
Data-Independent Acquisition-Based Proteome and Phosphoproteome Profiling Reveals Early Protein Phosphorylation and Dephosphorylation Events in Arabidopsis Seedlings upon Cold Exposure


Abstract
:1. Introduction
2. Results
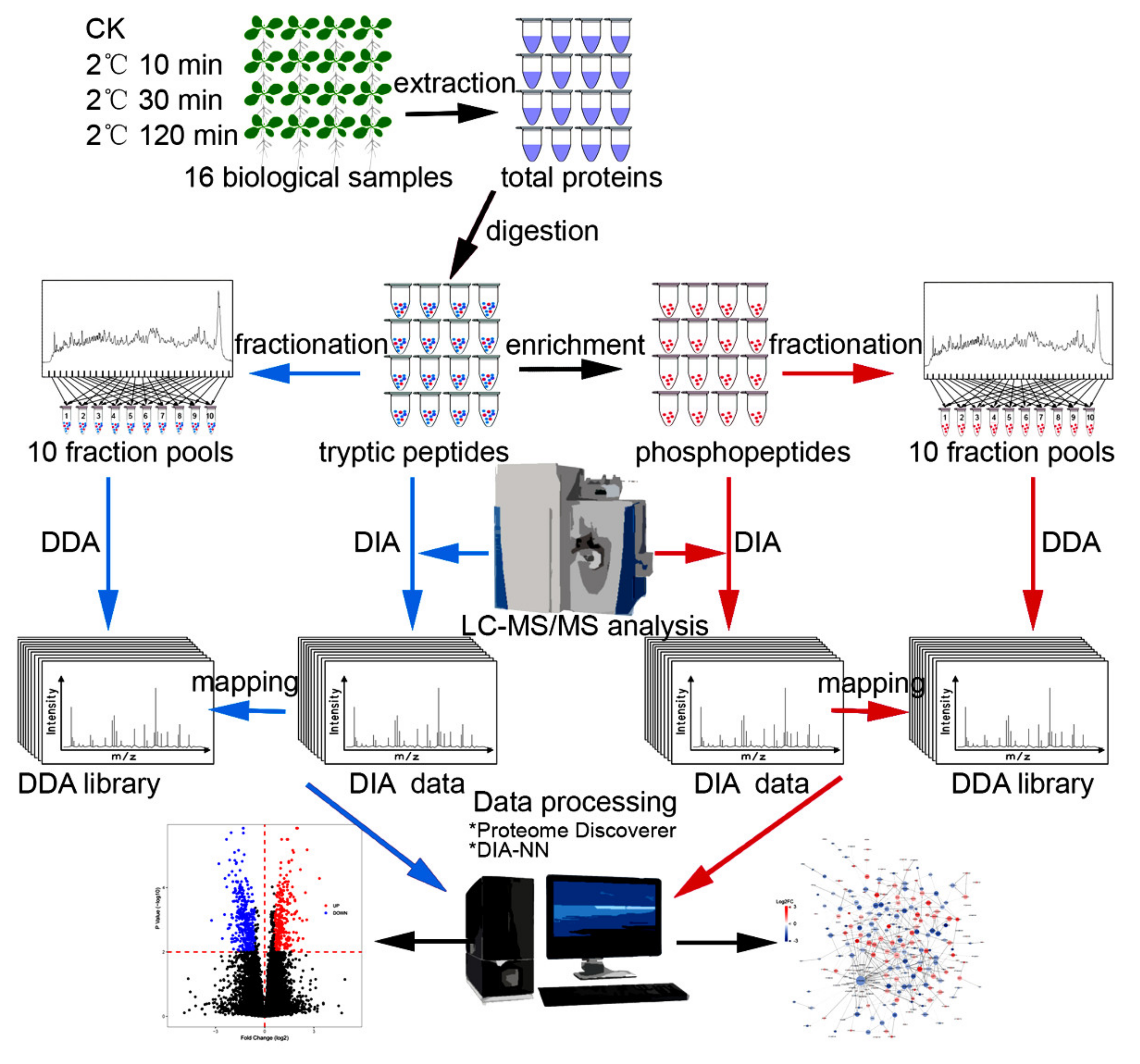
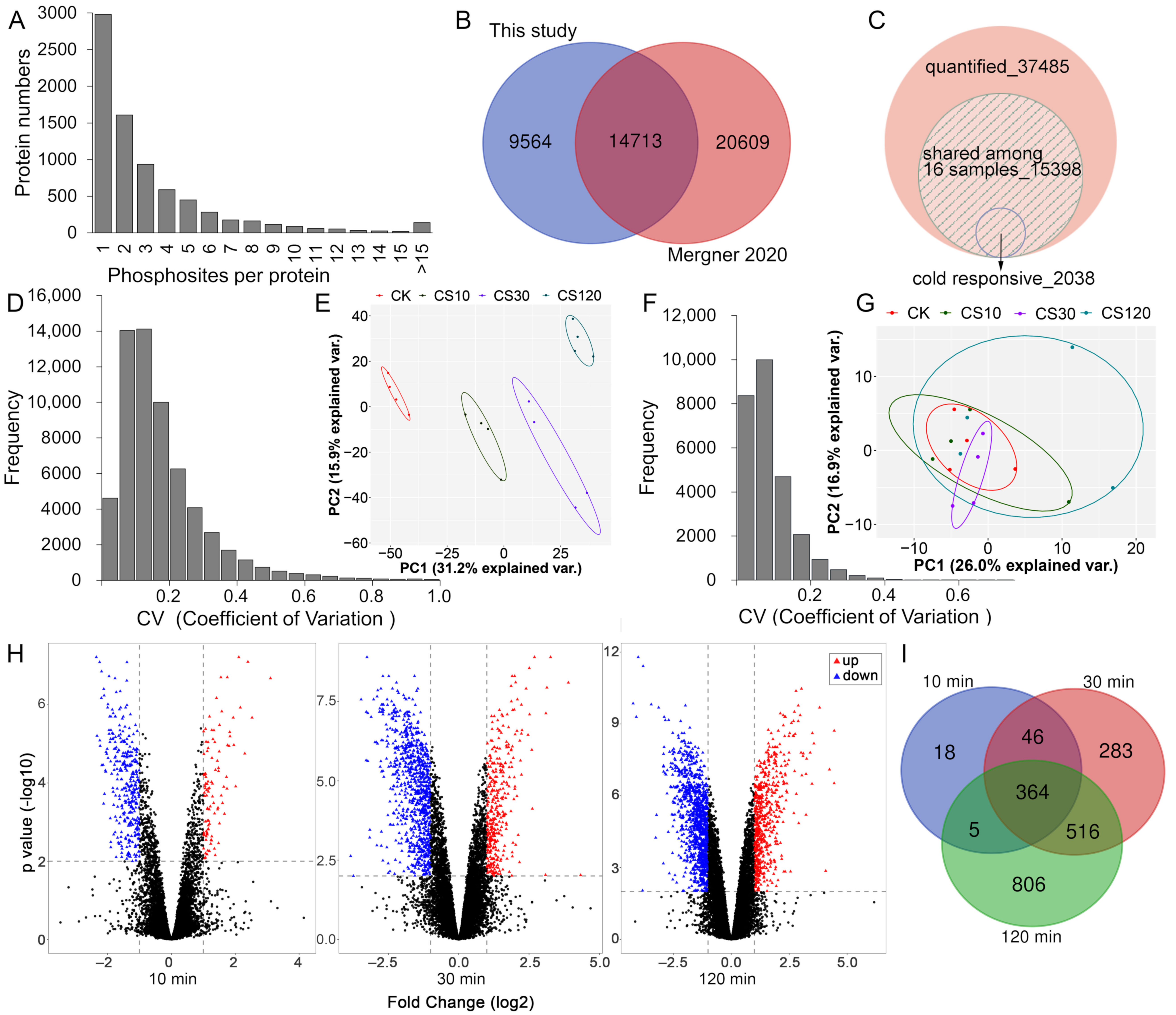
2.1. Proteome and Phosphoproteome Analyses of Arabidopsis Seedlings upon Short-Time Cold Stress
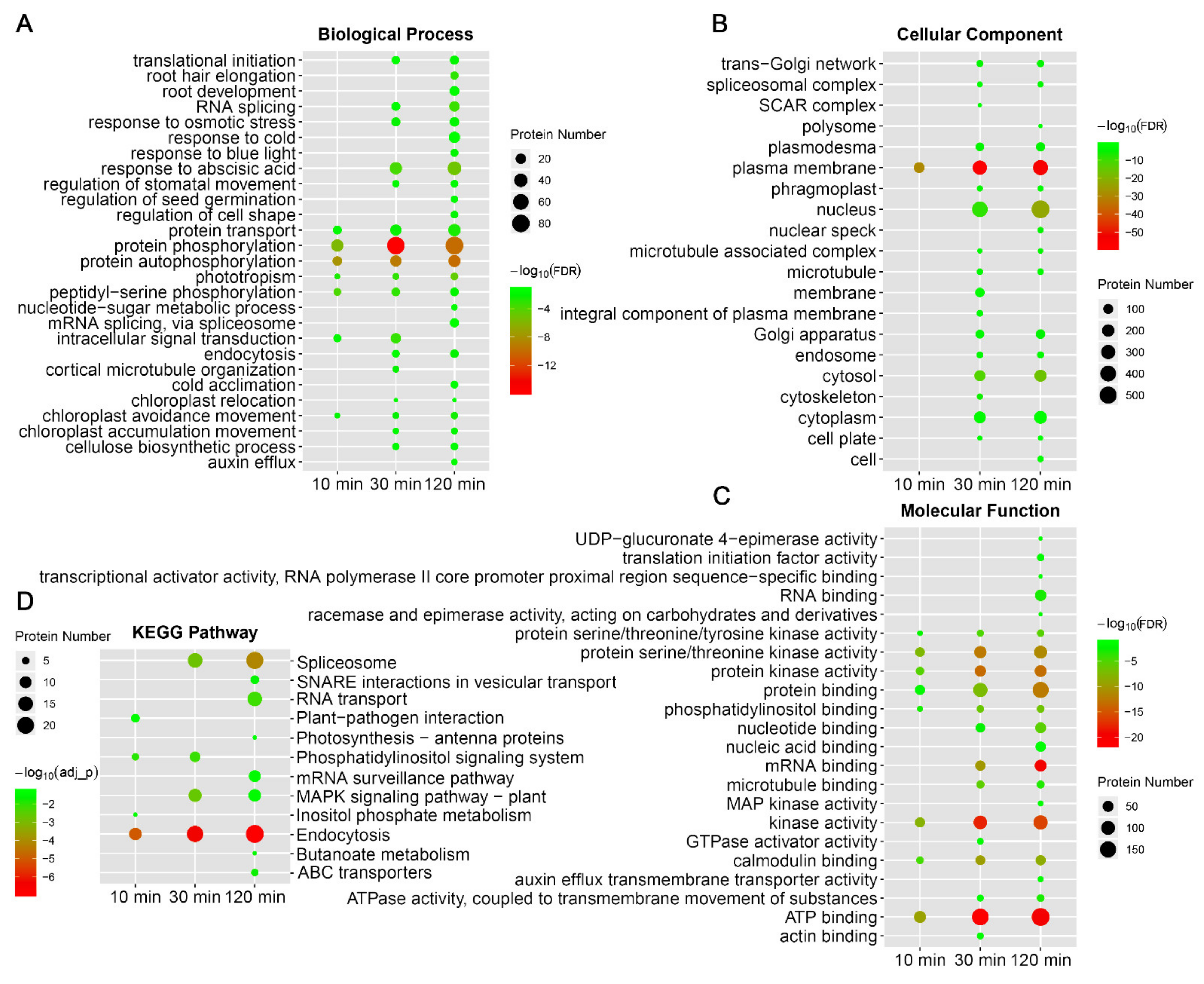
2.2. Identification of Early-Cold-Responsive Proteins
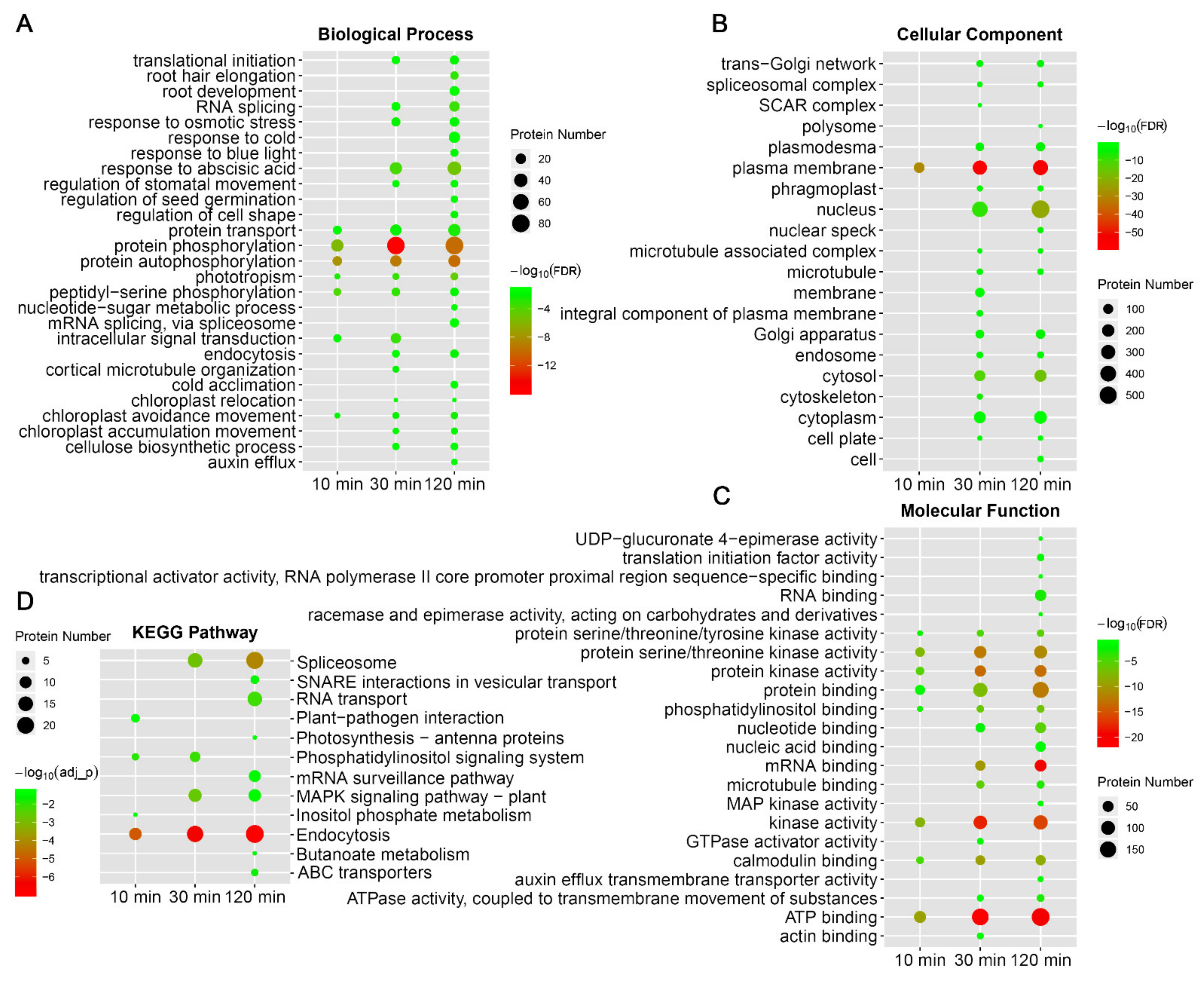
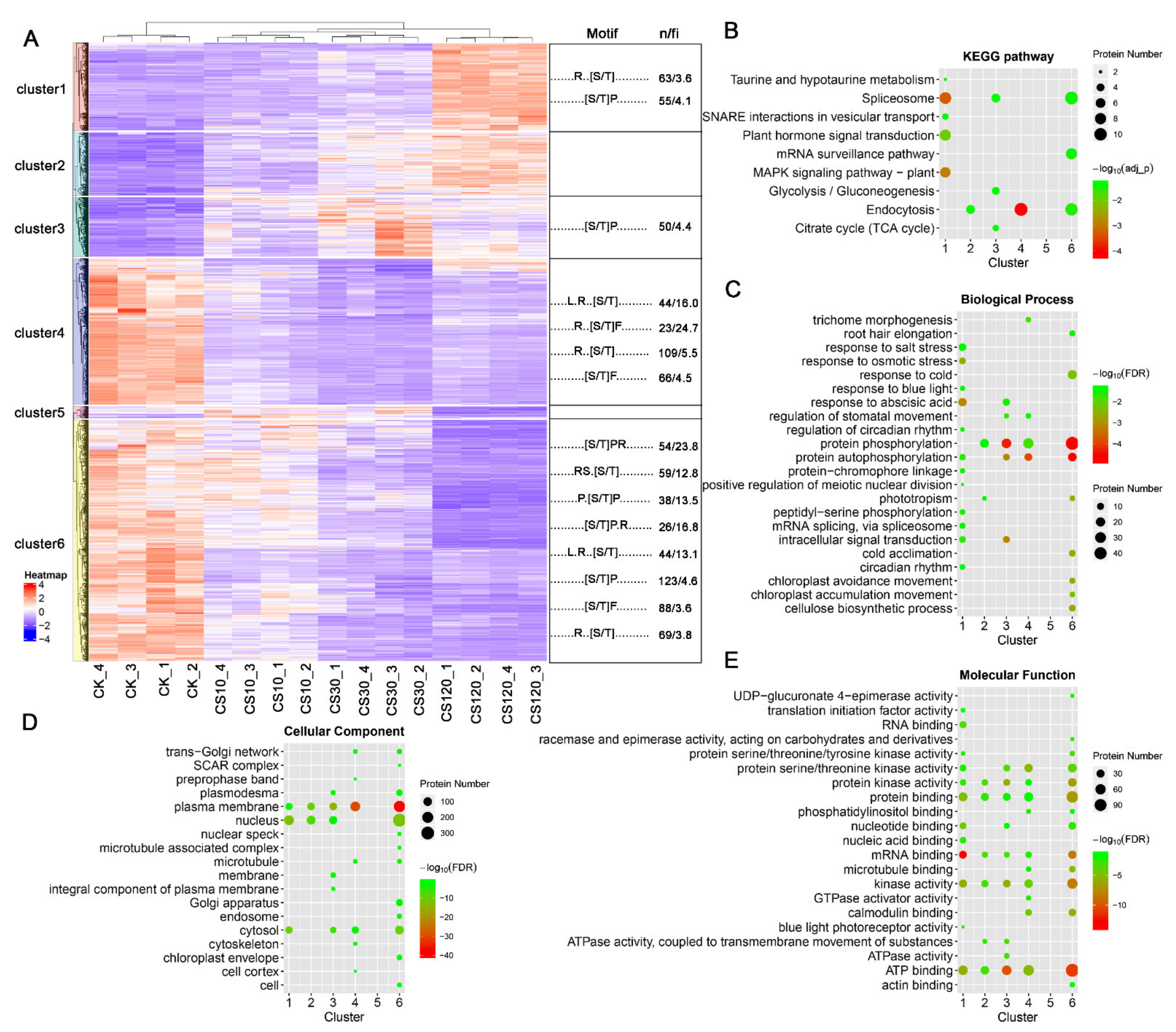
2.3. Protein Phosphorylation and Dephosphorylation Response Showed Distinct Patterns in Biological Processes or Pathways
2.4. Motif Analysis of Sequences Flanking p-Sites of Cold-Responsive Phosphopeptides
2.5. Cold Stimuli Induced Dynamic Protein Phosphorylation Modification
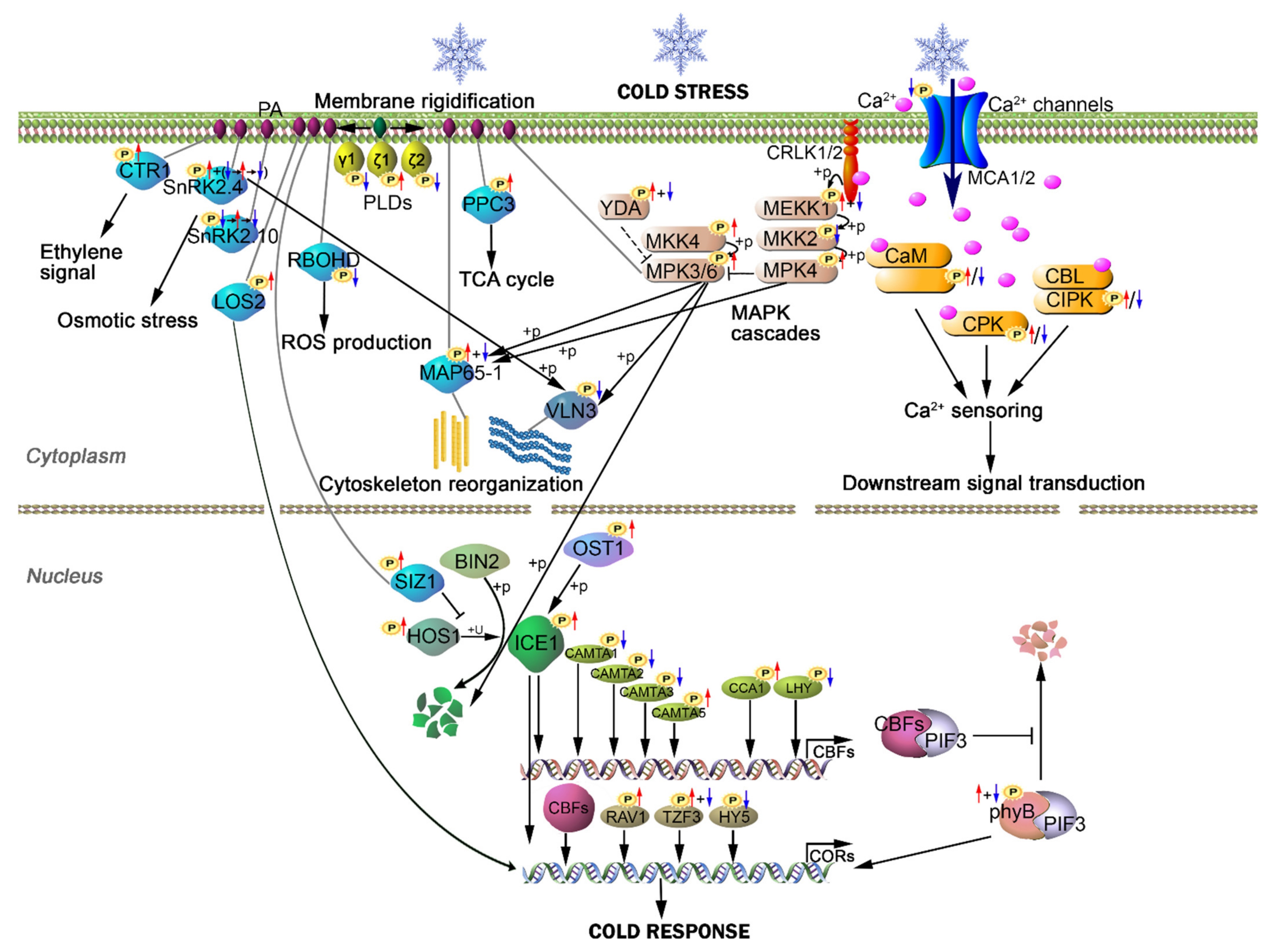
2.6. Cold-Induced Phosphorylation and Dephosphorylation in Phospholipid Signaling Proteins
2.7. Cold-Mediated Phosphorylation Modification in Cytoskeleton Proteins
2.8. Cold-Mediated Protein Phosphorylation in Calcium Signaling Proteins
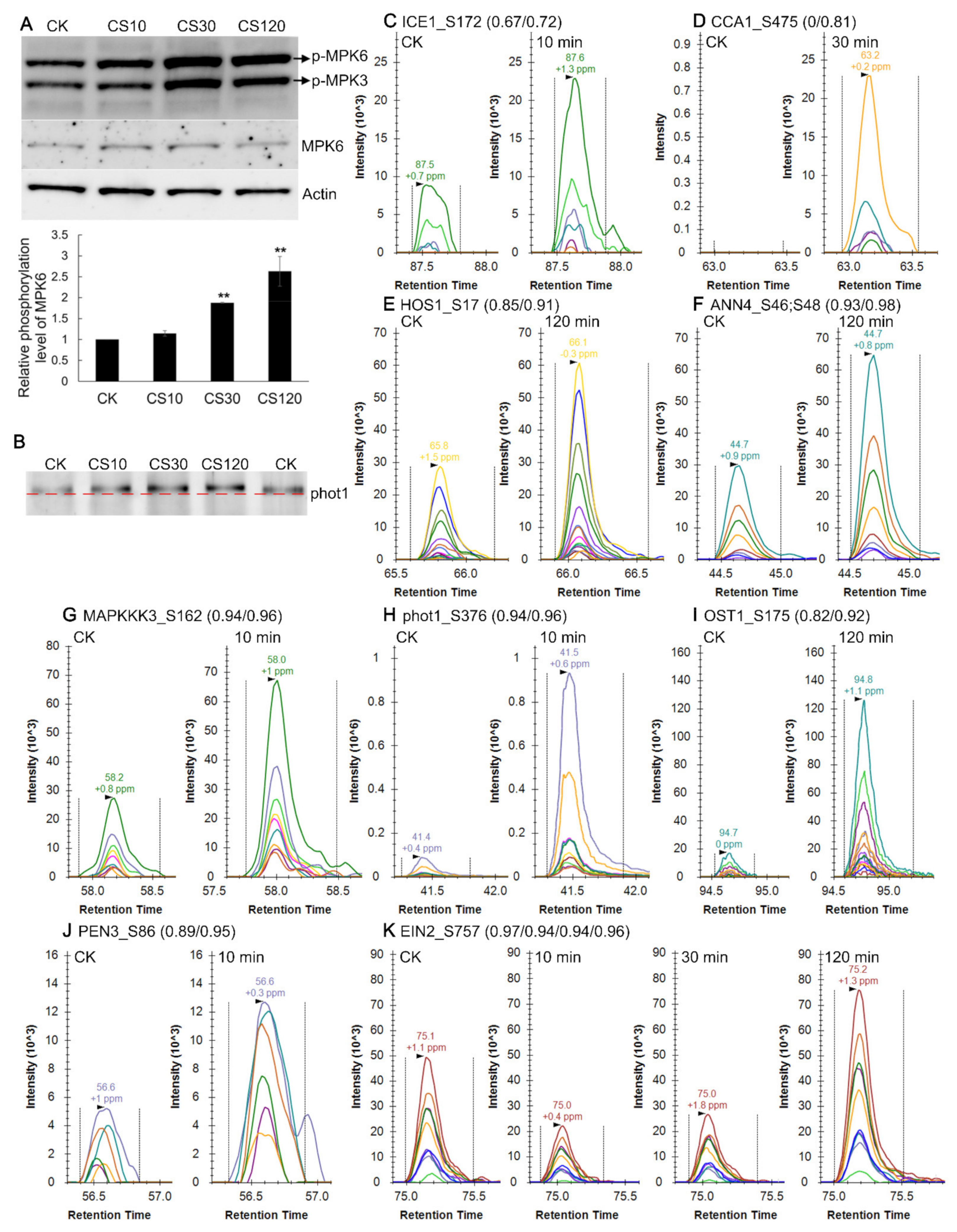
2.9. MAPK Cascades Response upon Cold Stress
2.10. Phosphorylation Modification of Transcription Factors in Early Cold Response
2.11. Phosphorylation Modification of Proteins Belonging to COR Genes in Early Cold Response
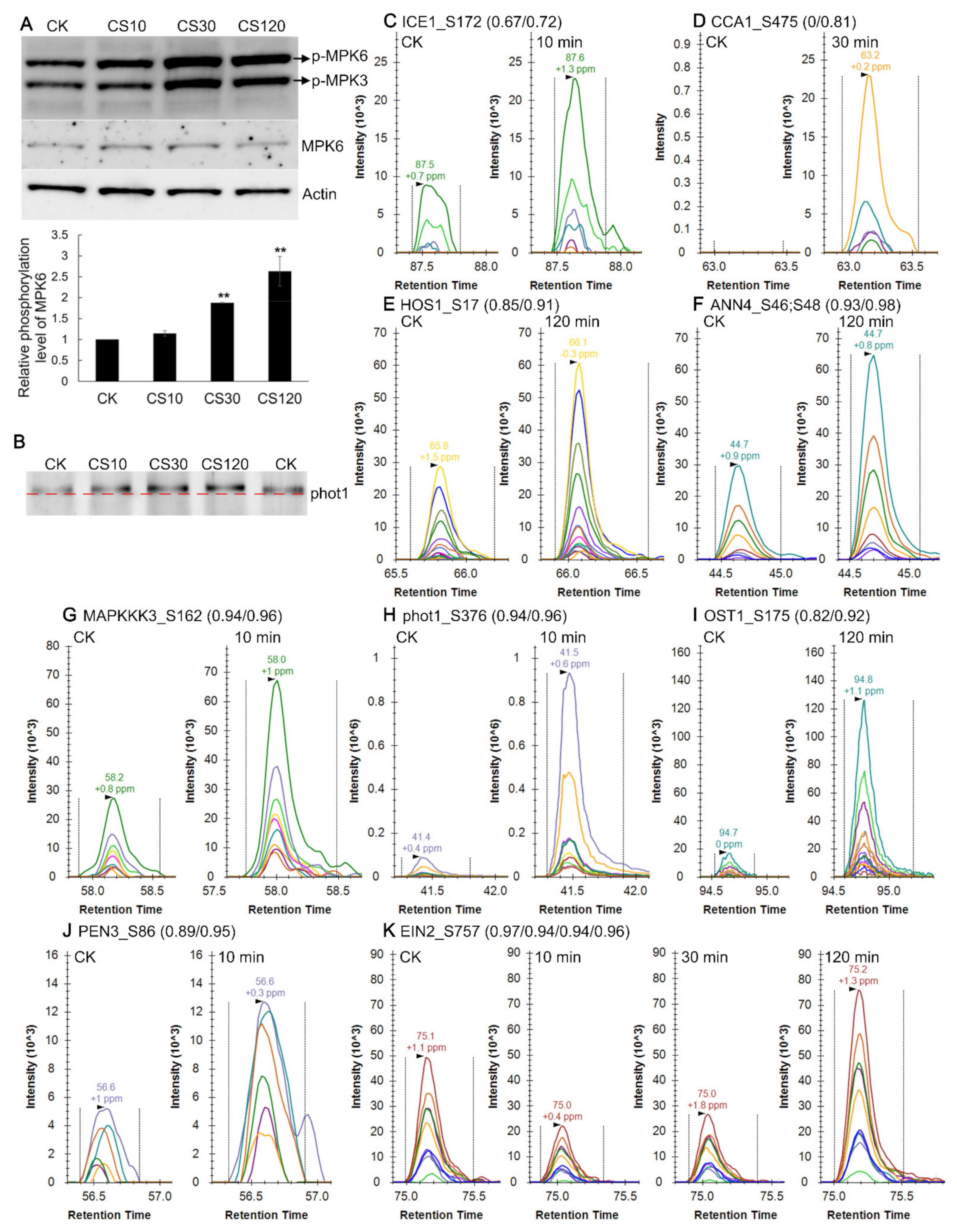
2.12. Validation of Cold-Induced Protein Phosphorylation Level Changes
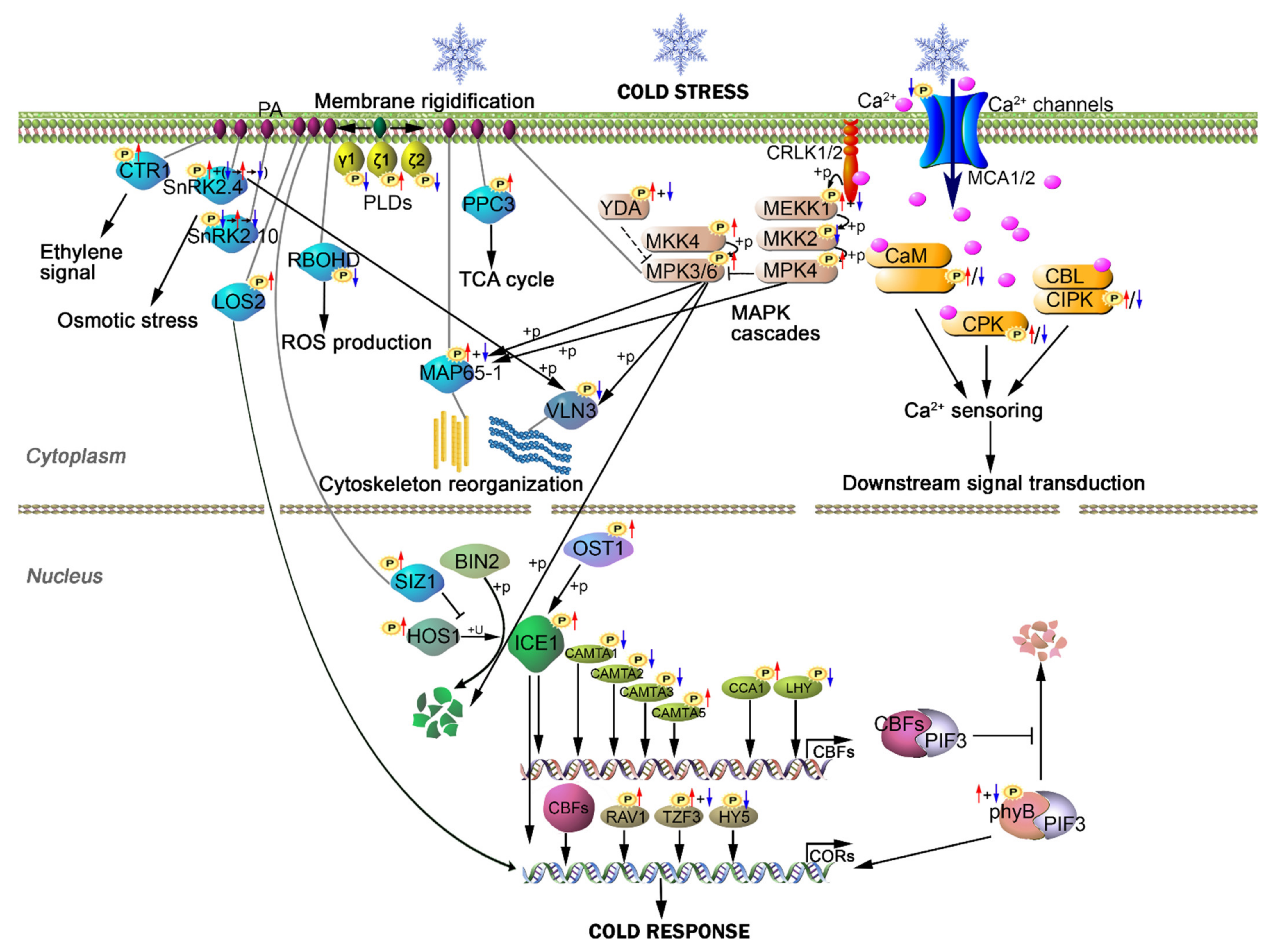
3. Discussion
4. Materials and Methods
4.1. Plant Growth and Cold Treatment
4.2. Protein Extraction and Peptides Preparation
4.3. Mass Spectrometric Acquisition
4.4. Mass Spectrometric Raw Data Analysis
4.5. Bioinformatic Analysis
4.6. Immunoblot Analysis
4.7. Parallel Reaction Monitoring
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Thomashow, M.F. PLANT COLD ACCLIMATION: Freezing Tolerance Genes and Regulatory Mechanisms. Annu. Rev. Plant Biol. 1999, 50, 571–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Shi, Y.; Yang, S. Advances and challenges in uncovering cold tolerance regulatory mechanisms in plants. New Phytol. 2019, 222, 1690–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Liu, D.; Chong, K. Cold signaling in plants: Insights into mechanisms and regulation. J. Integr. Plant Biol. 2018, 60, 745–756. [Google Scholar] [CrossRef] [Green Version]
- Gong, Z.; Xiong, L.; Shi, H.; Yang, S.; Herrera-Estrella, L.R.; Xu, G.; Chao, D.-Y.; Li, J.; Wang, P.-Y.; Qin, F.; et al. Plant abiotic stress response and nutrient use efficiency. Sci. China Life Sci. 2020, 63, 635–674. [Google Scholar] [CrossRef]
- Shi, Y.; Ding, Y.; Yang, S. Molecular Regulation of CBF Signaling in Cold Acclimation. Trends Plant Sci. 2018, 23, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Chinnusamy, V.; Ohta, M.; Kanrar, S.; Lee, B.-H.; Hong, X.; Agarwal, M.; Zhu, J.-K. ICE1: A regulator of cold-induced transcriptome and freezing tolerance inArabidopsis. Genes Dev. 2003, 17, 1043–1054. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Li, H.; Zhang, X.; Xie, Q.; Gong, Z.; Yang, S. OST1 Kinase Modulates Freezing Tolerance by Enhancing ICE1 Stability in Arabidopsis. Dev. Cell 2015, 32, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Wang, P.; Si, T.; Hsu, C.-C.; Wang, L.; Zayed, O.; Yu, Z.; Zhu, Y.; Dong, J.; Tao, W.A.; et al. MAP Kinase Cascades Regulate the Cold Response by Modulating ICE1 Protein Stability. Dev. Cell 2017, 43, 618–629.e5. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ding, Y.; Shi, Y.; Zhang, X.; Zhang, S.; Gong, Z.; Yang, S. MPK3- and MPK6-Mediated ICE1 Phosphorylation Negatively Regulates ICE1 Stability and Freezing Tolerance in Arabidopsis. Dev. Cell 2017, 43, 630–642.e4. [Google Scholar] [CrossRef]
- Ye, K.; Li, H.; Ding, Y.; Shi, Y.; Song, C.-P.; Gong, Z.; Yang, S. BRASSINOSTEROID-INSENSITIVE2 Negatively Regulates the Stability of Transcription Factor ICE1 in Response to Cold Stress in Arabidopsis. Plant Cell 2019, 31, 2682–2696. [Google Scholar] [CrossRef]
- Chinnusamy, V.; Zhu, J.; Zhu, J.-K. Cold stress regulation of gene expression in plants. Trends Plant Sci. 2007, 12, 444–451. [Google Scholar] [CrossRef]
- Mehrotra, S.; Verma, S.; Kumar, S.; Kumari, S.; Mishra, B.N. Transcriptional regulation and signalling of cold stress response in plants: An overview of current understanding. Environ. Exp. Bot. 2020, 180, 104243. [Google Scholar] [CrossRef]
