
Cytotoxic Aβ Protofilaments Are Generated in the Process of Aβ Fibril Disaggregation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
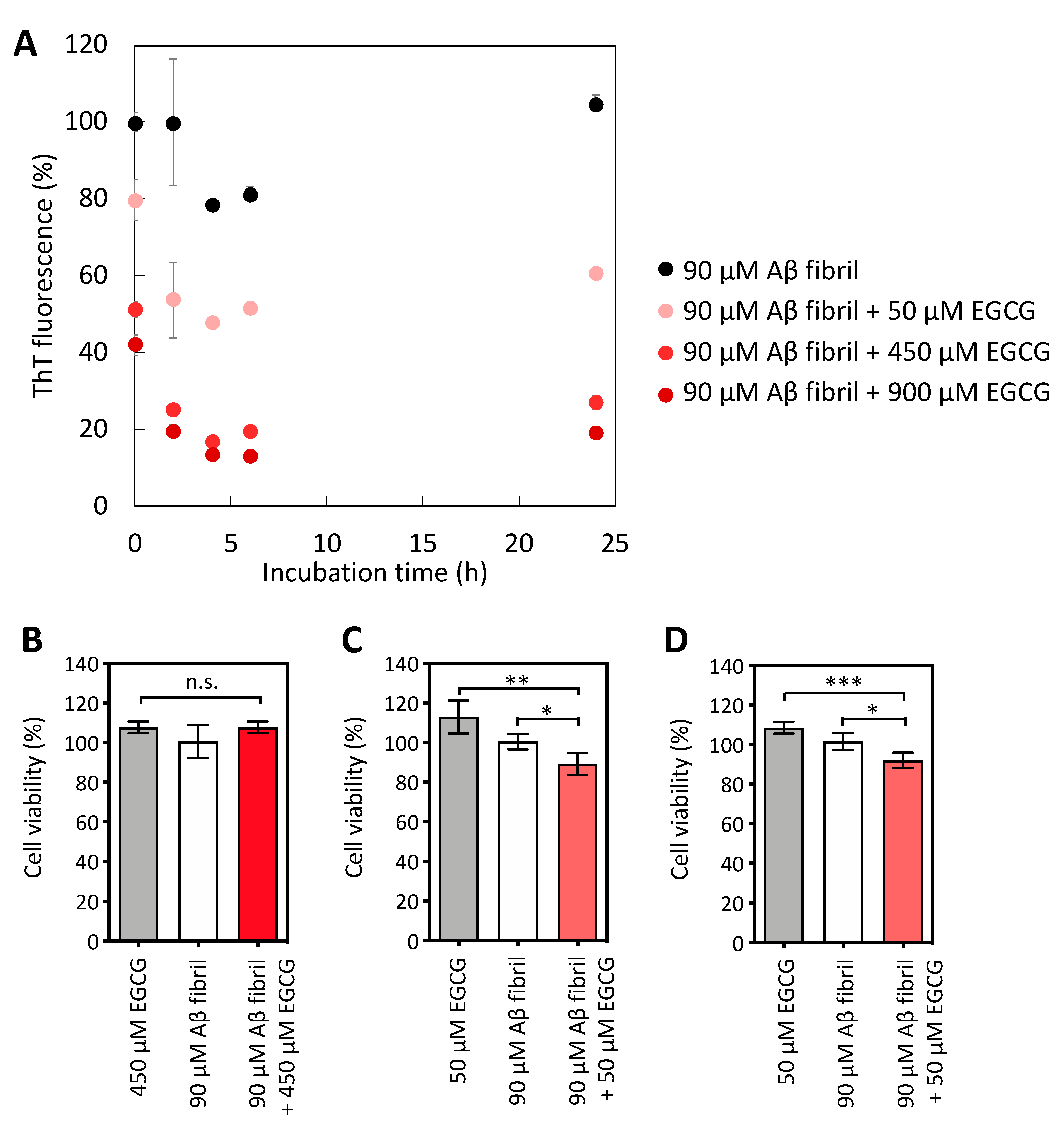
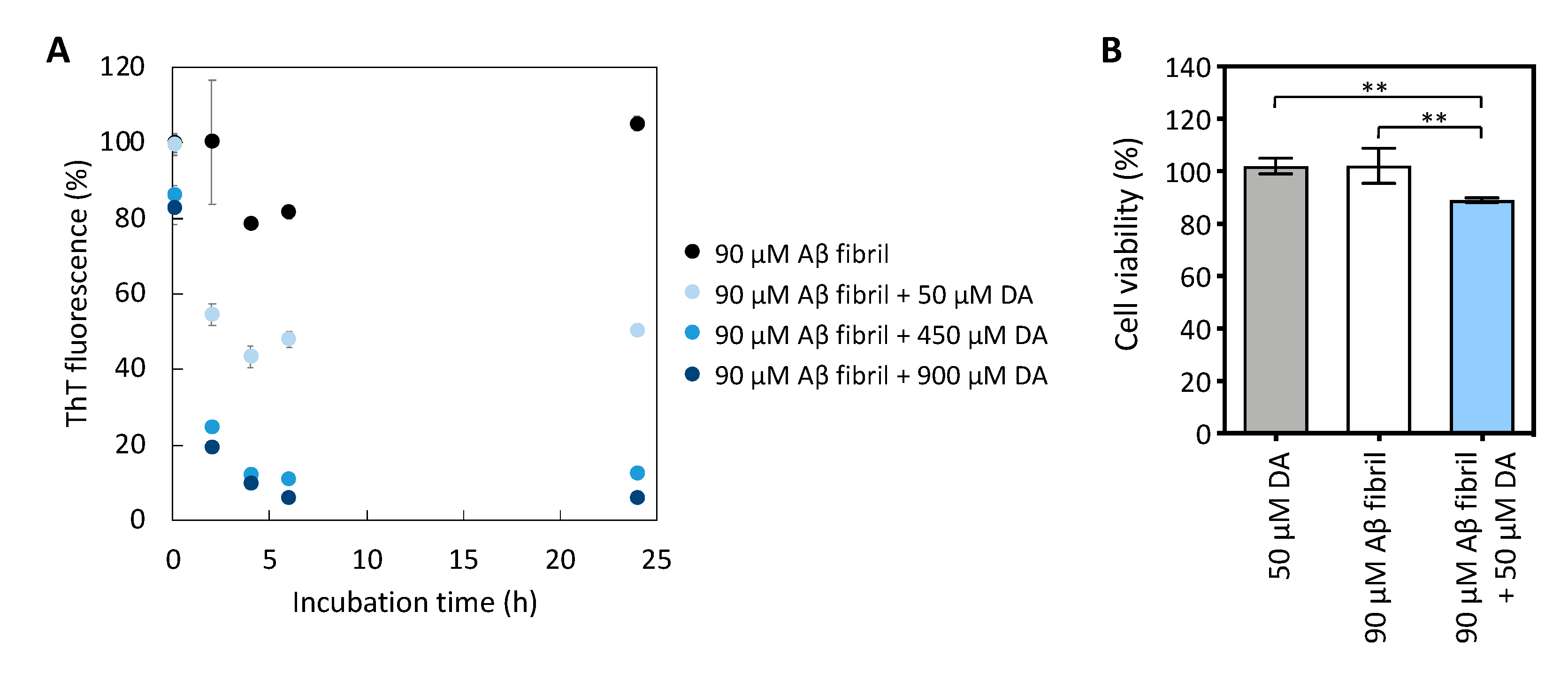
2.1. Mild Disaggregation of Aβ Fibrils by EGCG and DA to Produce Cytotoxic Aβ Aggregates
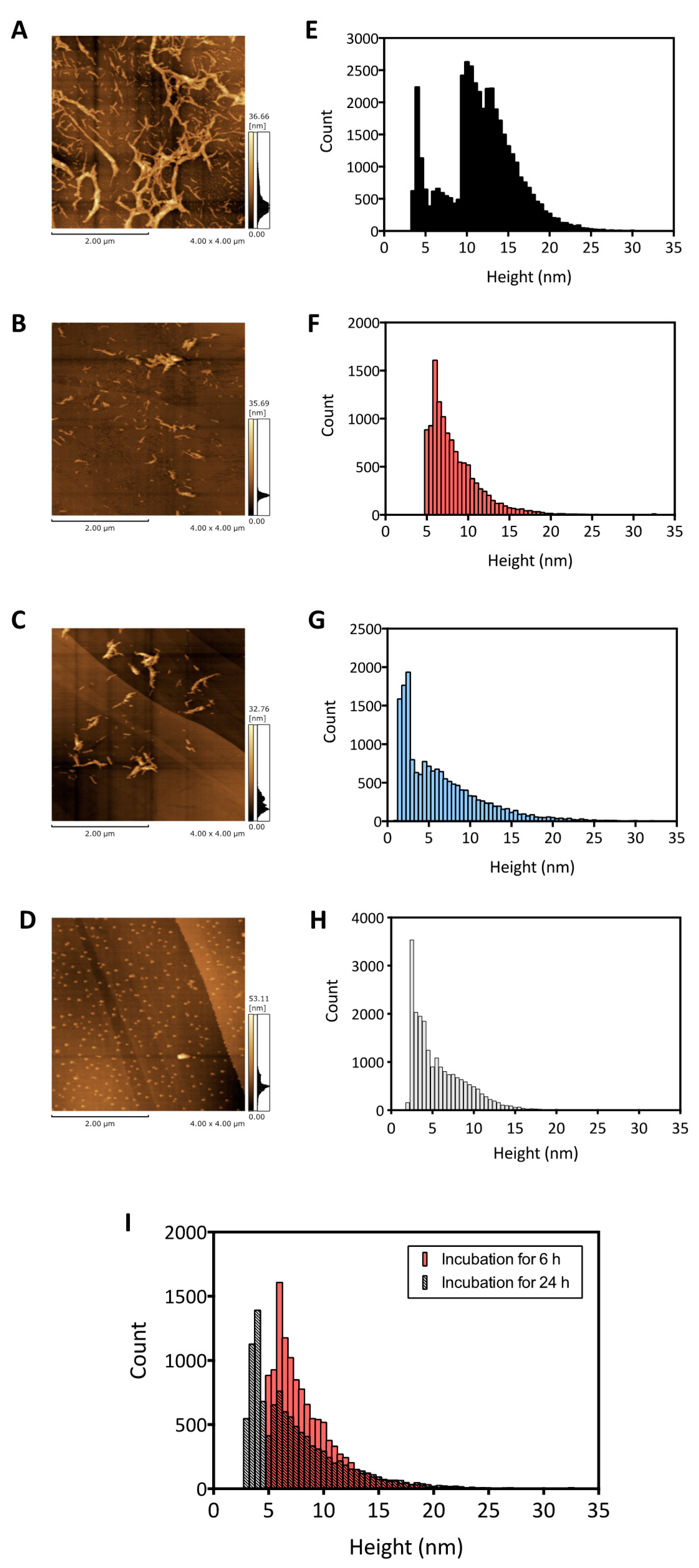
2.2. AFM Imaging Analysis of Disaggregated Aβ Fibrils
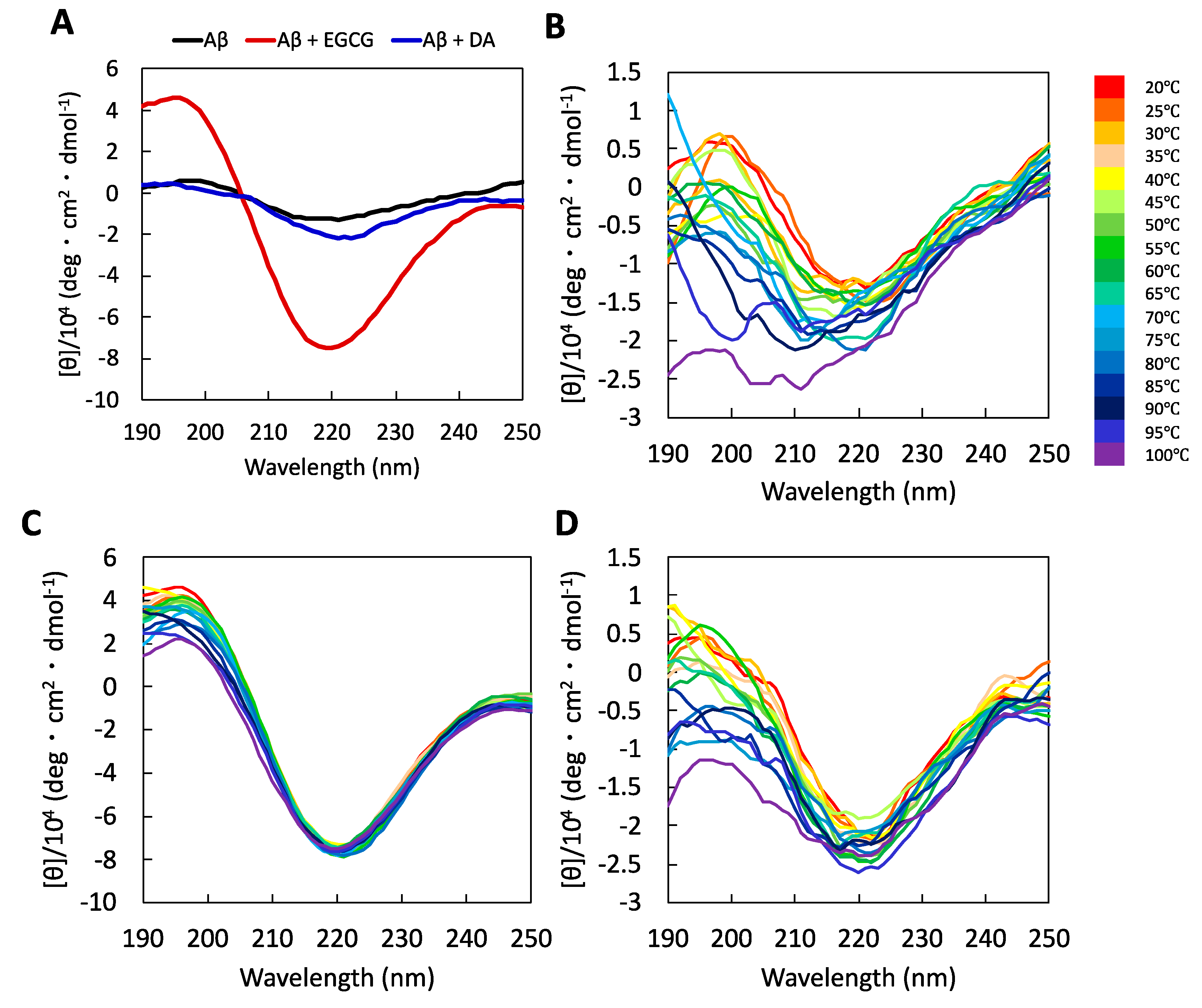
2.3. Characterization of Secondary Structures of the Aβ Protofibrils Produced by Disaggregation Using EGCG and DA
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Preparation of Aβ Fibrils and Disaggregated Aβ Solutions
4.3. ThT Fluorescence Measurement
4.4. MTS Cytotoxicity Assay
4.5. Imaging by AFM
4.6. CD Spectrometry Measurement
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Fukumoto, H.; Tokuda, T.; Kasai, T.; Ishigami, N.; Hidaka, H.; Kondo, M.; Allsop, D.; Nakagawa, M. High-molecular-weight beta-amyloid oligomers are elevated in cerebrospinal fluid of Alzheimer patients. FASEB J. 2010, 24, 2716–2726. [Google Scholar] [CrossRef]
- Lesne, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef]
- Tomiyama, T.; Nagata, T.; Shimada, H.; Teraoka, R.; Fukushima, A.; Kanemitsu, H.; Takuma, H.; Kuwano, R.; Imagawa, M.; Ataka, S.; et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann. Neurol 2008, 63, 377–387. [Google Scholar] [CrossRef]
