
Role of Receptors in Relation to Plaques and Tangles in Alzheimer’s Disease Pathology

 ,
,  ,
,
Abstract
:1. Introduction
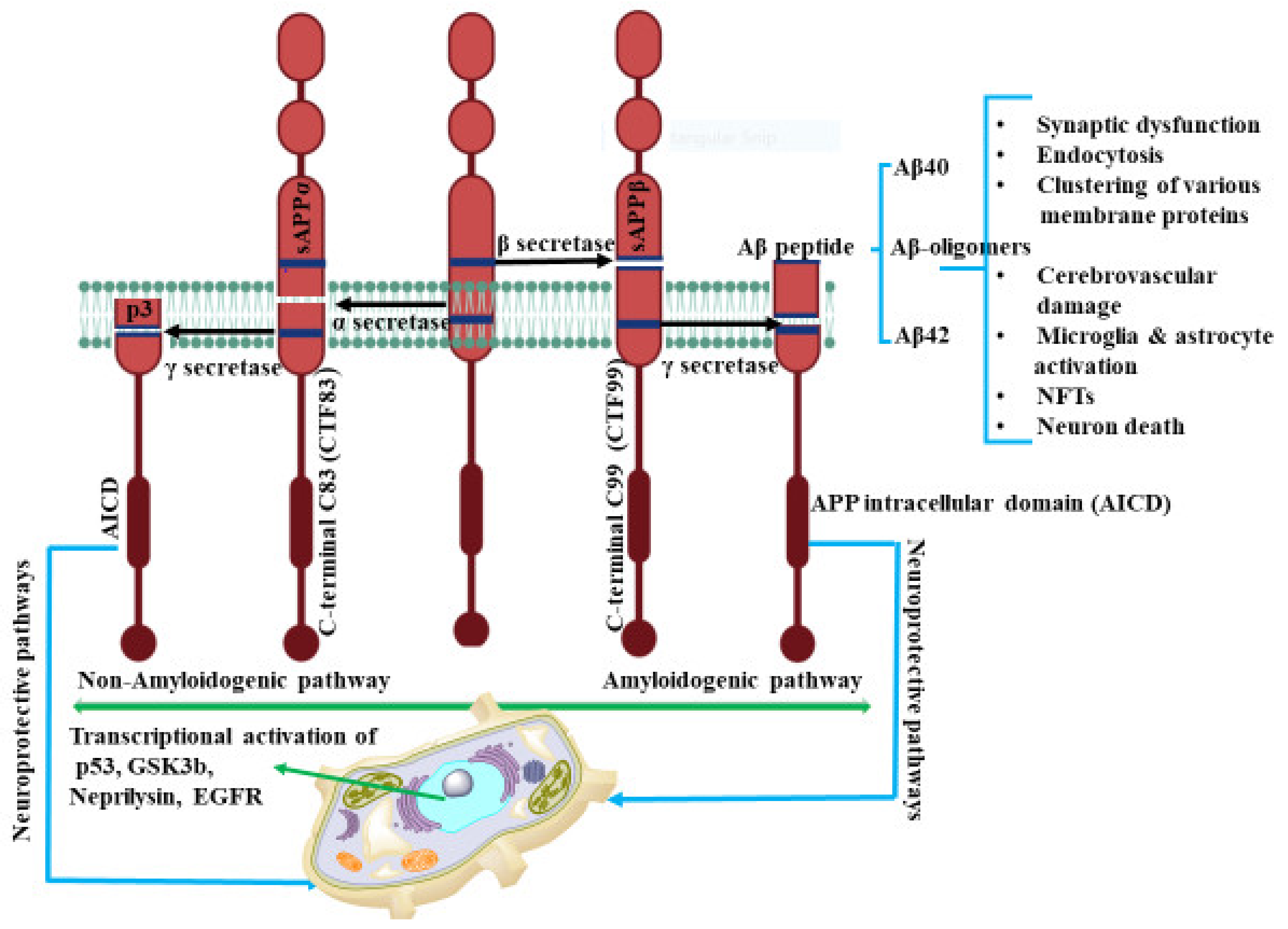
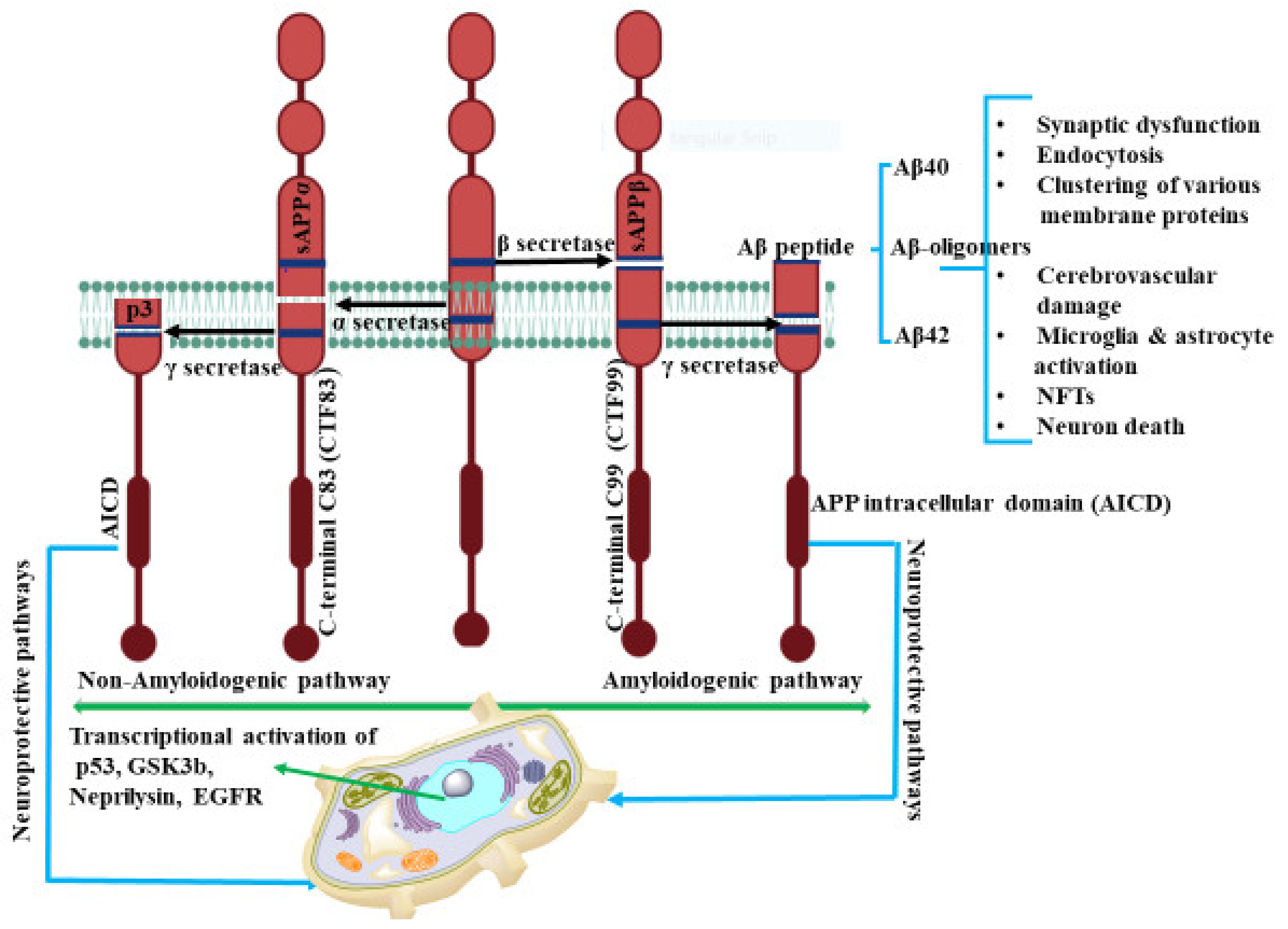
2. Amyloid Beta (Aβ) Formation
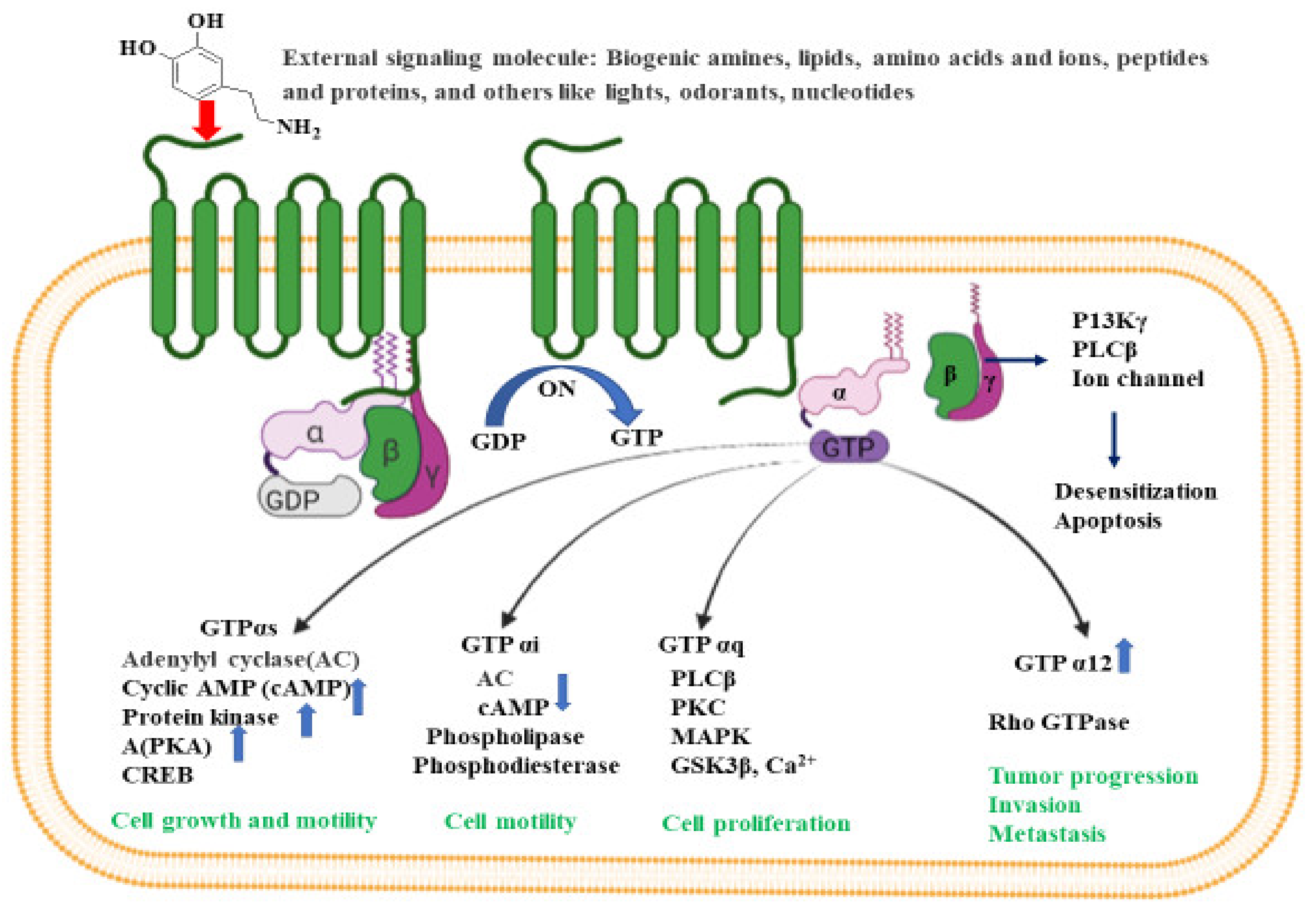
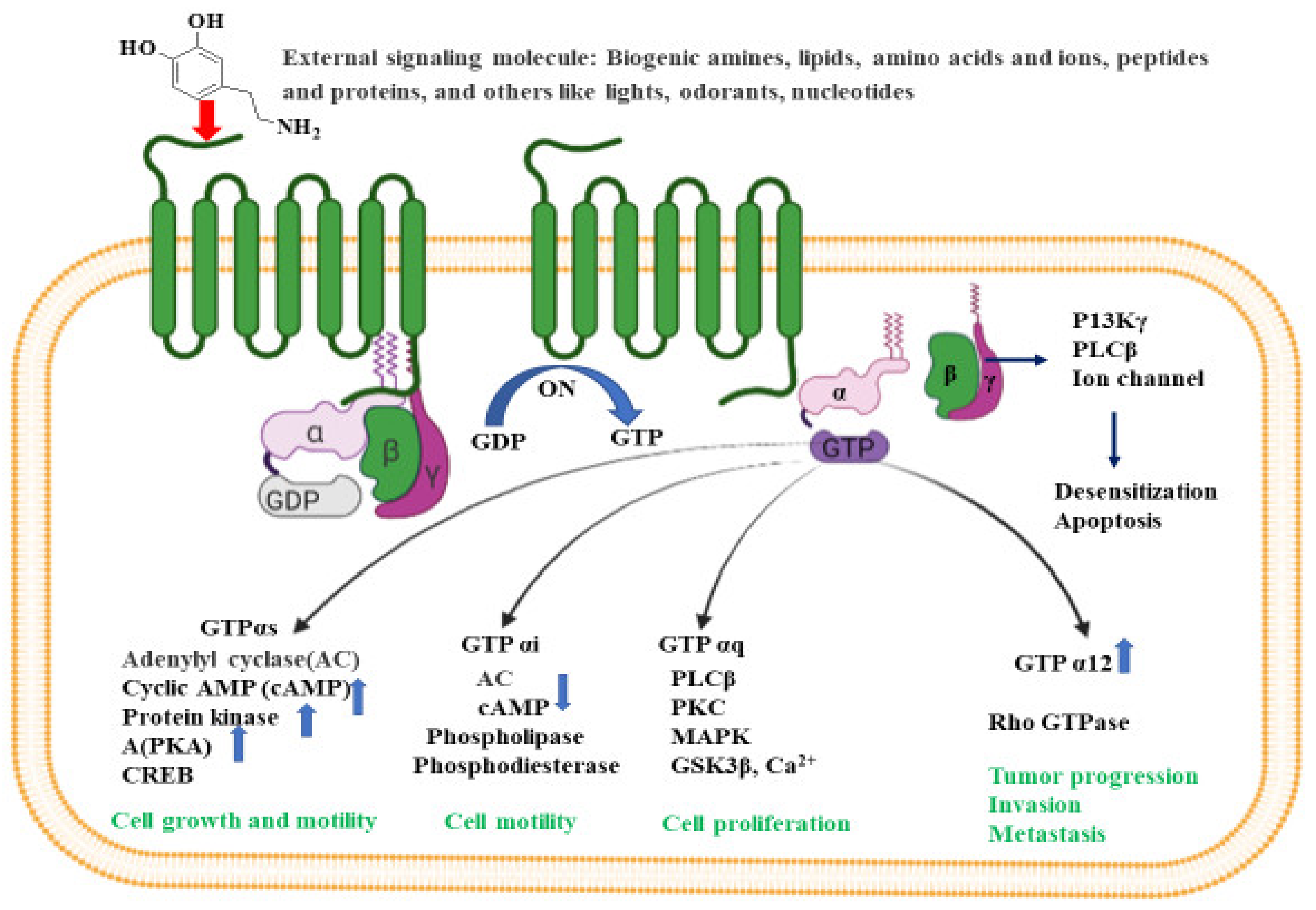
3. G-Protein-Coupled Receptors
4. N-Methyl D-Aspartate (NMDA) Receptors
5. α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid Receptor (AMPAR)
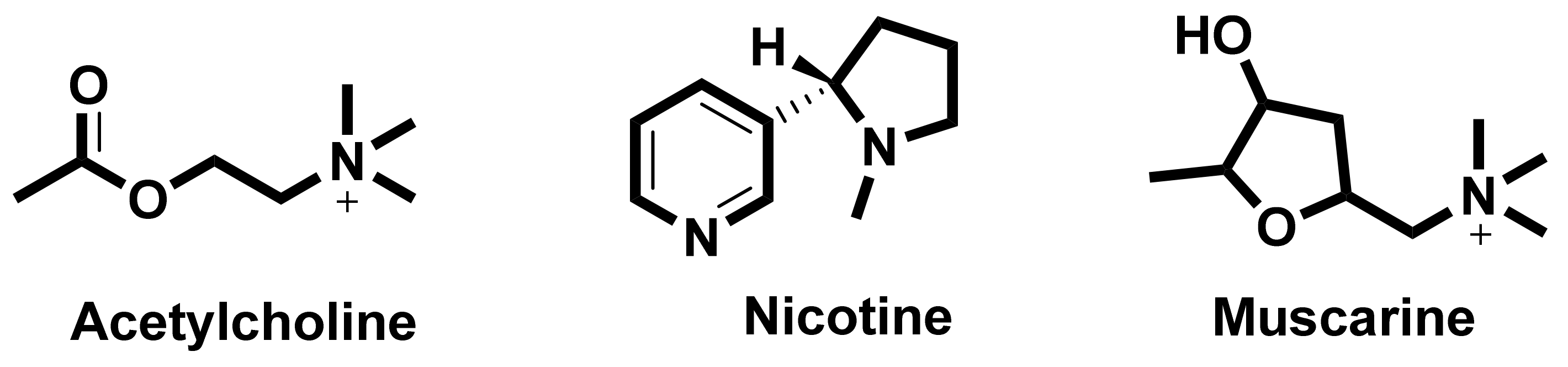
6. Cholinergic Receptors
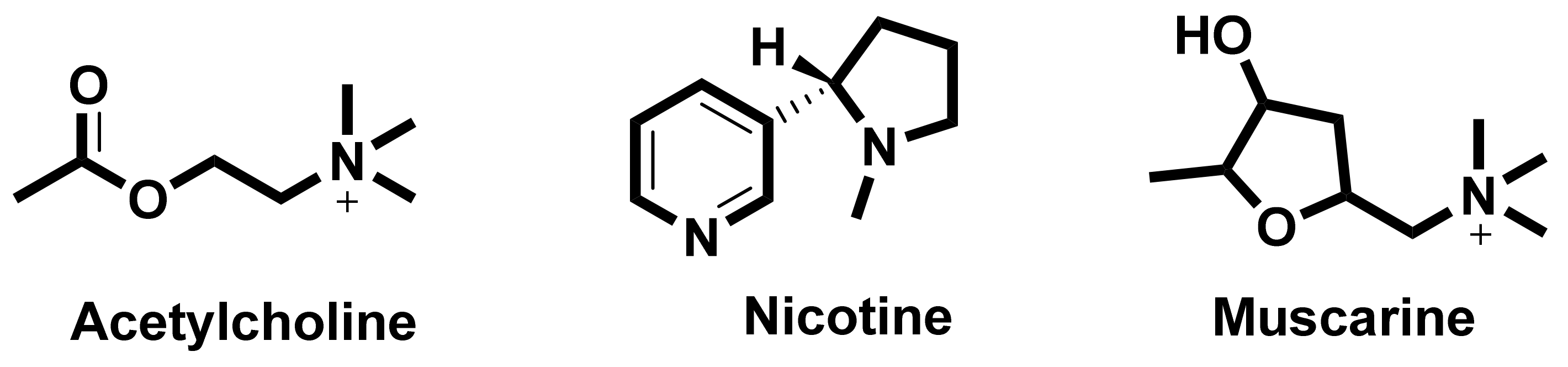
6.1. Nicotinic Acetylcholine Receptors (nAChRs)
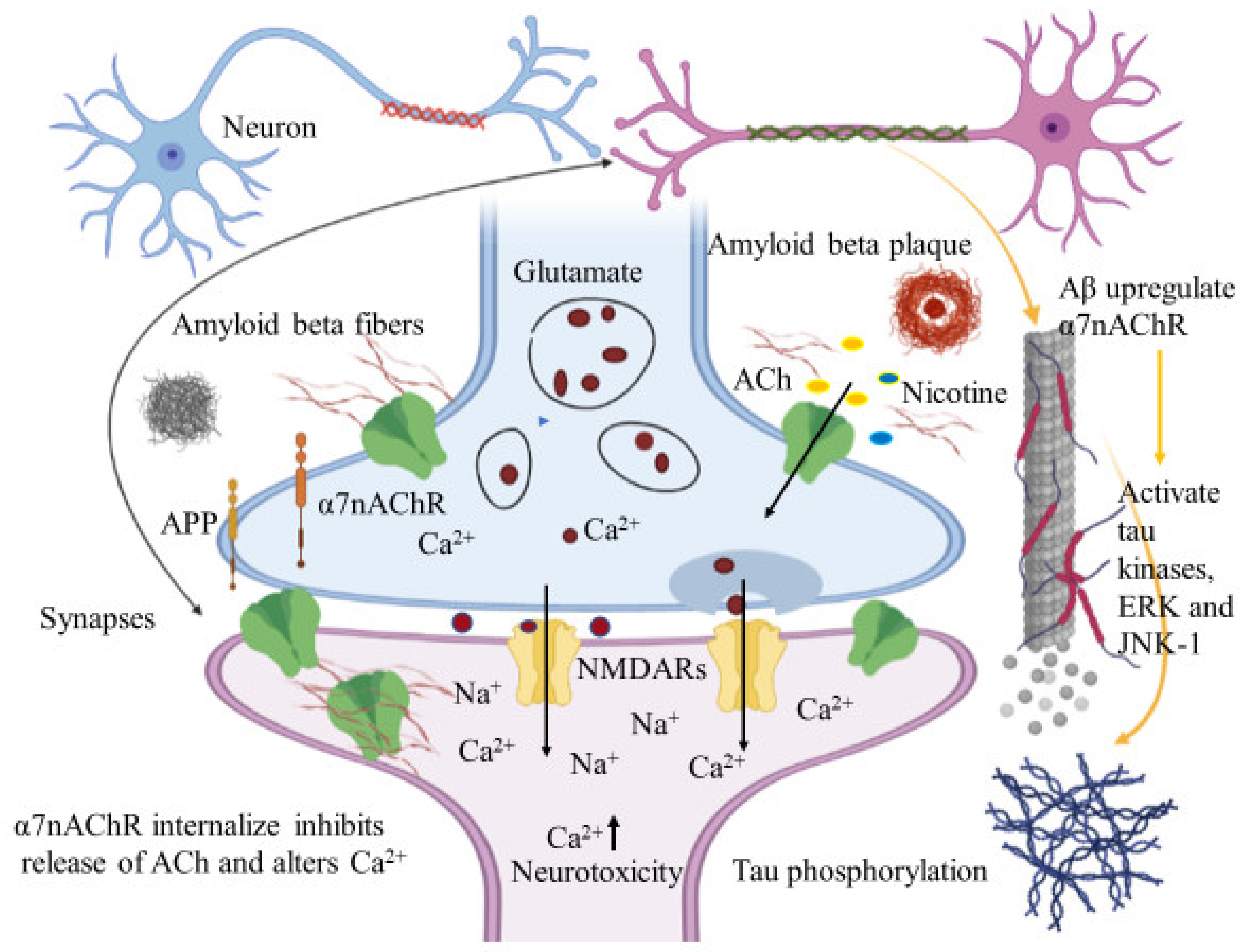
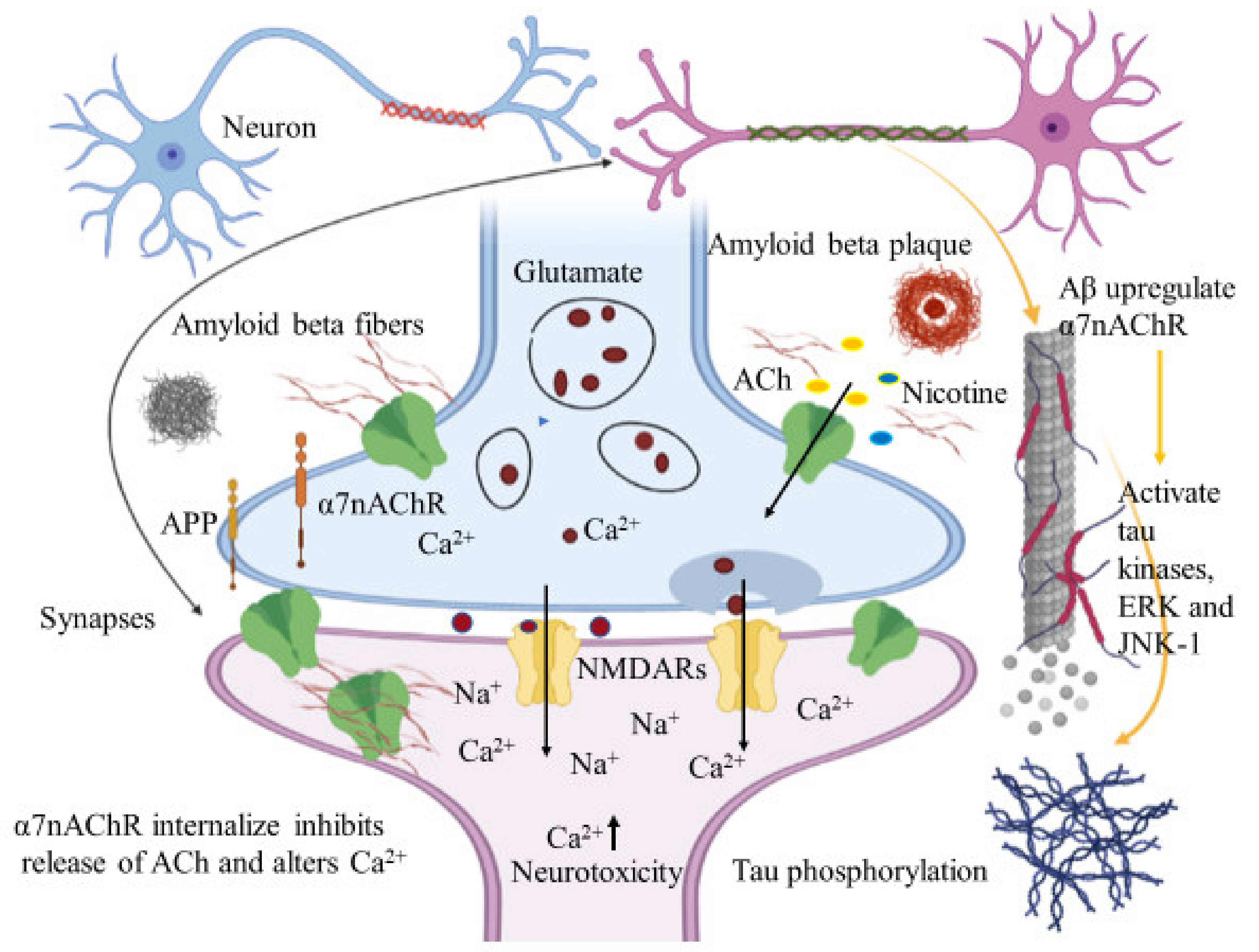
6.1.1. Interaction between Amyloid Beta and α7nAChRs
6.1.2. Allosteric Modulation of nAChRs
6.2. Muscarinic Acetylcholine Receptors
7. Gamma Amino Butyric Acid Receptors
Relation between Amyloid Beta, Tau Formation and GABAs
8. 5-Hydroxytryptamine Receptors
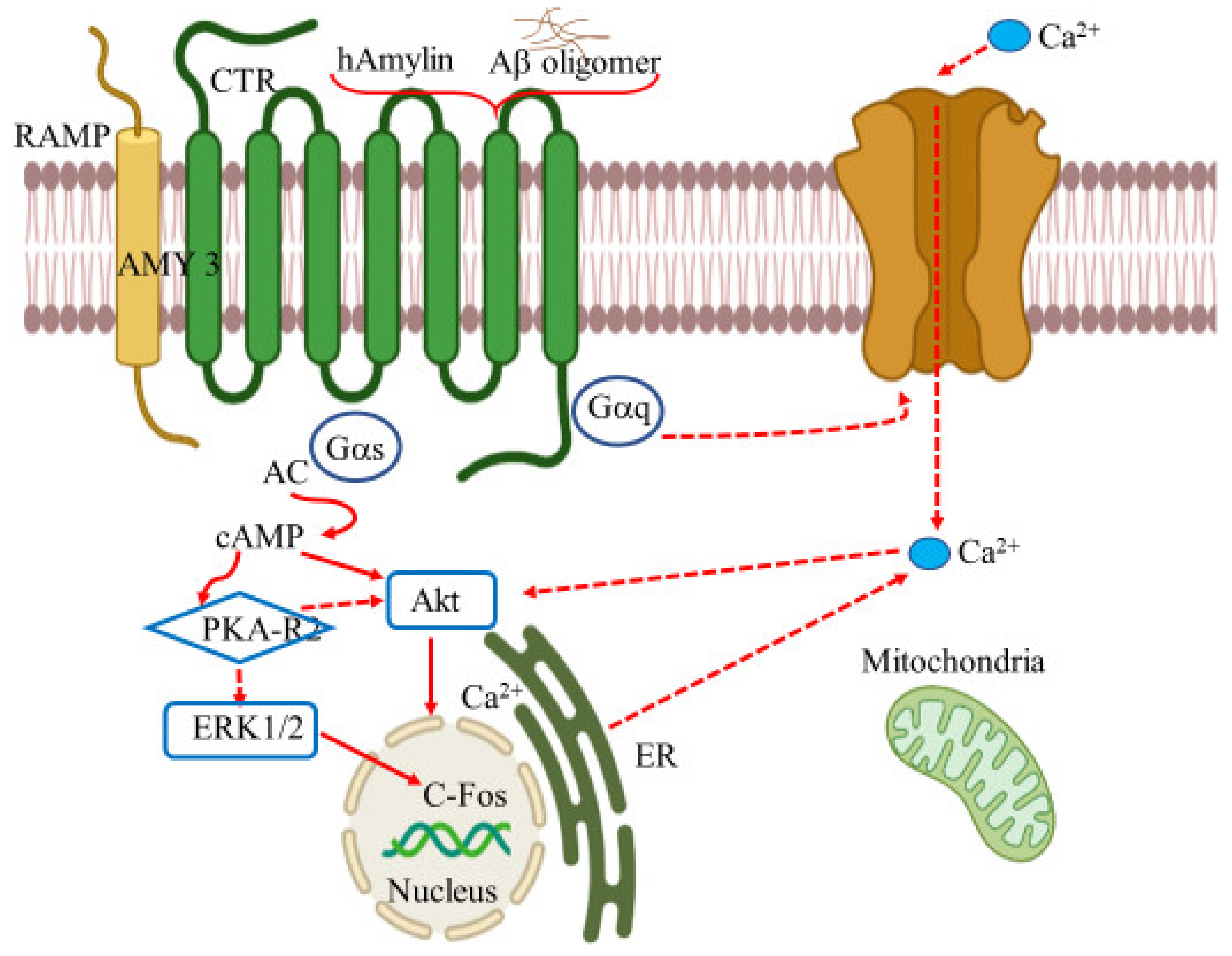
9. Amylin Receptors
10. Netrin Receptors
11. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| Aβ | Amyloid-β proteins |
| APP | Amyloid-β precursor protein |
| NFTs | Neurofibrillary tangles |
