
Synaptamide Improves Cognitive Functions and Neuronal Plasticity in Neuropathic Pain



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
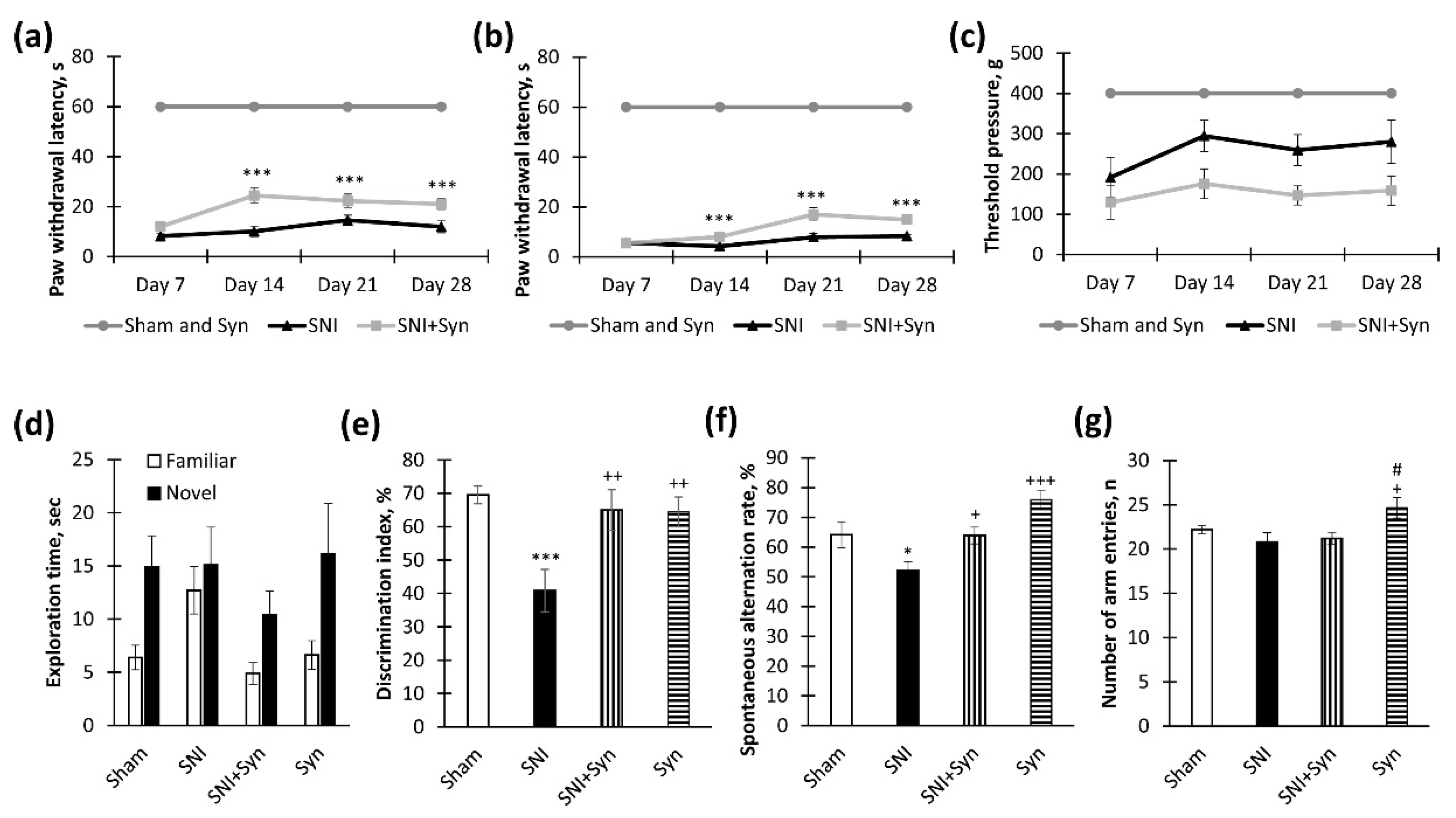
2.1. Synaptamide Improves Behavioral Parameters in Neuropathic Pain
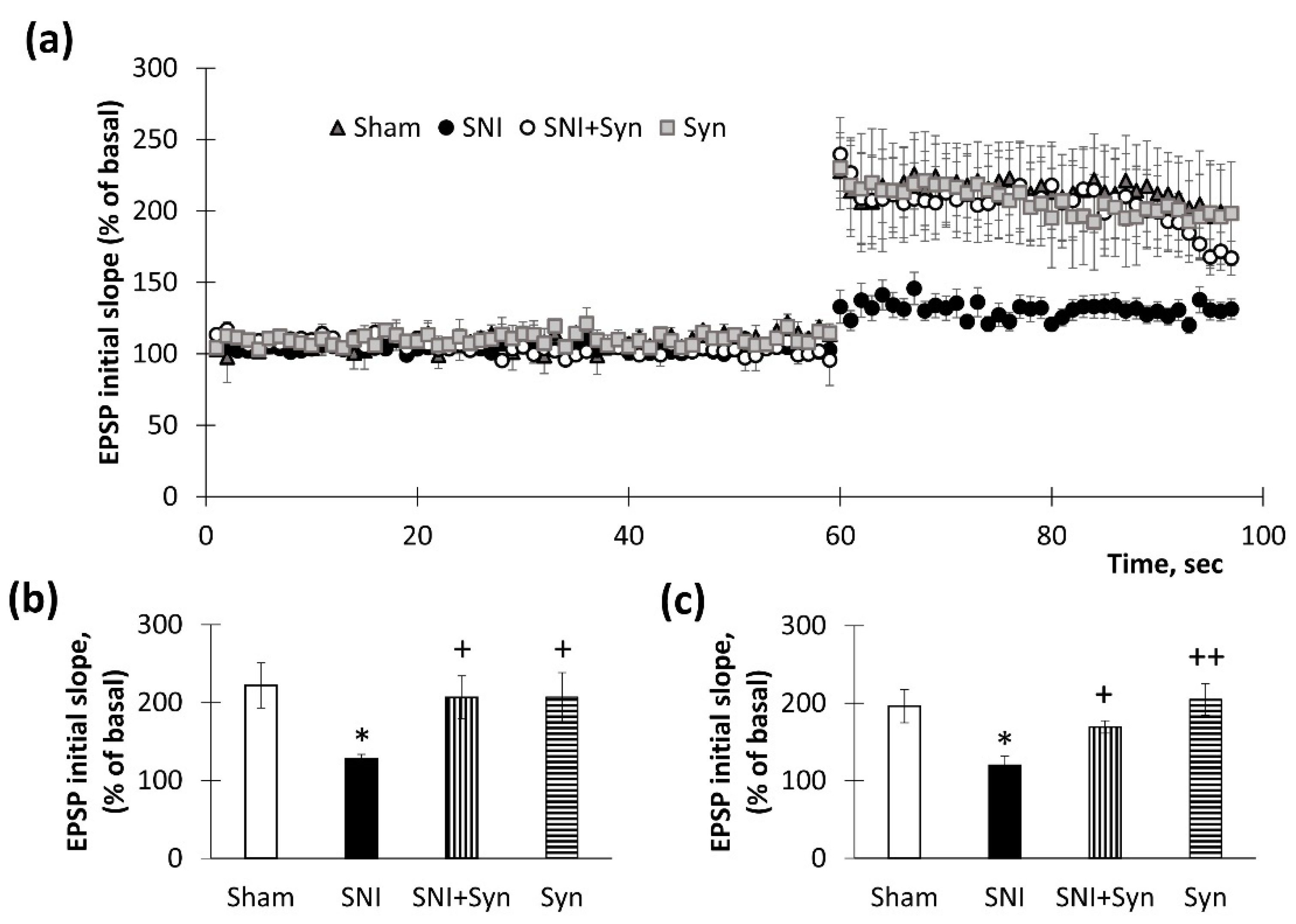
2.2. Synaptic Plasticity in the Hippocampus upon Synaptamide Administration in Neuropathic Pain
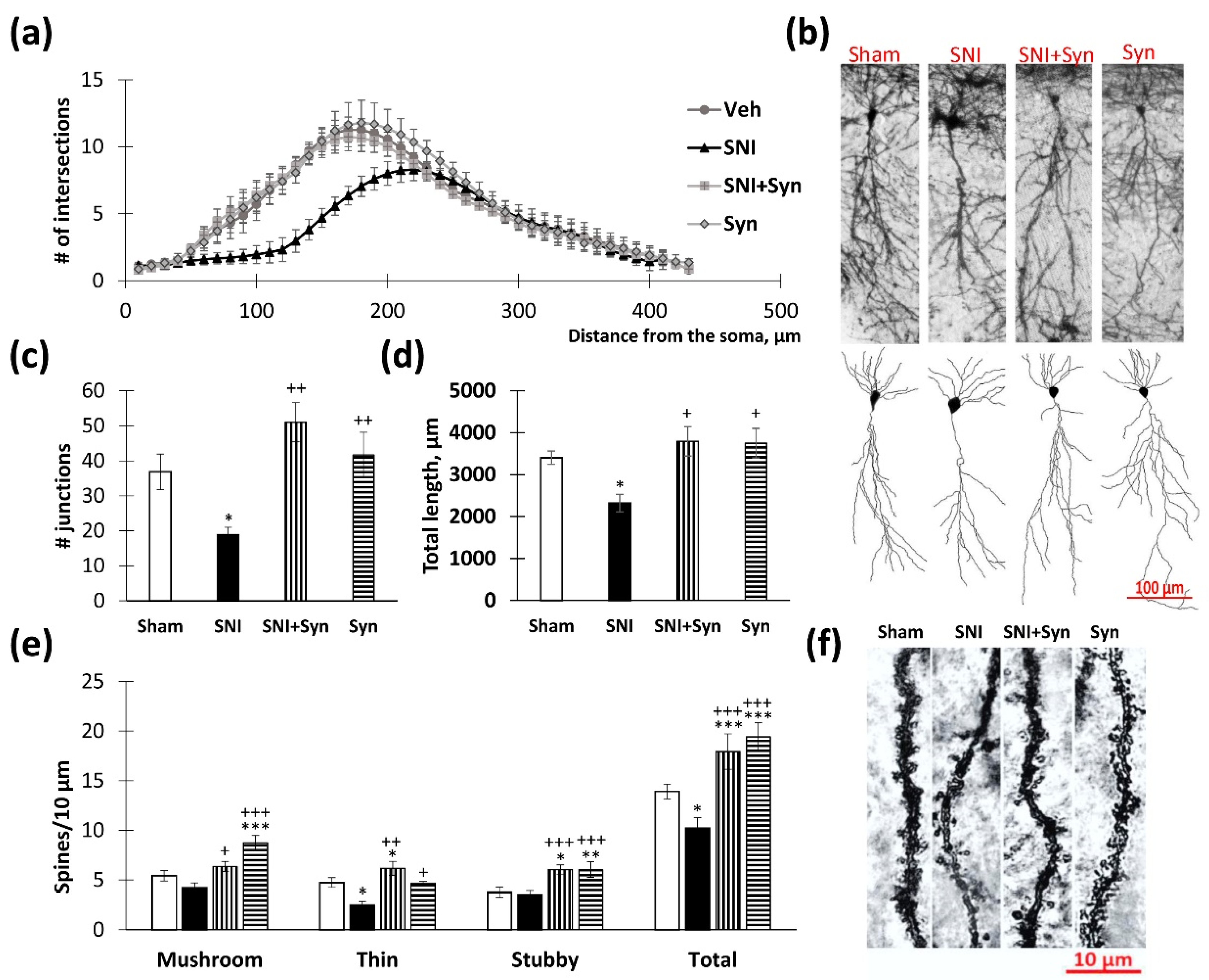
2.3. Neuronal Tree Morphology upon Synaptamide Administration in Neuropathic Pain
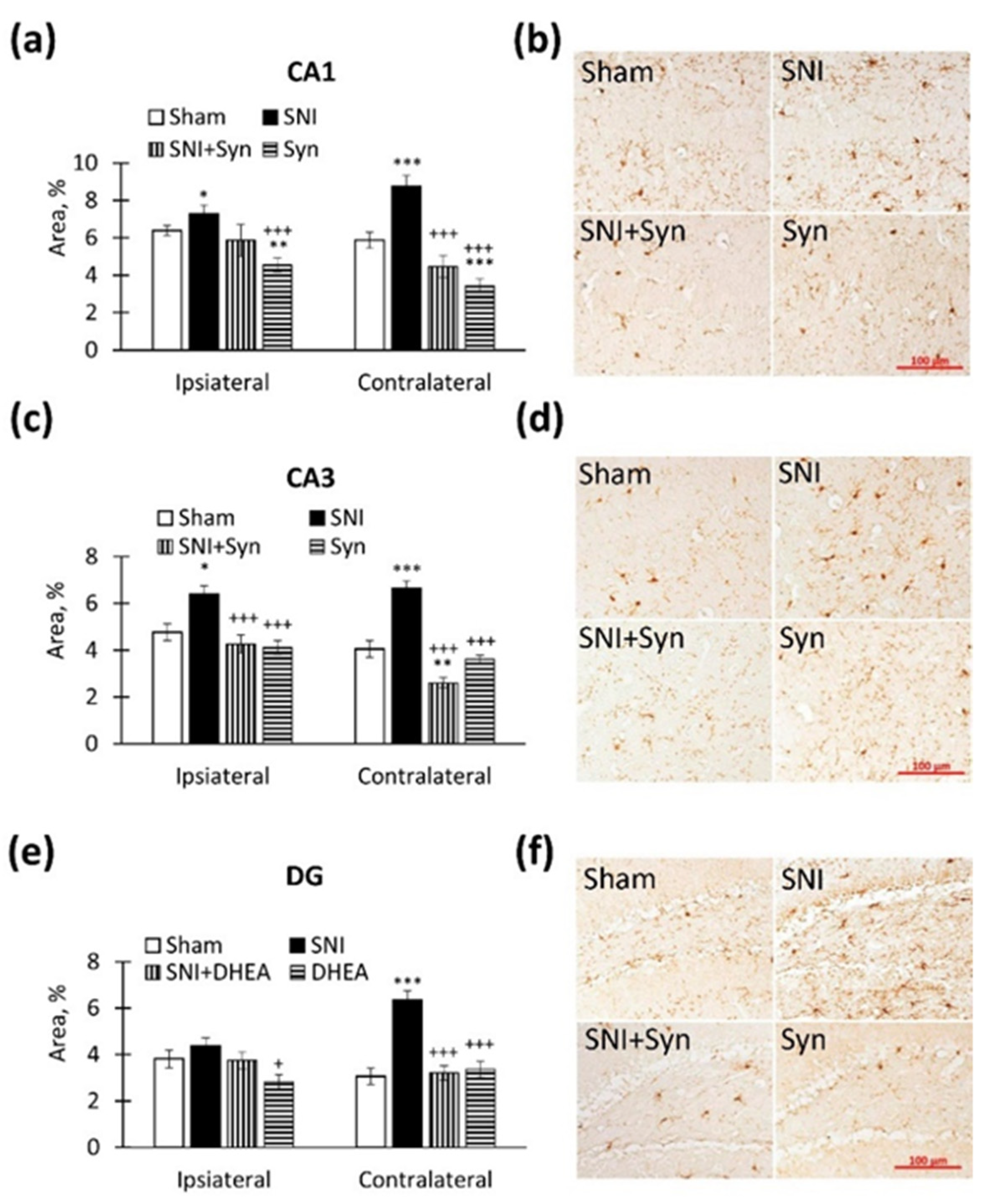
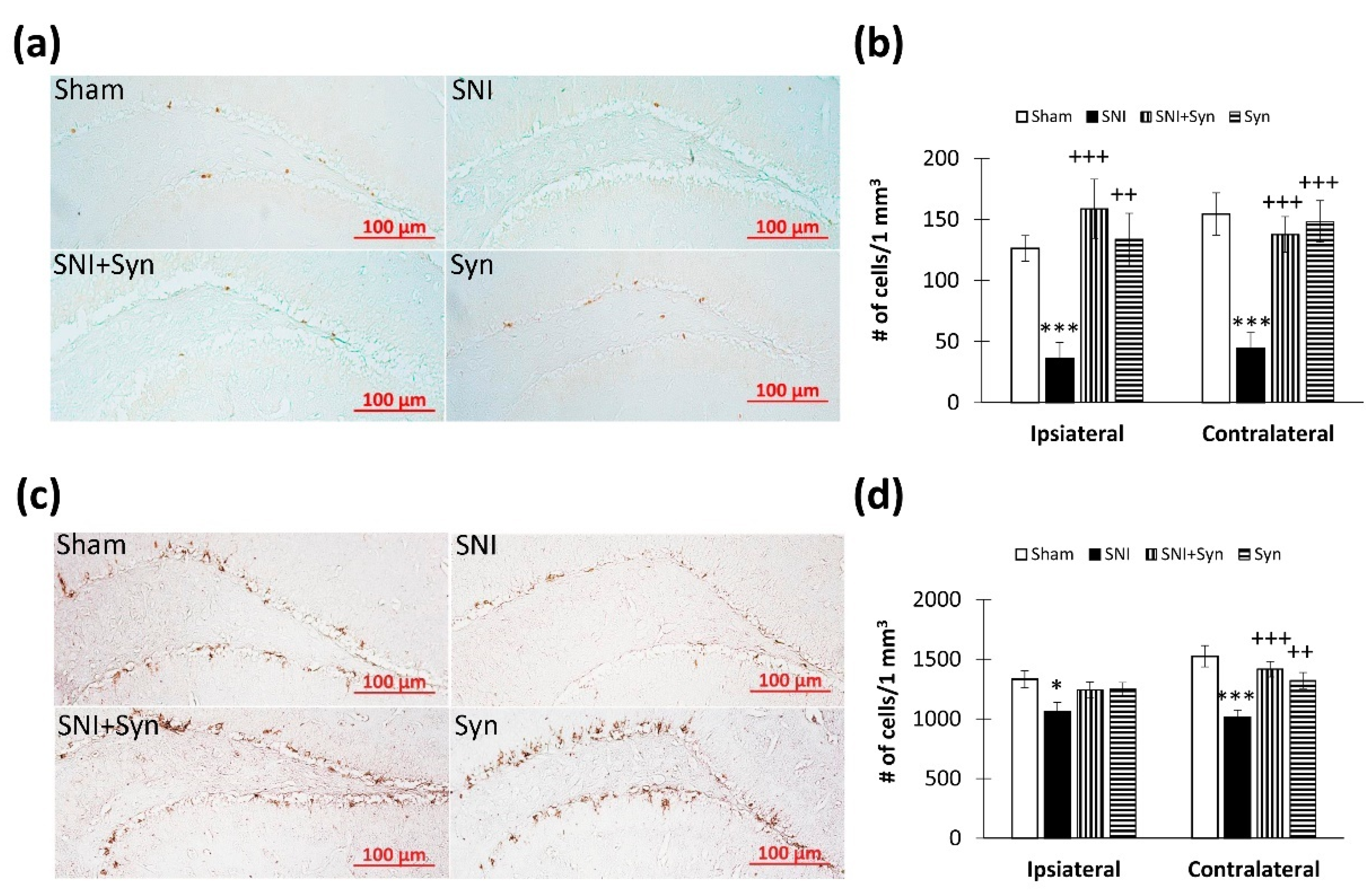
2.4. Microglial Activity within the Hippocampus in SNI and Synaptamide Treatment
2.5. Hippocampal Neurogenesis in SNI and Synaptamide Treatment
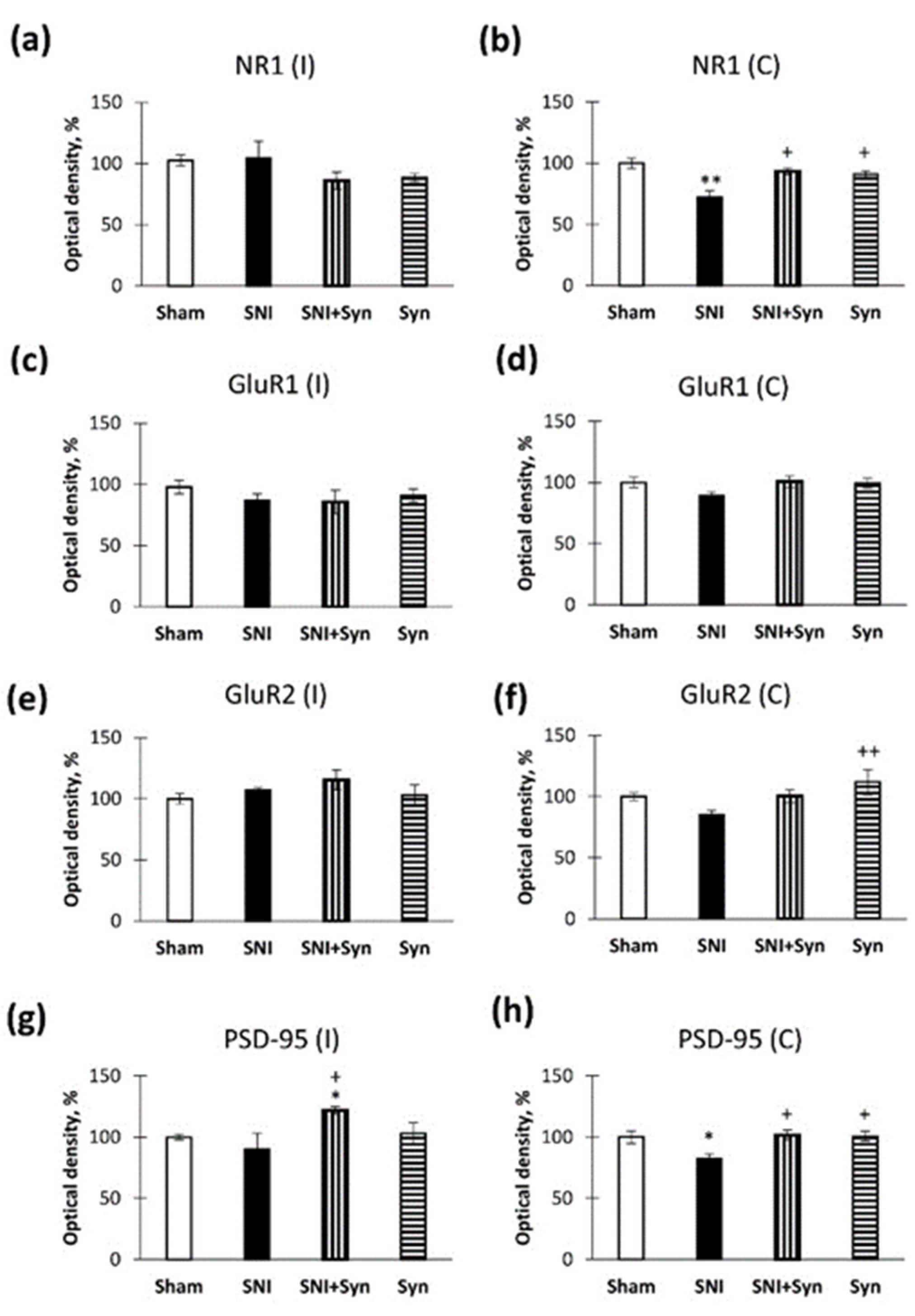
2.6. Neuropathic Pain and Treatment Alter the Hippocampal Level of Glutamate Receptors and PSD-95
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Surgery and Treatment
4.3. Synaptamide Preparation
4.4. Behavioral Tests
4.5. Thermal Allodynia
4.6. Mechanical Hyperalgesia
4.7. Y-Maze Testing
4.8. Novel Object Recognition Test
4.9. Golgi–Cox Staining
4.10. Sholl Analysis
4.11. Immunohistochemical Studies
4.12. ELISA
4.13. Electrophysiological Recordings
4.14. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed]
- Costigan, M.; Scholz, J.; Woolf, C.J. Neuropathic pain: A maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 2009, 32, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Aman, M.M.; Mahmoud, A.; Waghray-Penmetcha, T. Neuropathic Pain: Complex Regional Pain Syndrome (CRPS). In Chronic Pain Management in General and Hospital Practice; Springer: Singapore, 2021; pp. 225–247. [Google Scholar]
- Kocot-Kępska, M.; Zajączkowska, R.; Mika, J.; Wordliczek, J.; Dobrogowski, J.; Przeklasa-Muszyńska, A. Peripheral mechanisms of neuropathic pain—The role of neuronal and non-neuronal interactions and their implications for topical treatment of neuropathic pain. Pharmaceuticals 2021, 14, 77. [Google Scholar] [CrossRef]
- Seifert, F.; Maihöfner, C. Central mechanisms of experimental and chronic neuropathic pain: Findings from functional imaging studies. Cell. Mol. Life Sci. 2009, 66, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, L.; Silva, R.; Pinto-Ribeiro, F.; Pêgo, J.M.; Bessa, J.M.; Pertovaara, A.; Sousa, N.; Almeida, A. Neuropathic pain is associated with depressive behaviour and induces neuroplasticity in the amygdala of the rat. Exp. Neurol. 2008, 213, 48–56. [Google Scholar] [CrossRef]
