
Skin Aging, Cellular Senescence and Natural Polyphenols

Abstract
1. Introduction
2. Cellular Senescence in Skin Aging
2.1. Biomarkers of Cellular Senescence in the Skin
2.1.1. Senescence-Associated Ultrastructural Changes
- Hypertrophy and increased granularity. In senescent cells, both an increase in size and, if adherent cells, a flattening of the shape can be observed [3]. For these changes, the activation of the mammalian target of the rapamycin (mTOR) signaling pathway is responsible [53]. These morphological changes are easily detectable by light microscopy and quantified by flow cytometry (as an increase in forward scatter, FSC parameters). However, though in situ and in vivo quantification can be a problem, changes in plasma membrane protein expression represent a promising new biomarker of senescence [54]. The size increase by up to nine times was found in senescent fibroblasts [3]. The size increasing with other senescence markers was also confirmed in aged keratinocytes [55] and in a model of UVB-induced senescence in human melanocytes [56]. The senescent nuclei of skin cells also showed hypertrophy. In particular, the mean nuclear area of fibroblasts was shown to be 255 μm2 at early passage, compared to 293 μm2 at later passage [57]. The increase in granular content in senescent cells can be monitored by transmission electron microscopy as intracellular electron-dense particles [58]. However, cell granularity levels can also be conveniently detected by flow cytometry as an increase in the side scatter (SSC) parameter. The increase in granularity in senescent human fibroblasts is a result of the intracellular deposit formation, including lipofuscin in lysosomes and glycogen particles [59,60]. In a model of the UVB-promoted senescence of melanocytes, the cell population showing a high granularity was mostly growth-arrested at G2/M phase [61];
- Increase in lysosomal mass and SA-β-Gal staining. The increase in the lysosomal mass in senescent cells is associated with the accumulation of old lysosomes and increased lysosomal biogenesis. Accumulated lipofuscin may be in line with the impaired lysosomal turnover mechanism [62]. Lysosomal biogenesis is largely controlled by the transcription factor EB (TFEB), an effector protein within the mTOR signaling pathway that regulates multiple lysosomal proteins. During senescence, it tends to be up- or down-regulated, making it difficult to use as a marker of senescence [63,64]. Alternatively, the detection of lipofuscin content can be used as a biomarker of lysosome accumulation, either by its typical autofluorescence properties and fluorescence-based methods, or by selective staining with Sudan black B, allowing for detection in cells, tissues and body fluids [65]. The term lipofuscin originates from the Greek words “lipo” (fat) and “fuscus” (dark) [1]. In addition, it is referred to as “aging fluorophore” or “aging pigment”. It is an insoluble material that mainly consists of a highly oxidized and crosslinked substrate, which are proteins, lipids and sugars. Transition metals also bind to lipofuscin and increase its intracellular cytotoxicity through the catalysis of ROS formation by the Fenton reaction. Lipofuscin is also present in small amounts in the cytosol (about 1% of the total intracellular content), while its cytotoxicity is suppressed by the macroautophagy activity of the cell. It preferentially accumulates in postmitotic tissue cells, such as neurons or muscle cells, which do not divide and are therefore unable to dilute the products of their damage (in the sense of the so called “garbage catastrophe theory of aging”) [66]. However, lipofuscin has also been shown to accumulate during the replicative senescence of human fibroblasts [67]. Lipofuscin accumulation has been detected in the basal layers of the aged epidermis [68]. The phototoxicity of visible light has been linked to accumulated lipofuscin in skin cells due to oxidative damage in nucleic acids, lipids and proteins, generating premutagenic DNA lesions and releasing pro-inflammatory cytokines and metalloproteinases, consequently exaggerating cell damage and skin aging [69]. The increase in the size and shape of lysosomes is mostly associated with an increase in the activity of the lysosomal enzyme, senescence-associated b-galactosidase (SA-β-Gal). Since SA-β-Gal is upregulated in senescent cells, its residual activity can be monitored at suboptimal pH 6.0. It is the most widely used marker of senescence in culture and tissue samples [70]. However, factors such as confluence during cell culture may contribute to the detection of a false positive signal [71]. Furthermore, this assay requires active enzymatic SA-β-Gal activity, which is often lost in fixed or cryopreserved tissues [72,73]. In addition, non-specific SA-β-Gal activity was detected in the early passage of adult melanocytes proliferating in culture [21]. The available methodologies allow, depending on the properties of a specific synthetic b-galactosidase substrate, for the quantification of senescent cells in in vitro models or tissues of an aged organism using a combination of flow cytometry or spectrofluorimetry with high-content image analysis [74]. SA-β-Gal activity has been successfully confirmed in human fibroblasts and keratinocytes undergoing replicative senescence in vitro, in skin samples or in the cells isolated from aged individuals [75,76,77,78,79]. Furthermore, SA-β-Gal has been used to confirm premature senescence in cultured fibroblasts, keratinocytes and melanocytes cells in response to various stressors, including UV light [43,50,80,81,82,83], cigarette smoke [84,85], ionization radiation [86], oxidants [87,88,89] or anticancer drugs [90];
- Accumulation of mitochondria. Senescent cells usually have a higher number of mitochondria and also display organelle enlargement [91]. Highly elongated or enlarged giant mitochondria were observed in senescent human foreskin diploid fibroblasts, with their population doubling between 90 and 94 times [91]. However, the mitochondrial membrane potential is reduced, which is associated with an increased ROS production and the release of mitochondrial enzymes, such as endonuclease G [92,93]. This is mainly due to the reduced specific autophagy of mitochondria, mitophagy, causing old and dysfunctional mitochondria to accumulate [94]. Reduced mitochondrial scission and excessive fusion, which likely occur to compensate for the dysfunction of mitochondria in senescent cells and to protect them from apoptosis and mitophagy [95], contribute to mitochondria enlargement [96]. Dysfunctional mitochondria also represent a major source of elevated ROS production in senescent cells, another important hallmark of senescence [97]. There is also a strong link between ROS-related mitochondrial damage and photoaging. The repetitive UVA exposure was found to be accompanied by a rise in mitochondrial DNA mutations. In particular, the photoaged skin comprises up to 10-fold more frequent mitochondrial DNA mutations compared to sun-protected skin [98,99,100]. Moreover, mitochondrial DNA mutations are positively associated with matrix metalloproteinase-1 (MMP-1) levels without the related increase in MMP-1 specific tissue inhibitors [101];
- Nuclear changes. Senescent nuclei may contain so termed senescence-associated heterochromatin foci (SAHFs), the silent domains that co-localize with H3K9me3 and heterochromatin protein 1 (HP1) and may lock cells in a senescent state by transcriptionally repressing genes involved in cell proliferation [102]. The SAHFs can be visualized by staining with 4′, 6-diamidino-2-phenylindole (DAPI) and appear as fluorescent spots representing condensed chromatin domains that block certain genes required for proliferation [12]. The long-term monitoring of senescent cells in vitro revealed the progressive proteolysis of histones 3 and 4 without DNA loss. A reduced histone content was also observed in nevus melanocytes, as compared to neighboring non-senescent melanocytes and keratinocytes in vivo [103]. These studies confirm the dramatic structural changes in chromatin in senescent cells.SAHFs are also implicated in the downregulation of lamin B1, a structural protein of the nuclear lamina/membrane [22,104]. Lamin B1 has been shown to be downregulated in cells undergoing mainly replicative senescence and OIS and UV-induced senescence in vitro [26,103,105,106], and also decline during the chronological aging of human skin in vivo [105], in senescent melanocytes within human nevi [103] and in the UV-exposed mouse skin epidermis [51]. The destabilization of nuclear integrity leads to other changes, such as a loss of constitutive heterochromatin condensation and the formation of cytoplasmic chromatin fragments that contain epigenetic tags associated with DNA damage [103]. SAHFs production is thought to be a compensatory mechanism that maintains constitutive heterochromatin [107]. SAHFs, however, are not a universal marker of senescence, but are observed especially in the case of OIS [17]. Lamin B1 downregulation preferentially depends on p53 and p16, but is independent of other signaling pathways associated with senescence, such as p38 mitogen-activated protein kinases (MAPK), NF-κB and DNA damage response (DDR) [108].
2.1.2. Changes in Cyclin-Dependent Kinase Inhibitors (CDKIs) Expression
2.1.3. Changes in Apoptosis Resistance
2.1.4. Senescence-Associated Secretory Phenotype
2.1.5. Metabolism Changes
2.1.6. Proteostasis Changes
2.1.7. Endoplasmic Reticulum (ER) Stress
2.1.8. Persistent DNA Damage
2.1.9. Pigmentation Changes and Skin Cellular Aging
3. Natural Polyphenols against Skin Cellular Aging
4. Polyphenols as Anti-Aging Cosmeceuticals
5. Cellular Aging of Skin and COVID-19 Pandemic
6. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Höhn, A.; Weber, D.; Jung, T.; Ott, C.; Hugo, M.; Kochlik, B.; Kehm, R.; König, J.; Grune, T.; Castro, J.P. Happily (n)ever after: Aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. 2017, 11, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Thomas, K.; Chrysiis, M.; Wolter, J.M.; Daniel, S.P. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef]
- Pérez-Jiménez, J.; Neveu, V.; Vos, F.; Scalbert, A. Identification of the 100 richest dietary sources of polyphenols: An application of the Phenol-Explorer database. Eur. J. Clin. Nutr. 2010, 64 (Suppl. S3), S112–S120. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, P.B.; Ha, S.E.; Vetrivel, P.; Kim, H.H.; Kim, S.M.; Kim, G.S. Functions of polyphenols and its anticancer properties in biomedical research: A narrative review. Transl. Cancer Res. 2020, 9, 7619–7631. [Google Scholar] [CrossRef]
- Daglia, M. Polyphenols as antimicrobial agents. Curr. Opin. Biotechnol. 2012, 23, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Domaszewska-Szostek, A.; Puzianowska-Kuźnicka, M.; Kuryłowicz, A. Flavonoids in skin senescence prevention and treatment. Int. J. Mol. Sci. 2021, 22, 6814. [Google Scholar] [CrossRef]
- Lujambio, A. To clear, or not to clear (senescent cells)? That is the question. Insid. Cell 2016, 1, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Aravinthan, A. Cellular senescence: A hitchhiker’s guide. Hum. Cell 2015, 28, 51–64. [Google Scholar] [CrossRef]
- Campisi, J. The biology of replicative senescence. Eur. J. Cancer 1997, 33, 703–709. [Google Scholar] [CrossRef]
- Cristofalo, V.J.; Lorenzini, A.; Allen, R.G.; Torres, C.; Tresini, M. Replicative senescence: A critical review. Mech. Ageing Dev. 2004, 125, 827–848. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef]
- Petrova, N.V.; Velichko, A.K.; Razin, S.V.; Kantidze, O.L. Small molecule compounds that induce cellular senescence. Aging Cell 2016, 15, 999–1017. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Dimri, G.P.; Leet, X.; Basile, G.; Acosta, M.; Scortt, G.; Roskelley, C.; Medrano, E.E.; Linskensi, M.; Rubeljii, I.; Pereira-Smithii, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Cell Biol. 1995, 92, 9363–9367. [Google Scholar]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Floyd, R.A. Reactive oxygen species and protein oxidation in aging: A look back, a look ahead. Arch. Biochem. Biophys. 2002, 397, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Krutmann, J.; Schikowski, T.; Morita, A.; Berneburg, M. Environmentally-Induced (Extrinsic) Skin Aging: Exposomal Factors and Underlying Mechanisms. J. Investig. Dermatol. 2021, 141, 1096–1103. [Google Scholar] [CrossRef]
- Krutmann, J.; Bouloc, A.; Sore, G.; Bernard, B.A.; Passeron, T. The skin aging exposome. J. Dermatol. Sci. 2017, 85, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.S.; Dreesen, O. Biomarkers of cellular senescence and skin aging. Front. Genet. 2018, 9, 247. [Google Scholar] [CrossRef] [PubMed]
- Farage, M.A.; Miller, K.W.; Elsner, P.; Maibach, H.I. Intrinsic and extrinsic factors in skin ageing: A review. Int. J. Cosmet. Sci. 2008, 30, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, J.S.; Wanat, K.; Seykora, J. Skin: Basic Structure and Function; Elsevier: Amsterdam, The Netherlands, 2014; ISBN 9780123864567. [Google Scholar]
- Blume-Peytavi, U.; Kottner, J.; Sterry, W.; Hodin, M.W.; Griffiths, T.W.; Watson, R.E.B.; Hay, R.J.; Griffiths, C.E.M. Age-Associated Skin Conditions and Diseases: Current Perspectives and Future Options. Gerontologist 2016, 56, S230–S242. [Google Scholar] [CrossRef]
