
Structural Characterization of the Interaction of Hypoxia Inducible Factor-1 with Its Hypoxia Responsive Element at the −964G > A Variation Site of the HLA-G Promoter Region

Abstract
1. Introduction
2. Results and Discussion
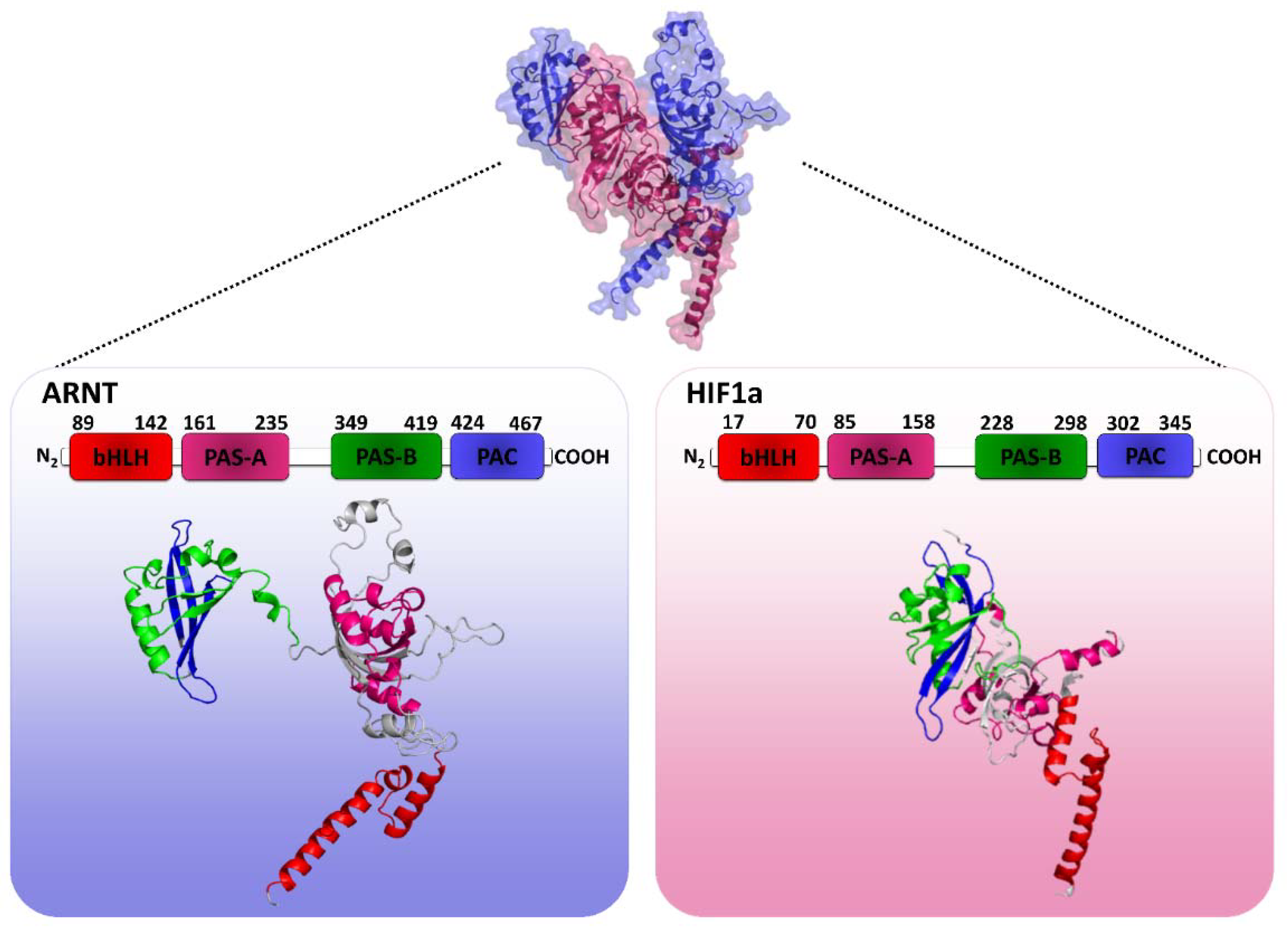
2.1. HIF1 Heterodimer 3D Structure Modeling
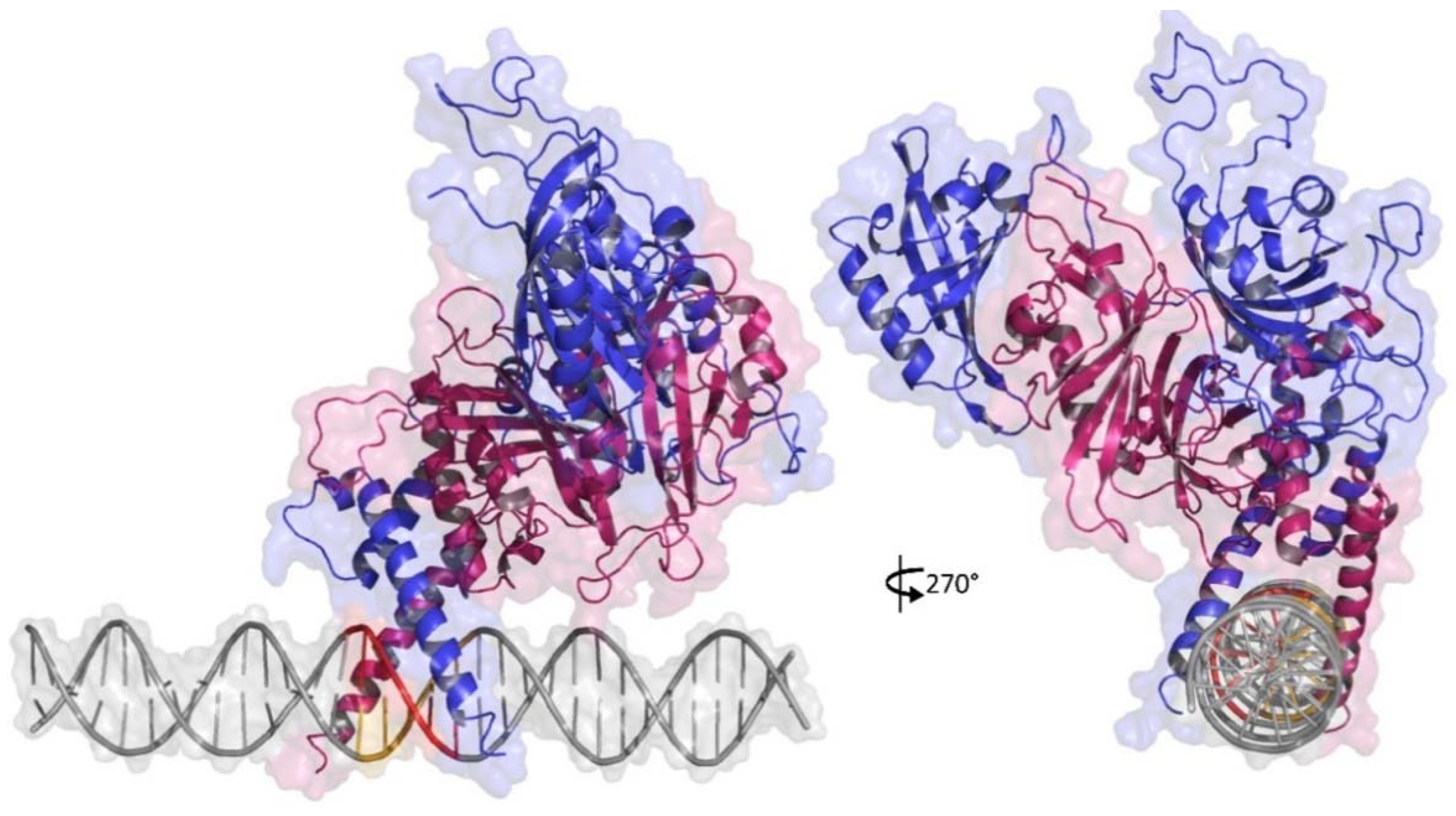
2.2. DNA Modeling
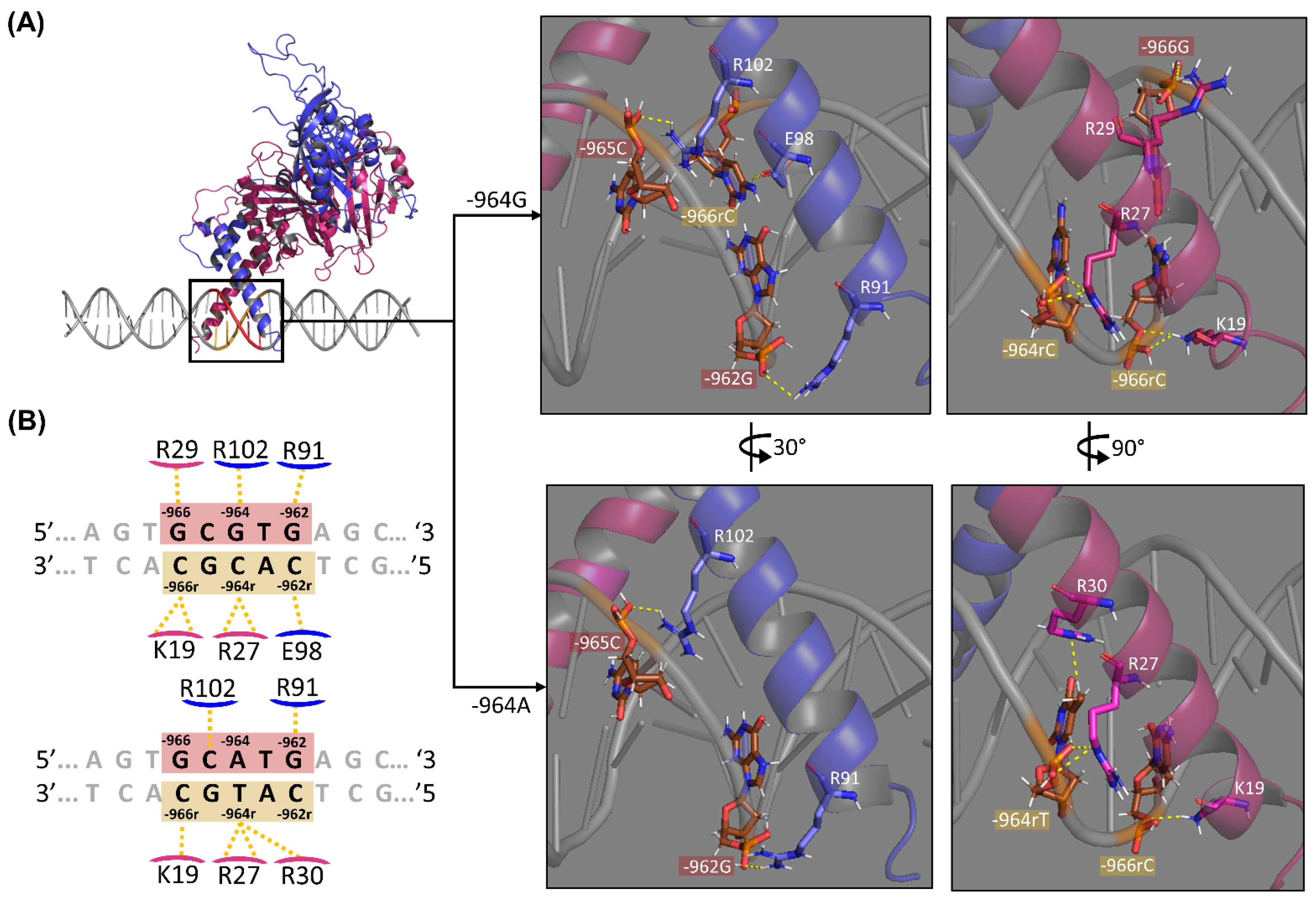
2.3. HIF1-HRE-964G/A Docking
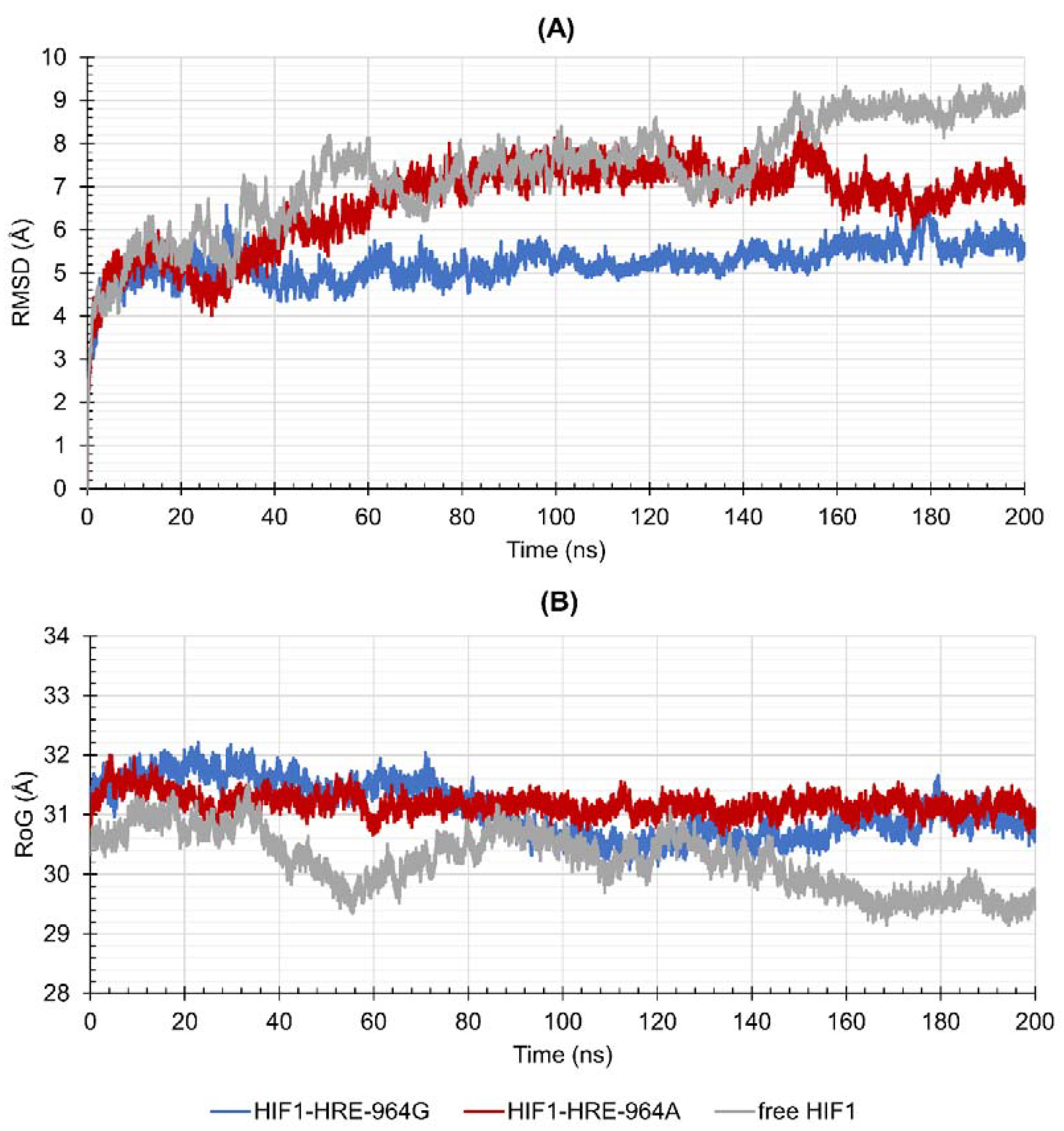
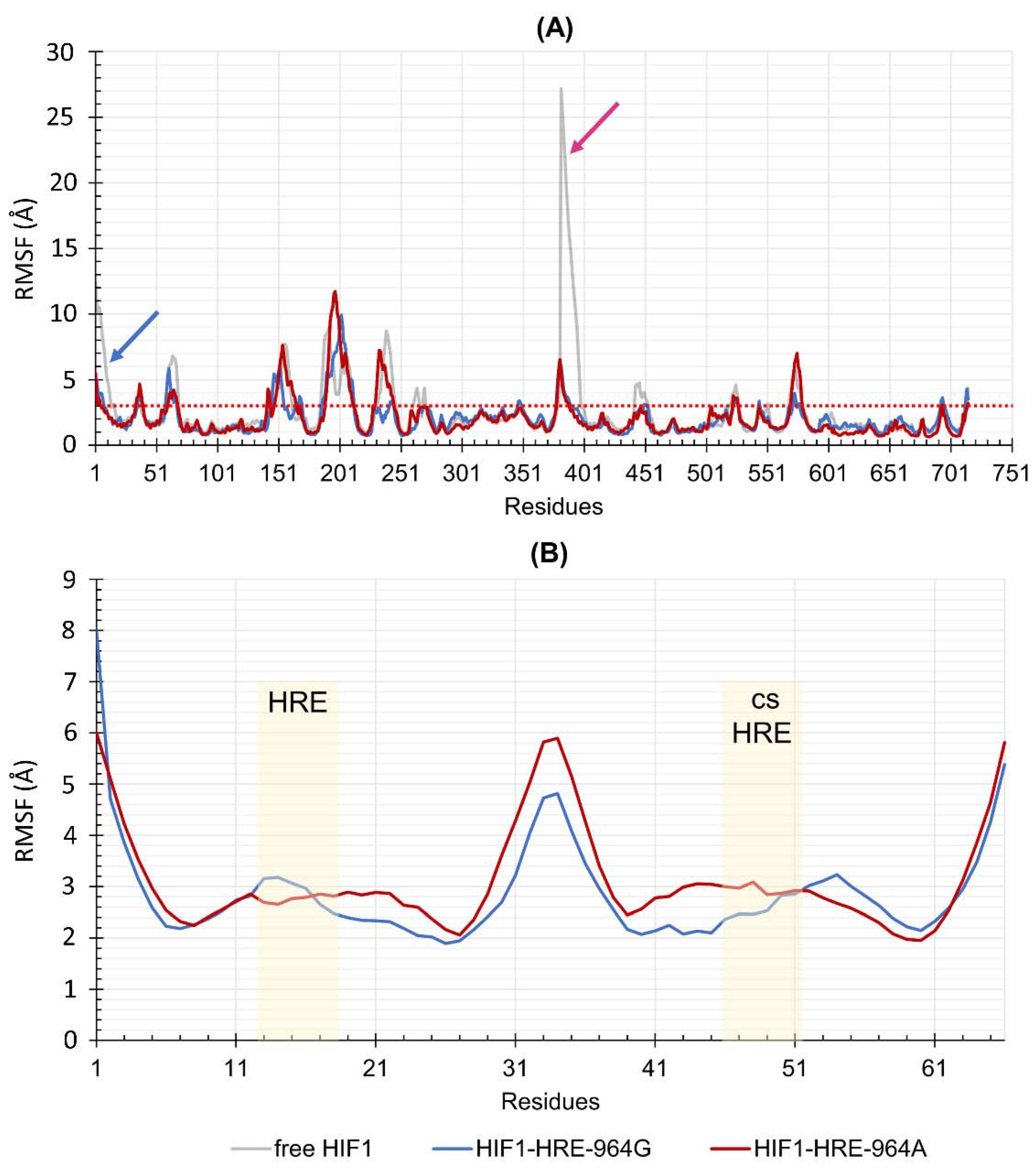
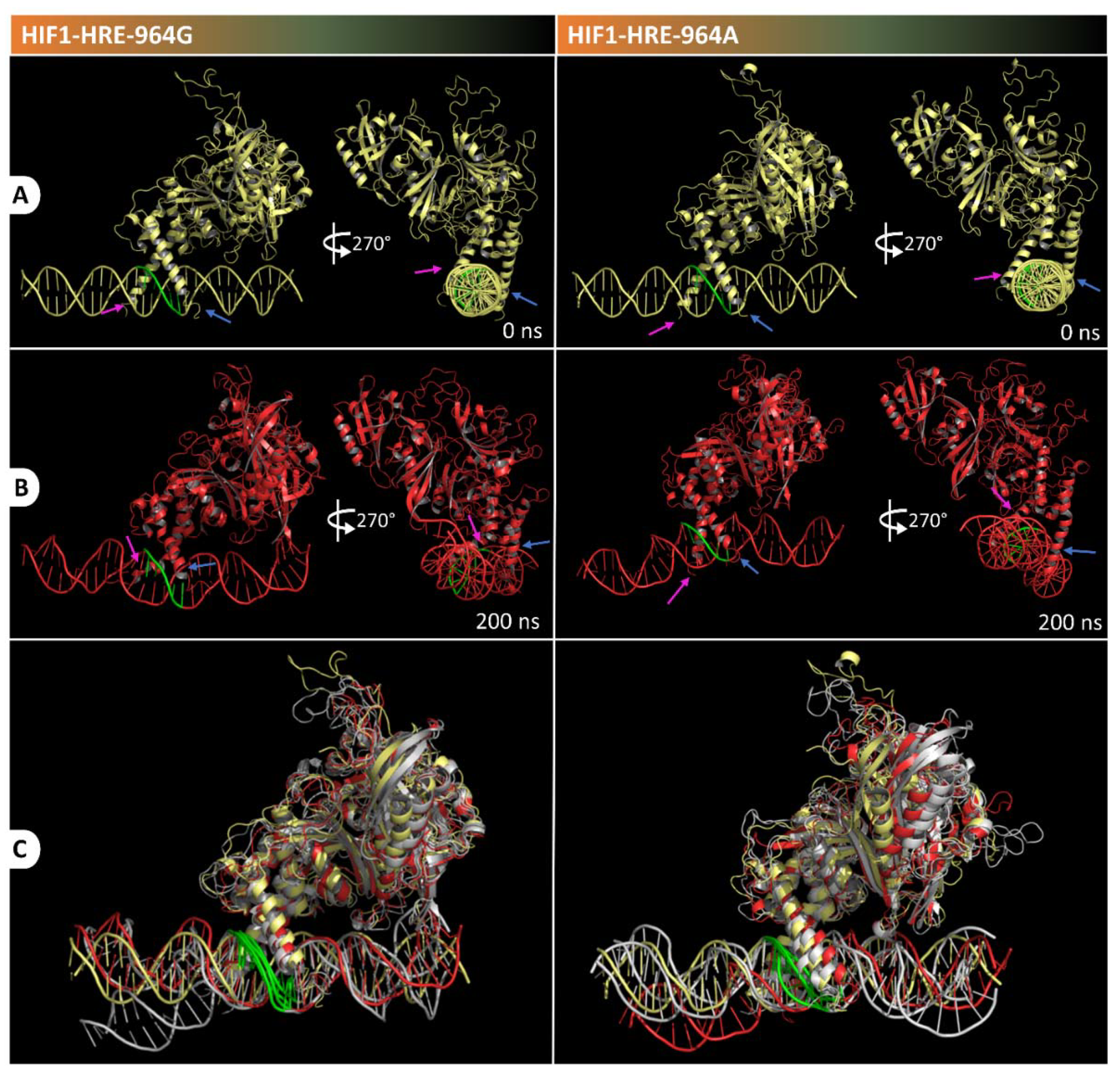
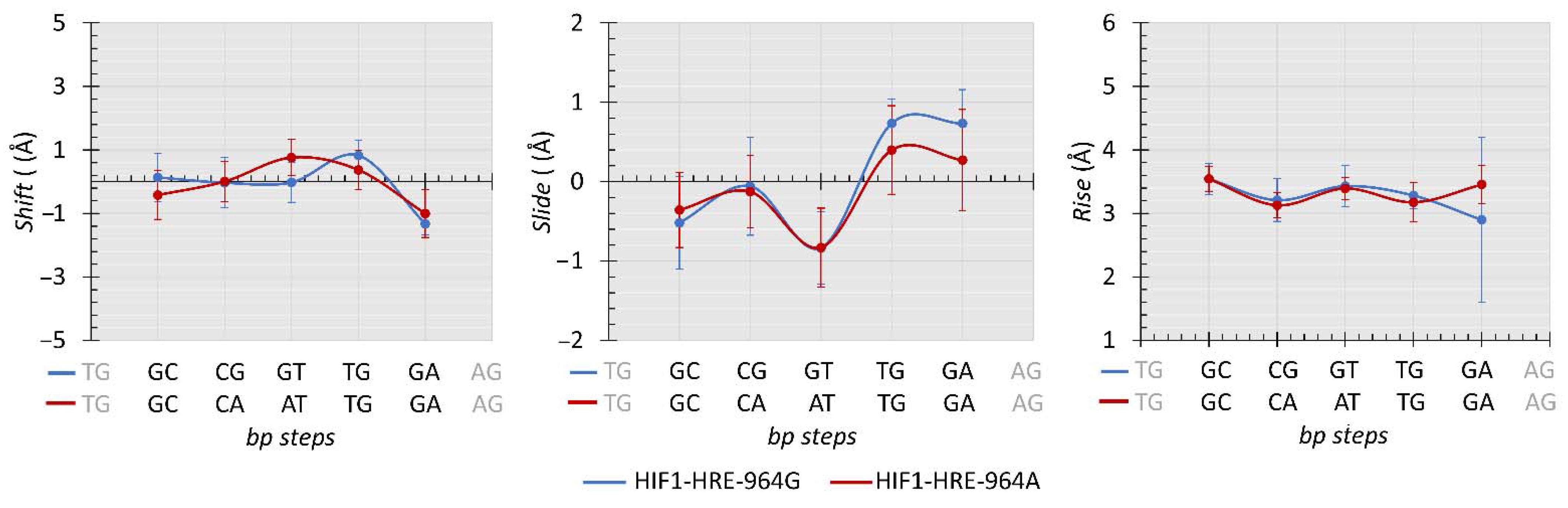
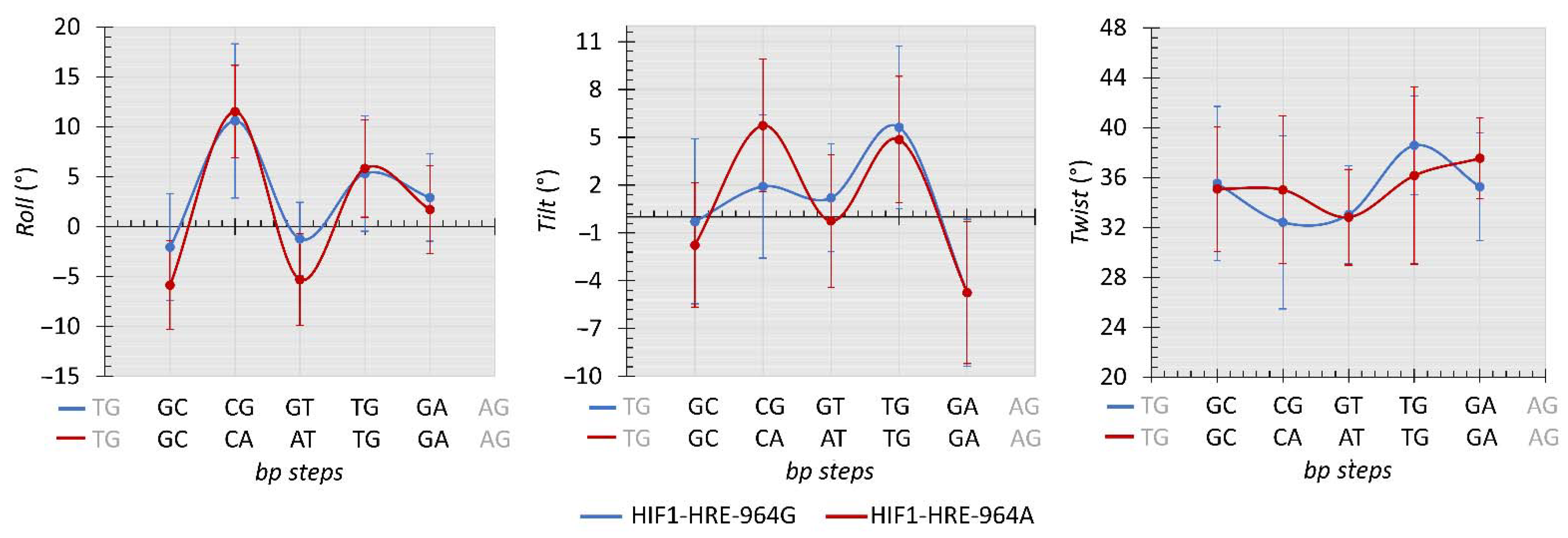
2.4. Protein-DNA Molecular Dynamic Simulation
3. Materials and Methods
3.1. HIF1 Dimer 3D Structure Modeling
3.2. DNA Modeling
3.3. HIF1-HRE-964G/A Molecular Docking
3.4. Molecular Dynamic Simulations
3.5. 3D Structure Visual Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dunwoodie, S.L. The Role of Hypoxia in Development of the Mammalian Embryo. Dev. Cell 2009, 17, 755–773. [Google Scholar] [CrossRef]
- Shao, Y.; Zhao, F.-Q. Emerging evidence of the physiological role of hypoxia in mammary development and lactation. J. Anim. Sci. Biotechnol. 2014, 5, 9. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Semenza, G.L. Role of hypoxia-inducible factors in breast cancer metastasis. Futur. Oncol. 2013, 9, 1623–1636. [Google Scholar] [CrossRef]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef]
- Benkert, P.; Künzli, M.; Schwede, T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009, 37, W510–W514. [Google Scholar] [CrossRef]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.D.M.; Espinosa, J.M.J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–5. [Google Scholar] [CrossRef]
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82. [Google Scholar] [CrossRef]
- Jiang, B.-H.; Rue, E.; Wang, G.L.; Roe, R.; Semenza, G.L. Dimerization, DNA Binding, and Transactivation Properties of Hypoxia-inducible Factor 1. J. Biol. Chem. 1996, 271, 17771–17778. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.-H.; Zheng, J.Z.; Leung, S.W.; Roe, R.; Semenza, G.L. Transactivation and Inhibitory Domains of Hypoxia-inducible Factor 1α. J. Biol. Chem. 1997, 272, 19253–19260. [Google Scholar] [CrossRef]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of hypoxia-inducible factor 1 is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef]