| CNS | Central nervous system |
| PNS | Peripheral nervous system |
| MTs | Microtubules |
| ROS | Reactive oxygen species |
| PHFs | Paired helical filaments |
| nAChRs | Nicotinic acetylcholine receptors |
| mAChRs | Muscarinic acetylcholine receptors |
| ACh | Acetylcholine |
| sAPPα | Soluble amyloid precursor protein–α |
| CTF | C-terminal fragment |
| AICD | APP intracellular domain |
| PI3K | Phosphatidylinositol 3 kinase |
| MAPK | Mitogen activated protein kinase |
| ERK | Extracellular signal regulated kinase |
| PKC | Protein kinase C |
| cAMP-PKA | Cyclic AMP–protein kinase A |
| LTD | Long-term depression |
| LTP | Long-term potentiation |
| CSF | Cerebrospinal fluid |
| BACE | β-Site APP cleaving enzyme |
| ADDLs | Amyloid-β-derived diffused ligands |
| NMDA | N-Methyl-D-aspartate |
| AβOs | Aβ oligomers |
| GPCRs | G-protein-coupled receptors |
| cAMP | Cyclic adenosine monophosphate |
| DAG | Diacylglycerol |
| IP3 | Inositol 1,4,5-triphosphate |
| GSK3β | Glycogen synthase kinase 3β |
| 5-HT | Serotonin; 5 hydroxytryptamine |
| NP | Norepinephrine |
| GAPs | GTPase activating proteins |
| AChE | Acetylcholinesterase |
| AChEi | Acetylcholinesterase inhibitors |
| α-BTX | α-Bungarotoxin |