- Choi, E.-H.; Man, M.-Q.; Xu, P.; Xin, S.; Liu, Z.; Crumrine, D.A.; Jiang, Y.J.; Fluhr, J.W.; Feingold, K.R.; Elias, P.M.; et al. Stratum Corneum Acidification Is Impaired in Moderately Aged Human and Murine Skin. J. Investig. Dermatol. 2007, 127, 2847–2856. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.-M.; Förl, M.; Winoto-Morbach, S.; Seite, S.; Schunck, M.; Proksch, E.; Schütze, S. Acid and neutral sphingomyelinase, ceramide synthase, and acid ceramidase activities in cutaneous aging. Exp. Dermatol. 2005, 14, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Kaya, G.; Tran, C.; Sorg, O.; Hotz, R.; Grand, D.; Carraux, P.; Didierjean, L.; Stamenkovic, I.; Saurat, J.-H. Hyaluronate Fragments Reverse Skin Atrophy by a CD44-Dependent Mechanism. PLoS Med. 2006, 3, e493. [Google Scholar] [CrossRef]
- Makrantonaki, E.; Zouboulis, C.C.; William, J. Cunliffe Scientific Awards. Characteristics and pathomechanisms of endogenously aged skin. Dermatology 2007, 214, 352–360. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Lovell, C.R.; Smolenski, K.A.; Duance, V.C.; Light, N.D.; Young, S.; Dyson, M. Type I and III collagen content and fibre distribution in normal human skin during ageing. Br. J. Dermatol. 1987, 117, 419–428. [Google Scholar] [CrossRef]
- Autio, P.; Risteli, J.; Haukipuro, K.; Risteli, L.; Oikarinen, A. Collagen synthesis in human skin in vivo: Modulation by aging, ultraviolet B irradiation and localization. Photodermatol. Photoimmunol. Photomed. 1994, 10, 212–216. [Google Scholar] [PubMed]
- Braverman, I.M.; Fonferko, E. Studies in cutaneous aging: I. The elastic fiber network. J. Investig. Dermatol. 1982, 78, 434–443. [Google Scholar] [CrossRef]
- Dawber, R.; Shuster, S.A.M. Scanning electron microscopy of dermal fibrous tissue networks in normal skin, solar elastosis and pseud0-xanth0ma elasticum. Br. J. Dermatol. 1971, 84, 130–134. [Google Scholar] [CrossRef]
- Kurban, R.S.; Kurban, A.K. Common skin disorders of aging: Diagnosis and treatment. Geriatrics 1993, 48, 30–31, 35–36, 39–42. [Google Scholar] [PubMed]
- Ressler, S.; Bartkova, J.; Niederegger, H.; Bartek, J.; Scharffetter-Kochanek, K.; Jansen-Dürr, P.; Wlaschek, M. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 2006, 5, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Waaijer, M.E.C.; Parish, W.E.; Strongitharm, B.H.; van Heemst, D.; Slagboom, P.E.; de Craen, A.J.M.; Sedivy, J.M.; Westendorp, R.G.J.; Gunn, D.A.; Maier, A.B. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell 2012, 11, 722–725. [Google Scholar] [CrossRef]
- Ghosh, K.; Capell, B.C. The Senescence-Associated Secretory Phenotype: Critical Effector in Skin Cancer and Aging. J. Investig. Dermatol. 2016, 136, 2133–2139. [Google Scholar] [CrossRef]
- Lee, Y.I.; Choi, S.; Roh, W.S.; Lee, J.H.; Kim, T.G. Cellular senescence and inflammaging in the skin microenvironment. Int. J. Mol. Sci. 2021, 22, 3849. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Birch-Machin, M.A.; Tindall, M.; Turner, R.; Haldane, F.; Rees, J.L. Mitochondrial DNA deletions in human skin reflect photo- rather than chronologic aging. J. Investig. Dermatol. 1998, 110, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. The role of cellular senescence in skin aging. J. Investig. Dermatol. Symp. Proc. 1998, 3, 1–5. [Google Scholar] [PubMed]
- Victorelli, S.; Lagnado, A.; Halim, J.; Moore, W.; Talbot, D.; Barrett, K.; Chapman, J.; Birch, J.; Ogrodnik, M.; Meves, A.; et al. Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. EMBO J. 2019, 38, e101982. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; Dimri, G.P.; Hara, E. Control of replicative senescence. In Handbook of the Biology of Aging; Academic Press: New York, NY, USA, 1996; pp. 121–149. [Google Scholar]
- McCart, E.A.; Thangapazham, R.L.; Lombardini, E.D.; Mog, S.R.; Panganiban, R.A.M.; Dickson, K.M.; Mansur, R.A.; Nagy, V.; Kim, S.-Y.; Selwyn, R.; et al. Accelerated senescence in skin in a murine model of radiation-induced multi-organ injury. J. Radiat. Res. 2017, 58, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Yi, Q.; Travers, J.B.; Spandau, D.F. UVB-induced Senescence in Human Keratinocytes Requires a Functional Insulin-like Growth Factor-1 Receptor and p53. Mol. Biol. Cell 2008, 19, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.S.; Ong, P.F.; Chojnowski, A.; Clavel, C.; Dreesen, O. Loss of lamin B1 is a biomarker to quantify cellular senescence in photoaged skin. Sci. Rep. 2017, 7, 15678. [Google Scholar] [CrossRef] [PubMed]
- Medrano, E.E.; Im, S.; Yang, F.; Abdel-Malek, Z.A. Ultraviolet B light induces G1 arrest in human melanocytes by prolonged inhibition of retinoblastoma protein phosphorylation associated with long-term expression of the p21Waf-1/SDI-1/Cip-1 protein. Cancer Res. 1995, 55, 4047–4052. [Google Scholar]
- Bent, E.H.; Gilbert, L.A.; Hemann, M.T. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev. 2016, 30, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Althubiti, M.; Lezina, L.; Carrera, S.; Jukes-Jones, R.; Giblett, S.M.; Antonov, A.; Barlev, N.; Saldanha, G.S.; Pritchard, C.A.; Cain, K.; et al. Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis. 2014, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Brégégère, F.; Soroka, Y.; Bismuth, J.; Friguet, B.; Milner, Y. Cellular senescence in human keratinocytes: Unchanged proteolytic capacity and increased protein load. Exp. Gerontol. 2003, 38, 619–629. [Google Scholar] [CrossRef]
- Martic, I.; Wedel, S.; Jansen-Dürr, P.; Cavinato, M. A new model to investigate UVB-induced cellular senescence and pigmentation in melanocytes. Mech. Ageing Dev. 2020, 190, 111322. [Google Scholar] [CrossRef]
- Mehta, I.S.; Figgitt, M.; Clements, C.S.; Kill, I.R.; Bridger, J.M. Alterations to nuclear architecture and genome behavior in senescent cells. Ann. N. Y. Acad. Sci. 2007, 1100, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.; Yoon, G.; Kang, H.T. A comparative analysis of the cell biology of senescence and aging. Cell. Mol. Life Sci. 2009, 66, 2503–2524. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Ott, C.; Hugo, M.; Jung, T.; Bulteau, A.-L.; Grune, T.; Höhn, A. Mitochondrial contribution to lipofuscin formation. Redox Biol. 2017, 11, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Robbins, E.; Levine, E.M.; Eagle, H. Morphologic changes accompanying senescence of cultured human diploid cells. J. Exp. Med. 1970, 131, 1211–1222. [Google Scholar] [CrossRef]
- Choi, S.-Y.; Bin, B.-H.; Kim, W.; Lee, E.; Lee, T.R.; Cho, E.-G. Exposure of human melanocytes to UVB twice and subsequent incubation leads to cellular senescence and senescence-associated pigmentation through the prolonged p53 expression. J. Dermatol. Sci. 2018, 90, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulou, E.; Tsimaratou, K.; Evangelou, K.; Fernandez Marcos, P.; Zoumpourlis, V.; Trougakos, I.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V. Specific lipofuscin staining as a novel biomarker to detect replicative and stress—Induced senescence. A method applicable in cryo—preserved and archival tissues. Aging (Albany NY) 2013, 5, 37–50. [Google Scholar] [CrossRef]
- Cho, S.; Hwang, E.S. Status of mTOR activity may phenotypically differentiate senescence and quiescence. Mol. Cells 2012, 33, 597–604. [Google Scholar] [CrossRef]
- Gianfranceschi, G.; Caragnano, A.; Piazza, S.; Manini, I.; Ciani, Y.; Verardo, R.; Toffoletto, B.; Finato, N.; Livi, U.; Beltrami, C.A.; et al. Critical role of lysosomes in the dysfunction of human Cardiac Stem Cells obtained from failing hearts. Int. J. Cardiol. 2016, 216, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Rizou, S.V.; Evangelou, K.; Myrianthopoulos, V.; Mourouzis, I.; Havaki, S.; Athanasiou, A.; Vasileiou, P.V.S.; Margetis, A.; Kotsinas, A.; Kastrinakis, N.G.; et al. A novel quantitative method for the detection of lipofuscin, the main by-product of cellular senescence, in fluids. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2019; Volume 1896, pp. 119–138. ISBN 9781493989317. [Google Scholar]
- Terman, A. Garbage catastrophe theory of aging: Imperfect removal of oxidative damage? Redox Rep. 2001, 6, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T.; Nilsson, E.; Döcke, W.D.; Brunk, U.T. Lipofuscin accumulation and ageing of fibroblasts. Gerontology 1995, 41 (Suppl. S2), 95–108. [Google Scholar] [CrossRef] [PubMed]
- Rübe, C.E.; Bäumert, C.; Schuler, N.; Isermann, A.; Schmal, Z.; Glanemann, M.; Mann, C.; Scherthan, H. Human skin aging is associated with increased expression of the histone variant H2A.J in the epidermis. NPJ Aging Mech. Dis. 2021, 7, 7. [Google Scholar] [CrossRef]
- Tonolli, P.N.; Baptista, M.S.; Chiarelli-Neto, O. Melanin, lipofuscin and the effects of visible light in the skin. J. Photochem. Photobiol. 2021, 7, 100044. [Google Scholar] [CrossRef]
- Gary, R.K.; Kindell, S.M. Quantitative assay of senescence-associated β-galactosidase activity in mammalian cell extracts. Anal. Biochem. 2005, 343, 329–334. [Google Scholar] [CrossRef]
- Holt, D.J.; Grainger, D.W. Senescence and quiescence induced compromised function in cultured macrophages. Biomaterials 2012, 33, 7497–7507. [Google Scholar] [CrossRef] [PubMed]
- Severino, J.; Allen, R.G.; Balin, S.; Balin, A.; Cristofalo, V.J. Is beta-galactosidase staining a marker of senescence in vitro and in vivo? Exp. Cell Res. 2000, 257, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, J.S. Cellular senescence: A promising strategy for cancer therapy. BMB Rep. 2019, 52, 35–41. [Google Scholar] [CrossRef]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Maier, A.B.; Westendorp, R.G.J.; Van Heemst, D. β-Galactosidase Activity as a Biomarker of Replicative Senescence during the Course of Human Fibroblast Cultures. Ann. N. Y. Acad. Sci. 2007, 1100, 323–332. [Google Scholar] [CrossRef]
- Tsai, C.-W.; Chiang, I.-N.; Wang, J.-H.; Young, T.-H. Chitosan delaying human fibroblast senescence through downregulation of TGF-β signaling pathway. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1852–1863. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Itzkovitz, S.; Cohen, A.; Rutenberg, A.; Berkovitz, R.; Yehezkel, S.; Shahar, H.; Selig, S.; Skorecki, K. Quantitative digital in situ senescence-associated β-galactosidase assay. BMC Cell Biol. 2011, 12, 16. [Google Scholar] [CrossRef]
- Zorin, V.; Zorina, A.; Smetanina, N.; Kopnin, P.; Ozerov, I.V.; Leonov, S.; Isaev, A.; Klokov, D.; Osipov, A.N. Diffuse colonies of human skin fibroblasts in relation to cellular senescence and proliferation. Aging (Albany NY) 2017, 9, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Wattanapitayakul, S.K.; Chularojmontri, L.; Schäfer-Korting, M. Ultraviolet B irradiation-induced keratinocyte senescence and impaired development of 3D epidermal reconstruct. Acta Pharm. 2021, 71, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, K.Y.; Lee, M.-K.; Jin, T.; Seo, S.J. Correction: Adiponectin Suppresses UVB-Induced Premature Senescence and hBD2 Overexpression in Human Keratinocytes. PLoS ONE 2016, 11, e0162738. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.-C.; Yang, J.-P.; Lee, J.-S.; Jeong, S.-H.; Dhong, E.-S.; Han, S.-K. Effects of Ultraviolet Irradiation on Cellular Senescence in Keratinocytes Versus Fibroblasts. J. Craniofac. Surg. 2019, 30, 270–275. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Leduc, C.; Verbeke, A.; Toussaint, O. UV, stress and aging. Dermato-Endocrinol. 2012, 4, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.Y.; Zhang, C.L.; Liu, X.C.; Qian, G.; Deng, D.Q. Effects of cigarette smoke extracts on the growth and senescence of skin fibroblasts In Vitro. Int. J. Biol. Sci. 2013, 9, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Kanaji, N.; Basma, H.; Nelson, A.; Farid, M.; Sato, T.; Nakanishi, M.; Wang, X.; Michalski, J.; Li, Y.; Gunji, Y.; et al. Fibroblasts that resist cigarette smoke-induced senescence acquire profibrotic phenotypes. Am. J. Physiol. Cell. Mol. Physiol. 2014, 307, L364–L373. [Google Scholar] [CrossRef] [PubMed]
- Loseva, O.; Shubbar, E.; Haghdoost, S.; Evers, B.; Helleday, T.; Harms-Ringdahl, M. Chronic low dose rate ionizing radiation exposure induces premature senescence in human fibroblasts that correlates with up regulation of proteins involved in protection against oxidative stress. Proteomes 2014, 2, 341–362. [Google Scholar] [CrossRef]
- Zdanov, S.; Remacle, J.; Toussaint, O. Establishment of H2O2-Induced Premature Senescence in Human Fibroblasts Concomitant with Increased Cellular Production of H2O2. Ann. N. Y. Acad. Sci. 2006, 1067, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Duan, J.; Zhang, Z.; Tong, T. Irreversible cellular senescence induced by prolonged exposure to H2O2 involves DNA-damage-and-repair genes and telomere shortening. Int. J. Biochem. Cell Biol. 2005, 37, 1407–1420. [Google Scholar] [CrossRef]
- Ido, Y.; Duranton, A.; Lan, F.; Weikel, K.A.; Breton, L.; Ruderman, N.B. Resveratrol Prevents Oxidative Stress-Induced Senescence and Proliferative Dysfunction by Activating the AMPK-FOXO3 Cascade in Cultured Primary Human Keratinocytes. PLoS ONE 2015, 10, e0115341. [Google Scholar] [CrossRef]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef]
- Yoon, Y.-S.; Yoon, D.-S.; Lim, I.K.; Yoon, S.-H.; Chung, H.-Y.; Rojo, M.; Malka, F.; Jou, M.-J.; Martinou, J.-C.; Yoon, G. Formation of elongated giant mitochondria in DFO-induced cellular senescence: Involvement of enhanced fusion process through modulation of Fis1. J. Cell. Physiol. 2006, 209, 468–480. [Google Scholar] [CrossRef]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef]
- Studencka, M.; Schaber, J. Senoptosis: Non-lethal DNA cleavage as a route to deep senescence. Oncotarget 2017, 8, 30656–30671. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Passos, J.F. Mitochondria: Are they causal players in cellular senescence? Biochim. Biophys. Acta-Bioenerg. 2015, 1847, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Dalle Pezze, P.; Nelson, G.; Otten, E.G.; Korolchuk, V.I.; Kirkwood, T.B.L.; von Zglinicki, T.; Shanley, D.P. Dynamic Modelling of Pathways to Cellular Senescence Reveals Strategies for Targeted Interventions. PLoS Comput. Biol. 2014, 10, e1003728. [Google Scholar] [CrossRef] [PubMed]