- Appelhoff, R.J.; Tian, Y.-M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential Function of the Prolyl Hydroxylases PHD1, PHD2, and PHD3 in the Regulation of Hypoxia-inducible Factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef]
- Berra, E. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1 in normoxia. EMBO J. 2003, 22, 4082–4090. [Google Scholar] [CrossRef]
- Salceda, S.; Caro, J. Hypoxia-inducible Factor 1α (HIF-1α) Protein Is Rapidly Degraded by the Ubiquitin-Proteasome System under Normoxic Conditions. J. Biol. Chem. 1997, 272, 22642–22647. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, O.; Levy, A.P.; Jiang, C.; Kaelin, W.G.; Goldberg, M.A. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10595–10599. [Google Scholar] [CrossRef]
- Lando, D. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef]
- Mahon, P.C. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef]
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 117, e207–e217. [Google Scholar] [CrossRef]
- Mouillot, G.; Marcou, C.; Zidi, I.; Guillard, C.; Sangrouber, D.; Carosella, E.D.; Moreau, P. Hypoxia Modulates HLA-G Gene Expression in Tumor Cells. Hum. Immunol. 2007, 68, 277–285. [Google Scholar] [CrossRef]
- Yaghi, L.; Poras, I.; Simoes, R.T.; Donadi, E.A.; Tost, J.; Daunay, A.; de Almeida, B.S.; Carosella, E.D.; Moreau, P. Hypoxia inducible factor-1 mediates the expression of the immune checkpoint HLA-G in glioma cells through hypoxia response element located in exon 2. Oncotarget 2016, 7, 63690–63707. [Google Scholar] [CrossRef]
- Le Gal, F.-A.; Riteau, B.; Sedlik, C.; Khalil-Daher, I.; Menier, C.; Dausset, J.; Guillet, J.-G.; Carosella, E.D.; Rouas-Freiss, N. HLA-G-mediated inhibition of antigen-specific cytotoxic T lymphocytes. Int. Immunol. 1999, 11, 1351–1356. [Google Scholar] [CrossRef]
