al. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat. Commun. 2015, 6, 10063. [Google Scholar] [CrossRef] [Green Version]
- Avdic, E.; Wang, R.; Li, D.X.; Tamma, P.D.; Shulder, S.E.; Carroll, K.C.; Cosgrove, S.E. Sustained impact of a rapid microarray-based assay with antimicrobial stewardship interventions on optimizing therapy in patients with Gram-positive bacteraemia. J. Antimicrob. Chemother. 2017, 72, 3191–3198. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Teng, C.B.; Cunningham, S.A.; Ihde, S.M.; Steckelberg, J.M.; Moriarty, J.P.; Shah, N.D.; Mandrekar, J.N.; Patel, R. Randomized Trial of Rapid Multiplex Polymerase Chain Reaction–Based Blood Culture Identification and Susceptibility Testing. Clin. Infect. Dis. 2015, 61, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, Z.; Liang, B.; Ye, F.; Gong, W. A review: Antimicrobial resistance data mining models and prediction methods study for pathogenic bacteria. J. Antibiot. 2021, 74, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Pesesky, M.W.; Hussain, T.; Wallace, M.; Patel, S.; Andleeb, S.; Burnham, C.A.D.; Dantas, G. Evaluation of machine learning and rules-based approaches for predicting antimicrobial resistance profiles in gram-negative bacilli from whole genome sequence data. Front. Microbiol. 2016, 7, 1887. [Google Scholar] [CrossRef] [PubMed]
- Mahfouz, N.; Ferreira, I.; Beisken, S.; von Haeseler, A.; Posch, A.E. Large-scale assessment of antimicrobial resistance marker databases for genetic phenotype prediction: A systematic review. J. Antimicrob. Chemother. 2020, 75, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Allesøe, R.; Joensen, K.G.; Cavaco, L.M.; Lund, O.; Aarestrup, F.M. PointFinder: A novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother. 2017, 72, 2764–2768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldgarden, M.; Brover, V.; Gonzalez-Escalona, N.; Frye, J.G.; Haendiges, J.; Haft, D.H.; Hoffmann, M.; Pettengill, J.B.; Prasad, A.B.; Tillman, G.E.; et al. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci. Rep. 2021, 11, 12728. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.; Beisken, S.; Lueftinger, L.; Weinmaier, T.; Klein, M.; Bacher, J.; Patel, R.; von Haeseler, A.; Posch, A.E. Species identification and antibiotic resistance prediction by analysis of whole-genome sequence data by use of ARESdb: An analysis of isolates from the unyvero lower respiratory tract infection trial. J. Clin. Microbiol. 2020, 58, e00273-20. [Google Scholar] [CrossRef] [Green Version]
- Drouin, A.; Giguère, S.; Déraspe, M.; Marchand, M.; Tyers, M.; Loo, V.G.; Bourgault, A.-M.; Laviolette, F.; Corbeil, J. Predictive computational phenotyping and biomarker discovery using reference-free genome comparisons. BMC Genom. 2016, 17, 754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aun, E.; Brauer, A.; Kisand, V.; Tenson, T.; Remm, M. A k-mer-based method for the identification of phenotype-associated genomic biomarkers and predicting phenotypes of sequenced bacteria. PLoS Comput. Biol. 2018, 14, e1006434. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.; Brettin, T.; Long, S.W.; Musser, J.M.; Olsen, R.J.; Olson, R.D.; Shukla, M.P.; Stevens, R.L.; Xia, F.F.-F.; Yoo, H.; et al. Developing an in silico minimum inhibitory concentration panel test for Klebsiella pneumoniae. Sci. Rep. 2018, 8, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lüftinger, L.; Májek, P.; Beisken, S.; Rattei, T.; Posch, A.E. Learning from Limited Data: Towards Best Practice Techniques for Antimicrobial Resistance Prediction From Whole Genome Sequencing Data. Front. Cell. Infect. Microbiol. 2021, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Drouin, A.; Letarte, G.; Raymond, F.; Marchand, M.; Corbeil, J.; Laviolette, F. Interpretable genotype-to-phenotype classifiers with performance guarantees. Sci. Rep. 2019, 9, 4071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genom. Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- ValizadehAslani, T.; Zhao, Z.; Sokhansanj, B.A.; Rosen, G.L. Amino Acid k-mer Feature Extraction for Quantitative Antimicrobial Resistance (AMR) Prediction by Machine Learning and Model Interpretation for Biological Insights. Biology 2020, 9, 365. [Google Scholar] [CrossRef] [PubMed]
- Tunstall, T.; Portelli, S.; Phelan, J.; Clark, T.G.; Ascher, D.B.; Furnham, N. Combining structure and genomics to understand antimicrobial resistance. Comput. Struct. Biotechnol. J. 2020, 18, 3377–3394. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y. A Fast Computation of Pairwise Sequence Alignment Scores between a Protein and a Set of Single-Locus Variants of Another Protein. In Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine, Orlando, FL, USA, 7–10 October 2012; Association for Computing Machinery: New York, NY, USA, 2012; pp. 414–417. [Google Scholar]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM Sigkdd International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016. [Google Scholar]
- Langendonk, R.F.; Neill, D.R.; Fothergill, J.L. The Building Blocks of Antimicrobial Resistance in Pseudomonas aeruginosa: Implications for Current Resistance-Breaking Therapies. Front. Cell. Infect. Microbiol. 2021, 11, 307. [Google Scholar] [CrossRef] [PubMed]