- Kamal, M.; Ishikawa, S.; Takahashi, F.; Suzuki, K.; Kamo, M.; Umezawa, T.; Shinozaki, K.; Kawamura, Y.; Uemura, M. Large-Scale Phosphoproteomic Study of Arabidopsis Membrane Proteins Reveals Early Signaling Events in Response to Cold. Int. J. Mol. Sci. 2020, 21, 8631. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-C.; Zhu, Y.; Arrington, J.V.; Paez, J.S.; Wang, P.; Zhu, P.; Chen, I.-H.; Zhu, J.-K.; Tao, W.A. Universal Plant Phosphoproteomics Workflow and Its Application to Tomato Signaling in Response to Cold Stress. Mol. Cell. Proteom. 2018, 17, 2068–2080. [Google Scholar] [CrossRef] [Green Version]
- Venable, J.D.; Dong, M.-Q.; Wohlschlegel, J.; Dillin, A.; Yates, J.R. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat. Methods 2004, 1, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.D.; Goodlett, D.R.; Masselon, C.D. Multiplexed and data-independent tandem mass spectrometry for global proteome profiling. Mass Spectrom. Rev. 2013, 33, 452–470. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent acquisition-based SWATH - MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, e8126. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Bernhardt, O.M.; Hogrebe, A.; Martinez-Val, A.; Verbeke, L.; Gandhi, T.; Kelstrup, C.D.; Reiter, L.; Olsen, J.V. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Yu, S.; Liu, S.; Tian, H.; Yu, C.; Tan, W.; Zhang, J.; Li, Z.; Jiang, F.; Duan, L. Data-Independent Acquisition Proteomics Unravels the Effects of Iron Ions on Coronatine Synthesis in Pseudomonas syringae pv. tomato DC 3000. Front. Microbiol. 2020, 11, 1362. [Google Scholar] [CrossRef]
- Wang, Y.; Sang, Z.; Xu, S.; Xu, Q.; Zeng, X.; Jabu, D.; Yuan, H. Comparative proteomics analysis of Tibetan hull-less barley under osmotic stress via data-independent acquisition mass spectrometry. GigaScience 2020, 9, giaa019. [Google Scholar] [CrossRef]
- Fan, K.-T.; Wang, K.-H.; Chang, W.-H.; Yang, J.-C.; Yeh, C.-F.; Cheng, K.-T.; Hung, S.-C.; Chen, Y.-R. Application of Data-Independent Acquisition Approach to Study the Proteome Change from Early to Later Phases of Tomato Pathogenesis Responses. Int. J. Mol. Sci. 2019, 20, 863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plieth, C.; Hansen, U.-P.; Knight, H.; Knight, M. Temperature sensing by plants: The primary characteristics of signal perception and calcium response. Plant J. 1999, 18, 491–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Gao, Z.; Liu, X.; Sun, D.; Tang, W. Transcriptional Profiling Reveals a Time-of-Day-Specific Role of REVEILLE 4/8 in Regulating the First Wave of Heat Shock–Induced Gene Expression in Arabidopsis. Plant Cell 2019, 31, 2353–2369. [Google Scholar] [CrossRef]
- Li, T.; Xu, S.; Oses-Prieto, J.; Putil, S.; Xu, P.; Wang, R.-J.; Li, K.H.; Maltby, D.A.; An, L.-H.; Burlingame, A.L.; et al. Proteomics Analysis Reveals Post-Translational Mechanisms for Cold-Induced Metabolic Changes in Arabidopsis. Mol. Plant 2011, 4, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Szymanski, J.; Levin, Y.; Savidor, A.; Breitel, D.; Chappell-Maor, L.; Heinig, U.; Töpfer, N.; Aharoni, A. Label-free deep shotgun proteomics reveals protein dynamics during tomato fruit tissues development. Plant J. 2017, 90, 396–417. [Google Scholar] [CrossRef]
- Tang, W.; Deng, Z.; Oses-Prieto, J.; Suzuki, N.; Zhu, S.; Zhang, X.; Burlingame, A.L.; Wang, Z.-Y. Proteomics Studies of Brassinosteroid Signal Transduction Using Prefractionation and Two-dimensional DIGE. Mol. Cell. Proteom. 2008, 7, 728–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.-L.; Chalkley, R.J.; Maynard, J.C.; Wang, W.; Ni, W.; Jiang, X.; Shin, K.; Cheng, L.; Savage, D.; Hühmer, A.F.R.; et al. Proteomic analysis reveals O-GlcNAc modification on proteins with key regulatory functions inArabidopsis. Proc. Natl. Acad. Sci. USA 2017, 114, E1536–E1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Zhang, Y.; Tan, J.; Chen, B.; Sun, L. Improved Integrated Whole Proteomic and Phosphoproteomic Profiles of Severe Acute Pancreatitis. J. Proteome Res. 2020, 19, 2471–2482. [Google Scholar] [CrossRef]
- Taus, T.; Köcher, T.; Pichler, P.; Paschke, C.; Schmidt, A.; Henrich, C.; Mechtler, K. Universal and Confident Phosphorylation Site Localization Using phosphoRS. J. Proteome Res. 2011, 10, 5354–5362. [Google Scholar] [CrossRef] [PubMed]
- Mergner, J.; Frejno, M.; List, M.; Papacek, M.; Chen, X.; Chaudhary, A.; Samaras, P.; Richter, S.; Shikata, H.; Messerer, M.; et al. Mass-spectrometry-based draft of the Arabidopsis proteome. Nature 2020, 579, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, In Vivo, and Site-Specific Phosphorylation Dynamics in Signaling Networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.-F.; Hsu, C.-C.; Hung, J.-N.; Wang, Y.-T.; Choong, W.-K.; Zeng, M.-Y.; Lin, P.-Y.; Hong, R.-W.; Sung, T.-Y.; Chen, Y.-J. Sequential Phosphoproteomic Enrichment through Complementary Metal-Directed Immobilized Metal Ion Affinity Chromatography. Anal. Chem. 2013, 86, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2019, 17, 41–44. [Google Scholar] [CrossRef]