- Boadas-Vaello, P.; Castany, S.; Homs, J.; Álvarez-Pérez, B.; Deulofeu, M.; Verdú, E. Neuroplasticity of ascending and descending pathways after somatosensory system injury: Reviewing knowledge to identify neuropathic pain therapeutic targets. Spinal Cord 2016, 54, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Nijs, J.; Meeus, M.; Versijpt, J.; Moens, M.; Bos, I.; Knaepen, K.; Meeusen, R. Brain-derived neurotrophic factor as a driving force behind neuroplasticity in neuropathic and central sensitization pain: A new therapeutic target? Expert Opin. Ther. Targets 2015, 19, 565–576. [Google Scholar] [CrossRef]
- Liu, M.G.; Chen, J. Roles of the hippocampal formation in pain information processing. Neurosci. Bull. 2009, 25, 237–266. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, N.; Wang, J.Y.; Chang, J.Y.; Woodward, D.J.; Luo, F. Ensemble encoding of nociceptive stimulus intensity in the rat medial and lateral pain systems. Mol. Pain 2011, 7, 64. [Google Scholar] [CrossRef]
- Auvray, M.; Myin, E.; Spence, C. The sensory-discriminative and affective-motivational aspects of pain. Neurosci. Biobehav. Rev. 2010, 34, 214–223. [Google Scholar] [CrossRef]
- Bushnell, M.C.; Čeko, M.; Low, L.A. Cognitive and emotional control of pain and its disruption in chronic pain. Nat. Rev. Neurosci. 2013, 14, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Cardoso-Cruz, H.; Lima, D.; Galhardo, V. Impaired spatial memory performance in a rat model of neuropathic pain is associated with reduced hippocampus-prefrontal cortex connectivity. J. Neurosci. 2013, 33, 2465–2480. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Zhou, L.J.; Wu, Y.; Li, F.; Shen, K.F.; Pang, R.P.; Wei, X.H.; Li, Y.Y.; Liu, X.G. Magnesium L-threonate prevents and restores memory deficits associated with neuropathic pain by inhibition of TNF-α. Pain Physician 2013, 16, E563–E575. [Google Scholar] [PubMed]
- Gui, W.S.; Wei, X.; Mai, C.L.; Murugan, M.; Wu, L.J.; Xin, W.J.; Zhou, L.J.; Liu, X.G. Interleukin-1β overproduction is a common cause for neuropathic pain, memory deficit, and depression following peripheral nerve injury in rodents. Mol. Pain 2016, 12, 1744806916646784. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Zhang, G.F.; Jia, M.; Xie, Z.M.; Yang, J.J.; Shen, J.C.; Zhou, Z.Q. Environmental enrichment improves pain sensitivity, depression-like phenotype, and memory deficit in mice with neuropathic pain: Role of NPAS4. Psychopharmacology 2019, 236, 1999–2014. [Google Scholar] [CrossRef]
- Qian, Y.; Xia, T.; Cui, Y.; Chu, S.; Ma, Z.; Gu, X. The role of CaMKII in neuropathic pain and fear memory in chronic constriction injury in rats. Int. J. Neurosci. 2019, 129, 146–154. [Google Scholar] [CrossRef]
- Tyrtyshnaia, A.; Manzhulo, I. Neuropathic pain causes memory deficits and dendrite tree morphology changes in mouse hippocampus. J. Pain Res. 2020, 13, 345–354. [Google Scholar] [CrossRef]
