
Doxorubicin Impairs Smooth Muscle Cell Contraction: Novel Insights in Vascular Toxicity

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
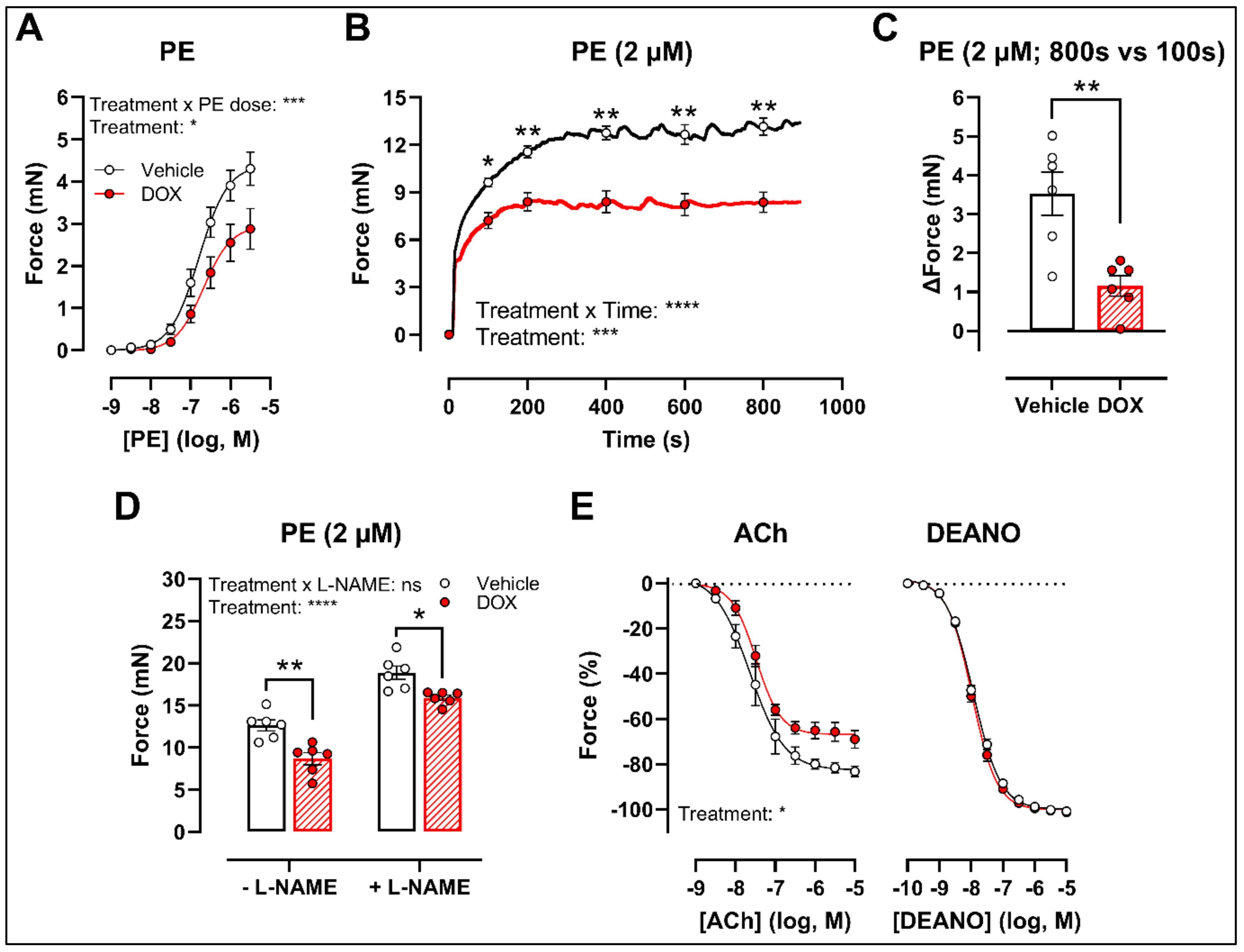
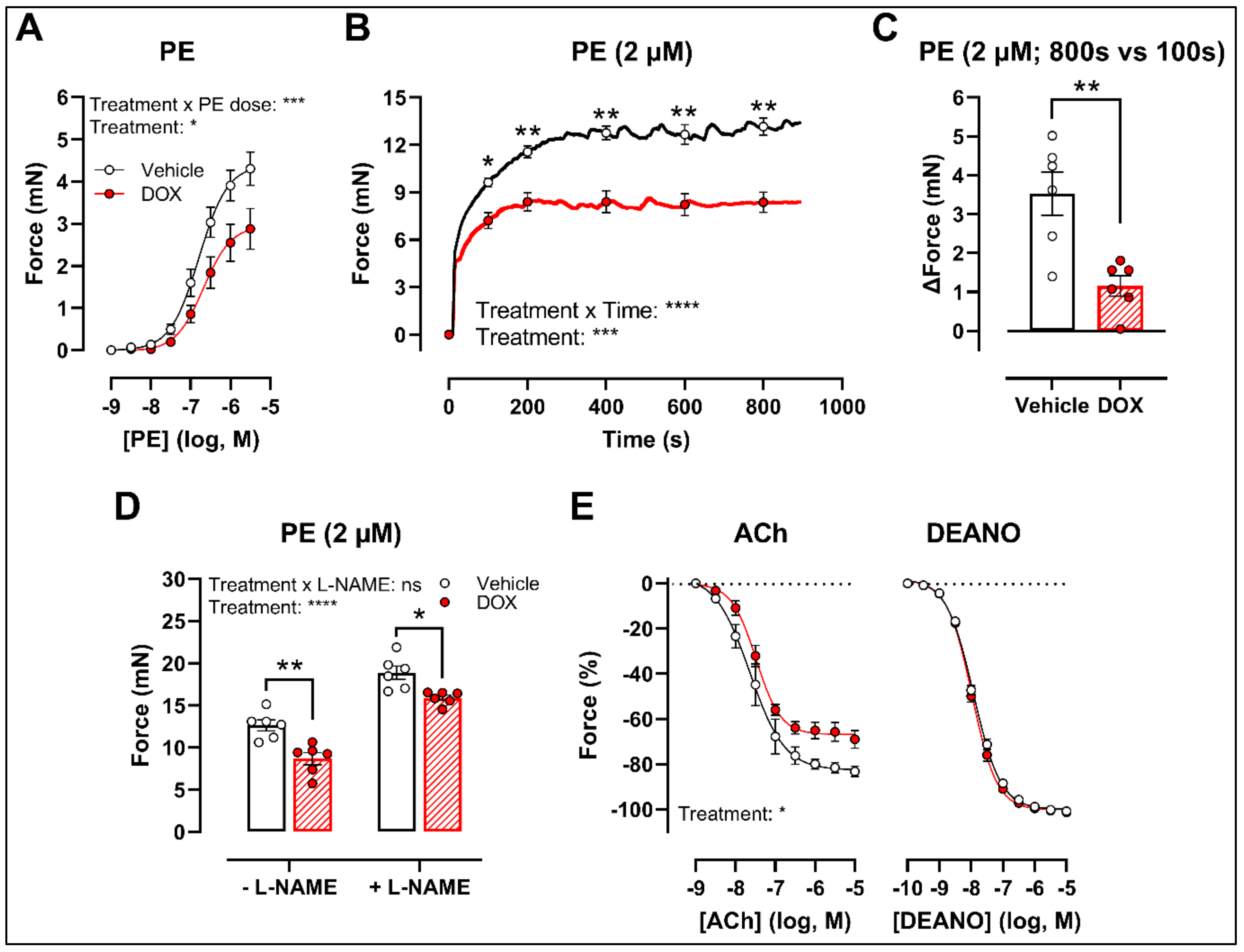
2.1. A Single Dose of DOX Impairs VSMC Contraction and Endothelium-Dependent Vasodilation
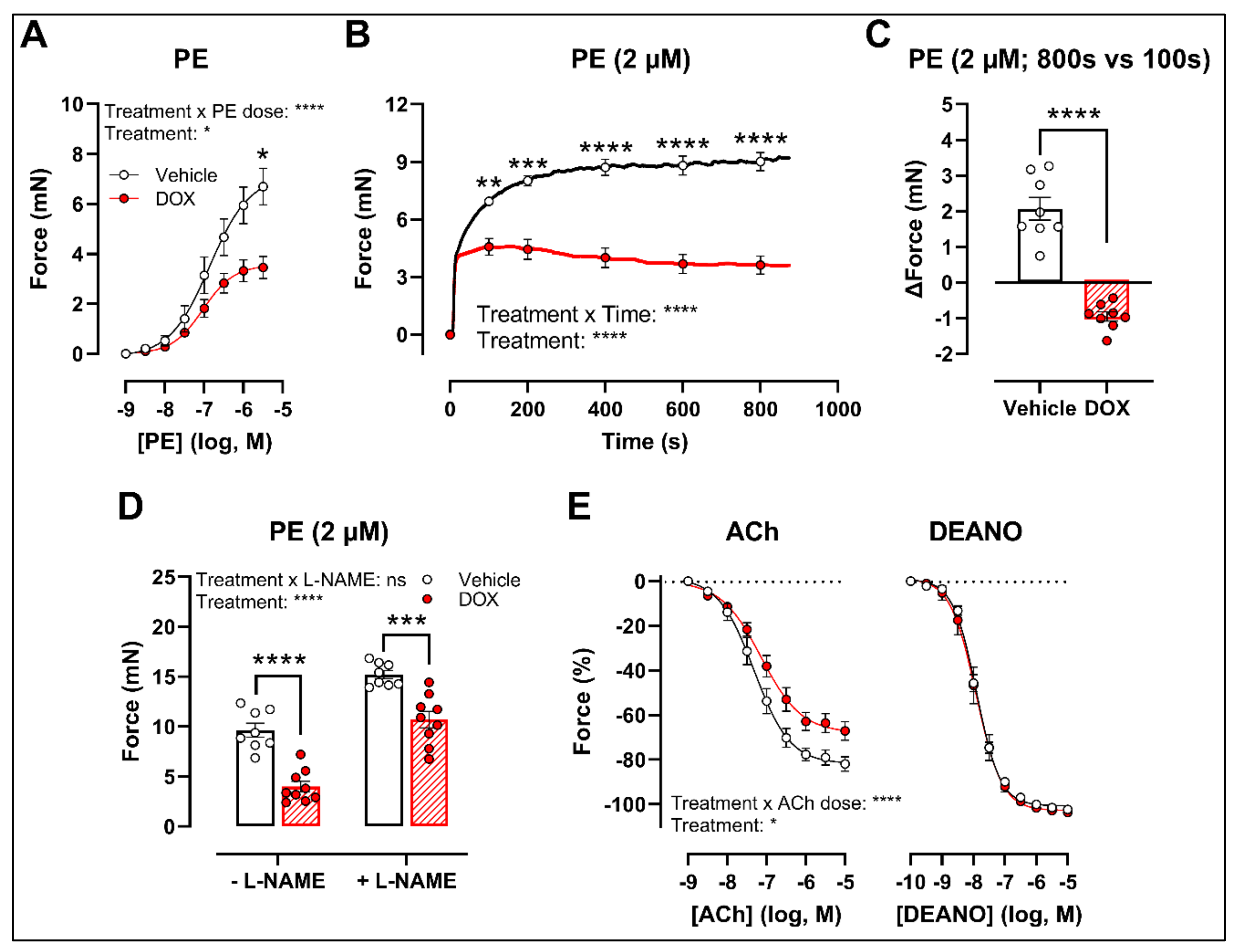
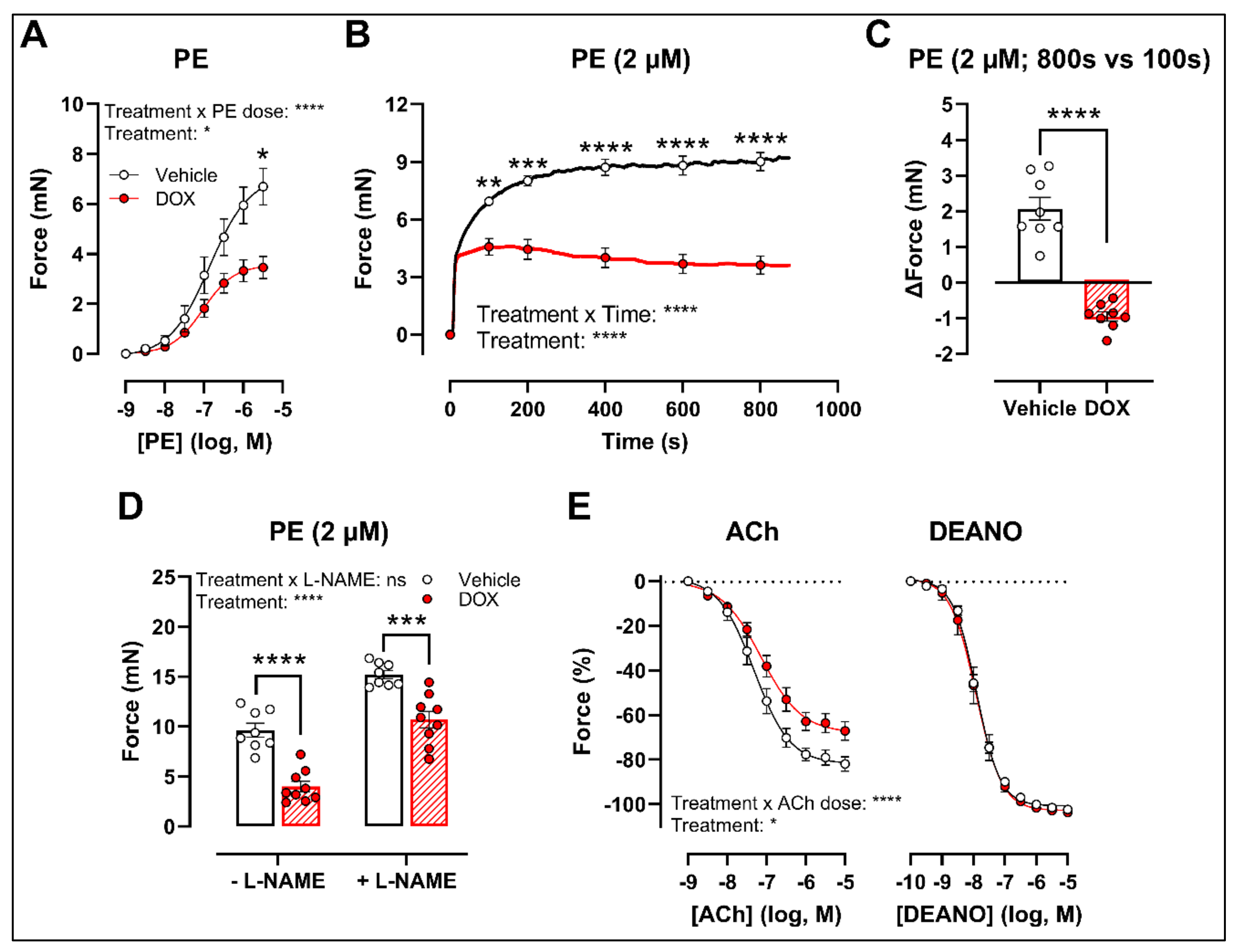
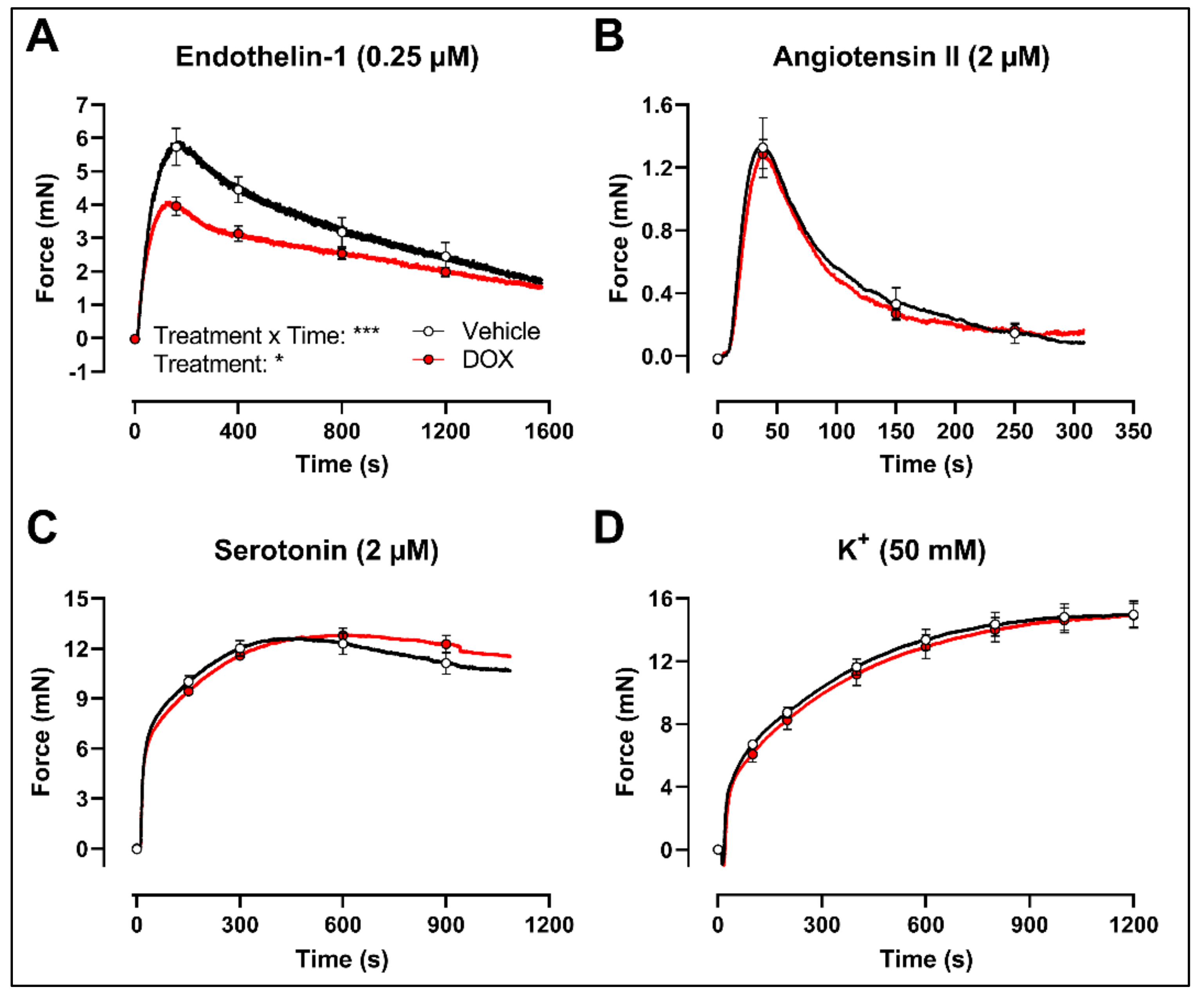
2.2. Ex Vivo Incubation of Aortic Rings with DOX Leads to Reduced VSMC Contraction and Impaired Endothelium-Dependent Relaxation
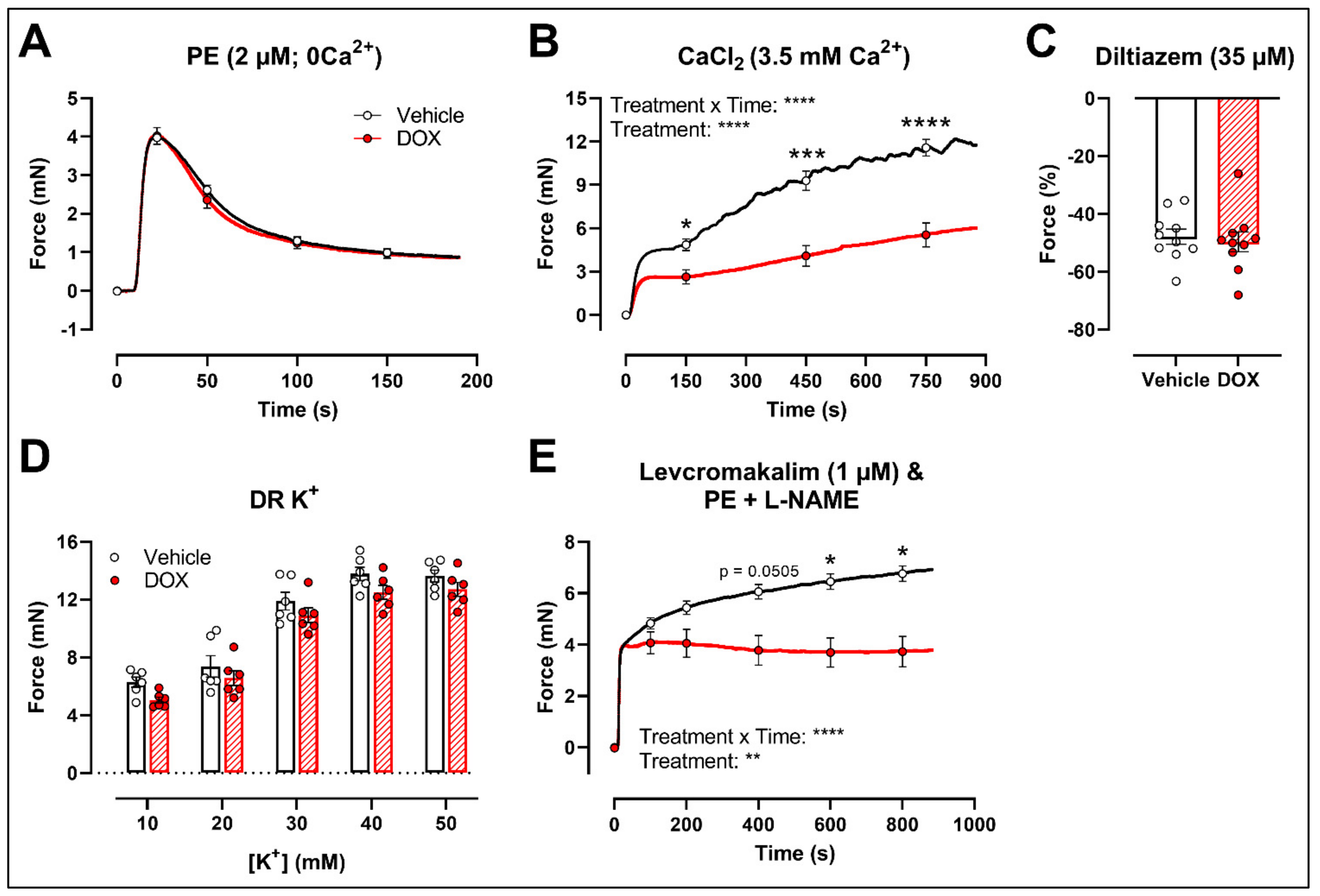
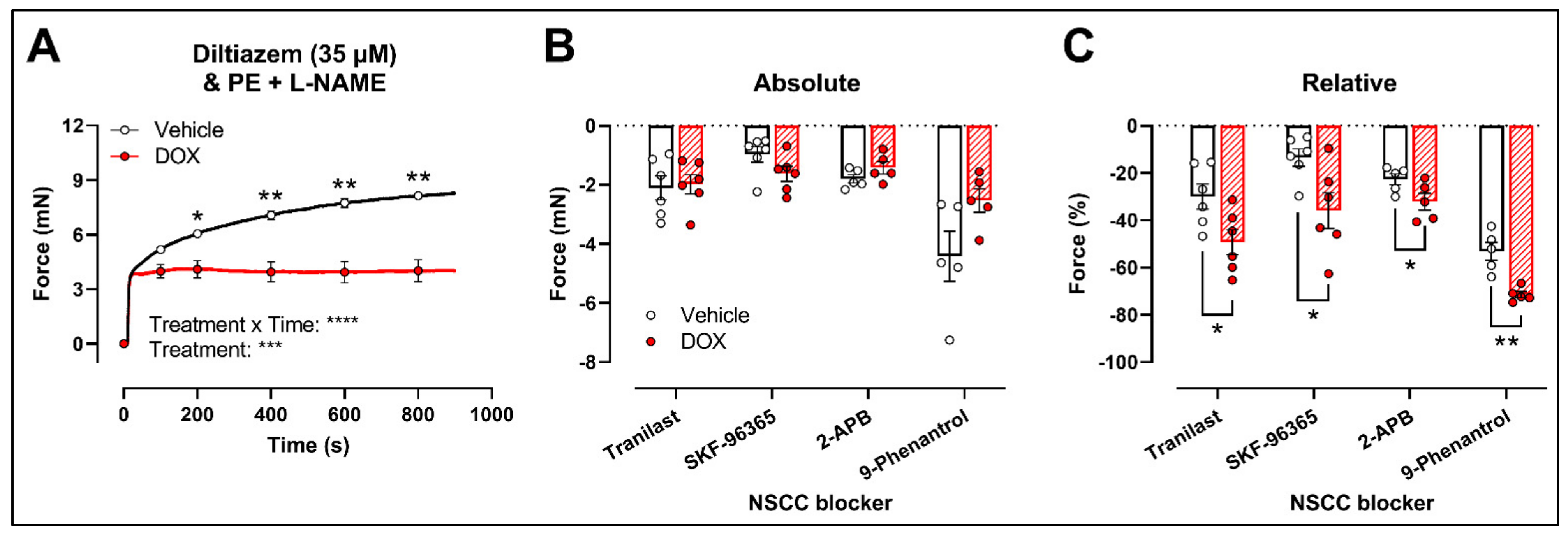
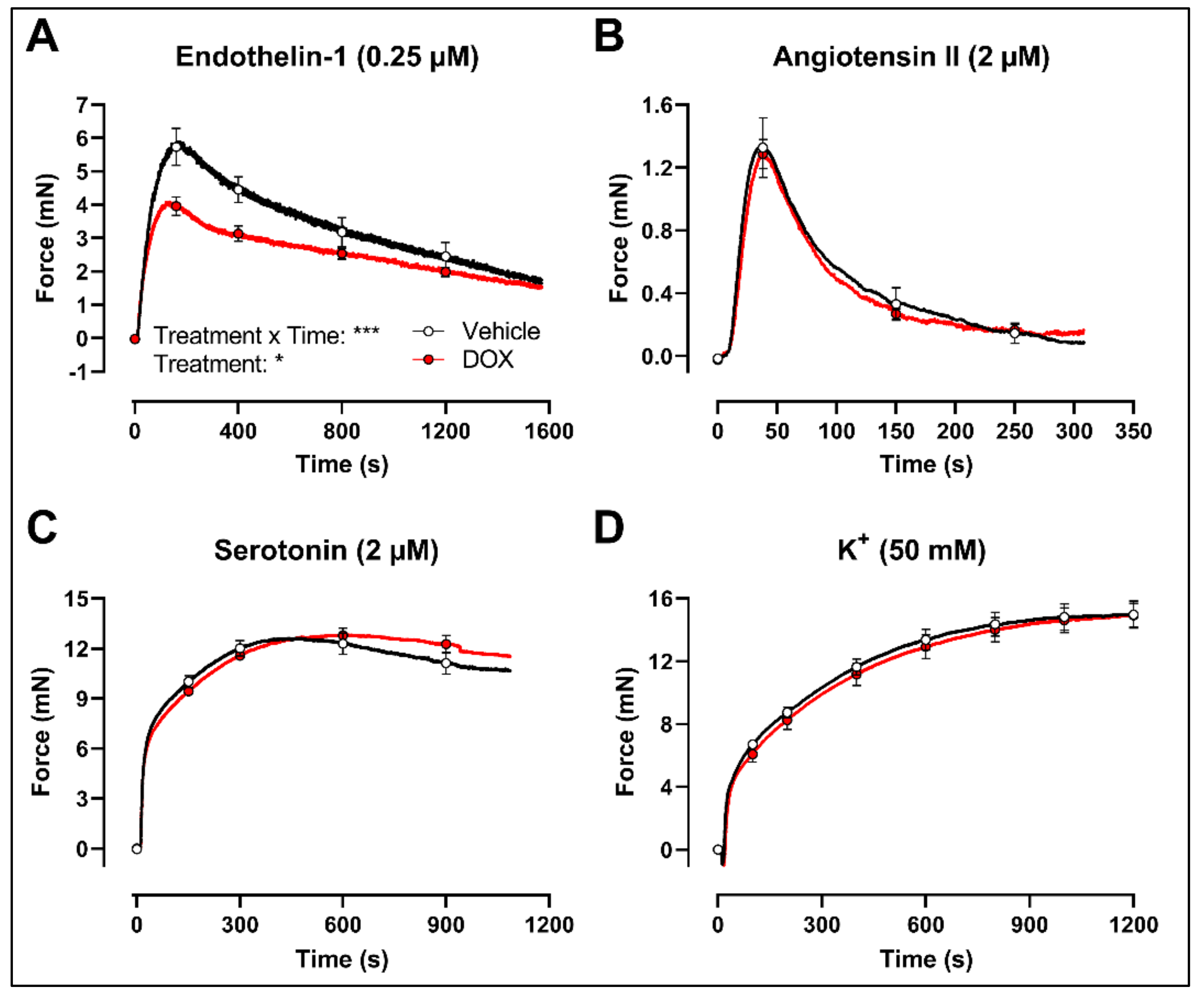
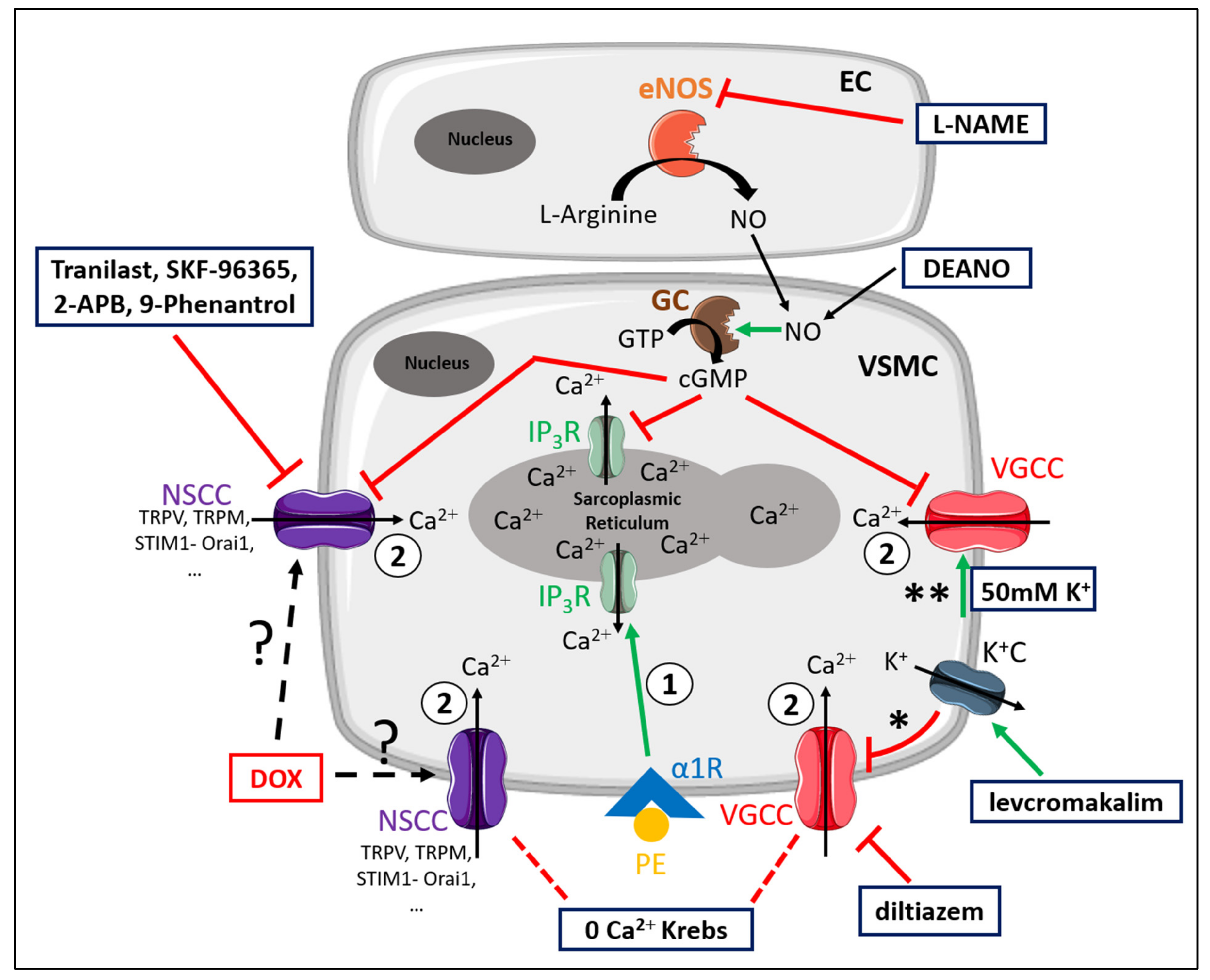
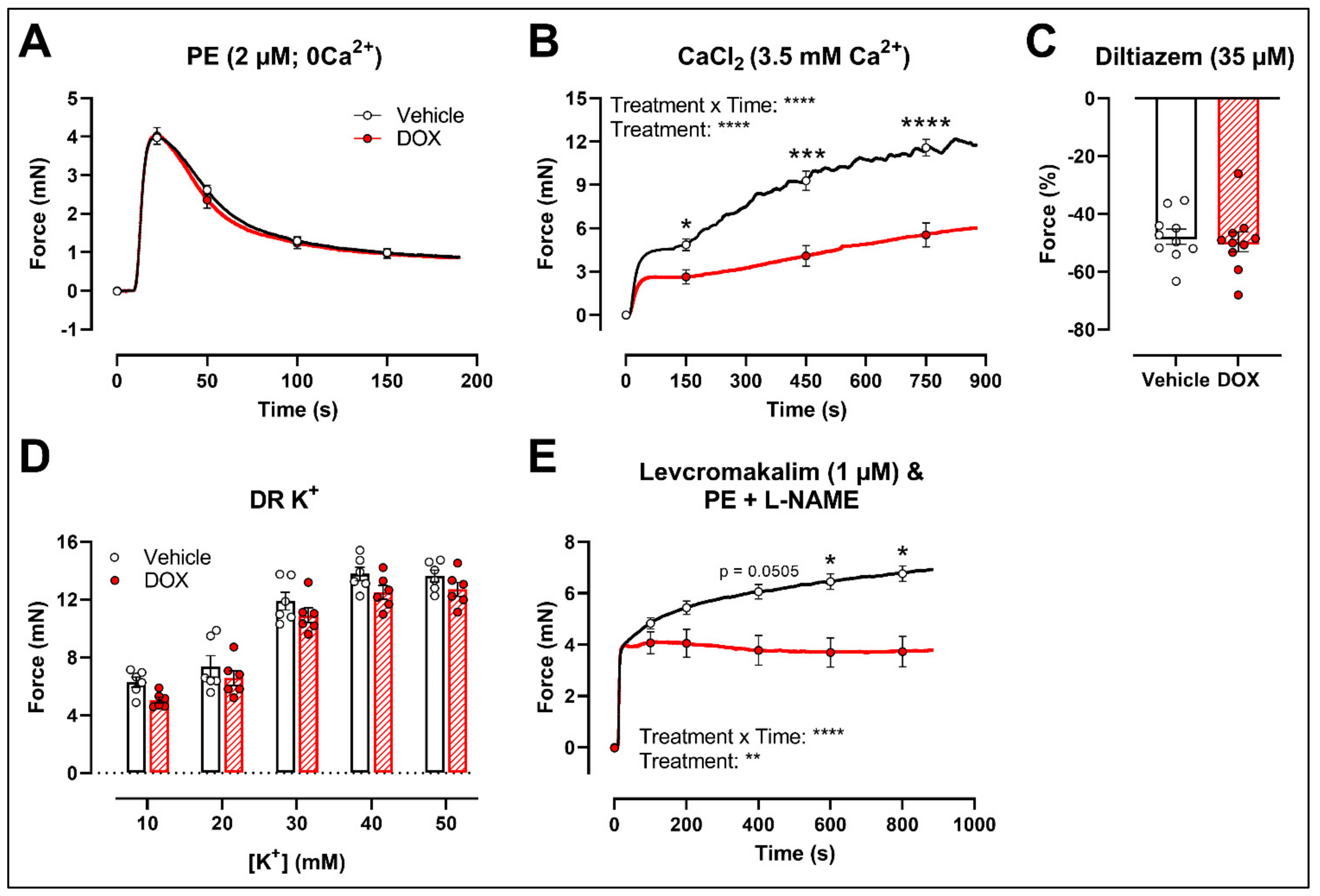
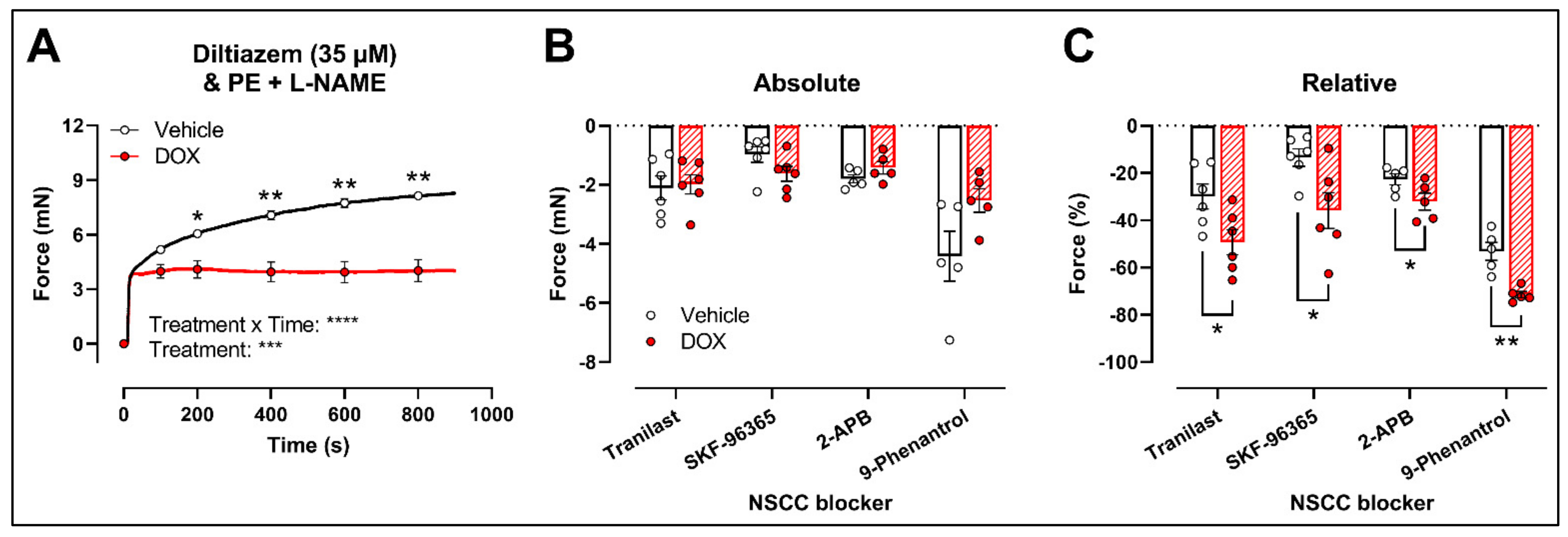
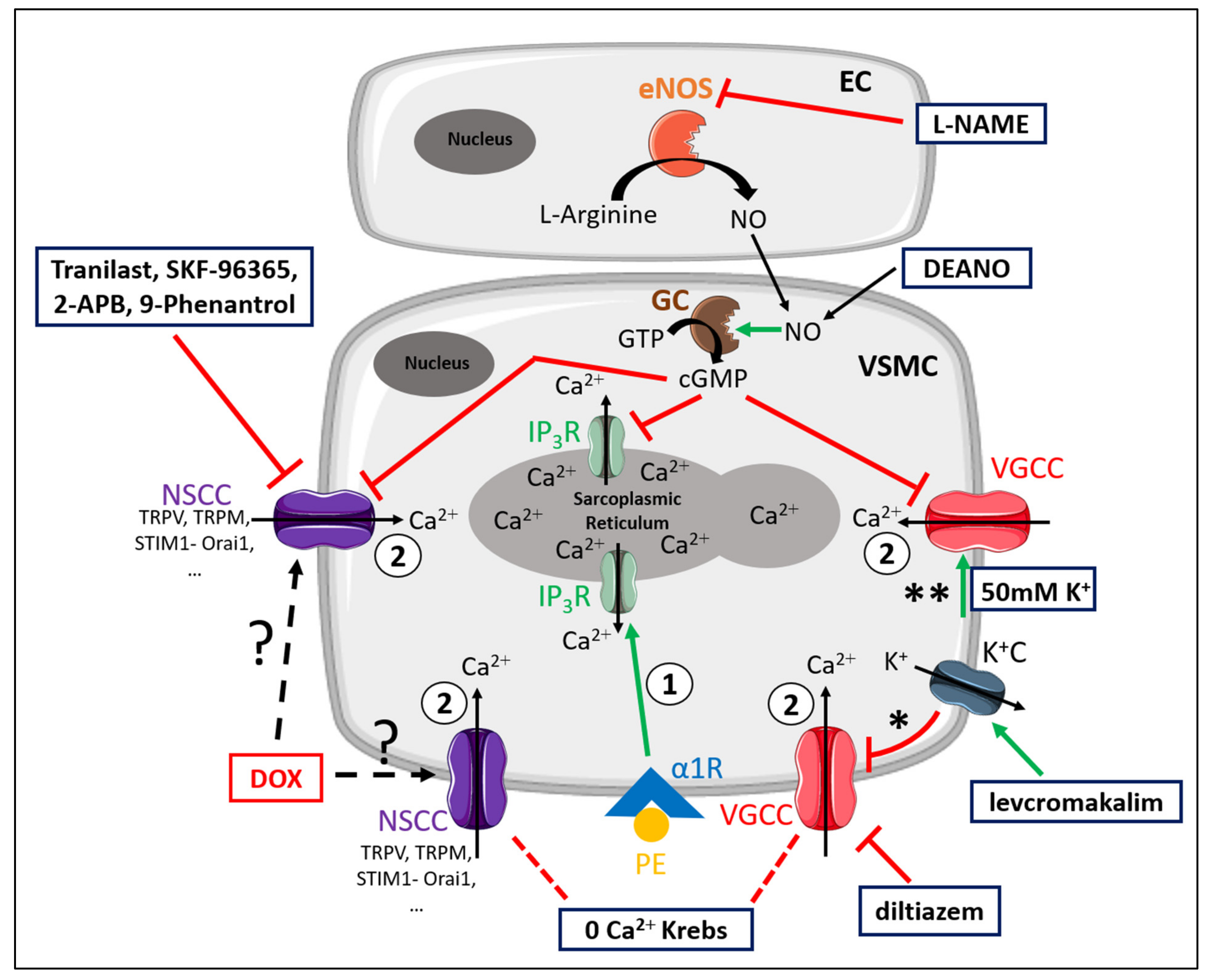
2.3. DOX Does Not Modulate Ca2+ Release from the Sarcoplasmic Reticulum or Ca2+ Influx through Voltage-Gated Calcium Channels
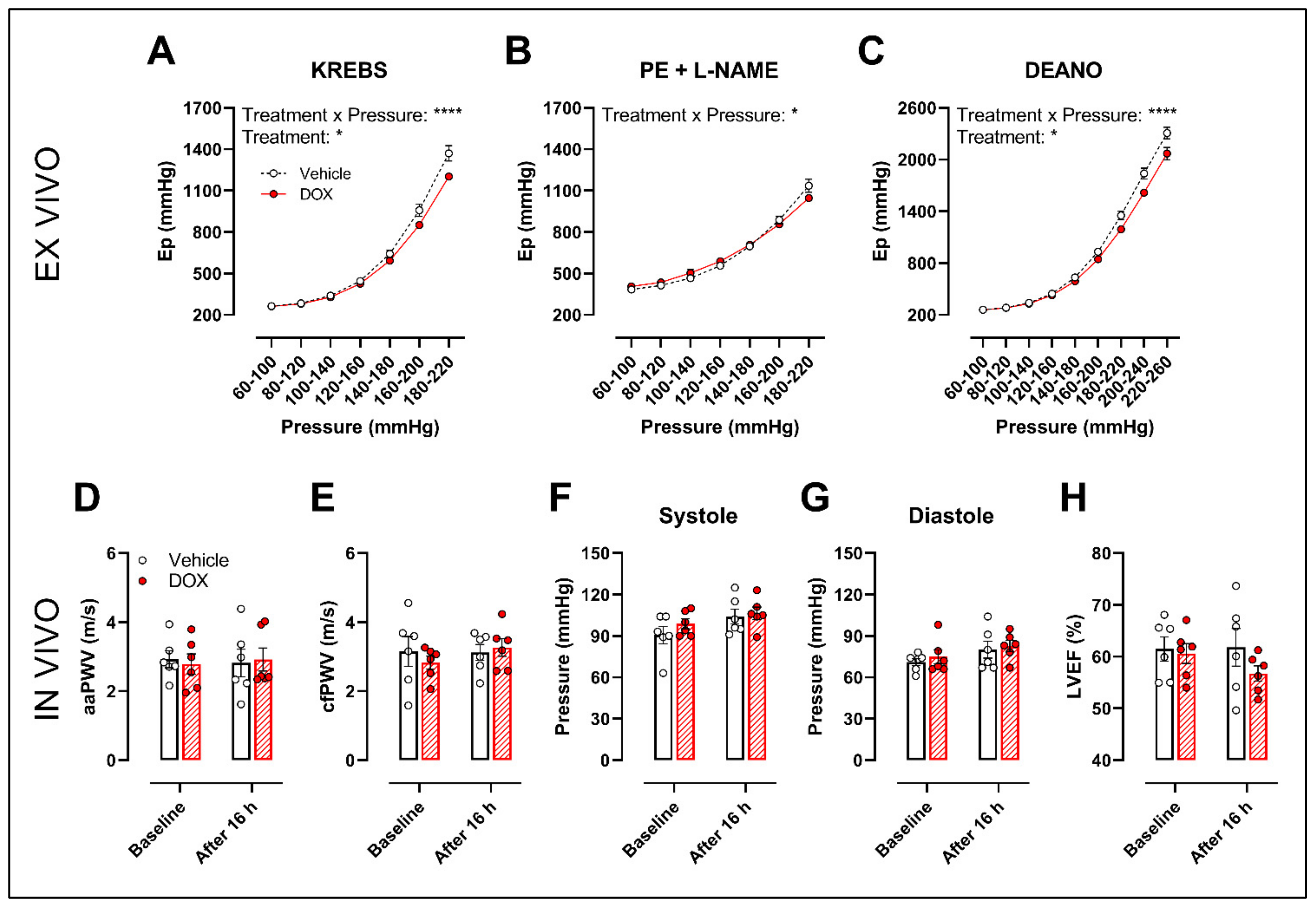
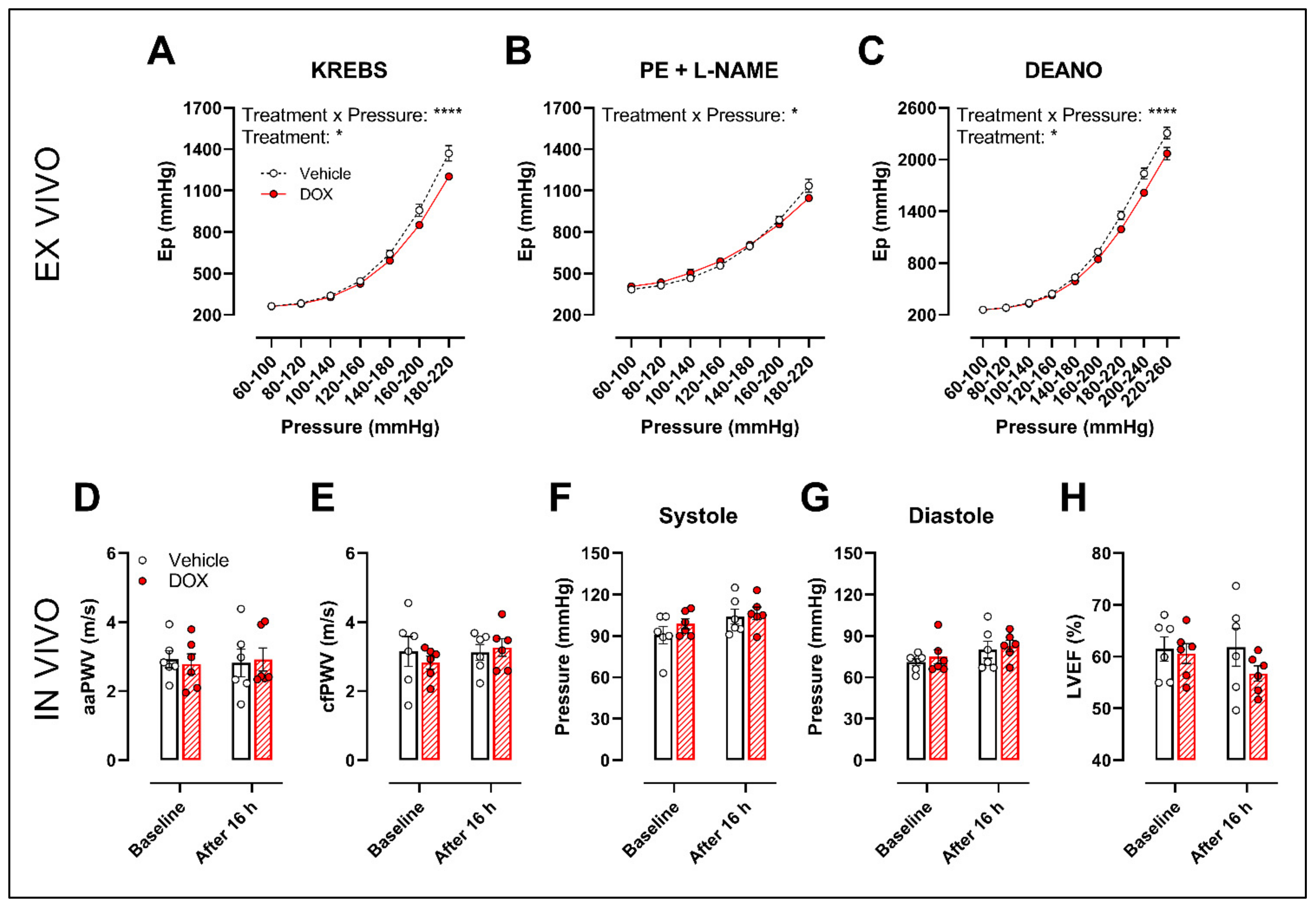
2.4. A Single Dose of DOX Decreases Ex Vivo Arterial Stiffness but Not In Vivo Arterial Stiffness and Blood Pressure
3. Discussion
4. Materials and Methods
4.1. Animals and Ethical Approval
4.2. DOX Treatment and Experimental Workflow
4.3. High-Frequency Ultrasound Imaging
4.4. Applanation Tonometry
4.5. Blood Pressure Evaluation
4.6. Evaluation of Vascular Reactivity
4.7. Evaluation of Arterial Stiffness in ROTSAC
4.8. Chemical Compounds
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharm. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Mitry, M.A.; Edwards, J.G. Doxorubicin induced heart failure: Phenotype and molecular mechanisms. Int. J. Cardiol. Heart Vasc. 2016, 10, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, J.; Picot, J.; Baxter, L.; Levitt, G.; Sullivan, I.; Clegg, A. Clinical and cost-effectiveness of cardioprotection against the toxic effects of anthracyclines given to children with cancer: A systematic review. Br. J. Cancer 2007, 96, 226–230. [Google Scholar] [CrossRef]