- Horuzsko, A.; Lenfant, F.; Munn, D.H.; Mellor, A.L. Maturation of antigen-presenting cells is compromised in HLA-G transgenic mice. Int. Immunol. 2001, 13, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Naji, A.; Menier, C.; Morandi, F.; Agaugué, S.; Maki, G.; Ferretti, E.; Bruel, S.; Pistoia, V.; Carosella, E.D.; Rouas-Freiss, N. Binding of HLA-G to ITIM-Bearing Ig-like Transcript 2 Receptor Suppresses B Cell Responses. J. Immunol. 2014, 192, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Gregori, S.; Magnani, C.F.; Roncarolo, M.G. Role of human leukocyte antigen-G in the induction of adaptive type 1 regulatory T cells. Hum. Immunol. 2009, 70, 966–969. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Sato, A. The HLA system: First of two parts. Engl. J. Med. Rev. 2000, 343, 702–709. [Google Scholar] [CrossRef]
- Lin, A.; Yan, W.H. Heterogeneity of HLA-G Expression in Cancers: Facing the Challenges. Front. Immunol. 2018, 9, 2164. [Google Scholar] [CrossRef]
- Kilburn, B.A.; Wang, J.; Duniec-Dmuchkowski, Z.M.; Leach, R.E.; Romero, R.; Armant, D.R. Extracellular Matrix Composition and Hypoxia Regulate the Expression of HLA-G and Integrins in a Human Trophoblast Cell Line1. Biol. Reprod. 2000, 62, 739–747. [Google Scholar] [CrossRef]
- Chang, C.-C.; Ferrone, S. HLA-G in melanoma: Can the current controversies be solved? Semin. Cancer Biol. 2003, 13, 361–369. [Google Scholar] [CrossRef]
- Wastowski, I.J.; Simões, R.T.; Yaghi, L.; Donadi, E.A.; Pancoto, J.T.; Poras, I.; Lechapt-Zalcman, E.; Bernaudin, M.; Valable, S.; Carlotti, C.G.; et al. Human Leukocyte Antigen-G Is Frequently Expressed in Glioblastoma and May Be Induced in Vitro by Combined 5-Aza-2′-Deoxycytidine and Interferon-γ Treatments. Am. J. Pathol. 2013, 182, 540–552. [Google Scholar] [CrossRef]
- Castelli, E.C.; Veiga-Castelli, L.C.; Yaghi, L.; Moreau, P.; Donadi, E.A. Transcriptional and Posttranscriptional Regulations of the HLA-G Gene. J. Immunol. Res. 2014, 2014, 734068. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Shon, A.M.; Ober, C. Evidence of balancing selection at the HLA-G promoter region. Hum. Mol. Genet. 2005, 14, 3619–3628. [Google Scholar] [CrossRef] [PubMed]
- Castelli, E.C.; Ramalho, J.; Porto, I.O.P.; Lima, T.H.A.; Felicio, L.P.; Sabbagh, A.; Donadi, E.A.; Mendes-Junior, C.T. Insights into HLA-G Genetics Provided by Worldwide Haplotype Diversity. Front. Immunol. 2014, 5, 476. [Google Scholar] [CrossRef] [PubMed]