- Steger, M.; Demichev, V.; Backman, M.; Ohmayer, U.; Ihmor, P.; Müller, S.; Ralser, M.; Daub, H. Time-resolved in vivo ubiquitinome profiling by DIA-MS reveals USP7 targets on a proteome-wide scale. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Wang, S.; Cai, Y.; Cheng, J.; Li, W.; Liu, Y.; Yang, H. motifeR: An Integrated Web Software for Identification and Visualization of Protein Posttranslational Modification Motifs. Proteomics 2019, 19, e1900245. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, K.J.; Friso, G.; Walther, D.; Schulze, W.X. Meta-Analysis of Arabidopsis thaliana Phospho-Proteomics Data Reveals Compartmentalization of Phosphorylation Motifs. Plant Cell 2014, 26, 2367–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Hsu, C.-C.; Du, Y.; Zhu, P.; Zhao, C.; Fu, X.; Zhang, C.; Paez, J.S.; Macho, A.P.; Tao, W.A.; et al. Mapping proteome-wide targets of protein kinases in plant stress responses. Proc. Natl. Acad. Sci. USA 2020, 117, 3270–3280. [Google Scholar] [CrossRef]
- Schwartz, D.; Gygi, S.P. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 2005, 23, 1391–1398. [Google Scholar] [CrossRef]
- Hou, Q.; Ufer, G.; Bartels, D. Lipid signalling in plant responses to abiotic stress. Plant Cell Environ. 2016, 39, 1029–1048. [Google Scholar] [CrossRef]
- Sicilia, C.B.; Silvestre, S.; Haslam, R.P.; Michaelson, L.V. Lipid remodelling: Unravelling the response to cold stress in Arabidopsis and its extremophile relative Eutrema salsugineum. Plant Sci. 2017, 263, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Welti, R.; Li, W.; Li, M.; Sang, Y.; Biesiada, H.; Zhou, H.E.; Rajashekar, C.B.; Williams, T.D.; Wang, X. Profiling membrane lipids in plant stress responses: Role of phospholipase Dα in freezing-induced lipid changes in arabidopsis. J. Biol. Chem. 2002, 277, 31994–32002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heilmann, I. Plant phosphoinositide signaling dynamics on demand. Biochim. Biophys. Acta 2016, 1861, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, I. Phosphoinositide signaling in plant development. Development 2016, 143, 2044–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokotylo, I.; Kravets, V.; Martinec, J.; Ruelland, E. The phosphatidic acid paradox: Too many actions for one molecule class? Lessons from plants. Prog. Lipid Res. 2018, 71, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, M.; Zhang, W.; Welti, R.; Wang, X. The plasma membrane–bound phospholipase Dδ enhances freezing tolerance in Arabidopsis thaliana. Nat. Biotechnol. 2004, 22, 427–433. [Google Scholar] [CrossRef]
- Rajashekar, C.; Zhou, H.-E.; Zhang, Y.; Li, W.; Wang, X. Suppression of phospholipase Dα1 induces freezing tolerance in Arabidopsis: Response of cold-responsive genes and osmolyte accumulation. J. Plant Physiol. 2006, 163, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Ruelland, E.; Cantrel, C.; Gawer, M.; Kader, J.-C.; Zachowski, A. Activation of Phospholipases C and D Is an Early Response to a Cold Exposure in Arabidopsis Suspension Cells. Plant Physiol. 2002, 130, 999–1007. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.-Y.; Xue, H.-W. Phosphatidic acid plays key roles regulating plant development and stress responses. J. Integr. Plant Biol. 2018, 60, 851–863. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, F.; Arisz, S.; Dekker, H.L.; Kramer, G.; de Koster, C.; Haring, M.A.; Munnik, T.; Testerink, C. Identification of novel candidate phosphatidic acid-binding proteins involved in the salt-stress response of Arabidopsis thaliana roots. Biochem. J. 2013, 450, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Testerink, C.; Dekker, H.L.; Lim, Z.-Y.; Johns, M.K.; Holmes, A.B.; de Koster, C.G.; Ktistakis, N.T.; Munnik, T. Isolation and identification of phosphatidic acid targets from plants. Plant J. 2004, 39, 527–536. [Google Scholar] [CrossRef]
- Lee, H.; Guo, Y.; Ohta, M.; Xiong, L.; Stevenson, B.; Zhu, J. LOS2, a genetic locus required for cold-responsive gene transcription encodes a bi-functional enolase. EMBO J. 2002, 21, 2692–2702. [Google Scholar] [CrossRef] [PubMed]
- Huo, C.; Zhang, B.; Wang, H.; Wang, F.; Liu, M.; Gao, Y.; Zhang, W.; Deng, Z.; Sun, D.; Tang, W. Comparative Study of Early Cold-Regulated Proteins by Two-Dimensional Difference Gel Electrophoresis Reveals a Key Role for Phospholipase Dα1 in Mediating Cold Acclimation Signaling Pathway in Rice. Mol. Cell. Proteom. 2016, 15, 1397–1411. [Google Scholar] [CrossRef] [Green Version]