- Saffarpour, S.; Janzadeh, A.; Rahimi, B.; Ramezani, F.; Nasirinezhad, F. Chronic nanocurcumin treatment ameliorates pain-related behavior, improves spatial memory, and reduces hippocampal levels of IL-1β and TNFα in the chronic constriction injury model of neuropathic pain. Psychopharmacology 2021, 238, 877–886. [Google Scholar] [CrossRef]
- Guida, F.; De Gregorio, D.; Palazzo, E.; Ricciardi, F.; Boccella, S.; Belardo, C.; Iannotta, M.; Infantino, R.; Formato, F.; Marabese, I.; et al. Behavioral, biochemical and electrophysiological changes in spared nerve injury model of neuropathic pain. Int. J. Mol. Sci. 2020, 21, 3396. [Google Scholar] [CrossRef]
- Rashid, M.A.; Katakura, M.; Kharebava, G.; Kevala, K.; Kim, H.Y. N-Docosahexaenoylethanolamine is a potent neurogenic factor for neural stem cell differentiation. J. Neurochem. 2013, 125, 869–884. [Google Scholar] [CrossRef]
- Kharebava, G.; Rashid, M.A.; Lee, J.W.; Sarkar, S.; Kevala, K.; Kim, H.Y. N-docosahexaenoylethanolamine regulates Hedgehog signaling and promotes growth of cortical axons. Biol. Open 2015, 4, 1660–1670. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Spector, A.A. N-Docosahexaenoylethanolamine: A neurotrophic and neuroprotective metabolite of docosahexaenoic acid. Mol. Aspects Med. 2018, 64, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Spector, A.A.; Xiong, Z.M. A synaptogenic amide N-docosahexaenoylethanolamide promotes hippocampal development. Prostaglandins Other Lipid Mediat. 2011, 96, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Huang, B.X.; Kwon, H.; Rashid, M.A.; Kharebava, G.; Desai, A.; Patnaik, S.; Marugan, J.; Kim, H.Y. Orphan GPR110 (ADGRF1) targeted by N-docosahexaenoylethanolamine in development of neurons and cognitive function. Nat. Commun. 2016, 7, 13123. [Google Scholar] [CrossRef]
- Park, T.; Chen, H.; Kevala, K.; Lee, J.W.; Kim, H.Y. N-Docosahexaenoylethanolamine ameliorates LPS-induced neuroinflammation via cAMP/PKA-dependent signaling. J. Neuroinflamm. 2016, 13, 284. [Google Scholar] [CrossRef]
- Paton, K.F.; Shirazi, R.; Vyssotski, M.; Kivell, B.M. N-docosahexaenoyl ethanolamine (synaptamide) has antinociceptive effects in male mice. Eur. J. Pain 2020, 24, 1990–1998. [Google Scholar] [CrossRef]
- Jensen, T.S.; Finnerup, N.B. Allodynia and hyperalgesia in neuropathic pain: Clinical manifestations and mechanisms. Lancet Neurol. 2014, 13, 924–935. [Google Scholar] [CrossRef]
- Xiong, B.; Zhang, W.; Zhang, L.; Huang, X.; Zhou, W.; Zou, Q.; Manyande, A.; Wang, J.; Tian, Y.; Tian, X. Hippocampal glutamatergic synapses impairment mediated novel-object recognition dysfunction in rats with neuropathic pain. Pain 2020, 161, 1824–1836. [Google Scholar] [CrossRef]
- Zhang, L.Q.; Zhang, W.; Li, T.; Yang, T.; Yuan, X.; Zhou, Y.; Zou, Q.; Yang, H.; Gao, F.; Tian, Y.; et al. GLP-1R activation ameliorated novel-object recognition memory dysfunction via regulating hippocampal AMPK/NF-κB pathway in neuropathic pain mice. Neurobiol. Learn. Mem. 2021, 182, 107463. [Google Scholar] [CrossRef]