- Svozil, D.; Kalina, J.; Omelka, M.; Schneider, B. DNA conformations and their sequence preferences. Nucleic Acids Res. 2008, 36, 3690–3706. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Potluri, N.; Lu, J.; Kim, Y.; Rastinejad, F. Structural integration in hypoxia-inducible factors. Nature 2015, 524, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 30. [Google Scholar] [CrossRef] [PubMed]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Fiser, A.; Do, R.K.G.; Šali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef]
- Frank-Kamenetskii, M.D. DNA topology. J. Mol. Struct. 1995, 336, 235–243. [Google Scholar] [CrossRef]
- Ussery, D.W. DNA Structure: A-, B- and Z-DNA Helix Families. In Encyclopedia of Life Sciences; John Wiley & Sons, Ltd.: Chichester, UK, 2002. [Google Scholar]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; Van Dijk, M.; De Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- De Vries, S.J.; Van Dijk, M.; Bonvin, A.M.J.J. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef]
- Graham, A.M.; Presnell, J.S. Hypoxia Inducible Factor (HIF) transcription factor family expansion, diversification, divergence and selection in eukaryotes. PLoS ONE 2017, 12, e0179545. [Google Scholar] [CrossRef]
- Schulte, K.W.; Green, E.; Wilz, A.; Platten, M.; Daumke, O. Structural Basis for Aryl Hydrocarbon Receptor-Mediated Gene Activation. Structure 2017, 25, 1025–1033.e3. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Su, X.; Potluri, N.; Kim, Y.; Rastinejad, F. NPAS1-ARNT and NPAS3-ARNT crystal structures implicate the bHLH-PAS family as multi-ligand binding transcription factors. Elife 2016, 5, e18790. [Google Scholar] [CrossRef] [PubMed]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of Oxygen Signaling at the Consensus HRE. Sci. Signal. 2005, 2005, re12. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided-Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Rohs, R.; Jin, X.; West, S.M.; Joshi, R.; Honig, B.; Mann, R.S. Origins of Specificity in Protein-DNA Recognition. Annu. Rev. Biochem. 2010, 79, 233–269. [Google Scholar] [CrossRef]