- Hafke, J.B.; Ehlers, K.; Föller, J.; Höll, S.-R.; Becker, S.; van Bel, A.J. Involvement of the Sieve Element Cytoskeleton in Electrical Responses to Cold Shocks. Plant Physiol. 2013, 162, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Mazars, C.; Thion, L.; Thuleau, P.; Graziana, A.; Knight, M.; Moreau, M.; Ranjeva, R. Organization of cytoskeleton controls the changes in cytosolic calcium of cold-shocked Nicotiana plumbaginifolia protoplasts. Cell Calcium 1997, 22, 413–420. [Google Scholar] [CrossRef]
- Wang, X.; Mao, T. Understanding the functions and mechanisms of plant cytoskeleton in response to environmental signals. Curr. Opin. Plant Biol. 2019, 52, 86–96. [Google Scholar] [CrossRef]
- Smertenko, A.; Chang, H.Y.; Sonobe, S.; Fenyk, S.I.; Weingartner, M.; Bogre, L.; Hussey, P. Control of the AtMAP65-1 interaction with microtubules through the cell cycle. J. Cell Sci. 2006, 119, 3227–3237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komis, G.; Illés, P.; Beck, M.; Šamaj, J. Microtubules and mitogen-activated protein kinase signalling. Curr. Opin. Plant Biol. 2011, 14, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lin, F.; Mao, T.; Nie, J.; Yan, M.; Yuan, M.; Zhang, W. Phosphatidic Acid Regulates Microtubule Organization by Interacting with MAP65-1 in Response to Salt Stress in Arabidopsis. Plant Cell 2012, 24, 4555–4576. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Renhu, N.; Naito, M.; Nakamura, A.; Shiba, H.; Yamamoto, T.; Suzaki, T.; Iida, H.; Miura, K. Ca2+-permeable mechanosensitive channels MCA1 and MCA2 mediate cold-induced cytosolic Ca2+ increase and cold tolerance in Arabidopsis. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Liu, Q.; Ding, Y.; Shi, Y.; Ma, L.; Wang, Y.; Song, C.; Wilkins, K.A.; Davies, J.M.; Knight, H.; Knight, M.R.; et al. The calcium transporter ANNEXIN1 mediates cold-induced calcium signaling and freezing tolerance in plants. EMBO J. 2021, 40, e104559. [Google Scholar] [CrossRef] [PubMed]
- Catalá, R.; Santos, E.; Alonso, J.M.; Ecker, J.; Zapater, J.M.M.; Salinas, J. Mutations in the Ca2+/H+ Transporter CAX1 Increase CBF/DREB1 Expression and the Cold-Acclimation Response in Arabidopsis. Plant Cell 2003, 15, 2940–2951. [Google Scholar] [CrossRef] [Green Version]
- Kidokoro, S.; Yoneda, K.; Takasaki, H.; Takahashi, F.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Different Cold-Signaling Pathways Function in the Responses to Rapid and Gradual Decreases in Temperature. Plant Cell 2017, 29, 760–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Park, S.; Gilmour, S.J.; Thomashow, M.F. Roles of CAMTA transcription factors and salicylic acid in configuring the low-temperature transcriptome and freezing tolerance of Arabidopsis. Plant J. 2013, 75, 364–376. [Google Scholar] [CrossRef]
- Almadanim, M.C.; Alexandre, B.; Rosa, M.G.; Sapeta, H.; Leitão, A.E.; Ramalho, J.C.; Lam, T.T.; Negrao, S.; Abreu, I.A.; Oliveira, M.M. Rice calcium-dependent protein kinase OsCPK17 targets plasma membrane intrinsic protein and sucrose-phosphate synthase and is required for a proper cold stress response. Plant Cell Environ. 2017, 40, 1197–1213. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Yang, G.; Khan, M.; Onodera, H.; Toki, S.; Yamaguchi, M. Over-expression of calcium-dependent protein kinase 13 and calreticulin interacting protein 1 confers cold tolerance on rice plants. Mol. Genet. Genom. 2007, 277, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Saijo, Y.; Hata, S.; Kyozuka, J.; Shimamoto, K.; Izui, K. Over-expression of a single Ca2+-dependent protein kinase confers both cold and salt/drought tolerance on rice plants. Plant J. 2000, 23, 319–327. [Google Scholar] [CrossRef]
- Teige, M.; Scheikl, E.; Eulgem, T.; Dóczi, R.; Ichimura, K.; Shinozaki, K.; Dangl, J.L.; Hirt, H. The MKK2 Pathway Mediates Cold and Salt Stress Signaling in Arabidopsis. Mol. Cell 2004, 15, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Tena, G.; Plotnikova, J.; Willmann, M.R.; Chiu, W.-L.; Gomez-Gomez, L.; Boller, T.; Ausubel, F.M.; Sheen, J. MAP kinase signalling cascade in Arabidopsis innate immunity. Nature 2002, 415, 977–983. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2016, 45, D1040–D1045. [Google Scholar] [CrossRef] [Green Version]
- Vogel, J.; Zarka, D.G.; Van Buskirk, H.A.; Fowler, S.G.; Thomashow, M.F. Roles of the CBF2 and ZAT12 transcription factors in configuring the low temperature transcriptome of Arabidopsis. Plant J. 2004, 41, 195–211. [Google Scholar] [CrossRef]