- Abdulmajeed, W.I.; Ibrahim, R.B.; Ishola, A.O.; Balogun, W.G.; Cobham, A.E.; Amin, A. Amitriptyline and phenytoin prevents memory deficit in sciatic nerve ligation model of neuropathic pain. J. Basic Clin. Physiol. Pharmacol. 2016, 27, 101–108. [Google Scholar] [CrossRef]
- Morel, V.; Etienne, M.; Wattiez, A.S.; Dupuis, A.; Privat, A.M.; Chalus, M.; Eschalier, A.; Daulhac, L.; Pickering, G. Memantine, a promising drug for the prevention of neuropathic pain in rat. Eur. J. Pharmacol. 2013, 721, 382–390. [Google Scholar] [CrossRef]
- Cook, S.C.; Wellman, C.L. Chronic stress alters dendritic morphology in rat medial prefrontal cortex. J. Neurobiol. 2004, 60, 236–248. [Google Scholar] [CrossRef]
- Zhu, C.; Xu, Q.; Mao, Z.; Lin, N. The Chinese medicine Wu-Tou decoction relieves neuropathic pain by inhibiting hippocampal microglia activation. Sci. Rep. 2018, 8, 12292. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, L.J.; Wang, J.; Li, D.; Ren, W.J.; Peng, J.; Wei, X.; Xu, T.; Xin, W.J.; Pang, R.P.; et al. TNF-α differentially regulates synaptic plasticity in the hippocampus and spinal cord by microglia-dependent mechanisms after peripheral nerve injury. J. Neurosci. 2017, 37, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Stratton, H.J.; Khanna, R. Sculpting Dendritic Spines during Initiation and Maintenance of Neuropathic Pain. J. Neurosci. 2020, 40, 7578–7589. [Google Scholar] [CrossRef] [PubMed]
- Jacobowitz, D.M.; Cole, J.T.; McDaniel, D.P.; Pollard, H.B.; Watson, W.D. Microglia activation along the corticospinal tract following traumatic brain injury in the rat: A neuroanatomical study. Brain Res. 2012, 1465, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Muccigrosso, M.M.; Godbout, J.P. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 2015, 96 Pt A, 29–41. [Google Scholar] [CrossRef]
- Zarruk, J.G.; Greenhalgh, A.D.; David, S. Microglia and macrophages differ in their inflammatory profile after permanent brain ischemia. Exp. Neurol. 2018, 301 Pt B, 120–132. [Google Scholar] [CrossRef]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of general and discriminating markers of differential microglia phenotypes. Front. Cell. Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Wang, Z.T.; Yu, G.; Wang, H.S.; Yi, S.P.; Su, R.B.; Gong, Z.H. Changes in VGLUT2 expression and function in pain-related supraspinal regions correlate with the pathogenesis of neuropathic pain in a mouse spared nerve injury model. Brain Res. 2015, 1624, 515–524. [Google Scholar] [CrossRef]
- Ultenius, C.; Linderoth, B.; Meyerson, B.A.; Wallin, J. Spinal NMDA receptor phosphorylation correlates with the presence of neuropathic signs following peripheral nerve injury in the rat. Neurosci. Lett. 2006, 399, 85–90. [Google Scholar] [CrossRef]