- Moya, B.; Juan, C.; Alberti, S.; Pérez, J.L.; Oliver, A. Benefit of Having Multiple ampD Genes for Acquiring β-Lactam Resistance without Losing Fitness and Virulence in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2008, 52, 3694–3700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, U.; Lee, C.-R. Distinct Roles of Outer Membrane Porins in Antibiotic Resistance and Membrane Integrity in Escherichia coli. Front. Microbiol. 2019, 10, 953. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-F.; Yan, J.-J.; Lei, H.-Y.; Teng, C.-H.; Wang, M.-C.; Tseng, C.-C.; Wu, J.-J. Loss of outer membrane protein C in Escherichia coli contributes to both antibiotic resistance and escaping antibody-dependent bactericidal activity. Infect. Immun. 2012, 80, 1815–1822. [Google Scholar] [CrossRef] [Green Version]
- Tenover, F.C.; Filpula, D.; Phillips, K.L.; Plorde, J.J. Cloning and sequencing of a gene encoding an aminoglycoside 6′-N-acetyltransferase from an R factor of Citrobacter diversus. J. Bacteriol. 1988, 170, 471–473. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, T.M. The Role of Unlabeled Data in Supervised Learning BT-Language, Knowledge, and Representation; Larrazabal, J.M., Miranda, L.A.P., Eds.; Springer: Dordrecht, The Netherlands, 2004; pp. 103–111. [Google Scholar]
- Simner, P.J.; Beisken, S.; Bergman, Y.; Posch, A.E.; Cosgrove, S.E.; Tamma, P.D. Cefiderocol Activity Against Clinical Pseudomonas aeruginosa Isolates Exhibiting Ceftolozane-Tazobactam Resistance. Open Forum Infect. Dis. 2021, 8, ofab311. [Google Scholar] [CrossRef]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 2017, 45, D535–D542. [Google Scholar] [CrossRef] [PubMed]
- Bethesda (MD): National Database of Antibiotic Resistant Organisms (NDARO), National Center for Biotechnology Information. Available online: https://www.ncbi.nlm.nih.gov/pathogens/antimicrobial-resistance/ (accessed on 1 December 2021).
- Karp, B.E.; Tate, H.; Plumblee, J.R.; Dessai, U.; Whichard, J.M.; Thacker, E.L.; Robertson Hale, K.; Wilson, W.; Friedman, C.R.; Griffin, P.M.; et al. National Antimicrobial Resistance Monitoring System: Two Decades of Advancing Public Health Through Integrated Surveillance of Antimicrobial Resistance. Foodborne Pathog. Dis. 2017, 14, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Kos, V.N.; Déraspe, M.; Mclaughlin, R.E.; Whiteaker, J.D.; Roy, P.H.; Alm, R.A.; Corbeil, J.; Gardner, H. The Resistome of Pseudomonas aeruginosa in Relationship to Phenotypic Susceptibility. Antimicrob. Agents Chemother. 2015, 59, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, P.N.A.; Peleg, A.Y.; Iredell, J.; Ingram, P.R.; Miyakis, S.; Stewardson, A.J.; Rogers, B.A.; McBryde, E.S.; Roberts, J.A.; Lipman, J.; et al. Meropenem versus piperacillin-tazobactam for definitive treatment of bloodstream infections due to ceftriaxone non-susceptible Escherichia coli and Klebsiella spp (the MERINO trial): Study protocol for a randomised controlled trial. Trials 2015, 16, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wayne, P. Performance Standards for Antimicrobial Susceptibility Testing, 29th ed.; CLSI supplement, M100; Wayne, P., Ed.; Clinical and Laboratory Standards Institute: Annapolis Junction, MD, USA, 2019. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef] [PubMed]
- Kokot, M.; Dlugosz, M.; Deorowicz, S. KMC 3: Counting and manipulating k-mer statistics. Bioinformatics 2017, 33, 2759–2761. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunnen, J.T.D.; Antonarakis, S.E. Mutation nomenclature extensions and suggestions to describe complex mutations: A discussion. Hum. Mutat. 2000, 15, 7–12. [Google Scholar] [CrossRef]
- Vis, J.K.; Vermaat, M.; Taschner, P.E.M.; Kok, J.N.; Laros, J.F.J. An efficient algorithm for the extraction of HGVS variant descriptions from sequences. Bioinformatics 2015, 31, 3751–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Lundberg, S.M.; Lee, S.-I. A Unified Approach to Interpreting Model Predictions. In Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; Curran Associates Inc.: Red Hook, NY, USA, 2017; pp. 4768–4777. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Májek, P.; Lüftinger, L.; Beisken, S.; Rattei, T.; Materna, A. Genome-Wide Mutation Scoring for Machine-Learning-Based Antimicrobial Resistance Prediction. Int. J. Mol. Sci. 2021, 22, 13049. https://doi.org/10.3390/ijms222313049
Májek P, Lüftinger L, Beisken S, Rattei T, Materna A. Genome-Wide Mutation Scoring for Machine-Learning-Based Antimicrobial Resistance Prediction. International Journal of Molecular Sciences. 2021; 22(23):13049. https://doi.org/10.3390/ijms222313049
Chicago/Turabian StyleMájek, Peter, Lukas Lüftinger, Stephan Beisken, Thomas Rattei, and Arne Materna. 2021. "Genome-Wide Mutation Scoring for Machine-Learning-Based Antimicrobial Resistance Prediction" International Journal of Molecular Sciences 22, no. 23: 13049. https://doi.org/10.3390/ijms222313049
APA StyleMájek, P., Lüftinger, L., Beisken, S., Rattei, T., & Materna, A. (2021). Genome-Wide Mutation Scoring for Machine-Learning-Based Antimicrobial Resistance Prediction. International Journal of Molecular Sciences, 22(23), 13049. https://doi.org/10.3390/ijms222313049