- Victorelli, S.; Passos, J.F. Reactive oxygen species detection in senescent cells. In Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1896, pp. 21–29. ISBN 9781493989317. [Google Scholar]
- Berneburg, M.; Gattermann, N.; Stege, H.; Grewe, M.; Vogelsang, K.; Ruzicka, T.; Krutmann, J. Chronically Ultraviolet-exposed Human Skin Shows a Higher Mutation Frequency of Mitochondrial DNA as Compared to Unexposed Skin and the Hematopoietic System. Photochem. Photobiol. 1997, 66, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Berneburg, M.; Krutmann, J. Mitochondrial DNA Deletions in Human Skin Reflect Photo-Rather than Chronologic Aging. J. Investig. Dermatol. 1998, 111, 709–710. [Google Scholar] [CrossRef]
- Berneburg, M.; Plettenberg, H.; Medve-König, K.; Pfahlberg, A.; Gers-Barlag, H.; Gefeller, O.; Krutmann, J. Induction of the Photoaging-Associated Mitochondrial Common Deletion In Vivo in Normal Human Skin. J. Investig. Dermatol. 2004, 122, 1277–1283. [Google Scholar] [CrossRef]
- Berneburg, M.; Plettenberg, H.; Krutmann, J. Photoaging of human skin. Photodermatol. Photoimmunol. Photomed. 2000, 16, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Mishima, Y.; Shimizu, M.; Suetake, I.; Takada, S. Interactions of HP1 Bound to H3K9me3 Dinucleosome by Molecular Simulations and Biochemical Assays. Biophys. J. 2018, 114, 2336–2351. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Dreesen, O.; Chojnowski, A.; Ong, P.F.; Zhao, T.Y.; Common, J.E.; Lunny, D.; Lane, E.B.; Lee, S.J.; Vardy, L.A.; Stewart, C.L.; et al. Lamin B1 fluctuations have differential effects on cellular proliferation and senescence. J. Cell Biol. 2013, 200, 605–617. [Google Scholar] [CrossRef]
- Sadaie, M.; Salama, R.; Carroll, T.; Tomimatsu, K.; Chandra, T.; Young, A.R.J.; Narita, M.; Pérez-Mancera, P.A.; Bennett, D.C.; Chong, H.; et al. Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev. 2013, 27, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Chandra, T.; Kirschner, K. Chromosome organisation during ageing and senescence. Curr. Opin. Cell Biol. 2016, 40, 161–167. [Google Scholar] [CrossRef]
- Freund, A.; Laberge, R.-M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Burd, C.E.; Sorrentino, J.A.; Clark, K.S.; Darr, D.B.; Krishnamurthy, J.; Deal, A.M.; Bardeesy, N.; Castrillon, D.H.; Beach, D.H.; Sharpless, N.E. Monitoring Tumorigenesis and Senescence In Vivo with a p16INK4a-Luciferase Model. Cell 2013, 152, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.T.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Wang, X.; Feng, Y.; Pan, L.; Wang, Y.; Xu, X.; Lu, J.; Huang, B. The proximal GC-rich region of p16INK4a gene promoter plays a role in its transcriptional regulation. Mol. Cell. Biochem. 2007, 301, 259–266. [Google Scholar] [CrossRef]
- Pan, K.; Chen, Y.; Roth, M.; Wang, W.; Wang, S.; Yee, A.S.; Zhang, X. HBP1-mediated transcriptional regulation of DNA methyltransferase 1 and its impact on cell senescence. Mol. Cell. Biol. 2013, 33, 887–903. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, W.; Sheng, L.; Zou, C.; Sun, H.; Zhang, C.; Liu, Y.; Shi, J.; Ma, E.; Yuan, L. Design, synthesis and biological evaluation of novel asperphenamate derivatives. Eur. J. Med. Chem. 2016, 110, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.-S.; Qian, Y.; Chen, X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell. Signal. 2010, 22, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Tan, D.; Wang, X.; Han, S.; Tan, J.; Zhao, Y.; Lu, J.; Huang, B. Histone deacetylase 3 represses p15INK4b and p21WAF1/cip1 transcription by interacting with Sp1. Biochem. Biophys. Res. Commun. 2006, 339, 165–171. [Google Scholar] [CrossRef]
- Koo, B.-H.; Kim, Y.; Je Cho, Y.; Kim, D.-S. Distinct roles of transforming growth factor-β signaling and transforming growth factor-β receptor inhibitor SB431542 in the regulation of p21 expression. Eur. J. Pharmacol. 2015, 764, 413–423. [Google Scholar] [CrossRef]
- Borrás, C.; Gómez-Cabrera, M.C.; Viña, J. The dual role of p53: DNA protection and antioxidant. Free Radic. Res. 2011, 45, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Rayess, H.; Wang, B.; Marilene, S.S.E. Cellular senescence and tumor supressor gene p 16. Int. J. Cancer 2013, 130, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Bringold, F.; Serrano, M. Tumor suppressors and oncogenes in cellular senescence. Exp. Gerontol. 2000, 35, 317–329. [Google Scholar] [CrossRef]
- Nassour, J.; Abbadie, C. A novel role for DNA single-strand breaks in senescence and neoplastic escape of epithelial cells. Mol. Cell. Oncol. 2016, 3, e1190885. [Google Scholar] [CrossRef]
- Ryu, Y.S.; Kang, K.A.; Piao, M.J.; Ahn, M.J.; Yi, J.M.; Bossis, G.; Hyun, Y.-M.; Park, C.O.; Hyun, J.W. Particulate matter-induced senescence of skin keratinocytes involves oxidative stress-dependent epigenetic modifications. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-Y.; Souroullas, G.P.; Diekman, B.O.; Krishnamurthy, J.; Hall, B.M.; Sorrentino, J.A.; Parker, J.S.; Sessions, G.A.; Gudkov, A.V.; Sharpless, N.E. Cells exhibiting strong p16 INK4 promoter activation in vivo display features of senescence. Proc. Natl. Acad. Sci. USA 2019, 116, 2603–2611. [Google Scholar] [CrossRef]
- Gervason, S.; Napoli, M.; Dreux-Zhiga, A.; Lazzarelli, C.; Garcier, S.; Briand, A.; Albouy, M.; Thepot, A.; Berthon, J.Y.; Filaire, E. Attenuation of negative effects of senescence in human skin using an extract from Sphingomonas hydrophobicum: Development of new skin care solution. Int. J. Cosmet. Sci. 2019, 41, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; Van Der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Satgunaseelan, L.; Chia, N.; Suh, H.; Virk, S.; Ashford, B.; Lum, T.; Ranson, M.; Clark, J.; Gupta, R. p16 expression in cutaneous squamous cell carcinoma of the head and neck is not associated with integration of high risk HPV DNA or prognosis. Pathology 2017, 49, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Eshkoor, S.A.; Ismail, P.; Rahman, S.A.; Oshkour, S.A. p16 gene expression in basal cell carcinoma. Arch. Med. Res. 2008, 39, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Kang, J.; Xia, J.; Li, Y.; Yang, B.; Chen, B.; Sun, W.; Song, X.; Xiang, W.; Wang, X.; et al. p53-related apoptosis resistance and tumor suppression activity in UVB-induced premature senescent human skin fibroblasts. Int. J. Mol. Med. 2008, 21, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Marthandan, S.; Menzel, U.; Priebe, S.; Groth, M.; Guthke, R.; Platzer, M.; Hemmerich, P.; Kaether, C.; Diekmann, S. Conserved genes and pathways in primary human fibroblast strains undergoing replicative and radiation induced senescence. Biol. Res. 2016, 49, 34. [Google Scholar] [CrossRef]
- Miyake, T.; Shimada, M.; Matsumoto, Y.; Okino, A. DNA Damage Response After Ionizing Radiation Exposure in Skin Keratinocytes Derived from Human-Induced Pluripotent Stem Cells. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Peters, G. Regulation of the INK4b–ARF–INK4a tumour suppressor locus: All for one or one for all. Nat. Rev. Mol. Cell Biol. 2006, 7, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, N.C.; Liu, T.; Cassidy, P.; Leachman, S.A.; Boucher, K.M.; Goodson, A.G.; Samadashwily, G.; Grossman, D. The p16INK4A tumor suppressor regulates cellular oxidative stress. Oncogene 2011, 30, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Waaijer, M.E.C.; Gunn, D.A.; Adams, P.D.; Pawlikowski, J.S.; Griffiths, C.E.M.; van Heemst, D.; Slagboom, P.E.; Westendorp, R.G.J.; Maier, A.B. P16INK4a Positive Cells in Human Skin Are Indicative of Local Elastic Fiber Morphology, Facial Wrinkling, and Perceived Age. J. Gerontol. Ser. A 2016, 71, 1022–1028. [Google Scholar] [CrossRef]
- Toussaint, O.; Medrano, E.E.; Von Zglinicki, T. Cellular and molecular mechanisms of stress-induced premature senescence (SIPS) of human diploid fibroblasts and melanocytes. Exp. Gerontol. 2000, 35, 927–945. [Google Scholar] [CrossRef]
- Singh, K.; Maity, P.; Krug, L.; Meyer, P.; Treiber, N.; Lucas, T.; Basu, A.; Kochanek, S.; Wlaschek, M.; Geiger, H.; et al. Superoxide anion radicals induce IGF-1 resistance through concomitant activation of PTP1B and PTEN. EMBO Mol. Med. 2015, 7, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.G.; Spandau, D.F.; Travers, J.B. Impact of Age and Insulin-Like Growth Factor-1 on DNA Damage Responses in UV-Irradiated Human Skin. Molecules 2017, 22, 356. [Google Scholar] [CrossRef]
- Li, J.-F.; Duan, H.-F.; Wu, C.-T.; Zhang, D.-J.; Deng, Y.; Yin, H.-L.; Han, B.; Gong, H.-C.; Wang, H.-W.; Wang, Y.-L. HGF accelerates wound healing by promoting the dedifferentiation of epidermal cells through-integrin/ILK pathway. Biomed. Res. Int. 2013, 2013, 470418. [Google Scholar] [CrossRef]
- Stagno, F.; Guglielmo, P.; Consoli, U.; Fiumara, P.; Russo, M.; Giustolisi, R. Successful Healing of Hydroxyurea-Related Leg Ulcers With Topical Granulocyte-Macrophage Colony-Stimulating Factor. Blood 1999, 94, 1479–1480. [Google Scholar] [CrossRef]
- Hirobe, T.; Furuya, R.; Hara, E.; Horii, I.; Tsunenaga, M.; Ifuku, O. Granulocyte-macrophage colony-stimulating factor (GM-CSF) controls the proliferation and differentiation of mouse epidermal melanocytes from pigmented spots induced by ultraviolet radiation B. Pigment Cell Res. 2004, 17, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Liu, C.; Chen, Z.; Blatchley, M.R.; Kim, D.; Zhou, J.; Xu, M.; Gerecht, S.; Fan, R. Senescent Cells with Augmented Cytokine Production for Microvascular Bioengineering and Tissue Repairs. Adv. Biosyst. 2019, 3, 1900089. [Google Scholar] [CrossRef]
- Hausmann, C.; Zoschke, C.; Wolff, C.; Darvin, M.E.; Sochorová, M.; Kováčik, A.; Wanjiku, B.; Schumacher, F.; Tigges, J.; Kleuser, B.; et al. Fibroblast origin shapes tissue homeostasis, epidermal differentiation, and drug uptake. Sci. Rep. 2019, 9, 2913. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.G.; Pereira-Smith, O.M. p53 Is Preferentially Recruited to the Promoters of Growth Arrest Genes p21 and GADD45 during Replicative Senescence of Normal Human Fibroblasts. Cancer Res. 2006, 66, 8356–8360. [Google Scholar] [CrossRef] [PubMed]
- Gilhar, A.; Ullmann, Y.; Karry, R.; Shalaginov, R.; Assy, B.; Serafimovich, S.; Kalish, R.S. Aging of Human Epidermis: Reversal of Aging Changes Correlates With Reversal of Keratinocyte Fas Expression and Apoptosis. J. Gerontol. Ser. A 2004, 59, B411–B415. [Google Scholar] [CrossRef] [PubMed]
- Morales-Ducret, C.J.; van de Rijn, M.; Smoller, B.R. Bcl-2 Expression in Melanocytic Nevi. Arch. Dermatol. Res. 1995, 131, 915–918. [Google Scholar] [CrossRef]
- Arck, P.C.; Overall, R.; Spatz, K.; Liezman, C.; Handjiski, B.; Klapp, B.F.; Birch-Machin, M.A.; Peters, E.M.; Arck, P.C.; Overall, R. Towards a “free radical theory of graying”: Melanocyte apoptosis in the aging human hair follicle is an indicator of oxidative stress induced tissue damage. FASEB J. 2006, 20, 1567–1569. [Google Scholar] [CrossRef]
- Haake, A.R.; Roublevskaia, I.; Cooklis, M. Apoptosis: A role in skin aging? J. Investig. Dermatol. Symp. Proc. 1998, 3, 28–35. [Google Scholar] [CrossRef]
- Haratake, A.; Uchida, Y.; Mimura, K.; Elias, P.M.; Holleran, W.M. Intrinsically aged epidermis displays diminished UVB-induced alterations in barrier function associated with decreased proliferation. J. Investig. Dermatol. 1997, 108, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Rochette, P.J.; Brash, D.E. Progressive apoptosis resistance prior to senescence and control by the anti-apoptotic protein BCL-xL. Mech. Ageing Dev. 2008, 129, 207–214. [Google Scholar] [CrossRef]
- Smalley, K.S.M.; Sondak, V.K.; Weber, J.S. c-KIT signaling as the driving oncogenic event in sub-groups of melanomas. Histol. Histopathol. 2009, 24, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Luca, M.; Gutman, M.; McConkey, D.J.; Langley, K.E.; Lyman, S.D.; Bar-Eli, M. Enforced c-KIT expression renders highly metastatic human melanoma cells susceptible to stem cell factor-induced apoptosis and inhibits their tumorigenic and metastatic potential. Oncogene 1996, 13, 2339–2347. [Google Scholar] [PubMed]
- Bhawan, J.; Andersen, W.; Lee, J.; Labadie, R.; Solares, G. Photoaging versus intrinsic aging: A morphologic assessment of facial skin*. J. Cutan. Pathol. 1995, 22, 154–159. [Google Scholar] [CrossRef]
- Han, A.; Chien, A.L.; Kang, S. Photoaging. Dermatol. Clin. 2014, 32, 291–299. [Google Scholar] [CrossRef]
- Gniadecki, R.; Hansen, M.; Wulf, H.C. Resistance of senescent keratinocytes to UV-induced apoptosis. Cell. Mol. Biol. 2000, 46, 121–127. [Google Scholar] [CrossRef]
- Chaturvedi, V.; Qin, J.-Z.; Stennett, L.; Choubey, D.; Nickoloff, B.J. Resistance to UV-induced apoptosis in human keratinocytes during accelerated senescence is associated with functional inactivation of p53. J. Cell. Physiol. 2004, 198, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Qin, Z.; Xia, W.; Shao, Y.; Voorhees, J.J.; Fisher, G.J. Matrix-Degrading Metalloproteinases in Photoaging. J. Investig. Dermatol. Symp. Proc. 2009, 14, 20–24. [Google Scholar] [CrossRef]
- Andrei, S.; Vera, G.; Ayellet, F.; Alex, S.; Michael, M.; Irit, Z.; Galit, S.; Naomi, G.; Varda, R. Change of the Death Pathway in Senescent Human Fibroblasts in Response to DNA Damage Is Caused by an Inability To Stabilize p53. Mol. Cell. Biol. 2001, 21, 1552–1564. [Google Scholar] [CrossRef]