- Kremer, L.C.; van Dalen, E.C.; Offringa, M.; Voute, P.A. Frequency and risk factors of anthracycline-induced clinical heart failure in children: A systematic review. Ann. Oncol. 2002, 13, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Schlitt, A.; Jordan, K.; Vordermark, D.; Schwamborn, J.; Langer, T.; Thomssen, C. Cardiotoxicity and oncological treatments. Dtsch. Arztebl. Int. 2014, 111, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Floyd, J.D.; Nguyen, D.T.; Lobins, R.L.; Bashir, Q.; Doll, D.C.; Perry, M.C. Cardiotoxicity of cancer therapy. J. Clin. Oncol. 2005, 23, 7685–7696. [Google Scholar] [CrossRef] [PubMed]
- Renu, K.; Abilash, V.G.; Tirupathi Pichiah, P.B.; Arunachalam, S. Molecular mechanism of doxorubicin-induced cardiomyopathy—An update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar] [CrossRef]
- Armstrong, G.T.; Oeffinger, K.C.; Chen, Y.; Kawashima, T.; Yasui, Y.; Leisenring, W.; Stovall, M.; Chow, E.J.; Sklar, C.A.; Mulrooney, D.A.; et al. Modifiable risk factors and major cardiac events among adult survivors of childhood cancer. J. Clin. Oncol. 2013, 31, 3673–3680. [Google Scholar] [CrossRef]
- Luu, A.Z.; Chowdhury, B.; Al-Omran, M.; Teoh, H.; Hess, D.A.; Verma, S. Role of Endothelium in Doxorubicin-Induced Cardiomyopathy. JACC Basic Transl. Sci. 2018, 3, 861–870. [Google Scholar] [CrossRef]
- Bar-Joseph, H.; Ben-Aharon, I.; Tzabari, M.; Tsarfaty, G.; Stemmer, S.M.; Shalgi, R. In vivo bioimaging as a novel strategy to detect doxorubicin-induced damage to gonadal blood vessels. PLoS ONE 2011, 6, e23492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parr, S.K.; Liang, J.; Schadler, K.L.; Gilchrist, S.C.; Steele, C.C.; Ade, C.J. Anticancer Therapy-Related Increases in Arterial Stiffness: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2020, 9, e015598. [Google Scholar] [CrossRef]
- Novo, G.; Di Lisi, D.; Manganaro, R.; Manno, G.; Lazzara, S.; Immordino, F.A.; Madaudo, C.; Carerj, S.; Russo, A.; Incorvaia, L.; et al. Arterial Stiffness: Effects of Anticancer Drugs Used for Breast Cancer Women. Front. Physiol. 2021, 12, 661464. [Google Scholar] [CrossRef] [PubMed]