- Salussolia, C.L.; Prodromou, M.L.; Borker, P.; Wollmuth, L.P. Arrangement of subunits in functional NMDA receptors. J. Neurosci. 2011, 31, 11295–11304. [Google Scholar] [CrossRef] [PubMed]
- Lachamp, P.; Balland, B.; Tell, F.; Baude, A.; Strube, C.; Crest, M.; Kessler, J.P. Early expression of AMPA receptors and lack of NMDA receptors in developing rat climbing fibre synapses. J. Physiol. 2005, 564 Pt 3, 751–763. [Google Scholar] [CrossRef]
- Wang, X.Q.; Zhong, X.L.; Li, Z.B.; Wang, H.T.; Zhang, J.; Li, F.; Zhang, J.Y.; Dai, R.P.; Xin-Fu, Z.; Li, C.Q.; et al. Differential roles of hippocampal glutamatergic receptors in neuropathic anxiety-like behavior after partial sciatic nerve ligation in rats. BMC Neurosci. 2015, 16, 14. [Google Scholar] [CrossRef]
- Goffer, Y.; Xu, D.; Eberle, S.E.; D’amour, J.; Lee, M.; Tukey, D.; Froemke, R.C.; Ziff, E.B.; Wang, J. Calcium-permeable AMPA receptors in the nucleus accumbens regulate depression-like behaviors in the chronic neuropathic pain state. J. Neurosci. 2013, 33, 19034–19044. [Google Scholar] [CrossRef]
- Luo, C.; Seeburg, P.H.; Sprengel, R.; Kuner, R. Activity-dependent potentiation of calcium signals in spinal sensory networks in inflammatory pain states. Pain 2008, 140, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, J.; Wu, Z.; Lin, Q.; Yue, Y.; Fang, L. Regulation of AMPA receptors in spinal nociception. Mol. Pain 2010, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wu, L.J.; Wang, H.; Zhang, X.; Vadakkan, K.I.; Kim, S.S.; Steenland, H.W.; Zhuo, M. Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J. Neurosci. 2008, 28, 7445–7453. [Google Scholar] [CrossRef] [PubMed]
- Won, S.; Incontro, S.; Nicoll, R.A.; Roche, K.W. PSD-95 stabilizes NMDA receptors by inducing the degradation of STEP61. Proc. Natl. Acad. Sci. USA 2016, 9, E4736–E4744. [Google Scholar] [CrossRef]
- Jeong, J.; Pandey, S.; Li, Y.; Badger, J.D., II; Lu, W.; Roche, K.W. PSD-95 binding dynamically regulates NLGN1 trafficking and function. Proc. Natl. Acad. Sci. USA 2019, 116, 12035–12044. [Google Scholar] [CrossRef]
- Husi, H.; Ward, M.A.; Choudhary, J.S.; Blackstock, W.P.; Grant, S.G. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 2000, 3, 661–669. [Google Scholar] [CrossRef]