- Tepper, C.G.; Seldin, M.F.; Mudryj, M. Fas-Mediated Apoptosis of Proliferating, Transiently Growth-Arrested, and Senescent Normal Human Fibroblasts. Exp. Cell Res. 2000, 260, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Yeo, E.J.; Hwang, Y.C.; Kang, C.M.; Choy, H.E.; Park, S.C. Reduction of UV-induced cell death in the human senescent fibroblasts. Mol. Cells 2000, 10, 415–422. [Google Scholar] [PubMed]
- Chen, Q.M.; Liu, J.; Merrett, J.B. Apoptosis or senescence-like growth arrest: Influence of cell-cycle position, p53, p21 and bax in H2O2 response of normal human fibroblasts. Biochem. J. 2000, 347, 543–551. [Google Scholar] [CrossRef]
- Wang, E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 1995, 55, 2284–2292. [Google Scholar] [PubMed]
- Marcotte, R.; Lacelle, C.; Wang, E. Senescent fibroblasts resist apoptosis by downregulating caspase-3. Mech. Ageing Dev. 2004, 125, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Papaconstantinou, J. Unifying Model of the Programmed (Intrinsic) and Stochastic (Extrinsic) Theories of Aging. Ann. N. Y. Acad. Sci. 1994, 719, 195–211. [Google Scholar] [CrossRef]
- Chaturvedi, V.; Qin, J.-Z.; Denning, M.F.; Choubey, D.; Diaz, M.O.; Nickoloff, B.J. Apoptosis in Proliferating, Senescent, and Immortalized Keratinocytes. J. Biol. Chem. 1999, 274, 23358–23367. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, X.; Lei, L.; Sun, L.; Jiao, A.; Zhu, K.; Xie, T.; Liu, H.; Zhang, X.; Su, Y.; et al. Age-Related Gene Alteration in Naïve and Memory T cells Using Precise Age-Tracking Model. Front. Cell Dev. Biol. 2021, 8, 1901. [Google Scholar] [CrossRef] [PubMed]
- Vukmanovic-Stejic, M.; Sandhu, D.; Seidel, J.A.; Patel, N.; Sobande, T.O.; Agius, E.; Jackson, S.E.; Fuentes-Duculan, J.; Suárez-Fariñas, M.; Mabbott, N.A.; et al. The Characterization of Varicella Zoster Virus–Specific T Cells in Skin and Blood during Aging. J. Investig. Dermatol. 2015, 135, 1752–1762. [Google Scholar] [CrossRef]
- Koguchi-Yoshioka, H.; Hoffer, E.; Cheuk, S.; Matsumura, Y.; Vo, S.; Kjellman, P.; Grema, L.; Ishitsuka, Y.; Nakamura, Y.; Okiyama, N.; et al. Skin T cells maintain their diversity and functionality in the elderly. Commun. Biol. 2021, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Friedman, K.M.; Prieto, P.A.; Devillier, L.E.; Gross, C.A.; Yang, J.C.; Wunderlich, J.R.; Rosenberg, S.A.; Dudley, M.E. Tumor-specific CD4+ Melanoma Tumor-infiltrating Lymphocytes. J. Immunother. 2012, 35, 400. [Google Scholar] [CrossRef]
- Pilkington, S.M.; Bulfone-Paus, S.; Griffiths, C.E.M.; Watson, R.E.B. Inflammaging and the Skin. J. Investig. Dermatol. 2021, 141, 1087–1095. [Google Scholar] [CrossRef]
- Neves, J.; Demaria, M.; Campisi, J.; Jasper, H. Of Flies, Mice, and Men: Evolutionarily Conserved Tissue Damage Responses and Aging. Dev. Cell 2015, 32, 9–18. [Google Scholar] [CrossRef]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF Induces Senescence and Apoptosis through Pathways Mediated by the Secreted Protein IGFBP7. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.W.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 Suppresses the Senescence-Associated Secretory Phenotype through Epigenetic Gene Regulation. PLoS ONE 2015, 10, e0116480. [Google Scholar] [CrossRef] [PubMed]
- Warnon, C.; Bouhjar, K.; Ninane, N.; Verhoyen, M.; Fattaccioli, A.; Fransolet, M.; de Rouvroit, C.L.; Poumay, Y.; Piel, G.; Mottet, D.; et al. HDAC2 and 7 down-regulation induces senescence in dermal fibroblasts. Aging (Albany NY) 2021, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.-M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef]
- Alimbetov, D.; Davis, T.; Brook, A.J.C.; Cox, L.S.; Faragher, R.G.A.; Nurgozhin, T.; Zhumadilov, Z.; Kipling, D. Suppression of the senescence-associated secretory phenotype (SASP) in human fibroblasts using small molecule inhibitors of p38 MAP kinase and MK2. Biogerontology 2016, 17, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Tiedje, C.; Ronkina, N.; Tehrani, M.; Dhamija, S.; Laass, K.; Holtmann, H.; Kotlyarov, A.; Gaestel, M. The p38/MK2-Driven Exchange between Tristetraprolin and HuR Regulates AU–Rich Element–Dependent Translation. PLoS Genet. 2012, 8, e1002977. [Google Scholar] [CrossRef] [PubMed]
- Niklander, S.; Bandaru, D.; Lambert, D.W.; Hunter, K.D. ROCK inhibition modulates the senescence-associated secretory phenotype (SASP) in oral keratinocytes. FEBS Open Bio. 2020, 10, 2740–2749. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.-J.; Kil, I.S.; Cho, E.-G. Extracellular Vesicles Derived from Senescent Fibroblasts Attenuate the Dermal Effect on Keratinocyte Differentiation. Int. J. Mol. Sci. 2020, 21, 1022. [Google Scholar] [CrossRef] [PubMed]
- Sha, J.; Arbesman, J.; Harter, M.L. Premature senescence in human melanocytes after exposure to solar UVR: An exosome and UV-miRNA connection. Pigment Cell Melanoma Res. 2020, 33, 671–684. [Google Scholar] [CrossRef]
- Narzt, M.-S.; Pils, V.; Kremslehner, C.; Nagelreiter, I.-M.; Schosserer, M.; Bessonova, E.; Bayer, A.; Reifschneider, R.; Terlecki-Zaniewicz, L.; Waidhofer-Söllner, P.; et al. Epilipidomics of Senescent Dermal Fibroblasts Identify Lysophosphatidylcholines as Pleiotropic Senescence-Associated Secretory Phenotype (SASP) Factors. J. Investig. Dermatol. 2021, 141, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.S.; Vukmanovic-Stejic, M.; Shih, B.B.; Trahair, H.; Subramanian, P.; Devine, O.P.; Glanville, J.; Gilroy, D.; Rustin, M.H.A.; Freeman, T.C.; et al. Recruitment of inflammatory monocytes by senescent fibroblasts inhibits antigen-specific tissue immunity during human aging. Nat. Aging 2021, 1, 101–113. [Google Scholar] [CrossRef]
- Kaur, A.; Ecker, B.L.; Douglass, S.M.; Kugel, C.H.; Webster, M.R.; Almeida, F.V.; Somasundaram, R.; Hayden, J.; Ban, E.; Ahmadzadeh, H.; et al. Remodeling of the collagen matrix in aging skin promotes melanoma metastasis and affects immune cell motility. Cancer Discov. 2019, 9, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Fisher, G.J. Role of Age-Associated Alterations of the Dermal Extracellular Matrix Microenvironment in Human Skin Aging: A Mini-Review. Gerontology 2015, 61, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.J.; Quan, T.; Purohit, T.; Shao, Y.; Cho, M.K.; He, T.; Varani, J.; Kang, S.; Voorhees, J.J. Collagen Fragmentation Promotes Oxidative Stress and Elevates Matrix Metalloproteinase-1 in Fibroblasts in Aged Human Skin. Am. J. Pathol. 2009, 174, 101–114. [Google Scholar] [CrossRef]
- Ezure, T.; Sugahara, M.; Amano, S. Senescent dermal fibroblasts negatively influence fibroblast extracellular matrix-related gene expression partly via secretion of complement factor D. BioFactors 2019, 45, 556–562. [Google Scholar] [CrossRef]
- Okazaki, M.; Yoshimura, K.; Uchida, G.; Harii, K. Correlation between age and the secretions of melanocyte-stimulating cytokines in cultured keratinocytes and fibroblasts. Br. J. Dermatol. Suppl. 2005, 153, 23–29. [Google Scholar] [CrossRef]
- Morita, A. Tobacco smoke causes premature skin aging. J. Dermatol. Sci. 2007, 48, 169–175. [Google Scholar] [CrossRef]
- Wlaschek, M.; Heinen, G.; Poswig, A.; Schwarz, A.; Krieg, T.; Scharffetter-Kochanek, K. UVA-induced autocrine stimulation of fibroblast-derived collagenase/mmp-1 by interrelated loops ofinterleukin–1 andinterleukin–6. Photochem. Photobiol. 1994, 59, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.K.; Damaghi, N.; Picart, S.D.; Markova, N.G.; Obayashi, K.; Okano, Y.; Masaki, H.; Grether-Beck, S.; Krutmann, J.; Smiles, K.A.; et al. UV-induced DNA damage initiates release of MMP-1 in human skin. Exp. Dermatol. 2008, 17, 1037–1044. [Google Scholar] [CrossRef]
- Bierman, J.C.; Laughlin, T.; Tamura, M.; Hulette, B.C.; Mack, C.E.; Sherrill, J.D.; Tan, C.Y.R.; Morenc, M.; Bellanger, S.; Oblong, J.E. Niacinamide mitigates SASP-related inflammation induced by environmental stressors in human epidermal keratinocytes and skin. Int. J. Cosmet. Sci. 2020, 42, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Wang, X.-J. TGFβ Signaling in Photoaging and UV-Induced Skin Cancer. J. Investig. Dermatol. 2021, 141, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Quan, T.; Shao, Y.; He, T.; Voorhees, J.J.; Fisher, G.J. Reduced Expression of Connective Tissue Growth Factor (CTGF/CCN2) Mediates Collagen Loss in Chronologically Aged Human Skin. J. Investig. Dermatol. 2010, 130, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Agius, E.; Lacy, K.E.; Vukmanovic-Stejic, M.; Jagger, A.L.; Papageorgiou, A.-P.; Hall, S.; Reed, J.R.; Curnow, S.J.; Fuentes-Duculan, J.; Buckley, C.D.; et al. Decreased TNF-α synthesis by macrophages restricts cutaneous immunosurveillance by memory CD4+ T cells during aging. J. Exp. Med. 2009, 206, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Taams, L.S.; van Amelsfort, J.M.R.; Tiemessen, M.M.; Jacobs, K.M.G.; de Jong, E.C.; Akbar, A.N.; Bijlsma, J.W.J.; Lafeber, F.P.J.G. Modulation of monocyte/macrophage function by human CD4+CD25+ regulatory T cells. Hum. Immunol. 2005, 66, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Tiemessen, M.M.; Jagger, A.L.; Evans, H.G.; van Herwijnen, M.J.C.; John, S.; Taams, L.S. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc. Natl. Acad. Sci. USA 2007, 104, 19446–19451. [Google Scholar] [CrossRef] [PubMed]
- Vukmanovic-Stejic, M.; Chambers, E.S.; Suárez-Fariñas, M.; Sandhu, D.; Fuentes-Duculan, J.; Patel, N.; Agius, E.; Lacy, K.E.; Turner, C.T.; Larbi, A.; et al. Enhancement of cutaneous immunity during aging by blocking p38 mitogen-activated protein (MAP) kinase–induced inflammation. J. Allergy Clin. Immunol. 2018, 142, 844–856. [Google Scholar] [CrossRef]
- Buchanan, J.P.; Peters, C.A.; Rasmussen, C.J.; Rothstein, G. Impaired expression of hematopoietic growth factors: A candidate mechanism for the hematopoietic defect of aging. Exp. Gerontol. 1996, 31, 135–144. [Google Scholar] [CrossRef]
- Pilkington, S.M.; Ogden, S.; Eaton, L.H.; Dearman, R.J.; Kimber, I.; Griffiths, C.E.M. Lower levels of interleukin-1β gene expression are associated with impaired Langerhans’ cell migration in aged human skin. Immunology 2018, 153, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Cumberbatch, M.; Dearman, R.J.; Kimber, I. Influence of ageing on Langerhans cell migration in mice: Identification of a putative deficiency of epidermal interleukin-1beta. Immunology 2002, 105, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.R.; Mälzer, J.N.; Domingo, C.; Jürchott, K.; Grützkau, A.; Babel, N.; Nienen, M.; Jelinek, T.; Niedrig, M.; Thiel, A. Low Thymic Activity and Dendritic Cell Numbers Are Associated with the Immune Response to Primary Viral Infection in Elderly Humans. J. Immunol. 2015, 195, 4699–4711. [Google Scholar] [CrossRef]
- Lee, H.-J.; Kim, T.-G.; Kim, S.H.; Park, J.Y.; Lee, M.; Lee, J.W.; Lee, S.H.; Lee, M.-G. Epidermal Barrier Function Is Impaired in Langerhans Cell-Depleted Mice. J. Investig. Dermatol. 2019, 139, 1182–1185. [Google Scholar] [CrossRef]
- Pilkington, S.M.; Dearman, R.J.; Kimber, I.; Griffiths, C.E.M. Langerhans cells express human β-defensin 3: Relevance for immunity during skin ageing. Br. J. Dermatol. 2018, 179, 1170–1171. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, G.R.; Almeida, P.P.; de Oliveira Santos, L.; Rodrigues, L.P.; de Carvalho, J.L.; Boroni, M. Hallmarks of Aging in Macrophages: Consequences to Skin Inflammaging. Cells 2021, 10, 1323. [Google Scholar] [CrossRef] [PubMed]
- Oblong, J.E.; Bowman, A.; Rovito, H.A.; Jarrold, B.B.; Sherrill, J.D.; Black, M.R.; Nelson, G.; Kimball, A.B.; Birch-Machin, M.A. Metabolic dysfunction in human skin: Restoration of mitochondrial integrity and metabolic output by nicotinamide (niacinamide) in primary dermal fibroblasts from older aged donors. Aging Cell 2020, 19, e13248. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.K.; Vousden, K.H. Metabolic regulation by p53. J. Mol. Med. 2011, 89, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Bittles, A.H.; Harper, N. Increased glycolysis in ageing cultured human diploid fibroblasts. Biosci. Rep. 1984, 4, 751–756. [Google Scholar] [CrossRef]
- Wiley, C.D.; Campisi, J. From Ancient Pathways to Aging Cells-Connecting Metabolism and Cellular Senescence. Cell Metab. 2016, 23, 1013–1021. [Google Scholar] [CrossRef]
- Windler, C.; Gey, C.; Seeger, K. Skin melanocytes and fibroblasts show different changes in choline metabolism during cellular senescence. Mech. Ageing Dev. 2017, 164, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.L.; Chin, T.; Tan, C.Y.R.; Rovito, H.A.; Quek, L.S.; Oblong, J.E.; Bellanger, S. Nicotinamide Metabolism Modulates the Proliferation/Differentiation Balance and Senescence of Human Primary Keratinocytes. J. Investig. Dermatol. 2019, 139, 1638–1647. [Google Scholar] [CrossRef] [PubMed]
- Prahl, S.; Kueper, T.; Biernoth, T.; Wöhrmann, Y.; Münster, A.; Fürstenau, M.; Schmidt, M.; Schulze, C.; Wittern, K.-P.; Wenck, H.; et al. Aging skin is functionally anaerobic: Importance of coenzyme Q10 for anti aging skin care. BioFactors 2008, 32, 245–255. [Google Scholar] [CrossRef]
- Kuehne, A.; Hildebrand, J.; Soehle, J.; Wenck, H.; Terstegen, L.; Gallinat, S.; Knott, A.; Winnefeld, M.; Zamboni, N. An integrative metabolomics and transcriptomics study to identify metabolic alterations in aged skin of humans in vivo. BMC Genom. 2017, 18, 169. [Google Scholar] [CrossRef] [PubMed]
- James, E.L.; Michalek, R.D.; Pitiyage, G.N.; de Castro, A.M.; Vignola, K.S.; Jones, J.; Mohney, R.P.; Karoly, E.D.; Prime, S.S.; Parkinson, E.K. Senescent Human Fibroblasts Show Increased Glycolysis and Redox Homeostasis with Extracellular Metabolomes That Overlap with Those of Irreparable DNA Damage, Aging, and Disease. J. Proteome Res. 2015, 14, 1854–1871. [Google Scholar] [CrossRef] [PubMed]