- Yersal, O.; Eryilmaz, U.; Akdam, H.; Meydan, N.; Barutca, S. Arterial Stiffness in Breast Cancer Patients Treated with Anthracycline and Trastuzumab-Based Regimens. Cardiol. Res. Pract. 2018, 2018, 5352914. [Google Scholar] [CrossRef] [Green Version]
- Chaosuwannakit, N.; D’Agostino, R., Jr.; Hamilton, C.A.; Lane, K.S.; Ntim, W.O.; Lawrence, J.; Melin, S.A.; Ellis, L.R.; Torti, F.M.; Little, W.C.; et al. Aortic stiffness increases upon receipt of anthracycline chemotherapy. J. Clin. Oncol. 2010, 28, 166–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, C.U.; Pfeffer, M.A.; Glynn, R.J.; Mitchell, G.F.; Taylor, J.O.; Hennekens, C.H. Increased pulse pressure and risk of heart failure in the elderly. JAMA 1999, 281, 634–639. [Google Scholar] [CrossRef]
- Sutton-Tyrrell, K.; Najjar, S.S.; Boudreau, R.M.; Venkitachalam, L.; Kupelian, V.; Simonsick, E.M.; Havlik, R.; Lakatta, E.G.; Spurgeon, H.; Kritchevsky, S.; et al. Elevated aortic pulse wave velocity, a marker of arterial stiffness, predicts cardiovascular events in well-functioning older adults. Circulation 2005, 111, 3384–3390. [Google Scholar] [CrossRef]
- Mitchell, G.F.; Vasan, R.S.; Keyes, M.J.; Parise, H.; Wang, T.J.; Larson, M.G.; D’Agostino, R.B., Sr.; Kannel, W.B.; Levy, D.; Benjamin, E.J. Pulse pressure and risk of new-onset atrial fibrillation. JAMA 2007, 297, 709–715. [Google Scholar] [CrossRef]
- Nagy, L.; Szabo, F.; Ivanyi, J.; Nemeth, L.; Kovacs, G.L.; Palatka, J.; Tarjan, J.; Toth, K.; Roth, E. A method for detection of doxorubicin-induced cardiotoxicity: Flow-mediated vasodilation of the brachial artery. Exp. Clin. Cardiol. 2001, 6, 87–92. [Google Scholar]
- Bosman, M.; Favere, K.; Neutel, C.H.G.; Jacobs, G.; De Meyer, G.R.Y.; Martinet, W.; Van Craenenbroeck, E.M.; Guns, P.D.F. Doxorubicin induces arterial stiffness: A comprehensive in vivo and ex vivo evaluation of vascular toxicity in mice. Toxicol. Lett. 2021, 346, 23–33. [Google Scholar] [CrossRef]
- He, H.; Wang, L.; Qiao, Y.; Zhou, Q.; Li, H.; Chen, S.; Yin, D.; Huang, Q.; He, M. Doxorubicin Induces Endotheliotoxicity and Mitochondrial Dysfunction via ROS/eNOS/NO Pathway. Front. Pharmacol. 