- Slattery, M.; Zhou, T.; Yang, L.; Dantas Machado, A.C.; Gordân, R.; Rohs, R. Absence of a simple code: How transcription factors read the genome. Trends Biochem. Sci. 2014, 39, 381–399. [Google Scholar] [CrossRef] [PubMed]
- Mandel-Gutfreund, Y.; Schueler, O.; Margalit, H. Comprehensive Analysis of Hydrogen Bonds in Regulatory Protein DNA-Complexes: In Search of Common Principles. J. Mol. Biol. 1995, 253, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Luscombe, N.M.; Thornton, J.M. Protein—DNA Interactions: Amino Acid Conservation and the Effects of Mutations on Binding Specificity. J. Mol. Biol. 2002, 2836, 991–1009. [Google Scholar] [CrossRef]
- Matthews, B.W. Protein-DNA interaction. No code for recognition. Nature 1988, 335, 294–295. [Google Scholar] [CrossRef] [PubMed]
- Pabo, C.O.; Sauer, R.T. Protein-DNA Recognition. Annu. Rev. Biochem. 1984, 53, 293–321. [Google Scholar] [CrossRef] [PubMed]
- Izumi, K. Disorders of Transcriptional Regulation: An Emerging Category of Multiple Malformation Syndromes. Mol. Syndromol. 2016, 7, 262–273. [Google Scholar] [CrossRef]
- Sagendorf, J.M.; Markarian, N.; Berman, H.M.; Rohs, R. DNAproDB: An expanded database and web-based tool for structural analysis of DNA–protein complexes. Nucleic Acids Res. 2019, 48, D277–D287. [Google Scholar] [CrossRef]
- Luscombe, N.M.; Austin, S.E.; Berman, H.M.; Thornton, J.M. An overview of the structures of protein-DNA complexes. Genome Biol. 2000, 1, 1–37. [Google Scholar] [CrossRef]
- Luscombe, N.M. Amino acid-base interactions: A three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 2001, 29, 2860–2874. [Google Scholar] [CrossRef] [PubMed]
- Bacsi, S.G.; Hankinson, O. Functional Characterization of DNA-binding Domains of the Subunits of the Heterodimeric Aryl Hydrocarbon Receptor Complex Imputing Novel and Canonical Basic Helix-Loop-Helix Protein-DNA Interactions. J. Biol. Chem. 1996, 271, 8843–8850. [Google Scholar] [CrossRef]
- Kolonko, M.; Greb-Markiewicz, B. BHLH–PAS proteins: Their structure and intrinsic disorder. Int. J. Mol. Sci. 2019, 20, 3653. [Google Scholar] [CrossRef]