- Wijeyesekera, A.; Selman, C.; Barton, R.H.; Holmes, E.; Nicholson, J.K.; Withers, D.J. Metabotyping of Long-Lived Mice using 1H NMR Spectroscopy. J. Proteome Res. 2012, 11, 2224–2235. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Mancuso, A.; Wellen, K.E.; Yang, X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013, 493, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, S.; Tanaka, H.; Hino, S.; Nakatsu, Y.; Igata, T.; Sakamoto, A.; Narita, M.; Nakao, M. Retinoblastoma protein promotes oxidative phosphorylation through upregulation of glycolytic genes in oncogene-induced senescent cells. Aging Cell 2015, 14, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Marmisolle, I.; Martínez, J.; Liu, J.; Mastrogiovanni, M.; Fergusson, M.M.; Rovira, I.I.; Castro, L.; Trostchansky, A.; Moreno, M.; Cao, L.; et al. Reciprocal regulation of acetyl-CoA carboxylase 1 and senescence in human fibroblasts involves oxidant mediated p38 MAPK activation. Arch. Biochem. Biophys. 2017, 613, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Quijano, C.; Cao, L.; Fergusson, M.M.; Romero, H.; Liu, J.; Gutkind, S.; Rovira, I.I.; Mohney, R.P.; Karoly, E.D.; Finkel, T. Oncogene-induced senescence results in marked metabolic and bioenergetic alterations. Cell Cycle 2012, 11, 1383–1392. [Google Scholar] [CrossRef]
- Kaplon, J.; Zheng, L.; Meissl, K.; Chaneton, B.; Selivanov, V.A.; Mackay, G.; van der Burg, S.H.; Verdegaal, E.M.E.; Cascante, M.; Shlomi, T.; et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 2013, 498, 109–112. [Google Scholar] [CrossRef]
- Korovila, I.; Hugo, M.; Castro, J.P.; Weber, D.; Höhn, A.; Grune, T.; Jung, T. Proteostasis, oxidative stress and aging. Redox Biol. 2017, 13, 550–567. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Park, S.-H.; Hartl, F.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Sander, C.S.; Chang, H.; Salzmann, S.; Müller, C.S.L.; Ekanayake-Mudiyanselage, S.; Elsner, P.; Thiele, J.J. Photoaging is Associated with Protein Oxidation in Human Skin In Vivo. J. Investig. Dermatol. 2002, 118, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Beedholm, R.; Clark, B.F.C.; Rattan, S.I.S. Mild heat stress stimulates 20S proteasome and its 11S activator in human fibroblasts undergoing aging in vitro. Cell Stress Chaperones 2004, 9, 49–57. [Google Scholar] [CrossRef]
- Sabath, N.; Levy-Adam, F.; Younis, A.; Rozales, K.; Meller, A.; Hadar, S.; Soueid-Baumgarten, S.; Shalgi, R. Cellular proteostasis decline in human senescence. Proc. Natl. Acad. Sci. USA 2020, 117, 31902–31913. [Google Scholar] [CrossRef]
- Widmer, R.; Ziaja, I.; Grune, T. Protein oxidation and degradation during aging: Role in skin aging and neurodegeneration. Free Radic. Res. 2006, 40, 1259–1268. [Google Scholar] [CrossRef]
- Hwang, J.S.; Hwang, J.S.; Chang, I.; Kim, S. Age-Associated Decrease in Proteasome Content and Activities in Human Dermal Fibroblasts: Restoration of Normal Level of Proteasome Subunits Reduces Aging Markers in Fibroblasts From Elderly Persons. J. Gerontol. Ser. A 2007, 62, 490–499. [Google Scholar] [CrossRef]
- Chondrogianni, N.; Stratford, F.L.L.; Trougakos, I.P.; Friguet, B.; Rivett, A.J.; Gonos, E.S. Central Role of the Proteasome in Senescence and Survival of Human Fibroblasts: Induction of a senescence-like phenotype upon its inhibition and resistance to stress upon its activation. J. Biol. Chem. 2003, 278, 28026–28037. [Google Scholar] [CrossRef] [PubMed]
- Petropoulos, I.; Conconi, M.; Wang, X.; Hoenel, B.; Brégégère, F.; Milner, Y.; Friguet, B. Increase of Oxidatively Modified Protein Is Associated With a Decrease of Proteasome Activity and Content in Aging Epidermal Cells. J. Gerontol. Ser. A 2000, 55, B220–B227. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.; Lewis, L.; Cristofalo, V.J. Proteasome inhibitors shorten replicative life span and induce a senescent-like phenotype of human fibroblasts. J. Cell. Physiol. 2006, 207, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.S.; Pahk, J.W.; Lee, J.S.; Shin, W.C.; Lee, S.Y.; Hong, E.K. Inonotus obliquus protects against oxidative stress-induced apoptosis and premature senescence. Mol. Cells 2011, 31, 423–429. [Google Scholar] [CrossRef]
- Liu, L.; Xie, H.; Chen, X.; Shi, W.; Xiao, X.; Lei, D.; Li, J. Differential response of normal human epidermal keratinocytes and HaCaT cells to hydrogen peroxide-induced oxidative stress. Clin. Exp. Dermatol. 2012, 37, 772–780. [Google Scholar] [CrossRef]
- Velarde, M.C.; Flynn, J.M.; Day, N.U.; Melov, S.; Campisi, J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging (Albany NY) 2012, 4, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Sakura, M.; Chiba, Y.; Kamiya, E.; Furukawa, A.; Kawamura, N.; Niwa, M.; Takeuchi, M.; Hosokawa, M. Spontaneous occurrence of photoageing-like phenotypes in the dorsal skin of old SAMP1 mice, an oxidative stress model. Exp. Dermatol. 2013, 22, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Han, J.; Jiang, C.; Zhang, Y. Biomarkers, oxidative stress and autophagy in skin aging. Ageing Res. Rev. 2020, 59, 101036. [Google Scholar] [CrossRef] [PubMed]
- Sitte, N.; Huber, M.; Grune, T.; Ladhoff, A.; Doecke, W.-D.; Von Zglinicki, T.; Davies, K.J.A. Proteasome inhibition by lipofuscin/ceroid during postmitotic aging of fibroblasts. FASEB J. 2000, 14, 1490–1498. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huang, X.; Wang, P.; Pan, Y.; Cao, D.; Liu, C.; Chen, A. The effects of HSP27 against UVB-induced photoaging in rat skin. Biochem. Biophys. Res. Commun. 2019, 512, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Eckhart, L.; Tschachler, E.; Gruber, F. Autophagic Control of Skin Aging. Front. Cell Dev. Biol. 2019, 7, 143. [Google Scholar] [CrossRef]
- Xu, S.; Cai, Y.; Wei, Y. mTOR signaling from cellular senescence to organismal aging. Aging Dis. 2014, 5, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; König, J.; Höhn, A.; Jung, T.; Grune, T. Macroautophagy is impaired in old murine brain tissue as well as in senescent human fibroblasts. Redox Biol. 2016, 10, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.-L.; Wang, P.; Huang, X.; Wang, Z.-Y.; Cao, D.; Liu, C.; Liu, Y.-Y.; Li, R.-L.; Chen, A.-J. Rapamycin Protects Skin Fibroblasts From UVA-Induced Photoaging by Inhibition of p53 and Phosphorylated HSP27. Front. Cell Dev. Biol. 2021, 9, 134. [Google Scholar] [CrossRef]
- Qin, D.; Ren, R.; Jia, C.; Lu, Y.; Yang, Q.; Chen, L.; Wu, X.; Zhu, J.; Guo, Y.; Yang, P.; et al. Rapamycin Protects Skin Fibroblasts from Ultraviolet B-Induced Photoaging by Suppressing the Production of Reactive Oxygen Species. Cell. Physiol. Biochem. 2018, 46, 1849–1860. [Google Scholar] [CrossRef]
- Kang, H.T.; Lee, K.B.; Kim, S.Y.; Choi, H.R.; Park, S.C. Autophagy Impairment Induces Premature Senescence in Primary Human Fibroblasts. PLoS ONE 2011, 6, e23367. [Google Scholar] [CrossRef] [PubMed]
- Höhn, A.; Sittig, A.; Jung, T.; Grimm, S.; Grune, T. Lipofuscin is formed independently of macroautophagy and lysosomal activity in stress-induced prematurely senescent human fibroblasts. Free Radic. Biol. Med. 2012, 53, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, K.; Shishido, M.; Fujimoto, K.; Hirota, Y.; Yo, K.; Gomi, T.; Tanaka, Y. Age-related disruption of autophagy in dermal fibroblasts modulates extracellular matrix components. Biochem. Biophys. Res. Commun. 2014, 443, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-F.; Gruber, F.; Ni, C.; Mildner, M.; Koenig, U.; Karner, S.; Barresi, C.; Rossiter, H.; Narzt, M.-S.; Nagelreiter, I.M.; et al. Suppression of Autophagy Dysregulates the Antioxidant Response and Causes Premature Senescence of Melanocytes. J. Investig. Dermatol. 2015, 135, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Ni, C.; Narzt, M.-S.; Nagelreiter, I.-M.; Zhang, C.F.; Larue, L.; Rossiter, H.; Grillari, J.; Tschachler, E.; Gruber, F. Autophagy deficient melanocytes display a senescence associated secretory phenotype that includes oxidized lipid mediators. Int. J. Biochem. Cell Biol. 2016, 81, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Xu, Z.; Xiao, Q.; Yang, Y.; Ying, J.; Xiang, L.; Zhang, C. Dysfunction of ATG7-dependent autophagy dysregulates the antioxidant response and contributes to oxidative stress-induced biological impairments in human epidermal melanocytes. Cell Death Discov. 2020, 6, 31. [Google Scholar] [CrossRef]
- Jeong, D.; Qomaladewi, N.P.; Lee, J.; Park, S.H.; Cho, J.Y. The Role of Autophagy in Skin Fibroblasts, Keratinocytes, Melanocytes, and Epidermal Stem Cells. J. Investig. Dermatol. 2020, 140, 1691–1697. [Google Scholar] [CrossRef]
- Chung, J.H. Photoaging in Asians. Photodermatol. Photoimmunol. Photomed. 2003, 19, 109–121. [Google Scholar] [CrossRef]
- Kang, H.Y.; Lee, J.W.; Papaccio, F.; Bellei, B.; Picardo, M. Alterations of the pigmentation system in the aging process. Pigment Cell Melanoma Res. 2021, 34, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Matos, L.; Gouveia, A.M.; Almeida, H. ER Stress Response in Human Cellular Models of Senescence. J. Gerontol. Ser. A 2015, 70, 924–935. [Google Scholar] [CrossRef]
- Druelle, C.; Drullion, C.; Desle, J.; Martin, N.; Saas, L.; Cormenier, J.; Malaquin, N.; Huot, L.; Slomianny, C.; Bouali, F.; et al. ATF6alpha regulates morphological changes associated with senescence in human fibroblasts. Oncotarget 2016, 7, 67699–67715. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A Review in the Theme: Cellular Mechanisms of Endoplasmic Reticulum Stress Signaling in Health and Disease. Am. J. Physiol. Physiol. 2014, 308, C415–C425. [Google Scholar] [CrossRef] [PubMed]
- Cormenier, J.; Martin, N.; Deslé, J.; Salazar-Cardozo, C.; Pourtier, A.; Abbadie, C.; Pluquet, O. The ATF6α arm of the Unfolded Protein Response mediates replicative senescence in human fibroblasts through a COX2/prostaglandin E2 intracrine pathway. Mech. Ageing Dev. 2018, 170, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Denoyelle, C.; Abou-Rjaily, G.; Bezrookove, V.; Verhaegen, M.; Johnson, T.M.; Fullen, D.R.; Pointer, J.N.; Gruber, S.B.; Su, L.D.; Nikiforov, M.A.; et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat. Cell Biol. 2006, 8, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Gülow, K.; Bienert, D.; Haas, I.G. BiP is feed-back regulated by control of protein translation efficiency. J. Cell Sci. 2002, 115, 2443–2452. [Google Scholar] [CrossRef]
- Boraldi, F.; Annovi, G.; Tiozzo, R.; Sommer, P.; Quaglino, D. Comparison of ex vivo and in vitro human fibroblast ageing models. Mech. Ageing Dev. 2010, 131, 625–635. [Google Scholar] [CrossRef] [PubMed]
- D’Adda Di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Zou, L. Single- and double-stranded DNA: Building a trigger of ATR-mediated DNA damage response. Genes Dev. 2007, 21, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. The ATM-mediated DNA-damage response: Taking shape. Trends Biochem. Sci. 2006, 31, 402–410. [Google Scholar] [CrossRef]
- Bekker-Jensen, S.; Lukas, C.; Kitagawa, R.; Melander, F.; Kastan, M.B.; Bartek, J.; Lukas, J. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J. Cell Biol. 2006, 173, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Falck, J.; Bartkova, J.; Bartek, J.; Lukas, J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat. Cell Biol. 2003, 5, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Turenne, G.A.; Paul, P.; Laflair, L.; Price, B.D. Activation of p53 transcriptional activity requires ATM’s kinase domain and multiple N-terminal serine residues of p53. Oncogene 2001, 20, 5100–5110. [Google Scholar] [CrossRef]
- Hewitt, G.; Jurk, D.; Marques, F.D.M.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef]
- Herbig, U.; Ferreira, M.; Condel, L.; Carey, D.; Sedivy, J.M. Cellular Senescence in Aging Primates. Science 2006, 311, 1257. [Google Scholar] [CrossRef] [PubMed]
- Waaijer, M.E.C.; Gunn, D.A.; van Heemst, D.; Slagboom, P.E.; Sedivy, J.M.; Dirks, R.W.; Tanke, H.J.; Westendorp, R.G.J.; Maier, A.B. Do senescence markers correlate in vitro and in situ within individual human donors? Aging (Albany NY) 2018, 10, 278–289. [Google Scholar] [CrossRef]
- Ikeda, H.; Aida, J.; Hatamochi, A.; Hamasaki, Y.; Izumiyama-Shimomura, N.; Nakamura, K.; Ishikawa, N.; Poon, S.S.; Fujiwara, M.; Tomita, K.; et al. Quantitative fluorescence in situ hybridization measurement of telomere length in skin with/without sun exposure or actinic keratosis. Hum. Pathol. 2014, 45, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Yamashita, R.; Ueda, M. Telomere length of the skin in association with chronological aging and photoaging. J. Dermatol. Sci. 2006, 43, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Koch, H.; Wittern, K.-P.; Bergemann, J. In Human Keratinocytes the Common Deletion Reflects Donor Variabilities Rather Than Chronologic Aging and can be Induced by Ultraviolet A Irradiation. J. Investig. Dermatol. 2001, 117, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Ortonne, J.-P. Pigmentary changes of the ageing skin. Br. J. Dermatol. 1990, 122, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Skoczyńska, A.; Budzisz, E.; Trznadel-Grodzka, E.; Rotsztejn, H. Melanin and lipofuscin as hallmarks of skin aging. Adv. Dermatol. Allergol. Dermatol. Alergol. 2017, 34, 97–103. [Google Scholar] [CrossRef]
- Wasmeier, C.; Hume, A.N.; Bolasco, G.; Seabra, M.C. Melanosomes at a glance. J. Cell Sci. 2008, 121, 3995–3999. [Google Scholar] [CrossRef] [PubMed]
- Wiriyasermkul, P.; Moriyama, S.; Nagamori, S. Membrane transport proteins in melanosomes: Regulation of ions for pigmentation. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183318. [Google Scholar] [CrossRef]