- Park, S.; Lee, C.; Doherty, C.J.; Gilmour, S.J.; Kim, Y.; Thomashow, M.F. Regulation of the Arabidopsis CBF regulon by a complex low-temperature regulatory network. Plant J. 2015, 82, 193–207. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Ding, Y.; Shi, Y.; Zhang, X.; Gong, Z.; Yang, S. The cbfs triple mutants reveal the essential functions of CBF s in cold acclimation and allow the definition of CBF regulons in Arabidopsis. New Phytol. 2016, 212, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Zhang, Z.; Xie, S.; Si, T.; Li, Y.; Zhu, J.-K. Mutational Evidence for the Critical Role of CBF Transcription Factors in Cold Acclimation in Arabidopsis. Plant Physiol. 2016, 171, 2744–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, M.A.; Farre, E.; Thomashow, M.F. CIRCADIAN CLOCK-ASSOCIATED 1 and LATE ELONGATED HYPOCOTYL regulate expression of the C-REPEAT BINDING FACTOR (CBF) pathway in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 7241–7246. [Google Scholar] [CrossRef] [Green Version]
- Gangappa, S.N.; Botto, J.F. The Multifaceted Roles of HY5 in Plant Growth and Development. Mol. Plant 2016, 9, 1353–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Huang, X.; Deng, X.W. The Photomorphogenic Central Repressor COP1: Conservation and Functional Diversification during Evolution. Plant Commun. 2020, 1, 100044. [Google Scholar] [CrossRef]
- Catalá, R.; Medina, J.; Salinas, J. Integration of low temperature and light signaling during cold acclimation response in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 16475–16480. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.-H.; Domijan, M.; Klose, C.; Biswas, S.; Ezer, D.; Gao, M.; Khattak, A.K.; Box, M.S.; Charoensawan, V.; Cortijo, S.; et al. Phytochromes function as thermosensors in Arabidopsis. Science 2016, 354, 886–889. [Google Scholar] [CrossRef] [Green Version]
- Legris, M.; Klose, C.; Burgie, E.S.; Rojas, C.C.R.; Neme, M.; Hiltbrunner, A.; Wigge, P.A.; Schäfer, E.; Vierstra, R.D.; Casal, J.J. Phytochrome B integrates light and temperature signals in Arabidopsis. Science 2016, 354, 897–900. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Shi, Y.; Peng, Y.; Jia, Y.; Yan, Y.; Dong, X.; Li, H.; Dong, J.; Li, J.; Gong, Z.; et al. Cold-Induced CBF–PIF3 Interaction Enhances Freezing Tolerance by Stabilizing the phyB Thermosensor in Arabidopsis. Mol. Plant 2020, 13, 894–906. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Zhao, L.; Ren, Y.; Yang, S.; Zhu, J.; Zhao, C. The transcription factor ICE1 functions in cold stress response by binding to the promoters ofCBFandCORgenes. J. Integr. Plant Biol. 2020, 62, 258–263. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Ohta, M.; Nakazawa, M.; Ono, M.; Hasegawa, P.M. ICE1 Ser403 is necessary for protein stabilization and regulation of cold signaling and tolerance. Plant J. 2011, 67, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Alsheikh, M.K.; Svensson, J.T.; Randall, S.K. Phosphorylation regulated ion-binding is a property shared by the acidic subclass dehydrins. Plant Cell Environ. 2005, 28, 1114–1122. [Google Scholar] [CrossRef]
- Candat, A.; Paszkiewicz, G.; Neveu, M.; Gautier, R.; Logan, D.C.; Avelange-Macherel, M.-H.; Macherel, D. The Ubiquitous Distribution of Late Embryogenesis Abundant Proteins across Cell Compartments in Arabidopsis Offers Tailored Protection against Abiotic Stress. Plant Cell 2014, 26, 3148–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maszkowska, J.; Debski, J.; Kulik, A.; Kistowski, M.; Bucholc, M.; Lichocka, M.; Klimecka, M.; Sztatelman, O.; Szymanska, K.; Dadlez, M. Phosphoproteomic analysis reveals that dehydrins ERD10 and ERD14 are phosphorylated by SNF1-related protein kinase 2.10 in response to osmotic stress. Plant. Cell Environ. 2018, 42, 931–946. [Google Scholar] [CrossRef] [PubMed]
- Bourmaud, A.; Gallien, S.; Domon, B. Parallel reaction monitoring using quadrupole-Orbitrap mass spectrometer: Principle and applications. Proteomics 2016, 16, 2146–2159. [Google Scholar] [CrossRef] [PubMed]
- Harashima, H.; Shinmyo, A.; Sekine, M. Phosphorylation of threonine 161 in plant cyclin-dependent kinase A is required for cell division by activation of its associated kinase. Plant J. 2007, 52, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Pribat, A.; Sormani, R.; Rousseau-Gueutin, M.; Julkowska, M.M.; Testerink, C.; Joubès, J.; Castroviejo, M.; Laguerre, M.; Meyer, C.; Germain, V.; et al. A novel class of PTEN protein in Arabidopsis displays unusual phosphoinositide phosphatase activity and efficiently binds phosphatidic acid. Biochem. J. 2011, 441, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Yoon, G.M.; Shemansky, J.M.; Lin, D.Y.; Ying, Z.I.; Chang, J.; Garrett, W.M.; Kessenbrock, M.; Groth, G.; Tucker, M.L.; et al. CTR1 phosphorylates the central regulator EIN2 to control ethylene hormone signaling from the ER membrane to the nucleus in Arabidopsis. Proc. Natl. Acad. Sci. USA 2012, 109, 19486–19491. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.-H.; Henderson, D.A.; Zhu, J.