| JNK-1 | c-Jun N-terminal kinase |
| VDCCs | Voltage-dependent calcium channels |
| GABA | γ-Aminobutyric acid |
| GAD | Glutamic acid decarboxylase |
| vGAT | Vesicular GABA transporter |
| KCC2 | Potassium chloride co-transporter 2 |
| CGN | Cerebellar granule neuron |
| ERK/mTOR | Extracellular-signal-regulated kinase/ mechanistic target of rapamycin |
| PP2A | Protein phosphatase 2A |
| Cdk5 | Cyclin-dependent kinase 5 |
| DG | Dentate gyrus |
| PLC | Phospholipase C |
| SSRI | Serotonin reuptake inhibitors |
| IAPP | Islet amyloid polypeptide |
| RAMP | Receptor activity modifying protein |
| CT | Calcitonin |
| T2DM | Type 2 diabetes mellitus |
| AMY3 | Amylin-3 receptor |
| CTR | Calcitonin receptor |
| HFNs | Human fetal neurons |
| TRPV4 | Transient receptor potential cation channel subfamily V member 4 |
| LTAD | Late-onset Alzheimer’s disease |
| RGP | Rabies virus glycoprotein |
| 3FTx | Three-finger toxin |
References
- Alzheimer Disease International. Available online: https://www.alzint.org/about/dementia-facts-figures/ (accessed on 23 May 2021).
- World Alzheimer Report 2018. Available online: https://www.alzint.org/u/WorldAlzheimerReport2018.pdf (accessed on 22 June 2021).