- Tarczewska, A.; Greb-Markiewicz, B. The significance of the intrinsically disordered regions for the functions of the bHLH transcription factors. Int. J. Mol. Sci. 2019, 20, 5036. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles that Govern the Folding of Protein Chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Uversky, V.N.; Dunker, A.K.; Kurgan, L. Introduction to intrinsically disordered proteins and regions. In Intrinsically Disordered Proteins; Elsevier: Oxford, UK, 2019; pp. 1–34. [Google Scholar] [CrossRef]
- Semenza, G.L.; Jiang, B.-H.; Leung, S.W.; Passantino, R.; Concordet, J.-P.; Maire, P.; Giallongo, A. Hypoxia Response Elements in the Aldolase A, Enolase 1, and Lactate Dehydrogenase A Gene Promoters Contain Essential Binding Sites for Hypoxia-inducible Factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef] [PubMed]
- Kolovos, P.; Knoch, T.A.; Grosveld, F.G.; Cook, P.R.; Papantonis, A. Enhancers and silencers: An integrated and simple model for their function. Epigenet. Chromatin 2012, 5, 1. [Google Scholar] [CrossRef]
- Weake, V.M.; Workman, J.L. Inducible gene expression: Diverse regulatory mechanisms. Nat. Rev. Genet. 2010, 11, 426–437. [Google Scholar] [CrossRef]
- Lu, X.J.; Olson, W.K. Resolving the discrepancies among nucleic acid conformational analyses. J. Mol. Biol. 1999, 285, 1563–1575. [Google Scholar] [CrossRef]
- Michael Gromiha, M.; Siebers, J.G.; Selvaraj, S.; Kono, H.; Sarai, A. Intermolecular and intramolecular readout mechanisms in protein-DNA recognition. J. Mol. Biol. 2004, 337, 285–294. [Google Scholar] [CrossRef]
- Sarai, A.; Mazur, J.; Nussinov, R.; Jernigan, R.L. Sequence dependence of DNA conformational flexibility. Biochemistry 1989, 28, 7842–7849. [Google Scholar] [CrossRef]
- Lavery, R.; Moakher, M.; Maddocks, J.H.; Petkeviciute, D.; Zakrzewska, K. Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 2009, 37, 5917–5929. [Google Scholar] [CrossRef]
- Djuranovic, D.; Hartmann, B. Conformational characteristics and correlations in crystal structures of nucleic acid oligonucleotides: Evidence for sub-states. J. Biomol. Struct. Dyn. 2003, 20, 771–788. [Google Scholar] [CrossRef] [PubMed]
- Olson, W.K.; Gorin, A.A.; Lu, X.-J.; Hock, L.M.; Zhurkin, V.B. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc. Natl. Acad. Sci. USA 1998, 95, 11163–11168. [Google Scholar] [CrossRef]