- Toro, C.; Deakin, J.F. NMDA receptor subunit NRI and postsynaptic protein PSD-95 in hippocampus and orbitofrontal cortex in schizophrenia and mood disorder. Schizophr. Res. 2005, 80, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Vickers, C.A.; Stephens, B.; Bowen, J.; Arbuthnott, G.W.; Grant, S.G.; Ingham, C.A. Neurone specific regulation of dendritic spines in vivo by post synaptic density 95 protein (PSD-95). Brain Res. 2006, 1090, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Pchitskaya, E.; Bezprozvanny, I. Dendritic Spines Shape Analysis-Classification or Clusterization? Perspective. Front. Synaptic Neurosci. 2020, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Rochefort, N.L.; Konnerth, A. Dendritic spines: From structure to in vivo function. EMBO Rep. 2012, 13, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Helm, M.S.; Dankovich, T.M.; Mandad, S.; Rammner, B.; Jähne, S.; Salimi, V.; Koerbs, C.; Leibrandt, R.; Urlaub, H.; Schikorski, T.; et al. A large-scale nanoscopy and biochemistry analysis of postsynaptic dendritic spines. Nat. Neurosci. 2021, 24, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Siri, S.O.; Rozés-Salvador, V.; de la Villarmois, E.A.; Ghersi, M.S.; Quassollo, G.; Pérez, M.F.; Conde, C. Decrease of Rab11 prevents the correct dendritic arborization, synaptic plasticity and spatial memory formation. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118735. [Google Scholar] [CrossRef] [PubMed]
- Spruston, N. Pyramidal neurons: Dendritic structure and synaptic integration. Nat. Rev. Neurosci. 2008, 9, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Milatovic, D.; Zaja-Milatovic, S.; Montine, K.S.; Shie, F.S.; Montine, T.J. Neuronal oxidative damage and dendritic degeneration following activation of CD14-dependent innate immune response in vivo. J. Neuroinflamm. 2004, 1, 20. [Google Scholar] [CrossRef]
- Deng, X.; Li, M.; Ai, W.; He, L.; Lu, D.; Patrylo, P.R.; Cai, H.; Luo, X.; Li, Z.; Yan, X. Lipolysaccharide-induced neuroinflammation is associated with Alzheimer-like amyloidogenic axonal pathology and dendritic degeneration in rats. Adv. Alzheimer Dis. 2014, 3, 78–93. [Google Scholar] [CrossRef]
- Meijerink, J.; Balvers, M.; Plastina, P.; Witkamp, R. Omega-3 polyunsaturated N-acylethanolamines: A link between diet and cellular biology. In The Endocannabinoidome; Academic Press: Cambridge, MA, USA, 2015; pp. 15–32. [Google Scholar]
- Park, T.; Chen, H.; Kim, H.Y. GPR110 (ADGRF1) mediates anti-inflammatory effects of N-docosahexaenoylethanolamine. J. Neuroinflamm. 2019, 16, 225. [Google Scholar] [CrossRef]
- Kim, Y.K.; Na, K.S.; Myint, A.M.; Leonard, B.E. The role of pro-inflammatory cytokines in neuroinflammation, neurogenesis and the neuroendocrine system in major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 277–284. [Google Scholar] [CrossRef]
- Sabariego, M.; Schönwald, A.; Boublil, B.L.; Zimmerman, D.T.; Ahmadi, S.; Gonzalez, N.; Leibold, C.; Clark, R.E.; Leutgeb, J.K.; Leutgeb, S. Time cells in the hippocampus are neither dependent on medial entorhinal cortex inputs nor necessary for spatial working memory. Neuron 2019, 102, 1235–1248.e5. [Google Scholar] [CrossRef]