- Lanctôt, K.L.; Amatniek, J.; Ancoli-Israel, S.; Arnold, S.E.; Ballard, C.; Cohen-Mansfield, J.; Ismail, Z.; Lyketsos, C.; Miller, D.S.; Musiek, E.; et al. Neuropsychiatric signs and symptoms of Alzheimer’s disease: New treatment paradigms. Alzheimer’s Dement. 2017, 3, 440–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reitz, C. Alzheimer’s disease and the amyloid cascade hypothesis: A critical review. Int. J. Alzheimer’s Dis. 2012, 2012, 369808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contestabile, A. The history of the cholinergic hypothesis. Behav. Brain Res. 2011, 221, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.X.; Iqbal, K. Hyperphosphorylation of microtubule-associated protein tau: A promising therapeutic target for Alzheimer disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.N. Simultaneous activation of Nrf2 and elevation of antioxidant compounds for reducing oxidative stress and chronic inflammation in human Alzheimer’s disease. Mech. Ageing Dev. 2016, 153, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.M.; Harrison, F.E. Shared Neuropathological Characteristics of Obesity, Type 2 Diabetes and Alzheimer’s Disease: Impacts on Cognitive Decline. Nutrients 2015, 7, 7332–7357. [Google Scholar] [CrossRef] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Thathiah, A.; De Strooper, B. The role of G protein-coupled receptors in the pathology of Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 73–87. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Lanoiselée, H.-M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.F.; Xu, T.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau protein hyperphosphorylation and aggregation in alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindwall, G.; Cole, R.D. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [CrossRef]
- Shin, R.W.; Iwaki, T.; Kitamoto, T.; Tateishi, J. Hydrated autoclave pretreatment enhances tau immunoreactivity in formalin-fixed normal and Alzheimer’s disease brain tissues. Lab. Investig. 1991, 64, 693–702. [Google Scholar]
- Pierre, M.; Nunez, J. Multisite phosphorylation of τ proteins from rat brain. Biochem. Biophys. Res. Commun. 1983, 115, 212–219. [Google Scholar] [CrossRef]
- Soeda, Y.; Takashima, A. New insights into drug discovery targeting tau protein. Front. Mol. Neurosci. 2020, 13, 590896. [Google Scholar] [CrossRef]
- Itzhaki, R.F. Corroboration of a Major Role for Herpes Simplex Virus Type 1 in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hueffer, K.; Khatri, S.; Rideout, S.; Harris, M.B.; Papke, R.L.; Stokes, C.; Schulte, M.K. Rabies virus modifies host behavior through a snake-toxin like region of its glycoprotein that inhibits neurotransmitter receptors in the CNS. Sci. Rep. 2017, 7, 12818. [Google Scholar] [CrossRef]
- Neri, P.; Bracci, L.; Rustici, M.; Santucci, A. Sequence Homology between HIV gp120, Rabies Virus Glycoprotein, and Snake Venom Neurotoxins. Arch. Virol. 1990, 114, 265–269. [Google Scholar] [CrossRef]
- Fulop, T.; Witkowski, J.M.; Larbi, A.; Khalil, A.; Herbein, G.; Frost, E.H. Does HIV Infection Contribute to Increased Beta-Amyloid Synthesis and Plaque Formation Leading to Neurodegeneration and Alzheimer’s Disease? J. Neurovirol. 2019, 25, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. Beta-amyloid and the amyloid hypothesis. Available online: https://www.alz.org/national/documents/topicsheet_betaamyloid.pdf (accessed on 23 May 2021).
- Hartmann, T.; Bieger, S.; Brühl, B.; Tienari, P.J.; Ida, N.; Allsop, D.; Roberts, G.W.; Masters, C.L.; Dotti, C.G.; Unsicker, K.; et al. Distinct sites of intracellular production for Alzheimer’s disease Aβ40/42 amyloid peptides. Nat. Med. 1997, 3, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.R.; O’Connor, K.; Tate, W.P.; Abraham, W.C. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog. Neurobiol. 2003, 70, 1–32. [Google Scholar] [CrossRef]
- Mills, J.; Reiner, P.B. Regulation of amyloid precursor protein cleavage. J. Neurochem. 1999, 72, 443–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solano, D.C.; Sironi, M.; Bonfini, C.; Solerte, S.B.; Govoni, S.; Racchi, M. Insulin regulates soluble amyloid precursor protein release via phosphatidyl inositol 3 kinase-dependent pathway. FASEB J. 2000, 14, 1015–1022. [Google Scholar] [CrossRef]
- Mills, J.; Charest, D.L.; Lam, F.; Beyreuther, K.; Ida, N.; Pelech, S.L.; Reiner, P.B. Regulation of amyloid precursor protein catabolism involves the mitogen-activated protein kinase signal transduction pathway. J. Neurosci. Res. 1997, 17, 9415–9422. [Google Scholar] [CrossRef]
- Skovronsky, D.M.; Moore, D.B.; Milla, M.E.; Doms, R.W.; Lee, V.M.Y. Protein kinase C-dependent α-secretase competes with β-secretase for cleavage of amyloid-β precursor protein in the trans-Golgi network. J. Biol. Chem. 2000, 275, 2568–2575. [Google Scholar] [CrossRef] [Green Version]
- Robert, S.J.; Zugaza, J.L.; Fischmeister, R.; Gardier, A.M.; Lezoualc’h, F. The human serotonin 5-HT4 receptor regulates secretion of nonamyloidogenic precursor protein. J. Biol. Chem. 2001, 276, 44881–44888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, A.; Sawmiller, D.; Tan, J. Restoring Soluble Amyloid Precursor Protein α Functions as a Potential Treatment for Alzheimer’s Disease. J. Neurosci. Res. 2017, 95, 973–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, A.; Furukawa, K.; Keller, J.N.; Mattson, M.P. Secreted form of beta-amyloid precursor protein shifts the frequency dependency for induction of LTD, and enhances LTP in hippocampal slices. Neuroreport 1997, 8, 2133–2137. [Google Scholar] [CrossRef] [PubMed]
- Sennvik, K.; Fastbom, J.; Blomberg, M.; Wahlund, L.O.; Winblad, B.; Benedikz, E. Levels of alpha- and beta-secretase cleaved amyloid precursor protein in the cerebrospinal fluid of Alzheimer’s disease patients. Neurosci. Lett. 2000, 278, 169–172. [Google Scholar] [CrossRef]
- Obregon, D.; Hou, H.; Deng, J.; Giunta, B.; Tian, J.; Darlington, D.; Shahaduzzaman, M.; Zhu, Y.; Mori, T.; Mattson, M.P.; et al. Soluble amyloid precursor protein-α modulates β-secretase activity and amyloid-β generation. Nat. Commun. 2012, 3, 777. [Google Scholar] [CrossRef] [PubMed]
- Nunan, J.; Small, D.H. Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Kimberly, W.T.; LaVoie, M.J.; Ostaszewski, B.L.; Ye, W.; Wolfe, M.S.; Selkoe, D.J. γ-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc. Natl. Acad. Sci. USA 2003, 100, 6382–6387. [Google Scholar] [CrossRef] [Green Version]
- Haass, C. Take five-BACE and the c-secretase quartet conduct Alzheimer’s amyloid β-peptide generation. EMBO J. 2004, 23, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Westaway, D.; Xia, W.; Carlson, G.; Diehl, T.; Levesque, G.; Johnson-Wood, K.; Lee, M.; Seubert, P.; Davis, A.; et al. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat. Med. 1997, 3, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Landmark Alzheimer’s drug approval confounds research community. Nature 2021, 594, 309–310. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, A.S.; Velasco, P.T.; Wood, M.; Viola, K.L.; Klein, W.L. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Umeda, T.; Ramser, E.M.; Yamashita, M.; Nakajima, K.; Mori, H.; Silverman, M.A.; Tomiyama, T. Intracellular amyloid β oligomers impair organelle transport and induce dendritic spine loss in primary neurons. Acta Neuropathol. Commun. 2015, 3, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalano, S.M.; Dodson, E.C.; Henze, D.A.; Joyce, J.G.; Krafft, G.A.; Kinney, G.G. The role of amyloid-beta derived diffusible ligands (ADDLs) in Alzheimer’s disease. Curr. Top. Med. Chem. 2006, 6, 597–608. [Google Scholar] [CrossRef]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, K.G.; Qian, Y.H. Alpha 7 nicotinic acetylcholine receptor and its effects on Alzheimer’s disease. Neuropeptides 2019, 73, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Bockaert, J.; Pin, J.P. Molecular tinkering of G protein-coupled receptors: An evolutionary success. EMBO J. 1999, 18, 1723–1729. [Google Scholar] [CrossRef] [Green Version]
- Hamm, H.E. The many faces of G protein signaling. J. Biol. Chem. 1998, 273, 669–672. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ning, Y.; Hedley, W.; Saunders, B.; Chen, Y.; Tindill, N.; Hannay, T.; Subramaniam, S. The molecule pages database. Nature 2002, 420, 716–717. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef] [Green Version]
- Urs, N.M.; Nicholls, P.J.; Caron, M.G. Integrated approaches to understanding antipsychotic drug action at GPCRs. Curr. Opin. Cell Biol. 2014, 27, 56–62. [Google Scholar] [CrossRef]
- Teng, L.; Zhao, J.; Wang, F.; Ma, L.; Pei, G. A GPCR/secretase complex regulates β-and γ-secretase specificity for Aβ production and contributes to AD pathogenesis. Cell Res. 2010, 20, 138–153. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.C. Role of GPCR signaling and calcium dysregulation in Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 101, 103414. [Google Scholar]
- Haque, M.; Kim, I.S.; Jakaria, M.; Akther, M.; Choi, D.K. Importance of GPCR-mediated microglial activation in Alzheimer’s disease. Front. Cell. Neurosci. 2018, 12, 258. [Google Scholar] [CrossRef] [PubMed]
- Suo, Z.; Wu, M.; Citron, B.A.; Wong, G.T.; Festoff, B.W. Abnormality of G-protein-coupled receptor kinases at prodromal and early stages of Alzheimer’s disease: An association with early beta-amyloid accumulation. J. Neurosci. 2004, 24, 3444–3452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, K.J.; Squires, K.E.; Hepler, J.R. Roles for regulator of G protein signaling proteins in synaptic signaling and plasticity. Mol. Pharmacol. 2016, 89, 273–286. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Hagino, Y.; Kasai, S.; Ikeda, K. Specific Roles of NMDA Receptor Subunits in Mental Disorders. Curr. Mol. Med. 2015, 15, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, H.; Singh, S.K.; Mancusso, R.; Gouaux, E. Subunit Arrangement and Function in NMDA Receptors. Nature 2005, 438, 185–192. [Google Scholar] [CrossRef]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, P.; Feng, J.; Wu, M. Dysfunction of NMDA receptors in Alzheimer’s disease. Neurol. Sci. 2016, 37, 1039–1047. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.C.; Zhao, J.; Li, S. Dysregulation of Synaptic and Extrasynaptic N-Methyl-d-Aspartate Receptors Induced by Amyloid-β. Neurosci. Bull. 2013, 29, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Rush, T.; Buisson, A. Reciprocal disruption of neuronal signaling and Aβ production mediated by extrasynaptic NMDA receptors: A downward spiral. Cell Tissue Res. 2014, 356, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat. Rev. Drug Discov. 2006, 5, 160–170. [Google Scholar] [CrossRef]
- Bleich, S.; Römer, K.; Wiltfang, J.; Kornhuber, J. Glutamate and the glutamate receptor system: A target for drug action. Int. J. Geriatr. Psychiatry 2003, 18, S33–S40. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Chen, H.S.; Zhang, D.; Lipton, S.A. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J. Neurosci. 2010, 30, 11246–11250. [Google Scholar] [CrossRef] [Green Version]
- Kuns, B.; Rosani, A.; Varghese, D. MemantineIn. In StatPearls; 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK500025/ (accessed on 23 May 2021).
- Dekundy, A. Coadministration of memantine with acetylcholinesterase inhibitors: Preclinical and clinical evidence. In Toxicology of Organophosphate & Carbamate Compounds; Academic Press: Cambridge, MA, USA, 2006; pp. 35–46. [Google Scholar]
- Robinson, D.M.; Keating, G.M. Memantine: A review of its use in Alzheimer’s disease. Drugs 2006, 66, 1515–1534. [Google Scholar] [CrossRef]
- Folch, J.; Busquets, O.; Ettcheto, M.; Sánchez-López, E.; Castro-Torres, R.D.; Verdaguer, E.; Garcia, M.L.; Olloquequi, J.; Casadesús, G.; Beas-Zarate, C.; et al. Memantine for the treatment of dementia: A review on its current and future applications. J. Alzheimer’s Dis. 2018, 62, 1223–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.S.; Lipton, S.A. Pharmacological implications of two distinct mechanisms of interaction of memantine with N-methyl-d-aspartate-gated channels. J. Pharmacol. Exp. Ther. 2005, 314, 961–997. [Google Scholar] [CrossRef] [Green Version]
- Henley, J.; Wilkinson, K. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 2016, 17, 337–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Shi, Y.; Jackson, A.C.; Bjorgan, K.; During, M.J.; Sprengel, R.; Seeburg, P.H.; Nicoll, R.A. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 2009, 62, 254–268. [Google Scholar] [CrossRef] [Green Version]
- Salpietro, V.; Dixon, C.L.; Guo, H.; Bello, O.D.; Vandrovcova, J.; Efthymiou, S.; Maroofian, R.; Heimer, G.; Burglen, L.; Valence, S.; et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat Commun. 2019, 10, 3094. [Google Scholar] [CrossRef] [Green Version]
- Guntupalli, S.; Widagdo, J.; Anggono, V. Amyloid-β-Induced Dysregulation of AMPA Receptor Trafficking. Neural Plast. 2016, 2016, 3204519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.H.; Savage, M.J.; Flood, D.G.; Thomas, J.M.; Levy, R.B.; Mahadomrongkul, V.; Shirao, T.; Aoki, C.; Huerta, P.T. AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3410–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Guo, O.; Huo, Y.; Wang, G.; Man, H.Y. Amyloid-β Induces AMPA Receptor Ubiquitination and Degradation in Primary Neurons and Human Brains of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Reinders, N.R.; Pao, Y.; Renner, M.C.; da Silva-Matos, C.M.; Lodder, T.R.; Malinow, R.; Kessels, H.W. Effects of Amyloid-β Require AMPAR Subunit GluA3. Proc. Natl. Acad. Sci. USA 2016, 113, E6526–E6534. [Google Scholar] [CrossRef] [Green Version]
- Carlson, A.B.; Kraus, G.P. Physiology, Cholinergic Receptors. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK526134/ (accessed on 6 September 2020).