- Savage, M.J.; Kalinina, J.; Wolfe, A.; Tugusheva, K.; Korn, R.; Cash-Mason, T.; Maxwell, J.W.; Hatcher, N.G.; Haugabook, S.J.; Wu, G.; et al. A sensitive abeta oligomer assay discriminates Alzheimer’s and aged control cerebrospinal fluid. J. Neurosci. 2014, 34, 2884–2897. [Google Scholar] [CrossRef]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Abeta oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Gremer, L.; Scholzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-beta(1–42) by cryo-electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef] [Green Version]
- Serpell, L.C. Alzheimer’s amyloid fibrils: Structure and assembly. Biochim. Biophys. Acta 2000, 1502, 16–30. [Google Scholar] [CrossRef] [Green Version]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 2003, 81, 678–699. [Google Scholar] [CrossRef]
- Lee, S.J.; Nam, E.; Lee, H.J.; Savelieff, M.G.; Lim, M.H. Towards an understanding of amyloid-beta oligomers: Characterization, toxicity mechanisms, and inhibitors. Chem. Soc. Rev. 2017, 46, 310–323. [Google Scholar] [CrossRef]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhu, M.; Manning-Bog, A.B.; Di Monte, D.A.; Fink, A.L. Dopamine and L-dopa disaggregate amyloid fibrils: Implications for Parkinson’s and Alzheimer’s disease. FASEB J. 2004, 18, 962–964. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Anti-amyloidogenic activity of tannic acid and its activity to destabilize Alzheimer’s beta-amyloid fibrils in vitro. Biochim. Biophys. Acta 2004, 1690, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Yoshiike, Y.; Takashima, A.; Hasegawa, K.; Naiki, H.; Yamada, M. Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: Implications for the prevention and therapeutics of Alzheimer’s disease. J. Neurochem. 2003, 87, 172–181. [Google Scholar] [CrossRef]
- Agrawal, N.; Skelton, A.A. 12-Crown-4 Ether Disrupts the Patient Brain-Derived Amyloid-beta-Fibril Trimer: Insight from All-Atom Molecular Dynamics Simulations. ACS Chem. Neurosci. 2016, 7, 1433–1441. [Google Scholar] [CrossRef]
- Lemkul, J.A.; Bevan, D.R. Destabilizing Alzheimer’s Abeta(42) protofibrils with morin: Mechanistic insights from molecular dynamics simulations. Biochemistry 2010, 49, 3935–3946. [Google Scholar] [CrossRef]
- Zhan, C.; Chen, Y.; Tang, Y.; Wei, G. Green Tea Extracts EGCG and EGC Display Distinct Mechanisms in Disrupting Abeta42 Protofibril. ACS Chem. Neurosci. 