- Hearing, V.J. Determination of melanin synthetic pathways. J. Investig. Dermatol. 2011, 131, E8–E11. [Google Scholar] [CrossRef]
- Raposo, G.; Marks, M.S. Melanosomes--dark organelles enlighten endosomal membrane transport. Nat. Rev. Mol. Cell Biol. 2007, 8, 786–797. [Google Scholar] [CrossRef]
- D’Alba, L.; Shawkey, M.D. Melanosomes: Biogenesis, Properties, and Evolution of an Ancient Organelle. Physiol. Rev. 2019, 99, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Prota, G.; Hu, D.-N.; Vincensi, M.R.; Mccormick, S.A.; Napolitano, A. Characterization of Melanins in Human Irides and Cultured Uveal Melanocytes From Eyes of Different Colors. Exp. Eye Res. 1998, 67, 293–299. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Medrano, E.E. Melanin Accumulation Accelerates Melanocyte Senescence by a Mechanism Involving p16INK4a/CDK4/pRB and E2F1. Ann. N. Y. Acad. Sci. 2000, 908, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Bastonini, E.; Kovacs, D.; Picardo, M. Skin pigmentation and pigmentary disorders: Focus on epidermal/dermal cross-talk. Ann. Dermatol. 2016, 28, 279–289. [Google Scholar] [CrossRef]
- Murase, D.; Hachiya, A.; Takano, K.; Hicks, R.; Visscher, M.O.; Kitahara, T.; Hase, T.; Takema, Y.; Yoshimori, T. Autophagy Has a Significant Role in Determining Skin Color by Regulating Melanosome Degradation in Keratinocytes. J. Investig. Dermatol. 2013, 133, 2416–2424. [Google Scholar] [CrossRef]
- Duval, C.; Cohen, C.; Chagnoleau, C.; Flouret, V.; Bourreau, E.; Bernerd, F. Key Regulatory Role of Dermal Fibroblasts in Pigmentation as Demonstrated Using a Reconstructed Skin Model: Impact of Photo-Aging. PLoS ONE 2014, 9, e114182. [Google Scholar]
- Kovacs, D.; Cardinali, G.; Aspite, N.; Cota, C.; Luzi, F.; Bellei, B.; Briganti, S.; Amantea, A.; Torrisi, M.R.; Picardo, M. Role of fibroblast-derived growth factors in regulating hyperpigmentation of solar lentigo. Br. J. Dermatol. 2010, 163, 1020–1027. [Google Scholar] [CrossRef]
- Murase, D.; Hachiya, A.; Amano, Y.; Ohuchi, A.; Kitahara, T.; Takema, Y. The Essential Role of p53 in Hyperpigmentation of the Skin via Regulation of Paracrine Melanogenic Cytokine Receptor Signaling. J. Biol. Chem. 2009, 284, 4343–4353. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Widlund, H.R.; Feige, E.; Lin, J.Y.; Wilensky, D.L.; Igras, V.E.; D’Orazio, J.; Fung, C.Y.; Schanbacher, C.F.; Granter, S.R.; et al. Central Role of p53 in the Suntan Response and Pathologic Hyperpigmentation. Cell 2007, 128, 853–864. [Google Scholar] [CrossRef]
- Yoon, J.E.; Kim, Y.; Kwon, S.; Kim, M.; Kim, Y.H.; Kim, J.H.; Park, T.J.; Kang, H.Y. Senescent fibroblasts drive ageing pigmentation: A potential therapeutic target for senile lentigo. Theranostics 2018, 8, 4620–4632. [Google Scholar] [CrossRef]
- Kovacs, D.; Bastonini, E.; Ottaviani, M.; Cota, C.; Migliano, E.; Dell’Anna, M.L.; Picardo, M. Vitiligo Skin: Exploring the Dermal Compartment. J. Investig. Dermatol. 2018, 138, 394–404. [Google Scholar] [CrossRef]
- Rani, S.; Bhardwaj, S.; Srivastava, N.; Sharma, V.L.; Parsad, D.; Kumar, R. Senescence in the lesional fibroblasts of non-segmental vitiligo patients. Arch. Dermatol. Res. 2017, 309, 123–132. [Google Scholar] [CrossRef]
- Reuter, J.; Merfort, I.; Schempp, C.M. Botanicals in Dermatology. Am. J. Clin. Dermatol. 2010, 11, 247–267. [Google Scholar] [CrossRef] [PubMed]
- Majidinia, M.; Karimian, A.; Alemi, F.; Yousefi, B.; Safa, A. Targeting miRNAs by polyphenols: Novel therapeutic strategy for aging. Biochem. Pharmacol. 2020, 173, 113688. [Google Scholar] [CrossRef] [PubMed]
- Menicacci, B.; Cipriani, C.; Margheri, F.; Mocali, A.; Giovannelli, L. Modulation of the Senescence-Associated Inflammatory Phenotype in Human Fibroblasts by Olive Phenols. Int. J. Mol. Sci. 2017, 18, 2275. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Park, H.; Kim, H.P. Effects of flavonoids on senescence-associated secretory phenotype formation from bleomycin-induced senescence in BJ fibroblasts. Biochem. Pharmacol. 2015, 96, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Choi, M. Anti-inflammatory and anti-aging effects of hydroxytyrosol on human dermal fibroblasts (HDFs). Biomed. Dermatol. 2018, 2, 21. [Google Scholar] [CrossRef]
- Nisticò, S.; Ehrlich, J.; Gliozzi, M.; Maiuolo, J.; Del Duca, E.; Muscoli, C.; Mollace, V. Telomere and telomerase modulation by bergamot polyphenolic fraction in experimental photoageing in human keratinocytes. J. Biol. Regul. Homeost. Agents 2015, 29, 723–728. [Google Scholar]
- Lee, T.H.; Do, M.H.; Oh, Y.L.; Cho, D.W.; Kim, S.H.; Kim, S.Y. Dietary Fermented Soybean Suppresses UVB-Induced Skin Inflammation in Hairless Mice via Regulation of the MAPK Signaling Pathway. J. Agric. Food Chem. 2014, 62, 8962–8972. [Google Scholar] [CrossRef]
- Magcwebeba, T.; Swart, P.; Swanevelder, S.; Joubert, E.; Gelderblom, W. Anti-inflammatory effects of aspalathus linearis and Cyclopia spp. Extracts in a UVB/Keratinocyte (HaCaT) model utilising interleukin-1-Accumulation as biomarker. Molecules 2016, 21, 1323. [Google Scholar] [CrossRef]
- Mao, G.-X.; Xing, W.-M.; Wen, X.-L.; Jia, B.-B.; Yang, Z.-X.; Wang, Y.-Z.; Jin, X.-Q.; Wang, G.-F.; Yan, J. Salidroside protects against premature senescence induced by ultraviolet B irradiation in human dermal fibroblasts. Int. J. Cosmet. Sci. 2015, 37, 321–328. [Google Scholar] [CrossRef]
- Britto, S.M.; Shanthakumari, D.; Agilan, B.; Radhiga, T.; Kanimozhi, G.; Prasad, N.R. Apigenin prevents ultraviolet-B radiation induced cyclobutane pyrimidine dimers formation in human dermal fibroblasts. Mutat. Res. Toxicol. Environ. Mutagenesis 2017, 821, 28–35. [Google Scholar] [CrossRef]
- Afaq, F.; Malik, A.; Syed, D.; Maes, D.; Matsui, M.S.; Mukhtar, H. Pomegranate fruit extract modulates UV-B-mediated phosphorylation of mitogen-activated protein kinases and activation of nuclear factor kappa B in normal human epidermal keratinocytes paragraph sign. Photochem. Photobiol. 2005, 81, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Kum, H.; Ryu, D.; Kim, M.; Jung, E.; Park, D. Protective effects of a new phloretin derivative against UVB-induced damage in skin cell model and human volunteers. Int. J. Mol. Sci. 2014, 15, 18919–18940. [Google Scholar] [CrossRef]
- Mantena, S.K.; Katiyar, S.K. Grape seed proanthocyanidins inhibit UV-radiation-induced oxidative stress and activation of MAPK and NF-kappaB signaling in human epidermal keratinocytes. Free Radic. Biol. Med. 2006, 40, 1603–1614. [Google Scholar] [CrossRef]
- Pacheco-Palencia, L.A.; Noratto, G.; Hingorani, L.; Talcott, S.T.; Mertens-Talcott, S.U. Protective effects of standardized pomegranate (Punica granatum L.) polyphenolic extract in ultraviolet-irradiated human skin fibroblasts. J. Agric. Food Chem. 2008, 56, 8434–8441. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Role of Reactive Oxygen Species in Biological Processes. Klin Wochenschr. 1991, 69, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Wei, J.; Zhao, C.; Li, G. Natural polyphenols targeting senescence: A novel prevention and therapy strategy for cancer. Int. J. Mol. Sci. 2020, 21, 684. [Google Scholar] [CrossRef] [PubMed]
- Farrukh, M.R.; Nissar, U.-A.; Kaiser, P.J.; Afnan, Q.; Sharma, P.R.; Bhushan, S.; Tasduq, S.A. Glycyrrhizic acid (GA) inhibits reactive oxygen Species mediated photodamage by blocking ER stress and MAPK pathway in UV-B irradiated human skin fibroblasts. J. Photochem. Photobiol. B Biol. 2015, 148, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.; Park, S.Y.; Lee, H.J.; Lee, T.Y.; Sun, Z.W.; Yi, T.H. Gallic acid regulates skin photoaging in UVB-exposed fibroblast and hairless mice. Phyther. Res. 2014, 28, 1778–1788. [Google Scholar] [CrossRef] [PubMed]
- Maruki-Uchida, H.; Kurita, I.; Sugiyama, K.; Sai, M.; Maeda, K.; Ito, T. The protective effects of piceatannol from passion fruit (Passiflora edulis) seeds in UVB-irradiated keratinocytes. Biol. Pharm. Bull. 2013, 36, 845–849. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.-H.; Jeong, G.-S. Fisetin inhibits TNF-α-induced inflammatory action and hydrogen peroxide-induced oxidative damage in human keratinocyte HaCaT cells through PI3K/AKT/Nrf-2-mediated heme oxygenase-1 expression. Int. Immunopharmacol. 2015, 29, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Huh, W.B.; Kim, J.-E.; Kang, Y.-G.; Park, G.; Lim, T.; Kwon, J.Y.; Song, D.S.; Jeong, E.H.; Lee, C.C.; Son, J.E.; et al. Brown Pine Leaf Extract and Its Active Component Trans-Communic Acid Inhibit UVB-Induced MMP-1 Expression by Targeting PI3K. PLoS ONE 2015, 10, e0128365. [Google Scholar] [CrossRef] [PubMed]
- Gurău, F.; Baldoni, S.; Prattichizzo, F.; Espinosa, E.; Amenta, F.; Procopio, A.D.; Albertini, M.C.; Bonafè, M.; Olivieri, F. Anti-senescence compounds: A potential nutraceutical approach to healthy aging. Ageing Res. Rev. 2018, 46, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, N.; Fujii, T.; Masaki, H.; Okubo, T.; Shimada, K.; Hashizume, R. Orange peel extract, containing high levels of polymethoxyflavonoid, suppressed UVB-induced COX-2 expression and PGE2 production in HaCaT cells through PPAR-γ activation. Exp. Dermatol. 2014, 23 (Suppl. S1), 18–22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yin, Z.; Ma, L.; Yin, Z.; Hu, Y.; Xu, Y.; Wu, D.; Permatasari, F.; Luo, D.; Zhou, B. The Protective Effect of Baicalin against UVB Irradiation Induced Photoaging: An In Vitro and In Vivo Study. PLoS ONE 2014, 9, e99703. [Google Scholar] [CrossRef] [PubMed]
- Sobiepanek, A.; Milner-Krawczyk, M.; Bobecka-Wesołowska, K.; Kobiela, T. The effect of delphinidin on the mechanical properties of keratinocytes exposed to UVB radiation. J. Photochem. Photobiol. B 2016, 164, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Ziemlewska, A.; Nizioł-Łukaszewska, Z.; Bujak, T.; Zagórska-Dziok, M.; Wójciak, M.; Sowa, I. Effect of fermentation time on the content of bioactive compounds with cosmetic and dermatological properties in Kombucha Yerba Mate extracts. Sci. Rep. 2021, 11, 18792. [Google Scholar] [CrossRef]
- Kanoi, R.; Loachan, P.; Das, S.; Rao, B.S.S. Mangiferin, a naturally occurring polyphenol, mitigates oxidative stress induced premature senescence in human dermal fibroblast cells. Mol. Biol. Rep. 2021, 48, 457–466. [Google Scholar] [CrossRef]
- Manosroi, J.; Chankhampan, C.; Kumguan, K.; Manosroi, W.; Manosroi, A. In vitro anti-aging activities of extracts from leaves of Ma Kiang (Cleistocalyx nervosum var. paniala). Pharm. Biol. 2015, 53, 862–869. [Google Scholar] [CrossRef]
- Gupta, V.K.; Kaur, R.; Singla, R.; Jaitak, V. Photoprotective, antioxidant screening and new ester from dry root extracts of Potentilla atrosanguinea (Himalayan cinquefoil). S. Afr. J. Bot. 2016, 103, 49–53. [Google Scholar] [CrossRef]
- Tito, A.; Carola, A.; Bimonte, M.; Barbulova, A.; Arciello, S.; de Laurentiis, F.; Monoli, I.; Hill, J.; Gibertoni, S.; Colucci, G.; et al. A tomato stem cell extract, containing antioxidant compounds and metal chelating factors, protects skin cells from heavy metal-induced damages. Int. J. Cosmet. Sci. 2011, 33, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Pastore, S.; Potapovich, A.; Kostyuk, V.; Mariani, V.; Lulli, D.; De Luca, C.; Korkina, L. Plant polyphenols effectively protect HaCaT cells from ultraviolet C-triggered necrosis and suppress inflammatory chemokine expression. Ann. N. Y. Acad. Sci. 2009, 1171, 305–313. [Google Scholar] [CrossRef]
- Joe, M.-J.; Kim, S.-N.; Choi, H.-Y.; Shin, W.-S.; Park, G.-M.; Kang, D.-W.; Kim, Y.K. The inhibitory effects of eckol and dieckol from Ecklonia stolonifera on the expression of matrix metalloproteinase-1 in human dermal fibroblasts. Biol. Pharm. Bull. 2006, 29, 1735–1739. [Google Scholar] [CrossRef]
- Heo, S.-J.; Ko, S.-C.; Cha, S.-H.; Kang, D.-H.; Park, H.-S.; Choi, Y.-U.; Kim, D.; Jung, W.-K.; Jeon, Y.-J. Effect of phlorotannins isolated from Ecklonia cava on melanogenesis and their protective effect against photo-oxidative stress induced by UV-B radiation. Toxicol. Vitr. 2009, 23, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.-H.; Ko, C.-I.; Kim, D.; Jeon, Y.-J. Protective effects of phlorotannins against ultraviolet B radiation in zebrafish (Danio rerio). Vet. Dermatol. 2012, 23, 51-e12. [Google Scholar] [CrossRef]
- So, M.J.; Cho, E.J. Phloroglucinol Attenuates Free Radical-induced Oxidative Stress. Prev. Nutr. Food Sci. 2014, 19, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Piao, M.J.; Ahn, M.J.; Kang, K.A.; Kim, K.C.; Zheng, J.; Yao, C.W.; Cha, J.W.; Hyun, C.L.; Kang, H.K.; Lee, N.H.; et al. Phloroglucinol inhibits ultraviolet B radiation-induced oxidative stress in the mouse skin. Int. J. Radiat. Biol. 2014, 90, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Jesumani, V.; Du, H.; Pei, P.; Aslam, M.; Huang, N. Comparative study on skin protection activity of polyphenol-rich extract and polysaccharide-rich extract from Sargassum vachellianum. PLoS ONE 2020, 15, e0227308. [Google Scholar] [CrossRef]
- Softa, M.; Percoco, G.; Lati, E.; Bony, P. Birch Sap (Betula alba) and Chaga Mushroom (Inonotus obliquus) Extracts Show Anti-Oxidant, Anti-Inflammatory and DNA Protection/Repair Activity In Vitro. J. Cosmet. Dermatol. Sci. Appl. 2019, 9, 188–205. [Google Scholar] [CrossRef]
- Lee, I.K.; Kim, Y.S.; Jang, Y.W.; Jung, J.Y.; Yun, B.S. New antioxidant polyphenols from the medicinal mushroom Inonotus obliquus. Bioorganic Med. Chem. Lett. 2007, 17, 6678–6681. [Google Scholar] [CrossRef]