- Cinalli, D.A., Jr.; Cohen, S.J.; Guthrie, K.; Stackman, R.W., Jr. Object recognition memory: Distinct yet complementary roles of the mouse CA1 and perirhinal cortex. Front. Mol. Neurosci. 2020, 13, 527543. [Google Scholar] [CrossRef] [PubMed]
- Umpierre, A.D.; Bystrom, L.L.; Ying, Y.; Liu, Y.U.; Worrell, G.; Wu, L.J. Microglial calcium signaling is attuned to neuronal activity in awake mice. eLife 2020, 9, e56502. [Google Scholar] [CrossRef] [PubMed]
- Ferro, A.; Auguste, Y.S.S.; Cheadle, L. Microglia, cytokines, and neural activity: Unexpected interactions in brain development and function. Front. Immunol. 2021, 12, 703527. [Google Scholar] [CrossRef] [PubMed]
- Lambert, K.G.; Buckelew, S.K.; Staffiso-Sandoz, G.; Gaffga, S.; Carpenter, W.; Fisher, J.; Kinsley, C.H. Activity-stress induces atrophy of apical dendrites of hippocampal pyramidal neurons in male rats. Physiol. Behav. 1998, 65, 43–49. [Google Scholar] [CrossRef]
- Manikandan, S.; Padma, M.K.; Srikumar, R.; Jeya Parthasarathy, N.; Muthuvel, A.; Sheela Devi, R. Effects of chronic noise stress on spatial memory of rats in relation to neuronal dendritic alteration and free radical-imbalance in hippocampus and medial prefrontal cortex. Neurosci. Lett. 2006, 399, 17–22. [Google Scholar] [CrossRef]
- Clark, I.A.; Vissel, B. Excess cerebral TNF causing glutamate excitotoxicity rationalizes treatment of neurodegenerative diseases and neurogenic pain by anti-TNF agents. J. Neuroinflamm. 2016, 13, 236. [Google Scholar] [CrossRef]
- Xu, N.; Tang, X.H.; Pan, W.; Xie, Z.M.; Zhang, G.F.; Ji, M.H.; Yang, J.J.; Zhou, M.T.; Zhou, Z.Q. Spared nerve injury increases the expression of microglia M1 markers in the prefrontal cortex of rats and provokes depression-like behaviors. Front. Neurosci. 2017, 11, 209. [Google Scholar] [CrossRef]
- Honjoh, K.; Nakajima, H.; Hirai, T.; Watanabe, S.; Matsumine, A. Relationship of inflammatory cytokines from M1-type microglia/macrophages at the injured site and lumbar enlargement with neuropathic pain after spinal cord injury in the CCL21 knockout (plt) mouse. Front. Cell. Neurosci. 2019, 13, 525. [Google Scholar] [CrossRef] [PubMed]
- Del Rey, A.; Yau, H.J.; Randolf, A.; Centeno, M.V.; Wildmann, J.; Martina, M.; Besedovsky, H.O.; Apkarian, V.A. Chronic neuropathic pain-like behavior correlates with IL-1β expression and disrupts cytokine interactions in the hippocampus. Pain 2011, 152, 2827–2835. [Google Scholar] [CrossRef]
- Ignatowski, T.A.; Covey, W.C.; Knight, P.R.; Severin, C.M.; Nickola, T.J.; Spengler, R.N. Brain-derived TNFalpha mediates neuropathic pain. Brain Res. 1999, 841, 70–77. [Google Scholar] [CrossRef]