- Tolstorukov, M.Y.; Colasanti, A.V.; McCandlish, D.; Olson, W.K.; Zhurkin, C.B. A Novel “Roll-and-Slide” mechanism of DNA Folding in Chromatin. Implications for nucleosome positioning. J. Mol. Biol. 2007, 371, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, R.E.; Chiu, T.K. Helix Bending as a Factor in Protein/DNA Recognition. Biopolym. Biospectrosc. Sect. 1997, 44, 361–403. [Google Scholar] [CrossRef]
- Gu, C.; Zhang, J.; Yang, Y.I.; Chen, X.; Ge, H.; Sun, Y.; Su, X.; Yang, L.; Xie, S.; Gao, Y.Q. DNA Structural Correlation in Short and Long Ranges. J. Phys. Chem. B 2015, 119, 13980–13990. [Google Scholar] [CrossRef]
- Curuksu, J.; Zacharias, M.; Lavery, R.; Zakrzewska, K. Local and global effects of strong DNA bending induced during molecular dynamics simulations. Nucleic Acids Res. 2009, 37, 3766–3773. [Google Scholar] [CrossRef] [PubMed]
- Lavery, R.; Sklenar, H. Defining the structure of irregular nucleic acids: Conventions and principles. J. Biomol. Struct. Dyn. 1989, 6, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Zhurkin, V.B.; Lysov, Y.P.; Florentiev, V.L.; Ivanov, V.I. Torsional flexibility of DNA as revealed by conformational analysis. Nucleic Acids Res. 1982, 10, 1811–1830. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. [20] VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef]
- Honorato, R.V.; Roel-Touris, J.; Bonvin, A.M.J.J. MARTINI-Based Protein-DNA Coarse-Grained HADDOCKing. Front. Mol. Biosci. 2019, 6, 102. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, M.; Van Dijk, A.D.J.; Hsu, V.; Rolf, B.; Bonvin, A.M.J.J. Information-driven protein-DNA docking using HADDOCK: It is a matter of flexibility. Nucleic Acids Res. 2006, 34, 3317–3325. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef]
- Maciaszczyk-Dziubinska, E.; Reymer, A.; Kumar, N.V.; Białek, W.; Mizio, K.; Tamás, M.J.; Wysocki, R. The ancillary N-terminal region of the yeast AP-1 transcription factor Yap8 contributes to its DNA binding specificity. Nucleic Acids Res. 2020, 48, 5426–5441. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 2.4; Schrödinger, LLC. Available online: https://pymol.org/2/ (accessed on 3 October 2021).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MQAPs | 4ZPK | HIF1 |