- Maurer, S.V.; Williams, L. The Cholinergic System Modulates Memory and Hippocampal Plasticity via Its Interactions with Non-Neuronal Cells. Front. Immunol. 2017, 8, 1489. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, D.; Wallace, T.L. A Review of the Cholinergic System and Therapeutic Approaches to Treat Brain Disorders. Curr. Top. Behav. Neurosci. 2020, 45, 1–28. [Google Scholar]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akıncıoğlu, H.; Gülçin, İ. Potent acetylcholinesterase inhibitors: Potential drugs for Alzheimer’s disease. Mini-Rev. Med. Chem. 2020, 20, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Rosengarten, B.; Paulsen, S.; Burr, O.; Kaps, M. Neurovascular coupling in Alzheimer patients: Effect of acetylcholine-esterase inhibitors. Neurobiol. Aging 2009, 30, 1918–1923. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, G.; Sancarlo, D.; Ruan, Q.; Yu, Z.; Panza, F.; Daniele, A.; Greco, A.; Seripa, D. Phytochemicals in the treatment of alzheimer’s disease: A systematic review. Curr. Drug Targets 2017, 18, 1487–1489. [Google Scholar] [CrossRef] [PubMed]
- Karlin, A.; Akabas, M.H. Toward a structural basis for the function of nicotinic Acetylcholine receptors and their cousins. Neuron 1995, 15, 1231–1244. [Google Scholar] [CrossRef] [Green Version]
- Hogg, R.C.; Raggenbass, M.; Bertrand, D. Nicotinic acetylcholine receptors: From structure to brain function. Rev. Physiol. Biochem. Pharmacol. 2003, 147, 1–46. [Google Scholar] [PubMed]
- Lykhmus, O.; Voytenko, L.; Koval, L. α7 Nicotinic acetylcholine receptor-specific antibody induces inflammation and amyloid β42 accumulation in the mouse brain to impair memory. PLoS ONE 2015, 10, e0122706. [Google Scholar] [CrossRef]
- Mitra, S.; Khatri, S.N.; Maulik, M.; Bult-Ito, A.; Schulte, M. Allosterism of Nicotinic Acetylcholine Receptors: Therapeutic Potential for Neuroinflammation Underlying Brain Trauma and Degenerative Disorders. Int. J. Mol. Sci. 2020, 21, 4918. [Google Scholar] [CrossRef]
- Sharma, G.; Vijayaraghavan, S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc. Natl. Acad. Sci. USA 2001, 98, 4148–4153. [Google Scholar] [CrossRef] [Green Version]
- De Jonge, W.J.; Ulloa, L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br. J. Pharmacol. 2007, 151, 915–929. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Wu, J. Nicotinic cholinergic mechanisms in Alzheimer’s disease. Int. Rev. Neurobiol. 2015, 124, 275–292. [Google Scholar]
- Liu, Q.; Xie, X.; Emadi, S.; Sierks, M.R.; Wu, J. A novel nicotinic mechanism underlies beta-amyloid induced neurotoxicity. Neuropharmacology 2015, 97, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Li, W.; Benedetti, N.J.; Li, D.H.S. Alpha 7 nicotinic acetylcholine receptors mediate beta-amyloid peptide-induced tau protein phosphorylation. J. Biol. Chem. 2003, 278, 31547–31553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Xie, X.; Lukas, R.J.; John, P.A.S.; Wu, J. A novel nicotinic mechanism underlies beta-amyloidinduced neuronal hyperexcitation. J. Neurosci. 2013, 33, 7253–7263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dineley, K.T.; Westerman, M.; Bui, D.; Bell, K.; Hsiao Ashe, K.; Sweatt, J.D. β-amyloid activates the mitogen-activated protein kinase cascade via hippocampal a7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer’s disease. J. Neurosci. 2001, 21, 4125–4133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.Y.; Lee, D.H.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. β-Amyloid1–42 binds to α7 nicotinic acetylcholine receptor with high affinity: Implications for Alzheimer’s disease pathology. J. Biol. Chem. 2000, 275, 5626–5632. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Hasegawa, K.; Yamada, M.; Naiki, H. Nicotine breaks down preformed Alzheimer’s β-amyloid fibrils in vitro. Biol. Psychiatry 2002, 52, 880–886. [Google Scholar] [CrossRef]
- Nordberg, A.; Hellström-Lindahl, E.; Lee, M.; Johnson, M.; Mousavi, M.; Hall, R.; Perry, E.; Bednar, I.; Court, J. Chronic nicotine treatment reduces β-amyloidosis in the brain of a mouse model of Alzheimer’s disease (APPsw). J. Neurochem. 2002, 81, 655–658. [Google Scholar] [CrossRef]
- Geula, C.; Mesulam, M. Special properties of cholinesterases in the cerebral cortex of Alzheimer’s disease. Brain Res. 1989, 498, 185–189. [Google Scholar] [CrossRef]
- Sberna, G.; Sáez-Valero, J.; Beyreuther, K.; Masters, C.L.; Small, D.H. The amyloid beta-protein of Alzheimer’s disease increases acetylcholinesterase expression by increasing intracellular calcium in embryonal carcinoma P19 cells. J. Neurochem. 1997, 69, 1177–1184. [Google Scholar] [CrossRef]
- Trang, A.; Khandhar, P.B. Physiology, Acetylcholinesterase. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK539735/ (accessed on 23 May 2021).
- Fodero, L.R.; Mok, S.S.; Losic, D.; Martin, L.L.; Aguilar, M.I.; Barrow, C.J.; Livett, B.G.; Small, D.H. α7-Nicotinic acetylcholine receptors mediate an Aβ1−42-induced increase in the level of acetylcholinesterase in primary cortical neurones. J. Neurochem. 2004, 88, 1186–1193. [Google Scholar] [CrossRef]
- Lazarevic-Pasti, T.; Leskovac, A.; Momic, T.; Petrovic, S.; Vasic, V. Modulators of acetylcholinesterase activity: From Alzheimer’s disease to anti-cancer drugs. Curr. Med. Chem. 2017, 24, 3283–3309. [Google Scholar] [CrossRef]
- Liu, Q.; Kawai, H.; Berg, D.K. Beta-amyloid peptide blocks the response of alpha 7-containing nicotinic receptors on hippocampal neurons. Proc. Natl. Acad. Sci. USA 2001, 98, 4734–4739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puzzo, D.; Privitera, L.; Leznik, E.; Fà, M.; Staniszewski, A.; Palmeri, A.; Arancio, O. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 2008, 28, 14537–14545. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2011, 11, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Shytle, R.D.; Mori, T.; Townsend, K.; Vendrame, M.; Sun, N.; Zeng, J.; Ehrhart, J.; Silver, A.A.; Sanberg, P.R.; Tan, J. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J. Neurochem. 2004, 89, 337–343. [Google Scholar] [CrossRef]
- Echeverria, V.; Yarkov, A.; Aliev, G. Positive modulators of the α7 nicotinic receptor against neuroinflammation and cognitive impairment in Alzheimer’s disease. Prog. Neurobiol. 2016, 144, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Maelicke, A. Allosteric modulation of nicotinic receptors as a treatment strategy for Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2000, 11, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Weltzin, M.M.; Schulte, M.K. Desformylflustrabromine Modulates α4β2 Neuronal Nicotinic Acetylcholine Receptor High- and Low-Sensitivity Isoforms at Allosteric Clefts Containing the β2 Subunit. J. Pharmacol. Exp. Ther. 2015, 354, 184–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razay, G.; Wilcock, G.K. Galantamine in Alzheimer’s disease. Expert. Rev. Neurother. 2008, 8, 9–17. [Google Scholar] [CrossRef]
- Echeverria, V.; Zeitlin, R. Cotinine: A potential new therapeutic agent against Alzheimer’s disease. CNS Neurosci. Ther. 2012, 18, 517–523. [Google Scholar] [CrossRef]
- Jiang, S.; Li, Y.; Zhang, C.; Zhao, Y.; Bu, G.; Xu, H.; Zhang, Y.W. M1 muscarinic acetylcholine receptor in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Abrams, P.; Andersson, K.E.; Buccafusco, J.J.; Chapple, C.; de Groat, W.C.; Fryer, A.D.; Kay, G.; Laties, A.; Nathanson, N.M.; Pasricha, P.J.; et al. Muscarinic receptors: Their distribution and function in body systems, and the implications for treating overactive bladder. Br. J. Pharmacol. 2006, 148, 565–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felder, C.C.; Bymaster, F.P.; Ward, J.; DeLapp, N. Therapeutic opportunities for muscarinic receptors in the central nervous system. J. Med. Chem. 2000, 43, 4333–4353. [Google Scholar] [CrossRef]
- Conn, P.J.; Jones, C.K.; Lindsley, C.W. Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol. Sci. 2009, 30, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudlak, M.; Tadi, P. Physiology, Muscarinic Receptor. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Volpicelli, L.A.; Allan, I.L. Muscarinic acetylcholine receptor subtypes in cerebral cortex and hippocampus. Prog. Brain Res. 2004, 145, 59–66. [Google Scholar] [PubMed]
- Ishii, M.; Kurachi, Y. Muscarinic acetylcholine receptors. Curr. Pharm. Des. 2006, 12, 3573–3581. [Google Scholar] [CrossRef]
- Miyakawa, T.; Yamada, M.; Duttaroy, A.; Wess, J. Hyperactivity and intact hippocampus-dependent learning in mice lacking the M1 muscarinic acetylcholine receptor. J. Neurosci. 2001, 21, 5239–5250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caccamo, A.; Oddo, S.; Billings, L.M.; Green, K.N.; Martinez-Coria, H.; Fisher, A.; LaFerla, F.M. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron 2006, 49, 671–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinoe, T.; Matsui, M.; Taketo, M.M.; Manabe, T. Modulation of synaptic plasticity by physiological activation of M1 muscarinic acetylcholine receptors in the mouse hippocampus. J. Neurosci. 2005, 25, 11194–11200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, A. Cholinergic treatments with emphasis on m1 muscarinic agonists as potential disease-modifying agents for Alzheimer’s disease. Neurotherapeutics 2008, 5, 433–442. [Google Scholar] [CrossRef]
- Lebois, E.P.; Thorn, C.; Edgerton, J.R.; Popiolek, M.; Xi, S. Muscarinic receptor subtype distribution in the central nervous system and relevance to aging and Alzheimer’s disease. Neuropharmacology 2018, 136, 362–373. [Google Scholar] [CrossRef]
- Caporaso, G.L.; Gandy, S.E.; Buxbaum, J.D.; Ramabhadran, T.V.; Greengard, P. Protein phosphorylation regulates secretion of Alzheimer beta/A4 amyloid precursor protein. Proc. Natl. Acad. Sci. USA 1992, 89, 3055–3059. [Google Scholar] [CrossRef] [Green Version]
- Buxbaum, J.D.; Oishi, M.; Chen, H.I.; Pinkas-Kramarski, R.; Jaffe, E.A.; Gandy, S.E.; Greengard, P. Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer beta/A4 amyloid protein precursor. Proc. Natl. Acad. Sci. USA 1992, 89, 10075–10078. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.A.; Fritz, J.J.; Wess, J.; Lah, J.J.; Levey, A.I. Deletion of M1 muscarinic acetylcholine receptors increases amyloid pathology in vitro and in vivo. J. Neurosci. 2010, 30, 4190–4196. [Google Scholar] [CrossRef] [Green Version]
- Clader, J.W.; Wang, Y. Muscarinic receptor agonists and antagonists in the treatment of Alzheimer’s disease. Curr. Pharm. Des. 2005, 11, 3353–3361. [Google Scholar] [CrossRef] [PubMed]
- English, B.A.; Webster, A.A. Acetylcholinesterase and its Inhibitors. In Primer on the Autonomic Nervous System; Academic Press: Cambridge, MA, USA, 2012; pp. 631–633. [Google Scholar]
- Hoskin, J.L.; Al-Hasan, Y.; Sabbagh, M.N. Nicotinic acetylcholine receptor agonists for the treatment of Alzheimer’s dementia: An update. Nicotine Tob. Res. 2019, 21, 370–376. [Google Scholar] [CrossRef]
- Pugle, M. What Is GABA?: A neurotransmitter known as gamma-aminobutyric acid. Available online: https://www.verywellhealth.com/gaba-5095143 (accessed on 22 June 2021).