2020, 11, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.E.; Duennwald, M.L.; Wang, H.; Chung, C.; Lopreiato, N.P.; Sweeny, E.A.; Knight, M.N.; Shorter, J. A synergistic small-molecule combination directly eradicates diverse prion strain structures. Nat. Chem. Biol. 2009, 5, 936–946. [Google Scholar] [CrossRef] [Green Version]
- Palhano, F.L.; Lee, J.; Grimster, N.P.; Kelly, J.W. Toward the molecular mechanism(s) by which EGCG treatment remodels mature amyloid fibrils. J. Am. Chem. Soc. 2013, 135, 7503–7510. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Kou, L.; Bin, Y.; Wan, L.; Xiang, J. Complicated function of dopamine in Abeta-related neurotoxicity: Dual interactions with Tyr(10) and SNK(26–28) of Abeta. J. Inorg. Biochem. 2016, 164, 119–128. [Google Scholar] [CrossRef]
- Meinhardt, J.; Sachse, C.; Hortschansky, P.; Grigorieff, N.; Fandrich, M. Abeta(1–40) fibril polymorphism implies diverse interaction patterns in amyloid fibrils. J. Mol. Biol. 2009, 386, 869–877. [Google Scholar] [CrossRef]
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fandrich, M. Cryo-EM structure and polymorphism of Abeta amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun. 2019, 10, 4760. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Rohou, A.; Lasker, K.; Yadav, J.K.; Schiene-Fischer, C.; Fandrich, M.; Grigorieff, N. Peptide dimer structure in an Abeta(1–42) fibril visualized with cryo-EM. Proc. Natl. Acad. Sci. USA 2015, 112, 11858–11863. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, R.; VanSchouwen, B.; Jafari, N.; Ni, X.; Ortega, J.; Melacini, G. Molecular Mechanism for the (-)-Epigallocatechin Gallate-Induced Toxic to Nontoxic Remodeling of Abeta Oligomers. J. Am. Chem. Soc. 2017, 139, 13720–13734. [Google Scholar] [CrossRef] [PubMed]
- Stroud, J.C.; Liu, C.; Teng, P.K.; Eisenberg, D. Toxic fibrillar oligomers of amyloid-beta have cross-beta structure. Proc. Natl. Acad. Sci. USA 2012, 109, 7717–7722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tofoleanu, F.; Brooks, B.R.; Buchete, N.V. Modulation of Alzheimer’s Abeta protofilament-membrane interactions by lipid headgroups. ACS Chem. Neurosci. 2015, 6, 446–455. [Google Scholar] [CrossRef]
- Chen, Y.; Li, X.; Zhan, C.; Lao, Z.; Li, F.; Dong, X.; Wei, G. A Comprehensive Insight into the Mechanisms of Dopamine in Disrupting Abeta Protofibrils and Inhibiting Abeta Aggregation. ACS Chem. Neurosci. 2021, 12, 4007–4019. [Google Scholar] [CrossRef]
- Singh, P.; Bhat, R. Binding of Noradrenaline to Native and Intermediate States during the Fibrillation of alpha-Synuclein Leads to the Formation of Stable and Structured Cytotoxic Species. ACS Chem. Neurosci. 2019, 10, 2741–2755. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef]