- Giansante, G.; Marte, A.; Romei, A.; Prestigio, C.; Onofri, F.; Benfenati, F.; Baldelli, P.; Valente, P. Presynaptic L-type Ca2+ channels increase glutamate release probability and excitatory strength in the hippocampus during chronic neuroinflammation. J. Neurosci. 2020, 40, 6825–6841. [Google Scholar] [CrossRef]
- Chang, P.K.; Khatchadourian, A.; McKinney, R.A.; Maysinger, D. Docosahexaenoic acid (DHA): A modulator of microglia activity and dendritic spine morphology. J. Neuroinflamm. 2015, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Shields, S.D.; Eckert, W.A., III; Basbaum, A.I. Spared nerve injury model of neuropathic pain in the mouse: A behavioral and anatomic analysis. J. Pain 2003, 4, 465–470. [Google Scholar] [CrossRef]
- Latyshev, N.A.; Ermolenko, E.V.; Kasyanov, S.P. Concentration and purification of polyunsaturated fatty acids from squid liver processing wastes. Eur. J. Lipid Sci. Technol. 2014, 116, 1608–1613. [Google Scholar] [CrossRef]
- Svetashev, V.I. Mild method for preparation of 4,4-dimethyloxazoline derivatives of polyunsaturated fatty acids for GC-MS. Lipids 2011, 46, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Bevins, R.A.; Besheer, J. Object recognition in rats and mice: A one-trial non-matching-to-sample learning task to study ‘recognition memory’. Nat. Protoc. 2006, 1, 1306–1311. [Google Scholar] [CrossRef]
- Sholl, D.A. Dendritic organization in the neurons of the visual and motor cortices of the cat. J. Anat. 1953, 87, 387–406. [Google Scholar]
- Bastian, T.W.; Duck, K.A.; Michalopoulos, G.C.; Chen, M.J.; Liu, Z.J.; Connor, J.R.; Lanier, L.M.; Sola-Visner, M.C.; Georgieff, M.K. Eltrombopag, a thrombopoietin mimetic, crosses the blood–brain barrier and impairs iron-dependent hippocampal neuron dendrite development. J. Thromb. Haemost. 2017, 15, 565–574. [Google Scholar] [CrossRef] [PubMed][Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyrtyshnaia, A.; Bondar, A.; Konovalova, S.; Manzhulo, I. Synaptamide Improves Cognitive Functions and Neuronal Plasticity in Neuropathic Pain. Int. J. Mol. Sci. 2021, 22, 12779. https://doi.org/10.3390/ijms222312779
Tyrtyshnaia A, Bondar A, Konovalova S, Manzhulo I. Synaptamide Improves Cognitive Functions and Neuronal Plasticity in Neuropathic Pain. International Journal of Molecular Sciences. 2021; 22(23):12779. https://doi.org/10.3390/ijms222312779
Chicago/Turabian StyleTyrtyshnaia, Anna, Anatoly Bondar, Sophia Konovalova, and Igor Manzhulo. 2021. "Synaptamide Improves Cognitive Functions and Neuronal Plasticity in Neuropathic Pain" International Journal of Molecular Sciences 22, no. 23: 12779. https://doi.org/10.3390/ijms222312779
APA StyleTyrtyshnaia, A., Bondar, A., Konovalova, S., & Manzhulo, I. (2021). Synaptamide Improves Cognitive Functions and Neuronal Plasticity in Neuropathic Pain. International Journal of Molecular Sciences, 22(23), 12779. https://doi.org/10.3390/ijms222312779