2019, 10, 1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duquaine, D.; Hirsch, G.A.; Chakrabarti, A.; Han, Z.; Kehrer, C.; Brook, R.; Joseph, J.; Schott, A.; Kalyanaraman, B.; Vasquez-Vivar, J.; et al. Rapid-onset endothelial dysfunction with adriamycin: Evidence for a dysfunctional nitric oxide synthase. Vasc. Med. 2003, 8, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Kalivendi, S.V.; Kotamraju, S.; Zhao, H.; Joseph, J.; Kalyanaraman, B. Doxorubicin-induced apoptosis is associated with increased transcription of endothelial nitric-oxide synthase. Effect of antiapoptotic antioxidants and calcium. J. Biol. Chem. 2001, 276, 47266–47276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, N.M.; Greufe, S.E.; Hydock, D.S.; Hayward, R. Doxorubicin-induced vascular dysfunction and its attenuation by exercise preconditioning. J. Cardiovasc. Pharmacol. 2013, 62, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Olukman, M.; Can, C.; Erol, A.; Oktem, G.; Oral, O.; Cinar, M.G. Reversal of doxorubicin-induced vascular dysfunction by resveratrol in rat thoracic aorta: Is there a possible role of nitric oxide synthase inhibition? Anadolu Kardiyol. Derg. 2009, 9, 260–266. [Google Scholar] [PubMed]
- Shen, B.; Ye, C.L.; Ye, K.H.; Zhuang, L.; Jiang, J.H. Doxorubicin-induced vasomotion and [Ca(2+)](i) elevation in vascular smooth muscle cells from C57BL/6 mice. Acta Pharmacol. Sin. 2009, 30, 1488–1495. [Google Scholar] [CrossRef]
- Fransen, P.; Van Hove, C.E.; Leloup, A.J.; Martinet, W.; De Meyer, G.R.; Lemmens, K.; Bult, H.; Schrijvers, D.M. Dissecting out the complex Ca2+-mediated phenylephrine-induced contractions of mouse aortic segments. PLoS ONE 2015, 10, e0121634. [Google Scholar] [CrossRef] [Green Version]
- van Langen, J.; Fransen, P.; Van Hove, C.E.; Schrijvers, D.M.; Martinet, W.; De Meyer, G.R.; Bult, H. Selective loss of basal but not receptor-stimulated relaxation by endothelial nitric oxide synthase after isolation of the mouse aorta. Eur. J. Pharmacol. 2012, 696, 111–119. [Google Scholar] [CrossRef]
- Fitch, R.M.; Vergona, R.; Sullivan, M.E.; Wang, Y.X. Nitric oxide synthase inhibition increases aortic stiffness measured by pulse wave velocity in rats. Cardiovasc. Res. 2001, 51, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Leloup, A.J.A.; Van Hove, C.E.; De Moudt, S.; De Keulenaer, G.W.; Fransen, P. Ex vivo aortic stiffness in mice with different eNOS activity. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1233–H1244. [Google Scholar] [CrossRef]