|---|---|---|
| PROCHECK–Ramachandran Plot (1) | ||
| Most favorable regions | 83.10 | 87.20 |
| Additional allowed regions | 15.70 | 11.10 |
| Generously allowed regions | 0.80 | 0.90 |
| Disallowed regions | 0.40 | 0.80 |
| ERRAT ARNT-HIF1/2a (2) | 66.67–58.43 | 76.16–70.86 |
| Verify-3D (3) | 41.77 | 68.87 |
| QMEAN (4) | −2.44 | −2.77 |
| Terms | HIF1-HRE-964G | HIF1-HRE-964A |
|---|---|---|
| HS (a.u.) | −163.53 (−152.70 ± 6.60) | −147.90 (−140.60 ± 4.60) |
| Evw (kcal/mol) | −84.98 (−87.70 ± 3.90 | −84.37 (−81.00 ± 3.90) |
| Eelec (kcal/mol) | −536.02 (−478.20 ± 38.90) | −435.78 (−425.80 ± 23.10) |
| Eair (kcal/mol) | 3.68 (12.10 ± 13.00) | 4.70 (7.80 ± 3.00) |
| Edesol(kcal/mol) | 28.29 (29.40 ± 2.40) | 23.15 (24.80 ± 1.50) |
| BSA (Å) | 2195.65 (2241.2 ± 122.40) | 2050.90 (2065.30 ± 86.90) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alves, C.C.; Donadi, E.A.; Giuliatti, S. Structural Characterization of the Interaction of Hypoxia Inducible Factor-1 with Its Hypoxia Responsive Element at the −964G > A Variation Site of the HLA-G Promoter Region. Int. J. Mol. Sci. 2021, 22, 13046. https://doi.org/10.3390/ijms222313046
Alves CC, Donadi EA, Giuliatti S. Structural Characterization of the Interaction of Hypoxia Inducible Factor-1 with Its Hypoxia Responsive Element at the −964G > A Variation Site of the HLA-G Promoter Region. International Journal of Molecular Sciences. 2021; 22(23):13046. https://doi.org/10.3390/ijms222313046
Chicago/Turabian StyleAlves, Cinthia C., Eduardo A. Donadi, and Silvana Giuliatti. 2021. "Structural Characterization of the Interaction of Hypoxia Inducible Factor-1 with Its Hypoxia Responsive Element at the −964G > A Variation Site of the HLA-G Promoter Region" International Journal of Molecular Sciences 22, no. 23: 13046. https://doi.org/10.3390/ijms222313046
APA StyleAlves, C. C., Donadi, E. A., & Giuliatti, S. (2021). Structural Characterization of the Interaction of Hypoxia Inducible Factor-1 with Its Hypoxia Responsive Element at the −964G > A Variation Site of the HLA-G Promoter Region. International Journal of Molecular Sciences, 22(23), 13046. https://doi.org/10.3390/ijms222313046