-K. TheArabidopsisCold-Responsive Transcriptome and Its Regulation by ICE1. Plant Cell 2005, 17, 3155–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Zhao, Y.; Li, Z.; Hsu, C.-C.; Liu, X.; Fu, L.; Hou, Y.-J.; Du, Y.; Xie, S.; Zhang, C.; et al. Reciprocal Regulation of the TOR Kinase and ABA Receptor Balances Plant Growth and Stress Response. Mol. Cell 2017, 69, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.; Oses-Prieto, J.A.; Kutschera, U.; Tseng, T.-S.; Hao, L.; Burlingame, A.L.; Wang, Z.-Y.; Briggs, W.R. Blue Light-Induced Proteomic Changes in Etiolated Arabidopsis Seedlings. J. Proteome Res. 2014, 13, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Mustilli, A.-C.; Merlot, S.; Vavasseur, A.; Fenzi, F.; Giraudat, J. Arabidopsis OST1 Protein Kinase Mediates the Regulation of Stomatal Aperture by Abscisic Acid and Acts Upstream of Reactive Oxygen Species Production. Plant Cell 2002, 14, 3089–3099. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Zhang, J.; Hsu, P.-K.; Ceciliato, P.H.O.; Zhang, L.; Dubeaux, G.; Munemasa, S.; Ge, C.; Zhao, Y.; Hauser, F.; et al. MAP3Kinase-dependent SnRK2-kinase activation is required for abscisic acid signal transduction and rapid osmotic stress response. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Li, Y.; Wang, Y.; Liu, X.; Ma, L.; Zhang, Z.; Mu, C.; Zhang, Y.; Peng, L.; Xie, S.; et al. Initiation and amplification of SnRK2 activation in abscisic acid signaling. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Liu, J.; Ming, Y.; Shi, Y.; Song, C.; Gong, Z.; Yang, S.; Ding, Y. Reciprocal regulation between the negative regulator PP2CG1 phosphatase and the positive regulator OST1 kinase confers cold response in Arabidopsis. J. Integr. Plant Biol. 2021, 63, 1568–1587. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jia, Y.; Ding, Y.; Shi, Y.; Li, Z.; Guo, Y.; Gong, Z.; Yang, S. Plasma Membrane CRPK1-Mediated Phosphorylation of 14-3-3 Proteins Induces Their Nuclear Import to Fine-Tune CBF Signaling during Cold Response. Mol. Cell 2017, 66, 117–128.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ding, Y.; Li, Z.; Shi, Y.; Wang, J.; Hua, J.; Gong, Z.; Zhou, J.-M.; Yang, S. PUB25 and PUB26 Promote Plant Freezing Tolerance by Degrading the Cold Signaling Negative Regulator MYB15. Dev. Cell 2019, 51, 222–235.e5. [Google Scholar] [CrossRef]
- Dong, C.-H.; Agarwal, M.; Zhang, Y.; Xie, Q.; Zhu, J.-K. The negative regulator of plant cold responses, HOS1, is a RING E3 ligase that mediates the ubiquitination and degradation of ICE1. Proc. Natl. Acad. Sci. USA 2006, 103, 8281–8286. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Jin, J.B.; Lee, J.; Yoo, C.Y.; Stirm, V.; Miura, T.; Ashworth, E.N.; Bressan, R.A.; Yun, D.-J.; Hasegawa, P.M. SIZ1-Mediated Sumoylation of ICE1 Controls CBF3/DREB1A Expression and Freezing Tolerance in Arabidopsis. Plant Cell 2007, 19, 1403–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartels, S.; Anderson, J.C.; Besteiro, M.A.G.; Carreri, A.; Hirt, H.; Buchala, A.; Metraux, J.-P.; Peck, S.C.; Ulm, R. MAP KINASE PHOSPHATASE1 and PROTEIN TYROSINE PHOSPHATASE1 Are Repressors of Salicylic Acid Synthesis and SNC1-Mediated Responses in Arabidopsis. Plant Cell 2009, 21, 2884–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartels, S.; Besteiro, M.A.G.; Lang, D.; Ulm, R. Emerging functions for plant MAP kinase phosphatases. Trends Plant Sci. 2010, 15, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Tamnanloo, F.; Damen, H.; Jangra, R.; Lee, J.S. MAP KINASE PHOSPHATASE1 Controls Cell Fate Transition during Stomatal Development. Plant Physiol. 2018, 178, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Xiong, L.; Lee, H.; Ishitani, M.; Tanaka, Y.; Stevenson, B.; Koiwa, H.; Bressan, R.A.; Hasegawa, P.M.; Zhu, J.-K. Repression of stress-responsive genes by FIERY2, a novel transcriptional regulator in Arabidopsis. Proc. Natl. Acad. Sci. USA 2002, 99, 10899–10904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Guo, G.; Zhang, M.; Liu, C.Y.; Hu, Q.; Lam, H.; Cheng, H.; Xue, Y.; Li, J.; Li, N. Stable Isotope Metabolic Labeling-based Quantitative Phosphoproteomic Analysis of Arabidopsis Mutants Reveals Ethylene-regulated Time-dependent Phosphoproteins and Putative Substrates of Constitutive Triple Response 1 Kinase. Mol. Cell. Proteom. 2013, 12, 3559–3582. [Google Scholar] [CrossRef] [Green Version]
- Escher, C.; Reiter, L.; MacLean, B.; Ossola, R.; Herzog, F.; Chilton, J.; MacCoss, M.J.; Rinner, O. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics 2012, 12, 1111–1121. [Google Scholar] [CrossRef] [Green Version]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef] [Green Version]