- Leloup, A.J.A.; Van Hove, C.E.; De Moudt, S.; De Meyer, G.R.Y.; De Keulenaer, G.W.; Fransen, P. Vascular smooth muscle cell contraction and relaxation in the isolated aorta: A critical regulator of large artery compliance. Physiol. Rep. 2019, 7, e13934. [Google Scholar] [CrossRef]
- Brozovich, F.V.; Nicholson, C.J.; Degen, C.V.; Gao, Y.Z.; Aggarwal, M.; Morgan, K.G. Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol. Rev. 2016, 68, 476–532. [Google Scholar] [CrossRef] [Green Version]
- Neumann-Raizel, H.; Shilo, A.; Lev, S.; Mogilevsky, M.; Katz, B.; Shneor, D.; Shaul, Y.D.; Leffler, A.; Gabizon, A.; Karni, R.; et al. 2-APB and CBD-Mediated Targeting of Charged Cytotoxic Compounds Into Tumor Cells Suggests the Involvement of TRPV2 Channels. Front. Pharmacol. 2019, 10, 1198. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Syed, A.U.; Prada, M.P.; Nystoriak, M.A.; Santana, L.F.; Nieves-Cintron, M.; Navedo, M.F. Calcium Channels in Vascular Smooth Muscle. Adv. Pharmacol. 2017, 78, 49–87. [Google Scholar] [CrossRef] [Green Version]
- Prevarskaya, N.; Zhang, L.; Barritt, G. TRP channels in cancer. Biochim. Biophys. Acta 2007, 1772, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Santoni, G.; Farfariello, V. TRP channels and cancer: New targets for diagnosis and chemotherapy. Endocr. Metab. Immune Disord. Drug Targets 2011, 11, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Fiorio Pla, A.; Gkika, D. Emerging role of TRP channels in cell migration: From tumor vascularization to metastasis. Front. Physiol. 2013, 4, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Luan, Y.; Yu, R.; Zhang, Z.; Zhang, J.; Wang, W. Transient receptor potential (TRP) channels, promising potential diagnostic and therapeutic tools for cancer. Biosci. Trends 2014, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azimi, I.; Roberts-Thomson, S.J.; Monteith, G.R. Calcium influx pathways in breast cancer: Opportunities for pharmacological intervention. Br. J. Pharmacol. 