- Govindpani, K.; Calvo-Flores Guzmán, B.; Vinnakota, C.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. Towards a better understanding of GABAergic remodeling in Alzheimer’s disease. Int. J. Mol. Sci. 2017, 18, 1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewett, B.E.; Sharma, S. Physiology, GABA. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK513311/ (accessed on 29 August 2020).
- Goetz, T.; Arslan, A.; Wisden, W.; Wulff, P. GABA(A) receptors: Structure and function in the basal ganglia. Prog. Brain Res. 2007, 160, 21–41. [Google Scholar] [PubMed] [Green Version]
- Olsen, R.W.; DeLorey, T.M. GABA Receptor Physiology and Pharmacology. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Siegel, G.J., Agranoff, B.W., Albers, R.W., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1999. Available online: https://www.ncbi.nlm.nih.gov/books/NBK28090/ (accessed on 11 July 2021).
- Terunuma, M. Diversity of structure and function of GABAB receptors: A complexity of GABAB-mediated signaling. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2018, 94, 390–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padgett, C.L.; Slesinger, P.A. GABAB receptor coupling to G-proteins and ion channels. Adv. Pharmacol. Sci. 2010, 58, 123–147. [Google Scholar]
- Tang, B.L. Amyloid Precursor Protein (APP) and GABAergic Neurotransmission. Cells 2019, 8, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinamarca, M.C.; Raveh, A.; Schneider, A.; Fritzius, T.; Früh, S.; Rem, P.D.; Stawarski, M.; Lalanne, T.; Turecek, R.; Choo, M.; et al. Complex formation of APP with GABA B receptors links axonal trafficking to amyloidogenic processing. Nat. Commun. 2019, 10, 1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, X.Q.; Yao, J.J.; Liu, D.D.; Ma, Q.; Mei. Aβ40 modulates GABAA receptor α6 subunit expression and rat cerebellar granule neuron maturation through the ERK/mTOR pathway. J. Neurochem. 2014, 128, 350–362. [Google Scholar] [CrossRef]
- Nykänen, N.P.; Kysenius, K.; Sakha, P.; Tammela, P.; Huttunen, H.J. Gamma-Aminobutyric acid type A GABAA receptor activation modulates tau phosphorylation. J. Biol. Chem. 2012, 287, 6743–6752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittington, R.A.; Bretteville, A.; Dickler, M.F.; Planel, E. Anesthesia and tau pathology. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 47, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Kullmann, D.M.; Ruiz, A.; Rusakov, D.M.; Scott, R.; Semyanov, A.; Walker, M.C. Presynaptic, extrasynaptic and axonal GABAA receptors in the CNS: Where and why? Prog. Biophys. Mol. Biol. 2005, 87, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Guo, Z.; Gearing, M.; Chen, G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s disease model. Nat. Commun. 2014, 5, 4159. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Flores Guzmán, B.; Vinnakota, C.; Govindpani, K.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. The GABAergic system as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2018, 146, 649–669. [Google Scholar] [CrossRef] [Green Version]
- Berumen, L.C.; Rodríguez, A.; Miledi, R.; García-Alcocer, G. Serotonin receptors in hippocampus. Sci. World J. 2012, 2012, 823493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, N.M.; Sharp, T. A review of central 5-HT receptors and their function. Neuropharmacology 1999, 38, 1083–1152. [Google Scholar] [CrossRef]
- Barnes, N.M.; Hales, T.G.; Lummis, S.C.; Peters, J.A. The 5-HT3 receptor—The relationship between structure and function. Neuropharmacology 2009, 56, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.E.; Nichols, C.D. Serotonin receptors. Chem. Rev. 2008, 108, 1614–1641. [Google Scholar] [CrossRef] [PubMed]
- Leysen, J.E. 5-HT2 receptors. Curr. Drug Targets-CNS Neurol. Disord. 2004, 3, 11–26. [Google Scholar] [CrossRef]
- Zhang, G.; Stackman, R.W., Jr. The role of serotonin 5-HT2A receptors in memory and cognition. Front. Pharmacol. 2015, 6, 225. [Google Scholar] [CrossRef] [Green Version]
- Hoyer, D.; Martin, G. 5-HT receptor classification and nomenclature: Towards a harmonization with the human genome. Neuropharmacology 1997, 36, 419–428. [Google Scholar] [CrossRef]
- Woods, S.; Clarke, N.N.; Layfield, R.; Fone, K.C. 5-HT (6) receptor agonists and antagonists enhance learning and memory in a conditioned emotion response paradigm by modulation of cholinergic and glutamatergic mechanisms. Br. J. Pharmacol. 2012, 167, 436–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay-Schmidt, A. The evolution of the serotonergic nervous system. Proc. Biol. Sci. 2000, 267, 1071–1079. [Google Scholar] [CrossRef]
- Meltzer, C.C.; Smith, G.; DeKosky, S.T.; Pollock, B.G.; Mathis, C.A.; Moore, R.Y.; Kupfer, D.J.; Reynolds, C.F. Serotonin in Aging, Late-Life Depression, and Alzheimer’s Disease: The Emerging Role of Functional Imaging. Neuropsychopharmacology 1998, 18, 407–430. [Google Scholar] [CrossRef] [Green Version]
- Cirrito, J.R.; Disabato, B.M.; Restivo, J.L.; Verges, D.K.; Goebel, W.D.; Sathyan, A.; Hayreh, D.; D’Angelo, G.; Benzinger, T.; Yoon, H.; et al. Serotonin signaling is associated with lower amyloid-β levels and plaques in transgenic mice and humans. Proc. Natl. Acad. Sci. USA 2011, 108, 14968–14973. [Google Scholar] [CrossRef] [Green Version]
- Manuel-Apolinar, L.; Rocha, L.; Pascoe, D.; Castillo, E.; Castillo, C.; Meneses, A. Modifications of 5-HT4 receptor expression in rat brain during memory consolidation. Brain Res. 2005, 1042, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Consolo, S.; Arnaboldi, S.; Giorgi, S.; Russi, G.; Ladinsky, H. 5-HT4 receptor stimulation facilitates acetylcholine release in rat frontal cortex. Neuroreport 1994, 5, 1230–1232. [Google Scholar] [CrossRef]
- Cachard-Chastel, M.; Lezoualc’h, F.; Dewachter, I.; Delomenie, C.; Croes, S.; Devijver, H.; Langlois, M.; Van Leuven, F.; Sicsic, S.; Gardier, A.M. 5-HT4 receptor agonists increase sAPPalpha levels in the cortex and hippocampus of male C57BL/6j mice. Br. J. Pharmacol. 2007, 150, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Cochet, M.; Donneger, R.; Cassier, E.; Gaven, F.; Lichtenthaler, S.F.; Marin, P.; Bockaert, J.; Dumuis, A.; Claeysen, S. 5-HT4 receptors constitutively promote the non-amyloidogenic pathway of APP cleavage and interact with ADAM10. ACS Chem. Neurosci. 2013, 4, 130–140. [Google Scholar] [CrossRef] [Green Version]
- Giannoni, P.; Gaven, F.; de Bundel, D.; Baranger, K.; Marchetti-Gauthier, E.; Roman, F.S.; Valjent, E.; Marin, P.; Bockaert, J.; Rivera, S.; et al. Early administration of RS 67333, a specific 5-HT4 receptor agonist, prevents amyloidogenesis and behavioral deficits in the 5XFAD mouse model of Alzheimer’s disease. Front. Aging Neurosci. 2013, 5, 96. [Google Scholar] [CrossRef] [PubMed]
- Moser, P.C.; Bergis, O.E.; Jegham, S.; Lochead, A.; Duconseille, E.; Terranova, J.P.; Caille, D.; Berque-Bestel, I.; Lezoualc’h, F.; Fischmeister, R.; et al. SL65.0155, a novel 5-hydroxytryptamine(4) receptor partial agonist with potent cognition-enhancing properties. J. Pharmacol. Exp. Ther. 2002, 302, 731–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieben, C.K.; Blokland, A.; Şık, A.; Sung, E.; van Nieuwenhuizen, P.; Schreiber, R. The selective 5-HT6 receptor antagonist Ro4368554 restores memory performance in cholinergic and serotonergic models of memory deficiency in the rat. Neuropsychopharmacology 2005, 30, 2169–2179. [Google Scholar] [CrossRef] [Green Version]
- Schaffhauser, H.; Mathiasen, J.R.; DiCamillo, A.; Huffman, M.J.; Lu, L.D.; McKenna, B.A.; Qian, J.; Marino, M.J. Dimebolin is a 5-HT6 antagonist with acute cognition enhancing activities. Biochem. Pharmacol. 2009, 78, 1035–1042. [Google Scholar] [CrossRef]
- Wilkinson, D.; Windfeld, K.; Colding-Jorgensen, E. Safety and efficacy of idalopirdine, a 5-HT6 receptor antagonist, in patients with moderate Alzheimer’s disease (LADDER): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2014, 13, 1092–1099. [Google Scholar] [CrossRef]
- Yun, H.M.; Park, K.R.; Kim, E.C.; Kim, S.; Hong, J.T. Serotonin 6 receptor controls Alzheimer’s disease and depression. Oncotarget 2015, 6, 26716–26728. [Google Scholar] [CrossRef]
- Schechter, L.E.; Dawson, L.A.; Harder, J.A. The potential utility of 5-HT1A receptor antagonists in the treatment of cognitive dysfunction associated with Alzheimer’s disease. Curr. Pharm. Des. 2002, 8, 139–145. [Google Scholar] [CrossRef]
- Lai, M.K.; Tsang, S.W.; Francis, P.T.; Esiri, M.M.; Keene, J.; Hope, T.; Chen, C.P.H. Reduced serotonin 5-HT1A receptor binding in the temporal cortex correlates with aggressive behavior in Alzheimer disease. Brain Res. 2003, 974, 82–87. [Google Scholar] [CrossRef]
- Millan, M.J.; Gobert, A.; Roux, S.; Porsolt, R.; Meneses, A.; Carli, M.; Di Cara, B.; Jaffard, R.; Rivet, J.M.; Lestage, P.; et al. The serotonin1A receptor partial agonist S15535 [4-(benzodioxan-5-yl)1-(indan-2-yl)piperazine] enhances cholinergic transmission and cognitive function in rodents: A combined neurochemical and behavioral analysis. J. Pharmacol. Exp. Ther. 2004, 311, 190–203. [Google Scholar] [CrossRef] [Green Version]