- Dorfer, V.; Pichler, P.; Stranzl, T.; Stadlmann, J.; Taus, T.; Winkler, S.; Mechtler, K. MS Amanda, a Universal Identification Algorithm Optimized for High Accuracy Tandem Mass Spectra. J. Proteome Res. 2014, 13, 3679–3684. [Google Scholar] [CrossRef]
- Shteynberg, D.; Nesvizhskii, A.; Moritz, R.L.; Deutsch, E. Combining Results of Multiple Search Engines in Proteomics. Mol. Cell. Proteom. 2013, 12, 2383–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Chen, T.; Wu, S.; Yang, C.; Bai, M.; Shu, K.; Li, K.; Zhang, G.; Jin, Z.; He, F.; et al. iProX: An integrated proteome resource. Nucleic Acids Res. 2019, 47, D1211–D1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Description | Fold 1 | Adjusted p Value | Fold | Adjusted p Value |
|---|---|---|---|---|---|
| 30 min | PRM | DIA | |||
| ATCG00710 | PSBH, Photosystem II reaction center protein H | 0.36 | 2.3 × 10−3 | 0.35 | 2.6 × 10−3 |
| AT1G20340 | PETE2, Cupredoxin superfamily protein | 0.43 | 8.5 × 10−5 | 0.4 | 3.1 × 10−3 |
| 120 min | |||||
| AT1G20340 | PETE2, Cupredoxin superfamily protein | 0.47 | 1.9 × 10−4 | 0.37 | 4.7 × 10−4 |
| AT4G35750 | SEC14 cytosolic factor family protein | 0.42 | 5.8 × 10−4 | 0.42 | 2.4 × 10−6 |
| ATCG00710 | PSBH, Photosystem II reaction center protein H | 0.46 | 4.0 × 10−3 | 0.47 | 8.2 × 10−3 |
| AT3G46640 | LUX, Homeodomain-like superfamily protein | 0.64 | 8.0 × 10−2 | 0.65 | 1.5 × 10−3 |
| AT4G12470 | AZI1, Azelaic acid induced 1 | 1.37 | 2.2 × 10−3 | 1.51 | 7.4 × 10−3 |
| AT5G14920 | GASA14, Gibberellin-regulated family protein | 1.61 | 7.1 × 10−3 | 1.83 | 7.4 × 10−3 |
| AT5G02160 | FIP, FTSH5 interacting protein | 2.02 | 1.3 × 10−3 | 2.18 | 4.7 × 10−4 |
| AGI | Name | Phosphopeptides 1 | p-Sites | Fold | adj_p | Fold | adj_p |
|---|---|---|---|---|---|---|---|
| PRM Data | DIA Data | ||||||
| MAPK cascade kinases | |||||||
| AT3G45640 | MPK3 | ICDFGLARPTSENDFMTEyVVTR | Y198 | 3.4 | 1.5 × 10−2 | 1.5 | 3.8 × 10−3 |
| AT4G01370 | MPK4 | TKSETDFMtEyVVTR2 | T201;Y203 | 14.1 | 7.6 × 10−5 | 5.3 | 7.3 × 10−7 |
| AT4G01370 | MPK4 | TKSETDFMtEYVVTR2 | T201 | 4.8 | 2.0 × 10−4 | 5.4 | 4.9 × 10−8 |
| AT2G43790 | MPK6 | VTSESDFMtEyVVTR | T221;Y223 | 2.3 | 1.9 × 10−2 | 1.9 | 2.1 × 10−1 |
| AT1G18150 | MPK8 | AAAAVASTLESEEADNGGGYsAR | S539 | 0.3 | 8.7 × 10−3 | 0.4 | 8.5 × 10−6 |
| AT1G18150 | MPK8 | HHAsLPR | S495 | 0.5 | 4.1 × 10−3 | 0.4 | 1.3 × 10−4 |
| AT3G18040 | MPK9 | SIASLVTtLESPPTSQHEGSDYR | T549 | 0.4 | 3.4 × 10−5 | 0.4 | 2.0 × 10−5 |
| AT3G18040 | MPK9 | SQLtTIYR | T51 | 0.2 | 4.3 × 10−4 | 0.3 | 8.1 × 10−8 |
| AT1G53510 | MPK18 | VAFNDTPTTVFWTDyVATR | Y189 | 2.6 | 5.5 × 10−2 | 2.9 | 2.7 × 10−6 |
| AT4G29810 | MKK2 | FLTQSGtFKDGDLR2 | T31 | 0.5 | 2.0 × 10−3 | 0.5 | 3.5 × 10−5 |
| AT1G51660 | MKK4 | ASPSQNRsPQNLHQLLPPPRPLSSSSSPTT | S344 | 2.3 | 2.5 × 10−3 | 2.4 | 9.9 × 10−4 |
| AT1G53570 | MAPKKK3 | LSGVVsLESSTGR | S162 | 3.1 | 1.3 × 10−2 | 3.9 | 1.2 × 10−5 |
| AT1G63700 | YDA | sPGSGGNFYTNSFFQEPSR | S794 | 2.1 | 7.4 × 10−5 | 2.5 | 2.3 × 10−4 |
| AT3G13530 | MAPKKK7 | TPsSVSGNELAR | S452 | 0.3 | 2.3 × 10−2 | 0.2 | 6.4 × 10−8 |
| AT4G08500 | MEKK1 | FKsFDLDK | S119 | 0.5 | 1.9 × 10−3 | 0.8 | 5.3 × 10−2 |
| AT4G24100 | MAP4K5 | GVsAWNFDVR | S352 | 2.1 | 2.6 × 10−4 | 2.2 | 2.7 × 10−5 |
| AT5G03730 | CTR1 | AStFLSSK | T704 | 12.2 | 1.7 × 10−4 | NA in CK | |
| AT1G08720 | EDR1 | HNTFLsSK | S823 | 4.1 | 1.0 × 10−4 | NA in CK | |
| AT3G06620 | RAF7 | NIGEGAPsWR | S59 | 0.2 | 1.8 × 10−2 | NA in CS120 | |
| AT2G31010 | RAF13 | KTMsLPsSPHAYR | S465;S468 | 0.0 | 2.5 × 10−1 | NA in CS120 | |
| AT1G79570 | RAF20 | TNsSLHEFGNK | S130 | 0.5 | 1.2 × 10−2 | 0.6 | 4.3 × 10−3 |
| AT2G35050 | RAF24 | RNtLVtGGVR | T1134;T1137 | 1.2 | 6.5 × 10−1 | 1.1 | 8.9 × 10−1 |
| AT5G57610 | RAF35 | ISGFDGMSsLGQPSYPNPHLQDR | S569 | 4.9 | 2.3 × 10−2 | NA in CK | |
| Other kinases | |||||||
| AT3G45780 | phot1 | ALsESTNLHPFMTK | S350 | 2.1 | 1.1 × 10−2 | 2.2 | 1.8 × 10−4 |
| AT3G45780 | phot1 | MsENVVPSGR | S376 | 10.6 | 8.4 × 10−5 | 7.4 | 7.6 × 10−10 |
| AT3G45780 | phot1 | MSENVVPsGR | S382 | 0.5 | 1.9 × 10−2 | 0.6 | 1.9 × 10−4 |
| AT5G58140 | phot2 | sLEIFNPSSGK | S22 | 2.8 | 9.1 × 10−5 | 3.9 | 3.6 × 10−7 |
| AT3G50500 | SnRK2.2 | SSVLHsQPKsTVGTPAYIAPEILLR | S173;S177 | 14.4 | 1.5 × 10−3 | 18.9 | 8.0 × 10−8 |
| AT3G50500 | SnRK2.2 | StVGTPAYIAPEILLR | T178 | 5.8 | 1.2 × 10−4 | 9.4 | 1.2 × 10−7 |
| AT4G40010 | SnRK2.7 | SSVLHsQPK2 | S154 | 5.4 | 1.1 × 10−4 | 4.3 | 3.6 × 10−10 |
| AT4G40010 | SnRK2.7 | StVGTPAYVAPEVLSR | T159 | 5.9 | 5.6 × 10−4 | NA in CK | |
| AT4G33950 | OST1 | sTVGTPAYIAPEVLLK | S175 | 12.3 | 1.3 × 10−6 | NA in CK | |
| AT4G33950 | OST1 | SSVLHsQPKsTVGTPAYIAPEVLLK | S171;S175 | 279.9 | 1.2 × 10−5 | NA in CK | |
| AT2G17290 | CPK6 | NsLNISMR2 | S536 | 2.3 | 6.1 × 10−3 | 1.7 | 1.8 × 10−2 |
| AT3G53930 | ATG1B | SSYGFsVER | S349 | 2.4 | 1.1 × 10−4 | NA in CK | |