2014, 171, 945–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosar, P.A.; Naziroglu, M.; Ovey, I.S.; Cig, B. Synergic Effects of Doxorubicin and Melatonin on Apoptosis and Mitochondrial Oxidative Stress in MCF-7 Breast Cancer Cells: Involvement of TRPV1 Channels. J. Membr. Biol. 2016, 249, 129–140. [Google Scholar] [CrossRef]
- Earley, S.; Brayden, J.E. Transient receptor potential channels in the vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef] [Green Version]
- Iwata, Y.; Katayama, Y.; Okuno, Y.; Wakabayashi, S. Novel inhibitor candidates of TRPV2 prevent damage of dystrophic myocytes and ameliorate against dilated cardiomyopathy in a hamster model. Oncotarget 2018, 9, 14042–14057. [Google Scholar] [CrossRef] [Green Version]
- Iwata, Y.; Matsumura, T. Blockade of TRPV2 is a Novel Therapy for Cardiomyopathy in Muscular Dystrophy. Int. J. Mol. Sci. 2019, 20, 3844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verigos, J.; Kordias, D.; Papadaki, S.; Magklara, A. Transcriptional Profiling of Tumorspheres Reveals TRPM4 as a Novel Stemness Regulator in Breast Cancer. Biomedicines 2021, 9, 1368. [Google Scholar] [CrossRef]
- Wynne, B.M.; Chiao, C.W.; Webb, R.C. Vascular Smooth Muscle Cell Signaling Mechanisms for Contraction to Angiotensin II and Endothelin-1. J. Am. Soc. Hypertens. 2009, 3, 84–95. [Google Scholar] [CrossRef] [Green Version]
- Bhaskaran, S.; Zaluski, J.; Banes-Berceli, A. Molecular interactions of serotonin (5-HT) and endothelin-1 in vascular smooth muscle cells: In vitro and ex vivo analyses. Am. J. Physiol. Cell Physiol. 2014, 306, C143–C151. [Google Scholar] [CrossRef] [Green Version]
- Kotamraju, S.; Konorev, E.A.; Joseph, J.; Kalyanaraman, B. Doxorubicin-induced apoptosis in endothelial cells and cardiomyocytes is ameliorated by nitrone spin traps and ebselen. Role of reactive oxygen and nitrogen species. J. Biol. Chem. 2000, 275, 33585–33592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Murata, T.; Yamawaki, H.; Yoshimoto, R.; Hori, M.; Sato, K.; Ozaki, H.; Karaki, H. Chronic effect of doxorubicin on vascular endothelium assessed by organ culture study. Life Sci. 2001, 69, 2685–2695. [Google Scholar] [CrossRef]