- Afshar, S.; Shahidi, S.; Rohani, A.H.; Asl, S.S.; Komaki, A. Protective effects of 5-HT1A receptor antagonist and 5-HT2A receptor agonist on the biochemical and histological features in a rat model of Alzheimer’s disease. J. Chem. Neuroanat. 2019, 96, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Buhot, M.C.; Martin, S.; Segu, L. Role of serotonin in memory impairment. Ann. Med. 2000, 32, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Mietlicki-Baase, E.G. Amylin-mediated control of glycemia, energy balance, and cognition. Physiol. Behav. 2016, 162, 130–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludvik, B.; Kautzky-Willer, A.; Prager, R.; Thomaseth, K.; Pacini, G. Amylin: History and overview. Diabet Med. 1997, 14, S9–S13. [Google Scholar] [CrossRef]
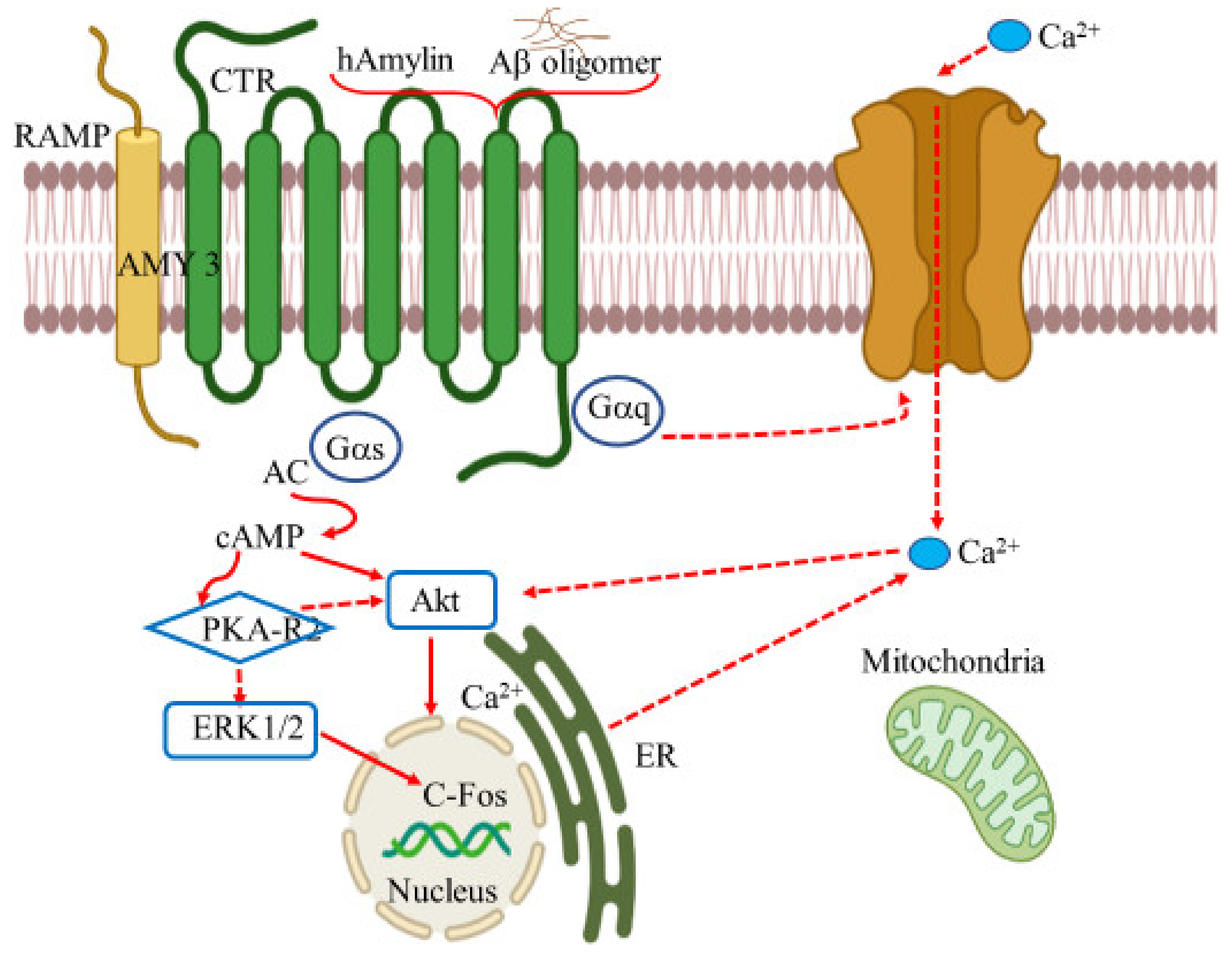
- Hay, D.L.; Christopoulos, G.; Christopoulos, A.; Sexton, P.M. Amylin receptors: Molecular composition and pharmacology. Biochem. Soc. Trans. 2004, 32, 865–867. [Google Scholar] [CrossRef]
- Qiu, W.Q. Amylin and its G-protein-coupled receptor: A probable pathological process and drug target for Alzheimer’s disease. Neuroscience 2017, 356, 44–51. [Google Scholar] [CrossRef]
- Grizzanti, J.; Corrigan, R.; Servizi, S.; Casadesus, G. Amylin Signaling in Diabetes and Alzheimer’s Disease: Therapy or Pathology? J. Neurol. Neuromed. 2019, 4, 12–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, W.; Patel, A.; Jhamandas, J.H. Amylin receptor: A common pathophysiological target in Alzheimer’s disease and diabetes mellitus. Front. Aging Neurosci. 2013, 5, 42. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Ruangkittisakul, A.; MacTavish, D.; Shi, J.Y.; Ballanyi, K.; Jhamandas, J.H. Amyloid β (Aβ) peptide directly activates amylin-3 receptor subtype by triggering multiple intracellular signaling pathways. J. Biol. Chem. 2012, 287, 18820–18830. [Google Scholar] [CrossRef] [Green Version]
- Kimura, R.; MacTavish, D.; Yang, J.; Westaway, D.; Jhamandas, J.H. Beta amyloid-induced depression of hippocampal long-term potentiation is mediated through the amylin receptor. J. Neurosci. 2012, 32, 17401–17406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhamandas, J.H.; Li, Z.; Westaway, D.; Yang, J.; Jassar, S.; MacTavish, D. Actions of β-amyloid protein on human neurons are expressed through the amylin receptor. Am. J. Pathol. 2011, 178, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Adler, B.L.; Yarchoan, M.; Hwang, H.M.; Louneva, N.; Blair, J.A.; Palm, R.; Smith, M.A.; Lee, H.G.; Arnold, S.E.; Casadesus, G. Effects of the amylin analogue pramlintide on Alzheimer’s disease pathogenesis and cognition. Neurobiol. Aging 2014, 35, 793–801. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, X.; Wallack, M.; Li, H.; Carreras, I.; Dedeoglu, A.; Hur, J.Y.; Zheng, H.; Li, H.; Fine, R.; et al. Intraperitoneal injection of the pancreatic peptide amylin potently reduces behavioral impairment and brain amyloid pathology in murine models of Alzheimer’s disease. Mol. Psychiatry 2015, 20, 252–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, W.Q.; Haihao, Z. Amylin and its analogs: A friend or foe for the treatment of Alzheimer’s disease? Front. Aging Neurosci. 2014, 6, 186. [Google Scholar] [CrossRef]
- Zhu, H.; Xue, X.; Wang, E.; Wallack, M.; Na, H.; Hooker, J.M.; Kowall, N.; Tao, Q.; Stein, T.D.; Wolozin, B.; et al. Amylin receptor ligands reduce the pathological cascade of Alzheimer’s disease. Neuropharmacology 2017, 119, 170–181. [Google Scholar] [CrossRef]
- Lim, Y.A.; Ittner, L.M.; Lim, Y.L.; Götz, J. Human but not rat amylin shares neurotoxic properties with Aβ42 in long-term hippocampal and cortical cultures. FEBS Lett. 2008, 582, 2188–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Yang, S.; Wang, C.; Zhang, J.; Huo, L.; Cheng, Y.; Wang, C.; Jia, Z.; Ren, L.; Kang, L.; et al. Multiple target of hAmylin on rat primary hippocampal neurons. Neuropharmacology 2017, 113, 241–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kefeli, U.; Ucuncu Kefeli, A.; Cabuk, D.; Isik, U.; Sonkaya, A.; Acikgoz, O.; Ozden, E.; Uygun, K. Netrin-1 in cancer: Potential biomarker and therapeutic target? Tumor Biol. 2017, 39, 1010428317698388. [Google Scholar] [CrossRef] [Green Version]
- Barallobre, M.J.; Pascual, M.; Del Río, J.A.; Soriano, E. The Netrin family of guidance factors: Emphasis on Netrin-1 signaling. Brain Res. Rev. 2005, 49, 22–47. [Google Scholar] [CrossRef]
- Moore, S.W.; Tessier-Lavigne, M.; Kennedy, T.E. Netrins and their receptors. Adv. Exp. Med. Biol. 2007, 621, 17–31. [Google Scholar]
- Llambi, F.; Causeret, F.; Bloch-Gallego, E.; Mehlen, P. Netrin-1 acts as a survival factor via its receptors UNC5H and DCC. EMBO J. 2001, 20, 2715–2722. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, P.; Mohamed, R.; Jayakumar, C.; Ramesh, G. Guidance cue netrin-1 and the regulation of inflammation in acute and chronic kidney disease. Mediat. Inflamm. 2014, 2014, 525891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, D.; Cole, S.J.; Cooper, H.M. Netrin-1: Diversity in development. Int. J. Biochem. Cell Biol. 2009, 41, 487–493. [Google Scholar] [CrossRef]
- Hong, K.; Hinck, L.; Nishiyama, M.; Poo, M.M.; Tessier-Lavigne, M.; Stein, E. A ligand-gated association between cytoplasmic domains of UNC5 and DCC family receptors converts netrin-induced growth cone attraction to repulsion. Cell 1999, 97, 927–941. [Google Scholar] [CrossRef] [Green Version]
- Boyer, N.P.; Gupton, S.L. Revisiting Netrin-1: One Who Guides (Axons). Front. Cell Neurosci. 2018, 12, 221. [Google Scholar] [CrossRef] [PubMed]
- Mille, F.; Llambi, F.; Guix, C.; Delloye-Bourgeois, C.; Guenebeaud, C.; Castro-Obregon, S.; Bredesen, D.E.; Thibert, C.; Mehlen, P. Interfering with multimerization of netrin-1 receptors triggers tumor cell death. Cell Death Differ. 2009, 16, 1344–1351. [Google Scholar] [CrossRef] [Green Version]
- Llambi, F.; Lourenço, F.C.; Gozuacik, D.; Guix, C.; Pays, L.; Del Rio, G.; Kimchi, A.; Mehlen, P. The dependence receptor UNC5H2 mediates apoptosis through DAP-kinase. EMBO J. 2005, 24, 1192–1201. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Toyama, Y.; Kusakari, S.; Nawa, M.; Matsuoka, M. An Alzheimer disease-linked rare mutation potentiates netrin receptor uncoordinated-5C-induced signaling that merges with amyloid β precursor protein signaling. J. Biol. Chem. 2016, 291, 12282–12293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Kang, S.S.; Wang, Z.; Ahn, E.H.; Xia, Y.; Liu, X.; Sandoval, I.M.; Manfredsson, F.P.; Zhang, Z.; Ye, K. Netrin-1 receptor UNC5C cleavage by active δ-secretase enhances neurodegeneration, promoting Alzheimer’s disease pathologies. Sci. Adv. 2021, 7, eabe4499. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, S.D.; Ruthazer, E.S.; Kennedy, T.E. Guiding synaptic plasticity: Novel roles for netrin-1 in synaptic plasticity and memory formation in the adult brain. J. Physiol. Adv. 2020, 599, 493–505. [Google Scholar] [CrossRef]
- Shabani, M.; Haghani, M.; Tazangi, P.E.; Bayat, M.; Moosavi, S.M.S.; Ranjbar, H. Netrin-1 improves the amyloid-β-mediated suppression of memory and synaptic plasticity. Brain Res. Bull. 2017, 131, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Zamani, E.; Parviz, M.; Roghani, M.; Mohseni-Moghaddam, P. Key mechanisms underlying netrin-1 prevention of impaired spatial and object memory in Aβ1-42 CA1-injected rats. Clin. Exp. Pharmacol. Physiol. 2019, 46, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Gorshkov, K.; Aguisanda, F.; Thorne, N.; Zheng, W. Astrocytes as targets for drug discovery. Drug Discov. Today 2018, 23, 673–680. [Google Scholar] [CrossRef]
- Finsterwald, C.; Dias, S.; Magistretti, P.J.; Lengacher, S. Ganglioside GM1 Targets Astrocytes to Stimulate Cerebral Energy Metabolism. Front. Pharmacol. 2021, 12, 653842. [Google Scholar] [CrossRef]
- Smit, T.; Deshayes, N.A.C.; Borchelt, D.R.; Kamphuis, W.; Middeldorp, J.; Hol, E.M. Reactive astrocytes as treatment targets in Alzheimer’s disease-Systematic review of studies using the APPswePS1dE9 mouse model. Glia 2021, 69, 1852–1881. [Google Scholar] [CrossRef]
- Lananna, B.V.; McKee, C.A.; King, M.W.; Del-Aguila, J.L.; Dimitry, J.M.; Farias, F.H.G.; Nadarajah, C.J.; Xiong, D.D.; Guo, C.; Cammack, A.J.; et al. Chi3l1/YKL-40 is controlled by the astrocyte circadian clock and regulates neuroinflammation and Alzheimer’s disease pathogenesis. Sci. Transl. Med. 2020, 12, eaax3519. [Google Scholar] [CrossRef]
- Shi, Y.; Andhey, P.S.; Ising, C.; Wang, K.; Snipes, L.L.; Boyer, K.; Lawson, S.; Yamada, K.; Qin, W.; Manis, M.; et al. Overexpressing low-density lipoprotein receptor reduces tau-associated neurodegeneration in relation to apoE-linked mechanisms. Neuron 2021, 109, 2413–2426. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Gao, J.; Kim, J.; Hong, C.; Kim, J.; Tontonoz, P. The E3 ubiquitin ligase Idol controls brain LDL receptor expression, ApoE clearance, and Aβ amyloidosis. Sci. Transl. Med. 2015, 7, 314ra184. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.J.; Yun, S.M.; Jo, C.; Jeong, J.; Park, M.H.; Han, C.; Koh, Y.H. Altered expression of Notch1 in Alzheimer’s disease. PLoS ONE 2019, 14, e0224941. [Google Scholar] [CrossRef]