| AT4G21390 | B120 | NTDTsVVVADLTK | S480 | 0.1 | 2.2 × 10−2 | NA in CS120 | |
| AT4G27300 | SD11 | NVPDISSsLSLR | S798 | 0.4 | 2.8 × 10−2 | NA in CS120 | |
| AT4G33080 | NDR6 | KLAFsTVGTPDYIAPEVLLK | S300 | 5.9 | 1.1 × 10−2 | NA in CK | |
| AT5G28290 | NEK3 | HRPVDLsANDTSR | S333 | 0.0 | 2.4 × 10−4 | NA in CS120 | |
| AT5G64940 | ATH13 | SVsIAGIFLPR | S31 | 0.1 | 2.1 × 10−3 | NA in CS120 | |
| AT1G68830 | STN7 | TVTEtIDEISDGRKtVWWNR | T541;T551 | 0.2 | 3.4 × 10−3 | NA in CS120 | |
| AT2G07180 | PBL17 | SVtLYEASSDSQGTR | T392 | 4.2 | 1.7 × 10−2 | NA in CK | |
| AT3G48750 | CDC2 | TFtHEVVTLWYR | T161 | 0.5 | 6.3 × 10−3 | 0.5 | 4.7 × 10−3 |
| AT1G30570 | HERK2 | LNtLAASTMGR | T498 | 0.2 | 1.7 × 10−3 | NA in CS120 | |
| Phosphatases | |||||||
| AT3G55270 | MKP1 | YVSKtPLSR | T474 | 9.4 | 2.2 × 10−5 | NA in CK | |
| AT4G21670 | CPL1 | DETALPVsSRPTDPR | S839 | 12.5 | 3.2 × 10−2 | NA in CK | |
| AT3G50110 | PEN3 | TDDIVPCPPGsSPR | S86 | 4.9 | 5.6 × 10−4 | 1.9 | 1.5 × 10−5 |
| Transcription factors | |||||||
| AT2G04880 | WRKY1 | VVTHNNMLDSEVDDKEGDANKtPQSSTLQSITK | T391 | 0.1 | 1.3 × 10−2 | NA in CS120 | |
| AT2G46830 | CCA1 | IsSNITDPWK | S475 | -- | -- | NA in CK | |
| AT3G16857 | ARR1 | SIFsFDNTK | S374 | 12.6 | 1.7 × 10−2 | NA in CK | |
| AT3G26744 | ICE1 | DLSSVPDFLsAR | S172 | 4.6 | 1.5 × 10−2 | NA in CK | |
| AT4G16110 | ARR2 | GPDsGTAAGGSNSDPFPANLR | S12 | 2.7 | 6.7 × 10−3 | NA in CK | |
| AT2G40620 | bZIP18 | LGSGSGsASDSAGPSAPR | S83 | 2.4 | 8.1 × 10−5 | 1.6 | 2.6 × 10−3 |
| AT3G09600 | RVE8 | VIsPQHELATLR | S157 | 2.6 | 7.2 × 10−4 | 2.3 | 2.8 × 10−6 |
| AT4G16150 | CAMTA5 | ETHEVHAAPAtPGNSYSSSItDHLSPK | T154;T164 | 14.6 | 1.9 × 10−3 | 5.0 | 5.5 × 10−8 |
| AT4G16150 | CAMTA5 | ETHEVHAAPAtPGNSYSSSITDHLSPK | T154 | 1.9 | 4.5 × 10−3 | 2.3 | 9.0 × 10−6 |
| E3 ligases | |||||||
| AT2G39810 | HOS1 | SIsLPTQPNYSSKPVQEALK | S17 | 2.1 | 2.8 × 10−4 | 2.3 | 4.2 × 10−7 |
| AT1G67900 | NPY8 | ALLAAHNIDPSNPNAAAFStTTSIAAPEDR | T549 | 0.3 | 3.1 × 10−3 | NA in CS120 | |
| AT2G23140 | PUB4 | SGPLAATTsAATR | S492 | 0.3 | 4.4 × 10−2 | NA in CS120 | |
| AT3G08020 | PHD finger protein | ATFGSVTQFPAASTsEGNHVDDK | S615 | 2.1 | 1.5 × 10−2 | NA in CK | |
| AT3G26730 | RING/U-boxprotein | NQTQsLsPPDVSR | S428;S430 | 2.0 | 2.9 × 10−2 | NA in CK | |
| AT3G60080 | BTL13 | TVsGLGIGMR | S282 | 0.3 | 1.0 × 10−2 | NA in CS120 | |
| AT5G15440 | EDL1 | RNsLLGGSENGPPPQK | S273 | 2.1 | 4.9 × 10−3 | NA in CK | |
| AT5G65683 | WAVH2 | FGFLSNPStPR | T50 | 0.1 | 1.2 × 10−2 | NA in CS120 | |
| Others | |||||||
| AT2G38750 | ANN4 | KAsKsFFVEDEER | S46;S48 | 2.2 | 9.3 × 10−4 | 1.9 | 1.5 × 10−4 |
| AT4G35920 | MCA1 | STsNVSSGHDLLSR | S278 | 0.4 | 6.0 × 10−4 | 0.4 | 2.6 × 10−5 |
| AT5G03280 | EIN2 | AAPTSNFTVGSDGPPsFR | S645 | 2.0 | 8.9 × 10−3 | 2.3 | 7.0 × 10−3 |
| AT5G03280 | EIN2 | SLSGEGGsGTGSLSR | S655 | 2.4 | 1.4 × 10−3 | 2.1 | 2.9 × 10−3 |
| AT5G03280 | EIN2 | TPGsIDSLYGLQR | S757 | 1.6 | 2.3 × 10−2 | 1.7 | 1.2 × 10−1 |
| AT1G20440 | COR47 | NNVPEHEtPTVATEESPATTTEVTDR | T14 | 0.3 | 1.1 × 10−3 | 0.4 | 1.5 × 10−3 |
| AT1G76180 | ERD14 | KKDETKPEEtPIASEFEQK | T46 | 0.1 | 7.0 × 10−6 | 0.2 | 4.2 × 10−7 |
| AT2G23120 | LEA protein | AYGAEGHQEPTPGLGGGSTDAPtPSGDAPAATTTDAK | T67 | 0.4 | 3.9 × 10−3 | 0.4 | 6.1 × 10−5 |
| AT3G57410 | VLN3 | AAALAALTSAFNSsSGR | S773 | 0.4 | 2.9 × 10−4 | 0.3 | 1.8 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, J.; Zhou, Z.; Feng, H.; Xing, J.; Niu, Y.; Deng, Z. Data-Independent Acquisition-Based Proteome and Phosphoproteome Profiling Reveals Early Protein Phosphorylation and Dephosphorylation Events in Arabidopsis Seedlings upon Cold Exposure. Int. J. Mol. Sci. 2021, 22, 12856. https://doi.org/10.3390/ijms222312856
Tan J, Zhou Z, Feng H, Xing J, Niu Y, Deng Z. Data-Independent Acquisition-Based Proteome and Phosphoproteome Profiling Reveals Early Protein Phosphorylation and Dephosphorylation Events in Arabidopsis Seedlings upon Cold Exposure. International Journal of Molecular Sciences. 2021; 22(23):12856. https://doi.org/10.3390/ijms222312856
Chicago/Turabian StyleTan, Jinjuan, Zhongjing Zhou, Hanqian Feng, Jiayun Xing, Yujie Niu, and Zhiping Deng. 2021. "Data-Independent Acquisition-Based Proteome and Phosphoproteome Profiling Reveals Early Protein Phosphorylation and Dephosphorylation Events in Arabidopsis Seedlings upon Cold Exposure" International Journal of Molecular Sciences 22, no. 23: 12856. https://doi.org/10.3390/ijms222312856
APA StyleTan, J., Zhou, Z., Feng, H., Xing, J., Niu, Y., & Deng, Z. (2021). Data-Independent Acquisition-Based Proteome and Phosphoproteome Profiling Reveals Early Protein Phosphorylation and Dephosphorylation Events in Arabidopsis Seedlings upon Cold Exposure. International Journal of Molecular Sciences, 22(23), 12856. https://doi.org/10.3390/ijms222312856