- Li, J.; Li, P.F.; Dietz, R.; von Harsdorf, R. Intracellular superoxide induces apoptosis in VSMCs: Role of mitochondrial membrane potential, cytochrome C and caspases. Apoptosis 2002, 7, 511–517. [Google Scholar] [CrossRef]
- Numaga-Tomita, T.; Shimauchi, T.; Oda, S.; Tanaka, T.; Nishiyama, K.; Nishimura, A.; Birnbaumer, L.; Mori, Y.; Nishida, M. TRPC6 regulates phenotypic switching of vascular smooth muscle cells through plasma membrane potential-dependent coupling with PTEN. FASEB J. 2019, 33, 9785–9796. [Google Scholar] [CrossRef] [Green Version]
- Petsophonsakul, P.; Furmanik, M.; Forsythe, R.; Dweck, M.; Schurink, G.W.; Natour, E.; Reutelingsperger, C.; Jacobs, M.; Mees, B.; Schurgers, L. Role of Vascular Smooth Muscle Cell Phenotypic Switching and Calcification in Aortic Aneurysm Formation. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1351–1368. [Google Scholar] [CrossRef] [PubMed]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Munoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef] [PubMed]
- Aston, W.J.; Hope, D.E.; Nowak, A.K.; Robinson, B.W.; Lake, R.A.; Lesterhuis, W.J. A systematic investigation of the maximum tolerated dose of cytotoxic chemotherapy with and without supportive care in mice. BMC Cancer 2017, 17, 684. [Google Scholar] [CrossRef] [Green Version]
- Young, R.C.; Ozols, R.F.; Myers, C.E. The anthracycline antineoplastic drugs. N. Engl. J. Med. 1981, 305, 139–153. [Google Scholar] [CrossRef]
- Hodjat, M.; Haller, H.; Dumler, I.; Kiyan, Y. Urokinase receptor mediates doxorubicin-induced vascular smooth muscle cell senescence via proteasomal degradation of TRF2. J. Vasc. Res. 2013, 50, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Di Lascio, N.; Stea, F.; Kusmic, C.; Sicari, R.; Faita, F. Non-invasive assessment of pulse wave velocity in mice by means of ultrasound images. Atherosclerosis 2014, 237, 31–37. [Google Scholar] [CrossRef]