- Kim, S.Y.; Go, K.C.; Song, Y.S.; Jeong, Y.S.; Kim, E.J.; Kim, B.J. Extract of the mycelium of T. matsutake inhibits elastase activity and TPA-induced MMP-1 expression in human fibroblasts. Int. J. Mol. Med. 2014, 34, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Si, H.; Jia, Z.; Liu, D. Dietary anti-aging polyphenols and potential mechanisms. Antioxidants 2021, 10, 283. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Qin, L.; Feng, R.; Hu, G.; Sun, H.; He, Y.; Zhang, R. Emerging senolytic agents derived from natural products. Mech. Ageing Dev. 2019, 181, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ma, L.; Su, M.; Zhou, Y.; Mao, K.; Li, C.; Peng, G.; Zhou, C.; Shen, B.; Dou, J. Baicalin induces cellular senescence in human colon cancer cells via upregulation of DEPP and the activation of Ras/Raf/MEK/ERK signaling. Cell Death Dis. 2018, 9, 217. [Google Scholar] [CrossRef]
- Lewinska, A.; Adamczyk-Grochala, J.; Bloniarz, D.; Olszowka, J.; Kulpa-Greszta, M.; Litwinienko, G.; Tomaszewska, A.; Wnuk, M.; Pazik, R. AMPK-mediated senolytic and senostatic activity of quercetin surface functionalized Fe3O4 nanoparticles during oxidant-induced senescence in human fibroblasts. Redox Biol. 2020, 28, 101337. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; He, Y.; Zhang, R.; Zheng, G.; Zhou, D. The curcumin analog EF24 is a novel senolytic agent. Aging (Albany NY) 2019, 11, 771–782. [Google Scholar] [CrossRef]
- Louis, A.; Petereit, F.; Lechtenberg, M.; Deters, A.; Hensel, A. Phytochemical Characterization of Rhododendron ferrugineum and In Vitro Assessment of an Aqueous Extract on Cell Toxicity. Planta Med. 2010, 76, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Wandrey, F.; Schmid, D.; Zülli, F. Senolytics: Eliminating “ zombie cells ” in the skin A novel anti-aging mechanism to combat senescent cells. HPC Today 2020, 15, 18–20. [Google Scholar]
- Chi, Y.S.; Kim, H.P. Suppression of cyclooxygenase-2 expression of skin fibroblasts by wogonin, a plant flavone from Scutellaria radix. Prostaglandins Leukot. Essent. Fat. Acids 2005, 72, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-S.; Cho, J.-G.; Hwang, E.-S.; Yang, J.-E.; Gao, W.; Fang, M.-Z.; Zheng, S.-D.; Yi, T.-H. Enhancement of Protective Effects of Radix Scutellariae on UVB-induced Photo Damage in Human HaCaT Keratinocytes. Appl. Biochem. Biotechnol. 2018, 184, 1073–1093. [Google Scholar] [CrossRef]
- Ferreira, M.S.; Magalhães, M.C.; Oliveira, R.; Sousa-Lobo, J.M.; Almeida, I.F. Trends in the use of botanicals in anti-aging cosmetics. Molecules 2021, 26, 3584. [Google Scholar] [CrossRef] [PubMed]
- Zillich, O.V.; Schweiggert-Weisz, U.; Eisner, P.; Kerscher, M. Polyphenols as active ingredients for cosmetic products. Int. J. Cosmet. Sci. 2015, 37, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Yutani, R.; Kikuchi, T.; Teraoka, R.; Kitagawa, S. Efficient Delivery and Distribution in Skin of Chlorogenic Acid and Resveratrol Induced by Microemulsion Using Sucrose Laurate. Chem. Pharm. Bull. 2014, 62, 274–280. [Google Scholar] [CrossRef]
- Abla, M.J.; Banga, A.K. Quantification of skin penetration of antioxidants of varying lipophilicity. Int. J. Cosmet. Sci. 2013, 35, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Di Mambro, V.M.; Fonseca, M.J. V Assays of physical stability and antioxidant activity of a topical formulation added with different plant extracts. J. Pharm. Biomed. Anal. 2005, 37, 287–295. [Google Scholar] [CrossRef] [PubMed]
- De Mello Costa, A.R.; Marquiafável, F.S.; de Oliveira Lima Leite Vaz, M.M.; Rocha, B.A.; Pires Bueno, P.C.; Amaral, P.L.M.; da Silva Barud, H.; Berreta-Silva, A.A. Quercetin-PVP K25 solid dispersions. J. Therm. Anal. Calorim. 2011, 104, 273–278. [Google Scholar] [CrossRef]
- Baby, A.R.; Haroutiounian-Filho, C.A.; Sarruf, F.D.; de Pinto, C.A.S.O.; Kaneko, T.M.; Velasco, M.V.R. Influence of Urea, Isopropanol, and Propylene Glycol on Rutin In Vitro Release from Cosmetic Semisolid Systems Estimated by Factorial Design. Drug Dev. Ind. Pharm. 2009, 35, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Zillich, O.V.; Schweiggert-Weisz, U.; Hasenkopf, K.; Eisner, P.; Kerscher, M. Release and in vitro skin permeation of polyphenols from cosmetic emulsions. Int. J. Cosmet. Sci. 2013, 35, 491–501. [Google Scholar] [CrossRef]
- Kitagawa, S.; Tanaka, Y.; Tanaka, M.; Endo, K.; Yoshii, A. Enhanced skin delivery of quercetin by microemulsion. J. Pharm. Pharmacol. 2009, 61, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, S.; Yoshii, K.; Morita, S.; Teraoka, R. Efficient Topical Delivery of Chlorogenic Acid by an Oil-in-Water Microemulsion to Protect Skin against UV-Induced Damage. Chem. Pharm. Bull. 2011, 59, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.C.; Rodrigues, D.; Sequeira, J.A.D.; Pereira, I.; Simões, A.; Costa, D.; Peixoto, D.; Costa, G.; Veiga, F. Nanotechnological breakthroughs in the development of topical phytocompounds-based formulations. Int. J. Pharm. 2019, 572, 118787. [Google Scholar] [CrossRef] [PubMed]
- Scognamiglio, I.; De Stefano, D.; Campani, V.; Mayol, L.; Carnuccio, R.; Fabbrocini, G.; Ayala, F.; La Rotonda, M.I.; De Rosa, G. Nanocarriers for topical administration of resveratrol: A comparative study. Int. J. Pharm. 2013, 440, 179–187. [Google Scholar] [CrossRef]
- Stan, M.S.; Chirila, L.; Popescu, A.; Radulescu, D.M.; Radulescu, D.E.; Dinischiotu, A. Essential Oil Microcapsules Immobilized on Textiles and Certain Induced Effects. Materials 2019, 12, 2029. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Martí, M.; Martínez, V.; Rubio, L.; Parra, J.L.; Coderch, L. Antioxidant cosmeto-textiles: Skin assessment. Eur. J. Pharm. Biopharm. 2013, 84, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-H.; Wu, Y.; Wei, H.-C.; Xu, Y.-Y.; Jia, L.-L.; Chen, J.; Yang, X.-S.; Dong, G.-H.; Gao, X.-H.; Chen, H.-D. Protective effects of green tea extracts on photoaging and photommunosuppression. Ski. Res. Technol. 2009, 15, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.-H.; Jung, E.Y.; Shin, K.-S.; Yu, K.-W.; Chang, U.J.; Suh, H.J. Tannase-converted green tea catechins and their anti-wrinkle activity in humans. J. Cosmet. Dermatol. 2013, 12, 137–143. [Google Scholar] [CrossRef]
- Chuarienthong, P.; Lourith, N.; Leelapornpisid, P. Clinical efficacy comparison of anti-wrinkle cosmetics containing herbal flavonoids. Int. J. Cosmet. Sci. 2010, 32, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Boo, Y.C. Human Skin Lightening Efficacy of Resveratrol and Its Analogs: From in Vitro Studies to Cosmetic Applications. Antioxidants 2019, 8, 332. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Jia, L.-L.; Zheng, Y.-N.; Xu, X.-G.; Luo, Y.-J.; Wang, B.; Chen, J.Z.S.; Gao, X.-H.; Chen, H.-D.; Matsui, M.; et al. Resveratrate protects human skin from damage due to repetitive ultraviolet irradiation. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Woon, C.G.; Jin, J.H.; Kyung, S.J.; Hwoon, B.J.; Mi, K.Y.; Chool, B.Y. Skin Anti-aging Effects of a Cream Containing Resveratryl Triacetate (RTA). J. Society Cosmet. Sci. Korea 2018, 44, 161–170. [Google Scholar] [CrossRef]
- to Brinke, A.S.; Janssens-Böcker, C.; Kerscher, M. Skin Anti-Aging Benefits of a 2% Resveratrol Emulsion. J. Cosmet. Dermatol. Sci. Appl. 2021, 11, 155–168. [Google Scholar] [CrossRef]
- Moyano-Mendez, J.R.; Fabbrocini, G.; De Stefano, D.; Mazzella, C.; Mayol, L.; Scognamiglio, I.; Carnuccio, R.; Ayala, F.; La Rotonda, M.I.; De Rosa, G. Enhanced antioxidant effect of trans-resveratrol: Potential of binary systems with polyethylene glycol and cyclodextrin. Drug Dev. Ind. Pharm. 2014, 40, 1300–1307. [Google Scholar] [CrossRef]
- Cornacchione, S.; Sadick, N.S.; Neveu, M.; Talbourdet, S.; Lazou, K.; Viron, C.; Renimel, I.; de Quéral, D.; Kurfurst, R.; Schnebert, S.; et al. In vivo skin antioxidant effect of a new combination based on a specific Vitis vinifera shoot extract and a biotechnological extract. J. Drugs Dermatol. 2007, 6, s8–s13. [Google Scholar] [PubMed]
- Sharif, A.; Akhtar, N.; Khan, M.S.; Menaa, A.; Menaa, B.; Khan, B.A.; Menaa, F. Formulation and evaluation on human skin of a water-in-oil emulsion containing Muscat hamburg black grape seed extract. Int. J. Cosmet. Sci. 2015, 37, 253–258. [Google Scholar] [CrossRef]
- Barnes, J.; Anderson, L.A.; Phillipson, J.D.; Newall, C.A. Herbal Medicines: A Guide for Healthcare Professionals, 2nd ed.; Pharmaceutical Press: London, UK; Chicago, IL, USA, 2002; ISBN 9780853694748. [Google Scholar]
- Akhtar, N.; Zaman, S.U.; Khan, B.A.; Amir, M.N.; Ebrahimzadeh, M.A. Calendula extract: Effects on mechanical parameters of human skin. Acta Pol. Pharm. 2011, 68, 693–701. [Google Scholar] [PubMed]
- Rakhmini, A.; Faridha, I.; Muchtar, S.V.; Patellongi, I.; Djawad, K.; Alam, G. Comparison of 10%, 20% and 40% Licorice Extract Cream as Skin Lightening Agent. Int. J. Med. Rev. Case Rep. 2018, 2, 4. [Google Scholar] [CrossRef]
- Stephens, T.J.; Sigler, M.L.; Herndon, J.H.; Dispensa, L.; Le Moigne, A. A placebo-controlled, double-blind clinical trial to evaluate the efficacy of Imedeen® Time Perfection® for improving the appearance of photodamaged skin. Clin. Cosmet. Investig. Dermatol. 2016, 9, 63–70. [Google Scholar] [CrossRef]
- Heinrich, U.; Moore, C.E.; De Spirt, S.; Tronnier, H.; Stahl, W. Green Tea Polyphenols Provide Photoprotection, Increase Microcirculation, and Modulate Skin Properties of Women. J. Nutr. 2011, 141, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Buonocore, D.; Lazzeretti, A.; Tocabens, P.; Nobile, V.; Cestone, E.; Santin, G.; Bottone, M.G.; Marzatico, F. Resveratrol-procyanidin blend: Nutraceutical and antiaging efficacy evaluated in a placebo-controlled, double-blind study. Clin. Cosmet. Investig. Dermatol. 2012, 5, 159–165. [Google Scholar] [CrossRef]
- Accorsi-Neto, A.; Haidar, M.; Simões, R.; Simões, M.; Soares-Jr, J.; Baracat, E. Effects of isoflavones on the skin of postmenopausal women: A pilot study. Clinics 2009, 64, 505–510. [Google Scholar] [CrossRef]
- Maan, A.A.; Nazir, A.; Khan, M.K.I.; Ahmad, T.; Zia, R.; Murid, M.; Abrar, M. The therapeutic properties and applications of Aloe vera: A review. J. Herb. Med. 2018, 12, 1–10. [Google Scholar] [CrossRef]
- Shelton, R.M. Aloe vera. Its chemical and therapeutic properties. Int. J. Dermatol. 1991, 30, 679–683. [Google Scholar] [CrossRef]
- Cho, S.; Lee, S.; Lee, M.-J.; Lee, D.H.; Won, C.-H.; Kim, S.M.; Chung, J.H. Dietary Aloe Vera Supplementation Improves Facial Wrinkles and Elasticity and It Increases the Type I Procollagen Gene Expression in Human Skin in vivo. Ann. Dermatol. 2009, 21, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Han, H.-S.; Shin, J.-S.; Myung, D.-B.; Ahn, H.S.; Lee, S.H.; Kim, H.J.; Lee, K.-T. Hydrangea serrata (Thunb.) Ser. Extract Attenuate UVB-Induced Photoaging through MAPK/AP-1 Inactivation in Human Skin Fibroblasts and Hairless Mice. Nutrients 2019, 11, 533. [Google Scholar] [CrossRef] [PubMed]
- Myung, D.-B.; Lee, J.-H.; Han, H.-S.; Lee, K.-Y.; Ahn, H.S.; Shin, Y.-K.; Song, E.; Kim, B.-H.; Lee, K.H.; Lee, S.H.; et al. Oral Intake of Hydrangea serrata (Thunb.) Ser. Leaves Extract Improves Wrinkles, Hydration, Elasticity, Texture, and Roughness in Human Skin: A Randomized, Double-Blind, Placebo-Controlled Study. Nutrients 2020, 12, 1588. [Google Scholar] [CrossRef] [PubMed]
- Shoji, T.; Masumoto, S.; Moriichi, N.; Ohtake, Y.; Kanda, T. Administration of apple polyphenol supplements for skin conditions in healthy women: A randomized, double-blind, placebo-controlled clinical trial. Nutrients 2020, 12, 1071. [Google Scholar] [CrossRef] [PubMed]
- Katz, D.L.; Doughty, K.; Ali, A. Cocoa and chocolate in human health and disease. Antioxid. Redox Signal. 2011, 15, 2779–2811. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.S.; Kim, J.R.; Park, G.Y.; Kim, J.E.; Lee, D.H.; Lee, K.W.; Chung, J.H. Cocoa flavanol supplementation influences skin conditions of photo-aged women: A 24-week double-blind, randomized, controlled trial. J. Nutr. 2016, 146, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Fam, V.W.; Holt, R.R.; Keen, C.L.; Sivamani, R.K.; Hackman, R.M. Prospective evaluation of mango fruit intake on facial wrinkles and erythema in postmenopausal women: A randomized clinical pilot study. Nutrients 2020, 12, 3381. [Google Scholar] [CrossRef] [PubMed]
- Park, S.E. Epidemiology, virology, and clinical features of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2; coronavirus disease-19). Pediatr. Infect. Vaccine 2020, 27, 1–10. [Google Scholar] [CrossRef]
- Bian, J.; Li, Z. Angiotensin-converting enzyme 2 (ACE2): SARS-CoV-2 receptor and RAS modulator. Acta Pharm. Sin. B 2021, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, L.; Zhang, Y.; Wang, X. An Investigation of the Expression of 2019 Novel Coronavirus Cell Receptor Gene ACE2 in a Wide Variety of Human Tissues. Infect. Dis. Poverty 2020, 9, 45. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, e105114. [Google Scholar] [CrossRef]
- Salamanna, F.; Maglio, M.; Landini, M.P.; Fini, M. Body Localization of ACE-2: On the Trail of the Keyhole of SARS-CoV-2. Front. Med. 2020, 7, 935. [Google Scholar] [CrossRef]
- Liao, X.; Xiao, J.; Li, S.-H.; Xiao, L.-L.; Cheng, B.; Fu, X.-B.; Cui, T.; Liu, H.-W. Critical role of the endogenous renin-angiotensin system in maintaining self-renewal and regeneration potential of epidermal stem cells. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2647–2656. [Google Scholar] [CrossRef]
- Recalcati, S. Cutaneous manifestations in COVID-19: A first perspective. J. Eur. Acad. Dermatol. Venereol. 2020, 34, e212–e213. [Google Scholar] [CrossRef]
- Xue, X.; Mi, Z.; Wang, Z.; Pang, Z.; Liu, H.; Zhang, F. High Expression of ACE2 on Keratinocytes Reveals Skin as a Potential Target for SARS-CoV-2. J. Investig. Dermatol. 2021, 141, 206–209. [Google Scholar] [CrossRef]