- Woo, H.N.; Park, J.S.; Gwon, A.R.; Arumugam, T.V.; Jo, D.G. Alzheimer’s disease and Notch signaling. Biochem. Biophys. Res. Commun. 2009, 390, 1093–1097. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef]
- Donner, L.; Toska, L.M.; Krüger, I.; Gröniger, S.; Barroso, R.; Burleigh, A.; Mezzano, D.; Pfeiler, S.; Kelm, M.; Gerdes, N.; et al. The collagen receptor glycoprotein VI promotes platelet-mediated aggregation of β-amyloid. Sci. Signal. 2020, 13, eaba9872. [Google Scholar] [CrossRef] [PubMed]
- Donner, L.; Fälker, K.; Gremer, L.; Klinker, S.; Pagani, G.; Ljungberg, L.U.; Lothmann, K.; Rizzi, F.; Schaller, M.; Gohlke, H.; et al. Platelets contribute to amyloid-β aggregation in cerebral vessels through integrin αIIbβ3-induced outside-in signaling and clusterin release. Sci. Signal. 2016, 9, ra52. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Skwarek-Maruszewska, A.; Horré, K.; Vandewyer, E.; Wolfs, L.; Snellinx, A.; Saito, T.; Radaelli, E.; Corthout, N.; Colombelli, J.; et al. Loss of GPR3 reduces the amyloid plaque burden and improves memory in Alzheimer’s disease mouse models. Sci. Transl. Med. 2015, 7, 309ra164. [Google Scholar] [CrossRef]
- Collins, B.E.; Greer, C.B.; Coleman, B.C.; Sweatt, J.D. Histone H3 lysine K4 methylation and its role in learning and memory. Epigenet. Chromatin 2019, 12, 7. [Google Scholar] [CrossRef]
- Cao, Q.; Wang, W.; Williams, J.B.; Yang, F.; Wang, Z.J.; Yan, Z. Targeting histone K4 trimethylation for treatment of cognitive and synaptic deficits in mouse models of Alzheimer’s disease. Sci. Adv. 2020, 6, eabc8096. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.N. | Potential Target | Therapeutic Approach & Observation | Conclusion | Ref. No. |
|---|---|---|---|---|
| 1. | Astrocytes | |||
| Reactive astrocytes Astrocytes are non-neuronal cells in CNS that are involved in regulating the neuronal health and blood-brain barrier (BBB) function. These astrocytic cells serve as a target for ganglioside GM1 for mediating its cerebral energy metabolism and neuroprotective effects. | Reactive astrocytes are closely linked with Aβ peptides and may regulate synaptic transmission and function of neuronal network, thereby resulting in impaired cognitive function in AD. Modifying the reactive astrocytes in the APPswePS1dE9 AD mouse model influences cognition and AD pathogenesis. | In AD, targeting reactive astrocytes characterized by enhanced intermediate filament proteins and cellular hypertrophy, to reduce astrogliosis is effective in ameliorating cognition. | [210,211,212] | |
| Chi3l1/YKL-40 YKL-40, a glycoprotein encoded by the Chi3l1 gene, is a human CSF biomarker of neuro inflammation, which is elevated in AD. | Deletion of Chi3l1 decreased amyloid plaque burden and increased periplaque expression of the microglial lysosomal marker CD68 in the APP/PS1 mouse model of AD. | Chi3l1/YKL-40 regulates glial activation, Aβ phagocytosis, and amyloid plaque deposition in mice and influences AD progression in humans, suggesting that the astrocyte circadian clock regulates neuro inflammation as induced by Chi3l1. | [212] | |
| 2. | Low-density lipoprotein receptor (LDLR) in relation to Apolipoprotein E (ApoE) | |||
| LDLR LDLR is an ApoE metabolic receptor with a key role in cholesterol metabolism. | In P301S tauopathy mice, over expression of LDLR in microglia cells down regulated ApoE levels, resulting in suppressed microglial activation. Likewise, the reduced level of ApoE and increased level of LDLR favors microglial catabolism over anabolism and enhances the oligodendrocyte progenitor cells (OPCs) along with preserving myelin integrity. | Raising levels of LDL protein significantly reduced ApoE level in mouse brain and improved tau pathology and neurodegeneration. | [213] | |
| Idol, an E3 ubiquitin ligase Idol is an E3 ubiquitin ligase that is transcriptionally regulated by LXRs (liver X receptors), targeting LDLR for degradation. | Idol is responsible for metabolism of brain ApoE and Aβ plaque biogenesis. The down regulation of Idol expression in APP/PS1 mouse model of AD increases brain LDLR, decreases ApoE, and reduces soluble and insoluble Aβ peptides and amyloid plaque burden thereby improving neuro inflammation. | LXR-Idol pathways play a significant role in modulating LDLR and ApoE protein expression in brain and may affect AD pathogenesis involving the removal of apolipoprotein E and amyloid beta in the brain. | [214] | |
| 3. | Notch Receptors Notch receptors are transmembrane proteins consisting of epidermal growth factor in extracellular domain with a key role in vascular development and angiogenesis. These proteins are highly expressed in the hippocampal area (region of synaptic plasticity) and depends upon γ-secretase for its proteolytic functioning. | Patients suffering from dementia had low plasma levels of soluble notch 1 receptor, compared to their healthy counterparts. Following amyloid beta treatment, the level of notch 1 protein and notch 1 mRNA level increased remarkably in human brain microvascular endothelial cells (HBMECs) and human iPSC-derived neuronal cells. | The levels of notch 1 receptor vary significantly in AD patients and are considered to be involved in AD pathogenesis and vascular dementia. | [215,216,217] |
| 4. | Integrin αIIbβ3 and Collagen receptor glycoprotein VI (GPVI) | Blocking the binding pathways and stimulating Aβ with integrin αIIbβ3 and GPVI may therapeutically reduce amyloid plaque formation mediated by platelet in cerebral vessels and brain parenchyma of AD patients. | Inhibition of integrin αIIbβ3 and GPVI on the surface of platelets may ameliorate vascular symptoms and cerebral amyloid angiopathy that contributes to dementia in AD patients. | [218,219] |
| 5. | GPR3 GPR3, the orphan G-protein coupled receptor, modulates the function of γ-secretase and the generation of Aβ peptide in the absence of Notch receptor proteolysis. | In four AD transgenic mouse models (a single APP transgenic model, a double APP/PS1transgenic model, and two App knock-in transgenic mouse models), the genetic deletion or loss of GPR3 decreased amyloid pathology in all of the models and alleviated cognitive deficits in the APP/PS1 mice. | GPR3 mediates the amyloidogenic proteolysis of APP. GPR3 removal significantly diminished the amyloid plaques and ameliorated memory in the transgenic AD mouse models. | [220] |
| 6. | Histone H3 lysine K4 trimethylation (H3K4me3) The transcriptional regulation of H3K4 methylation has been implicated in hippocampal and striatum-dependent memory formation in mice and human cognitive impairment. | Inhibition of H3K4-specific methyltransferases (catalyzed Histone H3K4me3 enzyme) in the P301S tau transgenic mouse model, significantly improved glutamatergic synaptic function and memory in PFC (pre-frontal cortex) pyramidal neurons. | Treatment of P301S mutant tau mouse model with a specific Sgk1 inhibitor, significantly reduced hyper phosphorylated tau protein in the frontal cortex and recovered the glutamatergic synaptic transmission in the mouse model, indicating the importance of H3K4me3-mediated Sgk1 up-regulation association of with AD-related pathologies. | [221,222] |
| Sgk1 (serum and glucocorticoid-regulated kinase 1) gene Sgk1 gene encodes serum and glucocorticoid-regulated kinase 1, and is highly expressed in PFC of AD patients. | Inhibition of the up-regulated levels of Sgk1 in P301S Tau model mice by the use of a specific Sgk1 inhibitor leads to the reduction of hyperphosphorylated tau protein, along with restoration of PFC glutamatergic synaptic function, and improvement of memory impairments in AD mice. | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, K.; Pradhan, S.; Duffy, L.K.; Yeasmin, S.; Bhattarai, N.; Schulte, M.K. Role of Receptors in Relation to Plaques and Tangles in Alzheimer’s Disease Pathology. Int. J. Mol. Sci. 2021, 22, 12987. https://doi.org/10.3390/ijms222312987
Sharma K, Pradhan S, Duffy LK, Yeasmin S, Bhattarai N, Schulte MK. Role of Receptors in Relation to Plaques and Tangles in Alzheimer’s Disease Pathology. International Journal of Molecular Sciences. 2021; 22(23):12987. https://doi.org/10.3390/ijms222312987
Chicago/Turabian StyleSharma, Kavita, Samjhana Pradhan, Lawrence K. Duffy, Sabina Yeasmin, Nirajan Bhattarai, and Marvin K. Schulte. 2021. "Role of Receptors in Relation to Plaques and Tangles in Alzheimer’s Disease Pathology" International Journal of Molecular Sciences 22, no. 23: 12987. https://doi.org/10.3390/ijms222312987
APA StyleSharma, K., Pradhan, S., Duffy, L. K., Yeasmin, S., Bhattarai, N., & Schulte, M. K. (2021). Role of Receptors in Relation to Plaques and Tangles in Alzheimer’s Disease Pathology. International Journal of Molecular Sciences, 22(23), 12987. https://doi.org/10.3390/ijms222312987