- Leloup, A.J.; Fransen, P.; Van Hove, C.E.; Demolder, M.; De Keulenaer, G.W.; Schrijvers, D.M. Applanation tonometry in mice: A novel noninvasive technique to assess pulse wave velocity and arterial stiffness. Hypertension 2014, 64, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- Fransen, P.; Van Hove, C.E.; van Langen, J.; Schrijvers, D.M.; Martinet, W.; De Meyer, G.R.; Bult, H. Contribution of transient and sustained calcium influx, and sensitization to depolarization-induced contractions of the intact mouse aorta. BMC Physiol. 2012, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Leloup, A.J.; Van Hove, C.E.; Kurdi, A.; De Moudt, S.; Martinet, W.; De Meyer, G.R.; Schrijvers, D.M.; De Keulenaer, G.W.; Fransen, P. A novel set-up for the ex vivo analysis of mechanical properties of mouse aortic segments stretched at physiological pressure and frequency. J. Physiol. 2016, 594, 6105–6115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bosman, M.; Krüger, D.N.; Favere, K.; Wesley, C.D.; Neutel, C.H.G.; Van Asbroeck, B.; Diebels, O.R.; Faes, B.; Schenk, T.J.; Martinet, W.; et al. Doxorubicin Impairs Smooth Muscle Cell Contraction: Novel Insights in Vascular Toxicity. Int. J. Mol. Sci. 2021, 22, 12812. https://doi.org/10.3390/ijms222312812
Bosman M, Krüger DN, Favere K, Wesley CD, Neutel CHG, Van Asbroeck B, Diebels OR, Faes B, Schenk TJ, Martinet W, et al. Doxorubicin Impairs Smooth Muscle Cell Contraction: Novel Insights in Vascular Toxicity. International Journal of Molecular Sciences. 2021; 22(23):12812. https://doi.org/10.3390/ijms222312812
Chicago/Turabian StyleBosman, Matthias, Dustin N. Krüger, Kasper Favere, Callan D. Wesley, Cédric H. G. Neutel, Birgit Van Asbroeck, Owen R. Diebels, Bart Faes, Timen J. Schenk, Wim Martinet, and et al. 2021. "Doxorubicin Impairs Smooth Muscle Cell Contraction: Novel Insights in Vascular Toxicity" International Journal of Molecular Sciences 22, no. 23: 12812. https://doi.org/10.3390/ijms222312812
APA StyleBosman, M., Krüger, D. N., Favere, K., Wesley, C. D., Neutel, C. H. G., Van Asbroeck, B., Diebels, O. R., Faes, B., Schenk, T. J., Martinet, W., De Meyer, G. R. Y., Van Craenenbroeck, E. M., & Guns, P.-J. D. F. (2021). Doxorubicin Impairs Smooth Muscle Cell Contraction: Novel Insights in Vascular Toxicity. International Journal of Molecular Sciences, 22(23), 12812. https://doi.org/10.3390/ijms222312812