- Tsukakoshi, K.; Yoshida, W.; Kobayashi, M.; Kobayashi, N.; Kim, J.; Kaku, T.; Iguchi, T.; Nagasawa, K.; Asano, R.; Ikebukuro, K.; et al. Esterification of PQQ Enhances Blood-Brain Barrier Permeability and Inhibitory Activity against Amyloidogenic Protein Fibril Formation. ACS Chem. Neurosci. 2018, 9, 2898–2903. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaku, T.; Tsukakoshi, K.; Ikebukuro, K. Cytotoxic Aβ Protofilaments Are Generated in the Process of Aβ Fibril Disaggregation. Int. J. Mol. Sci. 2021, 22, 12780. https://doi.org/10.3390/ijms222312780
Kaku T, Tsukakoshi K, Ikebukuro K. Cytotoxic Aβ Protofilaments Are Generated in the Process of Aβ Fibril Disaggregation. International Journal of Molecular Sciences. 2021; 22(23):12780. https://doi.org/10.3390/ijms222312780
Chicago/Turabian StyleKaku, Toshisuke, Kaori Tsukakoshi, and Kazunori Ikebukuro. 2021. "Cytotoxic Aβ Protofilaments Are Generated in the Process of Aβ Fibril Disaggregation" International Journal of Molecular Sciences 22, no. 23: 12780. https://doi.org/10.3390/ijms222312780
APA StyleKaku, T., Tsukakoshi, K., & Ikebukuro, K. (2021). Cytotoxic Aβ Protofilaments Are Generated in the Process of Aβ Fibril Disaggregation. International Journal of Molecular Sciences, 22(23), 12780. https://doi.org/10.3390/ijms222312780