- Novak, N.; Peng, W.; Naegeli, M.C.; Galvan, C.; Kolm-Djamei, I.; Brüggen, C.; Cabanillas, B.; Schmid-Grendelmeier, P.; Catala, A. SARS-CoV-2, COVID-19, skin and immunology—What do we know so far? Allergy Eur. J. Allergy Clin. Immunol. 2020, 76, 698–713. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.H. Aging of the skin barrier. Clin. Dermatol. 2019, 37, 336–345. [Google Scholar] [CrossRef]
- Sepe, S.; Rossiello, F.; Cancila, V.; Iannelli, F.; Matti, V.; Cicio, G.; Cabrini, M.; Marinelli, E.; Alabi, B.; di Lillo, A.; et al. DNA damage response at telomeres boosts the transcription of SARS-CoV-2 receptor ACE2 during aging. bioRxiv 2021. [Google Scholar] [CrossRef]
- Nehme, J.; Borghesan, M.; Mackedenski, S.; Bird, T.G.; Demaria, M. Cellular senescence as a potential mediator of COVID-19 severity in the elderly. Aging Cell 2020, 19, 1–14. [Google Scholar] [CrossRef]
- Bickler, S.W.; Cauvi, D.M.; Fisch, K.M.; Prieto, J.M.; Sykes, A.G.; Thangarajah, H.; Lazar, D.A.; Ignacio, R.C.; Gerstmann, D.R.; Ryan, A.F.; et al. Extremes of age are associated with differences in the expression of selected pattern recognition receptor genes and ACE2, the receptor for SARS-CoV-2: Implications for the epidemiology of COVID-19 disease. BMC Med. Genomics 2021, 14, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.A.; Kwok, S.; Berry, G.J.; Montine, T.J. Angiotensin-converting enzyme 2 (ACE2) expression increases with age in patients requiring mechanical ventilation. PLoS ONE 2021, 16, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dawn, A.G.; Edwards Dawn, M.; Yosipovitch, G. Psoriasis in the elderly. Aging Health 2007, 3, 611–623. [Google Scholar] [CrossRef]
- Tembhre, M.K.; Parihar, A.S.; Sharma, V.K.; Imran, S.; Bhari, N.; Lakshmy, R.; Bhalla, A. Enhanced expression of angiotensin-converting enzyme 2 in psoriatic skin and its upregulation in keratinocytes by interferon-γ: Implication of inflammatory milieu in skin tropism of SARS-CoV-2. Br. J. Dermatol. 2021, 184, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.G.; Murrell, D.F.; Garcet, S.; Navrazhina, K.; Lee, P.C.; Muscianisi, E.; Blauvelt, A. Secukinumab lowers expression of ACE2 in affected skin of patients with psoriasis. J. Allergy Clin. Immunol. 2021, 147, 1107–1109. [Google Scholar] [CrossRef]
- Radzikowska, U.; Ding, M.; Tan, G.; Zhakparov, D.; Peng, Y.; Wawrzyniak, P.; Wang, M.; Li, S.; Morita, H.; Altunbulakli, C.; et al. Distribution of ACE2, CD147, CD26, and other SARS-CoV-2 associated molecules in tissues and immune cells in health and in asthma, COPD, obesity, hypertension, and COVID-19 risk factors. Allergy 2020, 75, 2829–2845. [Google Scholar] [CrossRef] [PubMed]
- Schmader, K.E.; van der Horst, C.M.; Klotman, M.E. Epstein-Barr Virus and the Elderly Host. Rev. Infect. Dis. 1989, 11, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Church, T.M.; Swaminathan, S. Epstein-Barr Virus Lytic Replication Induces ACE2 Expression and Enhances SARS-CoV-2 Pseudotyped Virus Entry in Epithelial Cells. J. Virol. 2021, 95, e0019221. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.-J.; Hu, F.; Li, H.; Huang, H.-Y.; Wang, D.-W.; Liang, Y. A profiling analysis on the receptor ACE2 expression reveals the potential risk of different type of cancers vulnerable to SARS-CoV-2 infection. Ann. Transl. Med. 2020, 8, 481. [Google Scholar] [CrossRef] [PubMed]
- Chai, P.; Yu, J.; Ge, S.; Jia, R.; Fan, X. Genetic alteration, RNA expression, and DNA methylation profiling of coronavirus disease 2019 (COVID-19) receptor ACE2 in malignancies: A pan-cancer analysis. J. Hematol. Oncol. 2020, 13, 43. [Google Scholar] [CrossRef]
- Winkler, T.; Ben-David, U. Elevated expression of ACE2 in tumor-adjacent normal tissues of cancer patients. bioRxiv 2020. [Google Scholar] [CrossRef]
- Grzegrzolka, J.; Swiatko, K.; Pula, B.; Zamirska, A.; Olbromski, M.; Bieniek, A.; Szepietowski, J.; Rys, J.; Dziegiel, P.; Podhorska-Okolow, M. ACE and ACE2 expression in normal and malignant skin lesions. Folia Histochem. Cytobiol. 2013, 51, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, L.; Li, M.; Wang, X. The SARS-CoV-2 host cell receptor ACE2 correlates positively with immunotherapy response and is a potential protective factor for cancer progression. Comput. Struct. Biotechnol. J. 2020, 18, 2438–2444. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Yu, Y.; Trimpert, J.; Benthani, F.; Mairhofer, M.; Richter-Pechanska, P.; Wyler, E.; Belenki, D.; Kaltenbrunner, S.; Pammer, M.; et al. Virus-induced senescence is driver and therapeutic target in COVID-19. Nature 2021, 599, 283–289. [Google Scholar] [CrossRef]









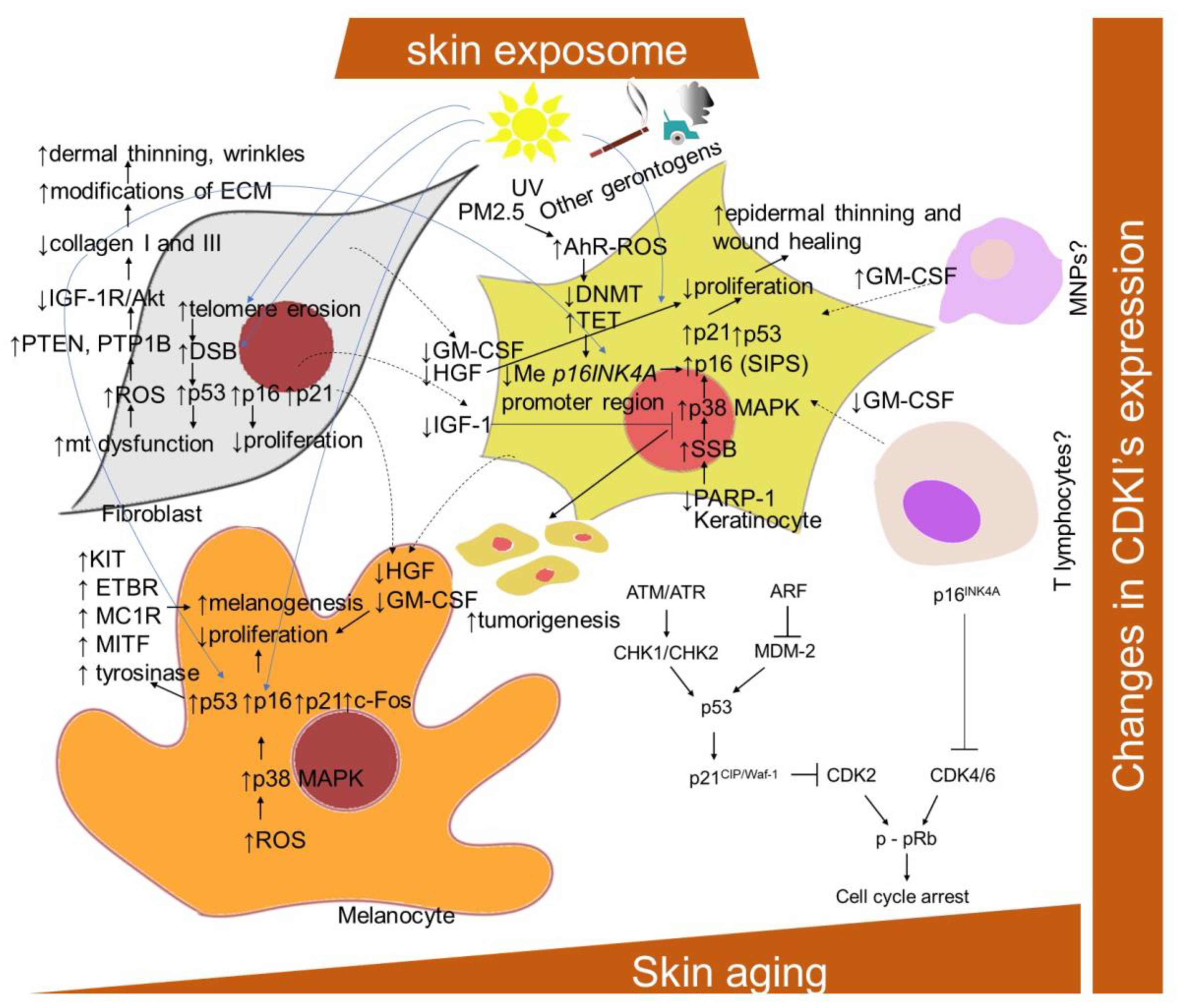{kind=link}
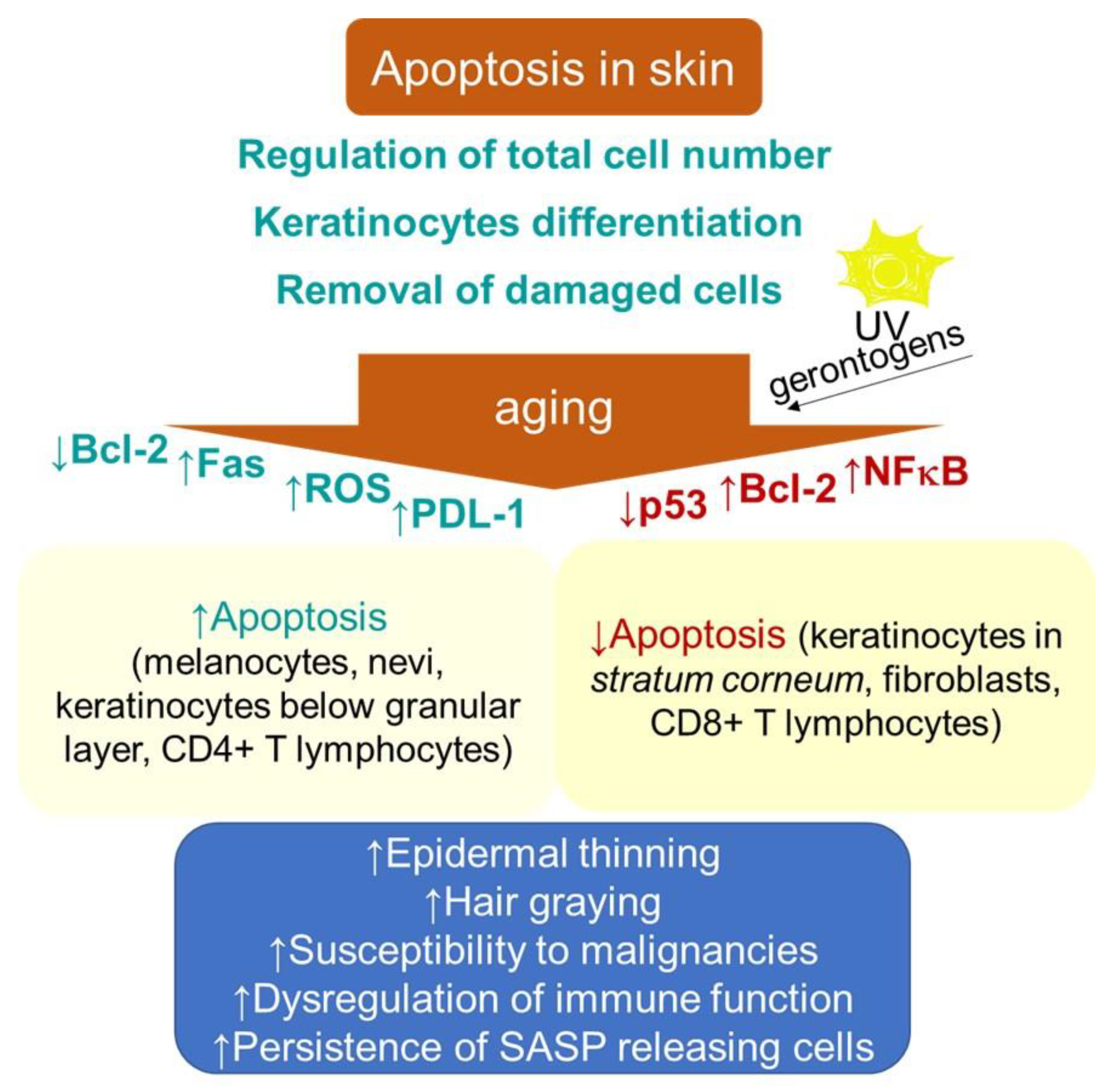{kind=link}
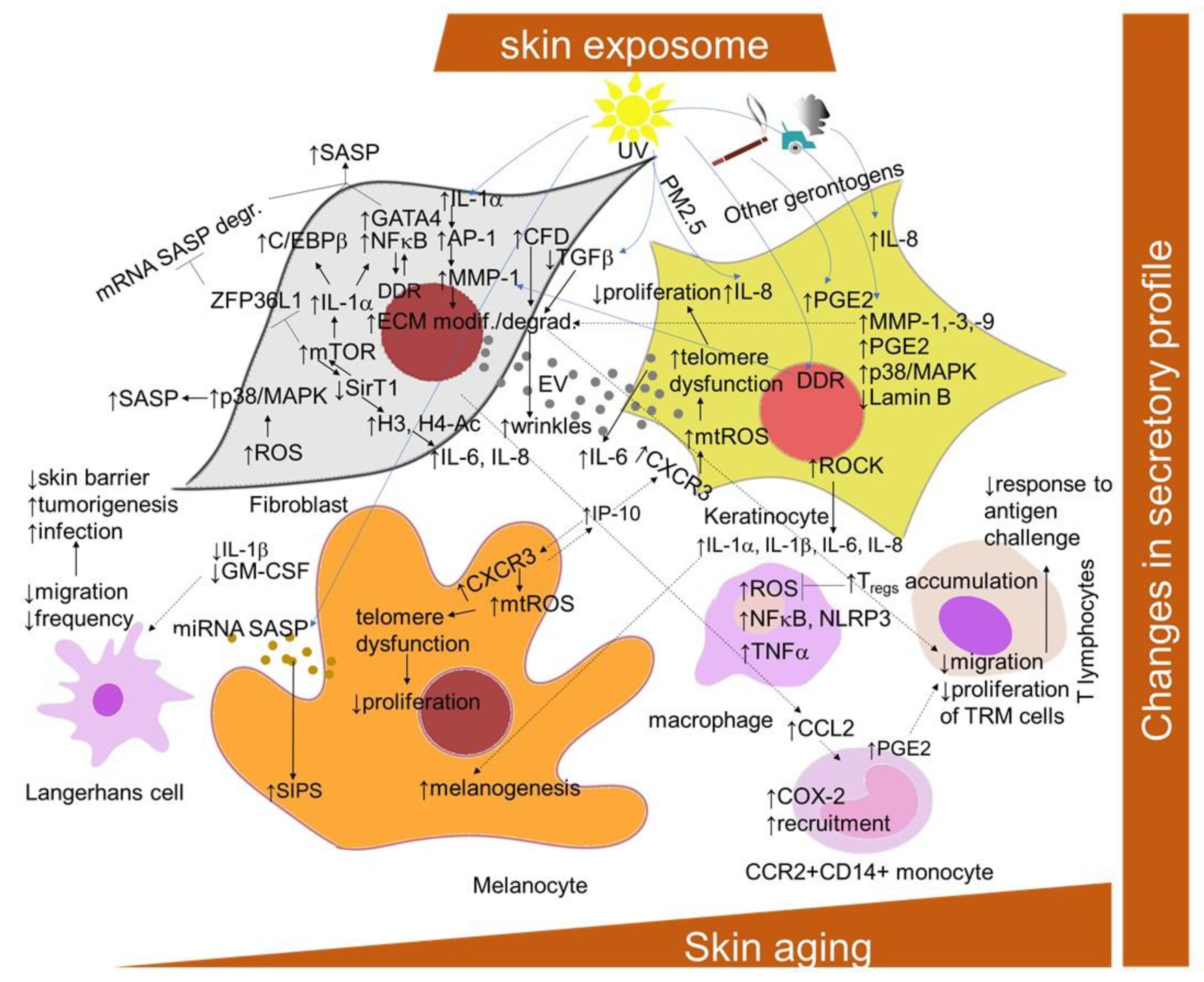{kind=link}
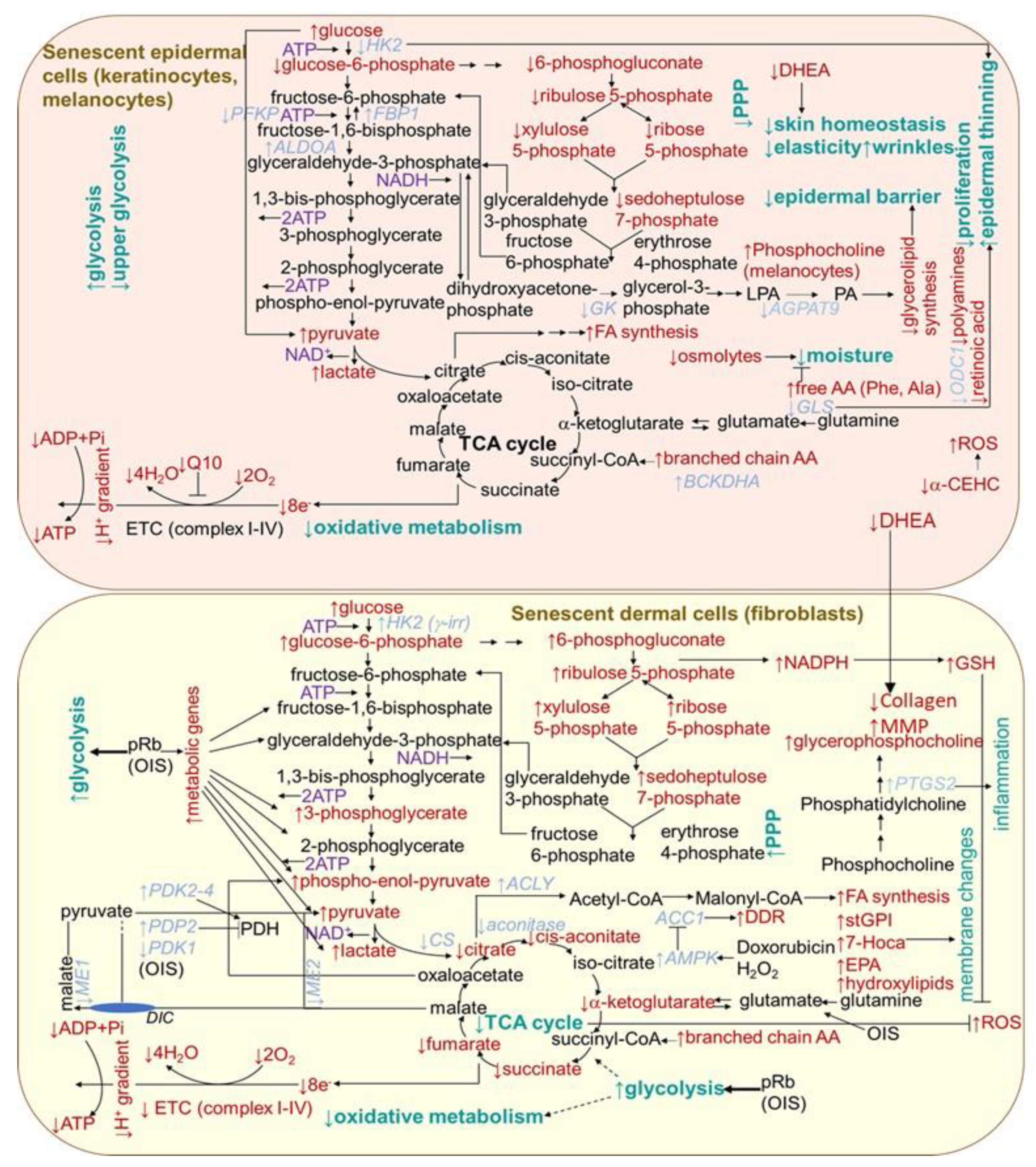{kind=link}
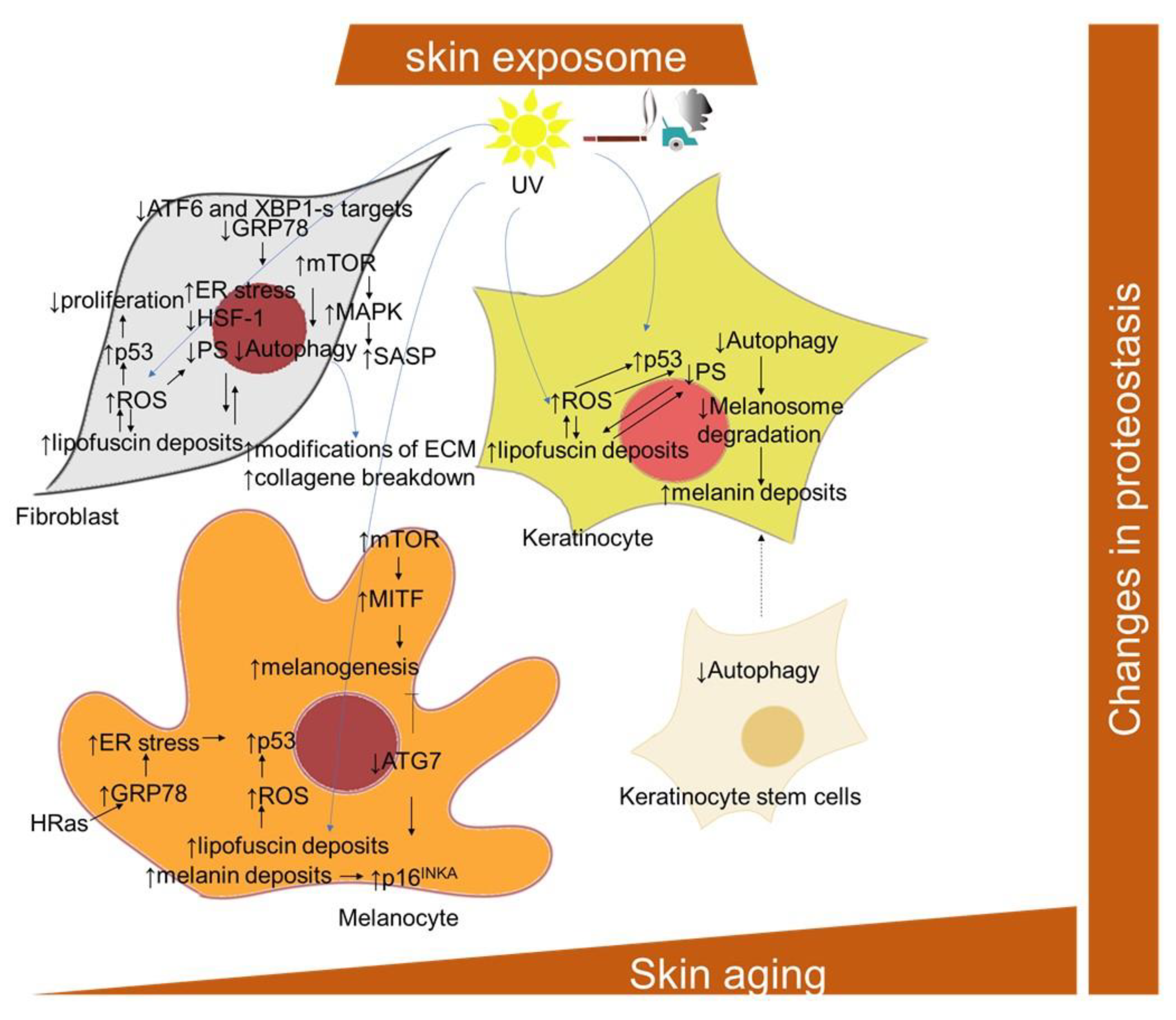{kind=link}
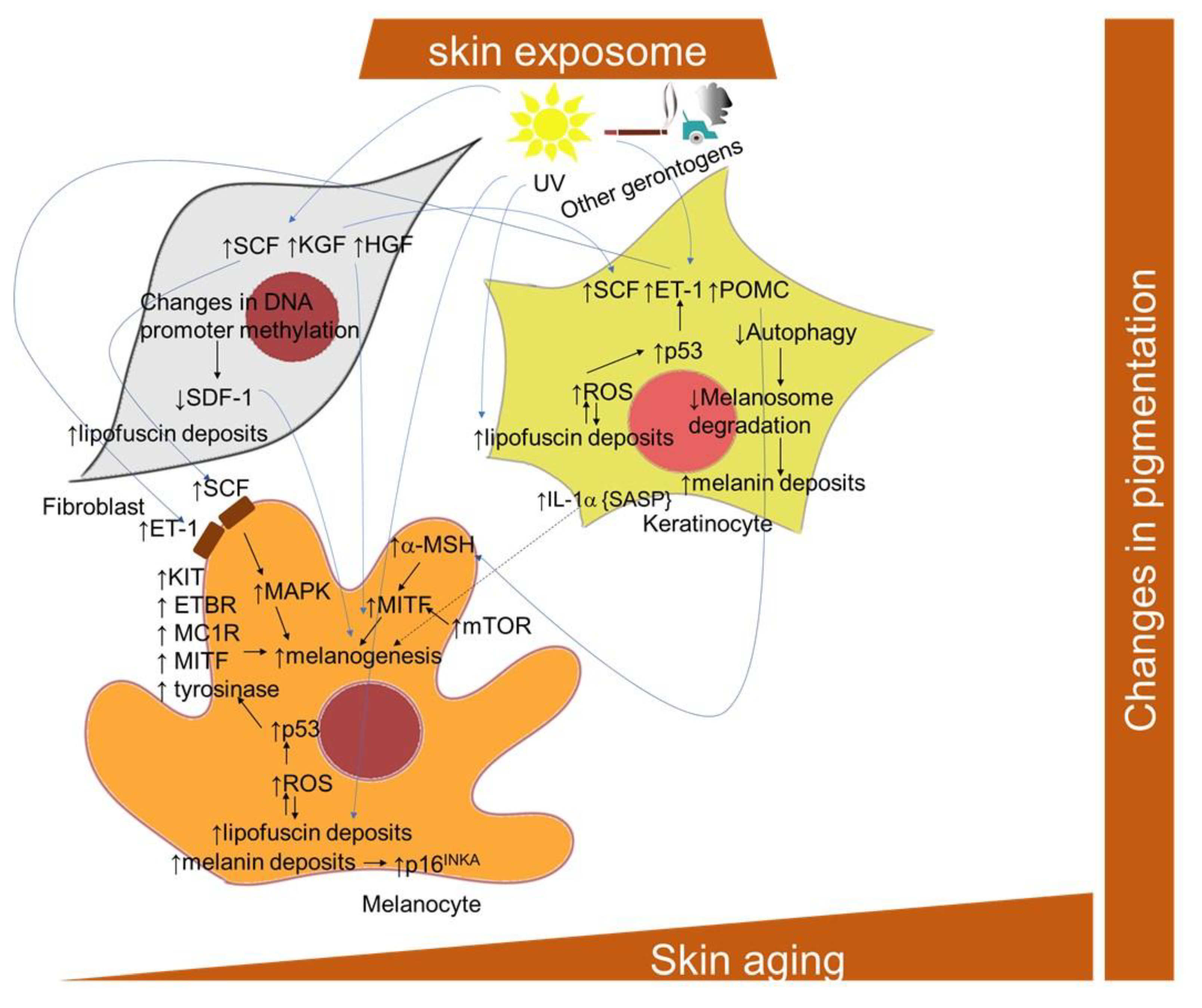{kind=link}
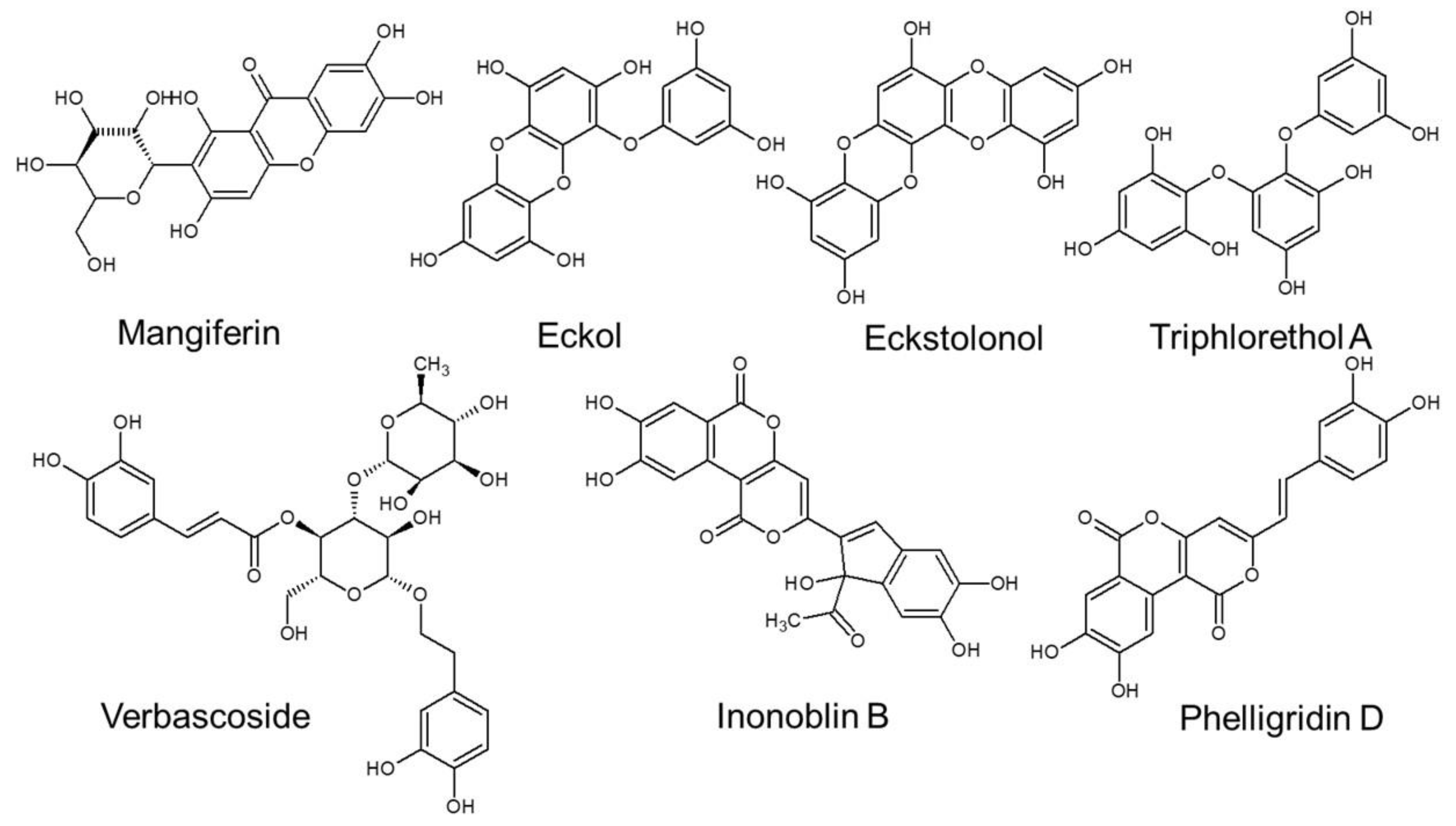{kind=link}
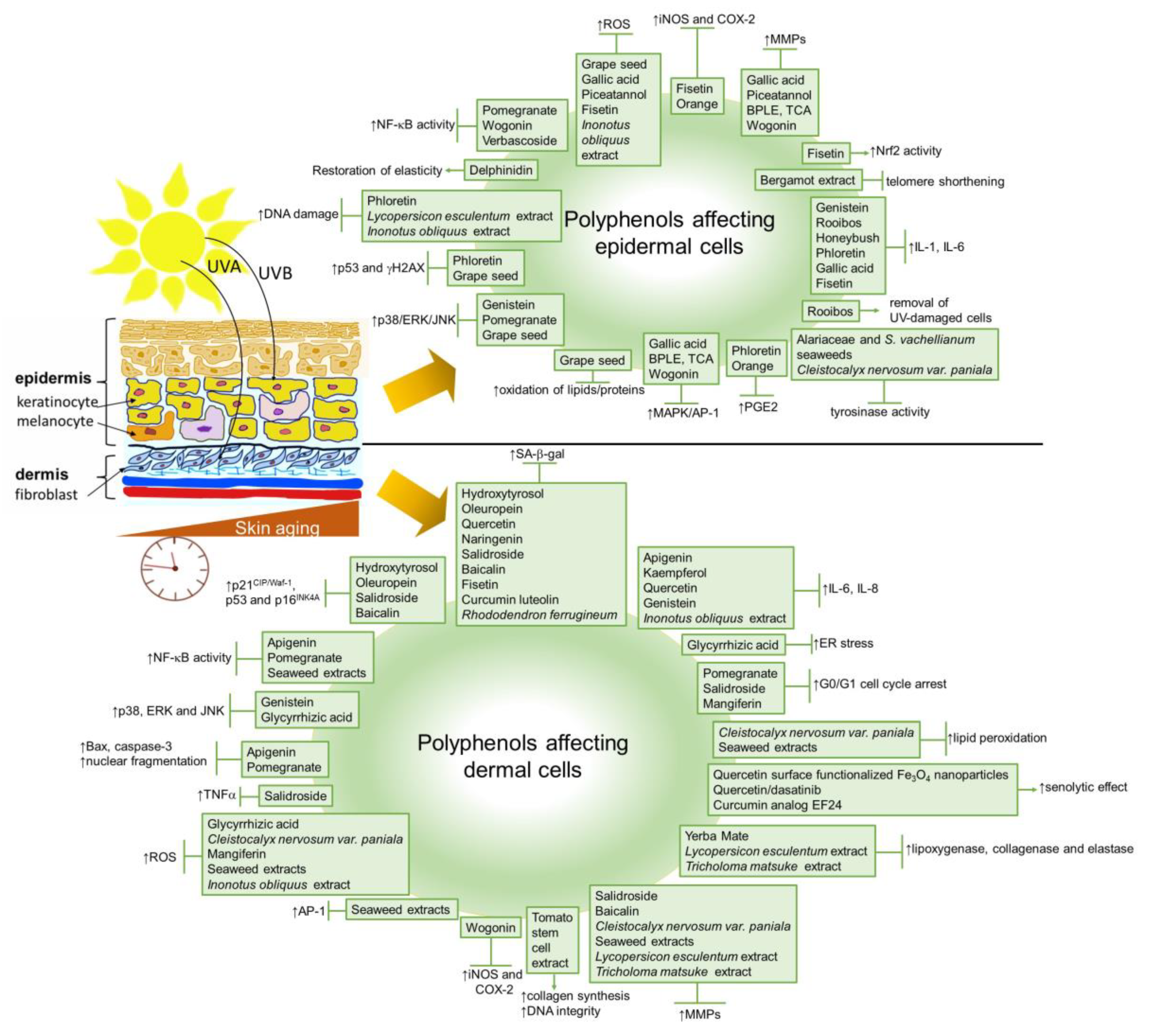{kind=link}
| Polyphenol | Type of Skin Cells | Assay Conditions | Effect | Reference |
|---|---|---|---|---|
| Hydroxytyrosol and Oleuropein | neonatal human dermal fibroblasts | 1 μM hydroxytyrosol or 10 μM oleuropein | reduced SA-β-Gal-positive cell number and p16INK4A protein expression | [299] |
| Apigenin | human foreskin fibroblasts | 10 or 20 μM for 24 h co-treated with bleomycin | decreased expression of IL-6, IL-8 and IL-1β mRNA; inhibited NF-κB activity | [300] |
| NHDF | 15 mM 1 h before and after UVB-exposure | downregulated NER expression; inhibited nuclear fragmentation and Bax and caspase-3 expression | [175] | |
| Kaempferol | human foreskin fibroblasts | 10 or 20 μM for 24 h co-treated with bleomycin | decreased expression of IL-6, IL-8 and IL-1β mRNA | [300] |
| Quercetin | human foreskin fibroblasts | 10 or 20 μM for 24 h co-treated with bleomycin | decreased expression of IL-6, IL-8 and IL-1β mRNA; reduced SA-β-Gal | [300] |
| Naringenin | human foreskin fibroblasts | 10 or 20 μM for 24 h co-treated with bleomycin | reduced SA-β-Gal | [300] |
| Bergamot polyphenol fraction | HaCaT | UVB-exposed | modulation of IL-1β; restored telomere length and telomerase activity | [302] |
| Genistein | NHDF and keratinocytes co-culture | 10 mM for 72 h after UVB exposure | inhibited IL-6 production; inhibited phosphorylation of p38, ERK and JNK | [303] |
| Rooibos methanolic and aqueous extracts | HaCaT | sub-lethal concentrations (0.05–0.55 mg/mL) for 24 h after UVB exposure | inhibited viability and proliferation facilitating the removal of accumulating icIL-1a | [304] |
| Honeybush aqueous extracts | HaCaT | 0.10–0.79 mg/mL for 24 h after UVB exposure | inhibited icIL-1a accumulation; increased caspase-3 activity in damaged cells (with opposing effect found for methanolic extract) | [304] |
| Pomegranate fruit extract | NHEK | 10–40 mg/mL for 24 h before UVB exposure | inhibited phosphorylation of ERK1 and 2, JNK1 and 2 and p38; inhibited phosphorylation of IκBα and IKKα; inhibited translocation of NF-κB/p65 | [307] |
| SKU-1064 skin fibroblasts | 5–60 mg/L for 2 h after UVB exposure | reduced activation of NF-κB; downregulation of caspase-3; increased G0/G1 phase arrest associated with DNA repair | [310] | |
| Phloretin | HaCaT | 50–200 mg/mL 12 h after UVB exposure | decreased DNA damage; reduced phosphorylation of p53 and γ-H2AX; inhibited IL-6 and prostaglandin E2 | [308] |
| Salidroside | NHDF | 1–10 mM for 24 h before UVB exposure | recovered viability; decreased SA-β-Gal-positive cells; relieved G1/G0 cell cycle arrest; suppressed p21CIP/Waf1 and p16INK4A expression; reduced MMP-1 activity; reduced IL-6 and TNF-α production | [305] |
| Grape seed proanthocyanidins | NHEK | 10–50 mM for 3–6 h before UVB exposure | inhibited intracellular release of H2O2; inhibited photo-oxidative damage of lipids and proteins; inhibited oxidative DNA damage; inhibited phosphorylation of ERK1 and 2, JNK and p38 | [309] |
| Glycyrrhizic acid | Hs68 foreskin fibroblasts | 10–25 mM for 16 h before UVB exposure | reduced ROS levels; restored Ca2+ levels; inhibited ER stress; reduced phosphorylation of p38 and JNK | [313] |
| Gallic acid | NDHF, HaCaT | 0.1–10 mM for 24 h after UVB exposure | decreased IL-6; decreased MMP-1 levels; decreased ROS production; suppressed phosphorylation of AP-1 | [314] |
| Piceatannol | NHEK | 0–2 mg/mL for 24 h before UVB exposure | suppressed ROS generation; reduced MMP-1 induction | [315] |
| Fisetin | HaCaT | 1–20 mM for 12 h cotreated with H2O2 (500 mM) or pre-treatment for 6 h before TNF-α stimulation | reduced ROS production; inhibited IL-1β and IL-6 production; decreased iNOS and COX-2 expression; increased Nrf2-mediated HO-1 expression | [316] |
| Brown pine leaf extract (BPLE) andtrans-communic acid (TCA) | HaCaT, reconstructed human skin models | BPLE (5, 10 μg/mL) and TCA (5, 10 μM) for 1 h before UVB exposure | inhibited MMP-1 expression; suppressed AP-1 expression; inhibited Akt and PI3K phosphorylation | [317] |
| Orange peel extract | HaCaT | 0.1–10 mg/mL prior to UVB exposure | suppressed COX-2 and PGE2 expression; activation of PPAR-γ | [319] |
| Wogonin | NIH/3T3 mouse skin fibroblasts | TPA, IL-1β and TNF-α and 10–100 mM wogonin for up to 2 h | decreased COX-2 and iNOS expression | [345] |
| HaCaT | 0.1–10 mM for 72 h after UVB exposure | inhibited MMP-1 and IL-6; blocked MAPK/AP-1 and NF-κB pathways | [346] | |
| Baicalin | human skin samples | 6.25–25 mg/mL after UVB exposure | decreased number of SA-β-Gal-positive cells; reduced G0/G1-phase cells; decreased expression of p16INK4A, p21CIP/Waf1 and p53; decrease in γ-H2AX levels; decreased expression of MMP-1 and MMP-3 | [320] |
| Delphinidin | HaCaT | 5 or 10 µM before or after UVB exposure | restored elastic properties | [321] |
| Extracts from yerba mate | HaCaT, BJ fibroblasts | 100–1000 µg/mL extracts | enhanced viability; inhibited activity of lipoxygenase, collagenase and elastase enzymes | [322] |
| Extracts from leaves of Cleistocalyx nervosum var. paniala | human skin fibroblasts mushroom tyrosinase | 0.1 mg/ml | inhibition of MMP-2, ROS scavenging, lipid peroxidation inhibition, tyrosinase inhibition effect | [324] |
| Mangiferin | human dermal fibroblasts | 10 μM/50 μM; 2 h followed by addition of H2O2 (10 μM) | decreased ROS production, stabilized mitochondrial membrane potential and decreased the number of cell cycle arrested cells | [323] |
| Extracts from three species of seaweeds Alariaceae, Eisenia bicyclis, Ecklonia cava and Ecklonia stolonifera; eckol, dieckol, eckstolonol, triphlorethol-A and phloroglucinol | human dermal fibroblasts; HeLa cells transfected with the NF-κB or AP-1 luciferase reporter plasmid DNA; mushroom tyrosinase; B16F10 mouse melanoma cells; Zebrafish embryos; male 7-week-old Balb/c mice | 10 μg/mL extracts before treatment with TNF-α (10 ng/mL); exposure to UVB (50 mJ/cm2) + phlorotannins (0.5–250 μM); zebrafish embryos preincubated with 50 μM phlorotannins for 1 h; phloroglucinol (10 or 50 mg/mL) applied to dorsal skin plus UVB (30 or 60 mJ/cm2) | inhibited MMP-1; blocked AP-1 and NF-κB reporter activities; inhibition of tyrosinase, melanogenesis and DNA damage; reduction in ROS, NO, biomarkers of oxidative damage, cell death and hyperpigmentation in vivo; reduction in number of mast cells and increase in the epidermal and dermal thickness | [328,329,330] |
| Phloroglucinnol | human WI-38 fibroblasts | 10, 25, 50, or 100 μg/mL phloroglucinol for 24 h after tratment with 50 μM H2O2 for 60 min | decrease in MDA in prematurely senescent cells and viability increase | [331] |
| Polyphenol-rich extract from the seaweed Sargassum vachellianum | free radical scavenging, anti-tyrosinase activity and moisture absorption and retention assay | 200–1000 μg/mL | potential in scavenging OH radical, and effective absorption of the UVB and UVA rays | [333] |
| Polyphenol-rich root extracts from Potentilla atrosanguinea | determination of total phenol content; free radical scavenging activity | dried aqueous-methanolic (H2O/MeOH) crude extract and ethyl acetate (EtOAc), n-butanol (n-BuOH), as well as aqueous (H2O) fractions of roots were evaluated (200 μg/mL) | H2O/MeOH crude extract showed highest antioxidant of DPPH radical scavenging, O2.− scavenging and Cu2+ reducing activity; photoprotective agents in sunscreen preparation; effective natural antioxidant | [325] |
| Extract from tomato stem cell (Lycopersicon esculentum) | murin fibroblasts NIH-3T3; HaCaT | different concentration of the extract for 12 h or 2 h and/or CuSO4 for 30 min | reduced heavy metal-induced toxicity, restored DNA integrity under heavy metal stress; decreased collagen degradation and renewed collagen synthesis | [326] |
| Verbascoside | HaCaT | 100 or 200 μmol/L added 2 min before UVC irradiation (20 min, 1.8 J/cm2) | decreased AP-1 and NF-κB and decreased level of proinflammatory mediators | [327] |
| Extract from the parasitic mushroom Inonotus obliquus | skin fibroblasts, keratinocytes or reconstructed epidermis | 2% aqueous extract added 2 h before UV irradiation (UV-A (5 J/cm2) + UV-B (100 mJ/cm2) | reduced ROS formation, reduced quantity of pro-inflammatory cytokines and increased DNA repair activity | [334] |
| Extract of the mycelium of Tricholoma matsuke | human skin fibroblasts | 0.1–100 μg/mL for 72 h and 24 h treatment in μcombination with TPA | decreased elastase activity, reduced the MMPs level | [336] |
| Quercetin surface functionalized Fe3O4 nanoparticles | senescent human foreskin fibroblasts BJ; senescence induced by 100 μM H2O2 for 2 h | treatment with 5 μg/mL for 24 h | decreased number of stress-induced senescent cells; promoted AMPK activity; reduced IL-8 and IFN-β | [340] |
| Quercetin/dasatinib | senescent MEFs from Ercc1−/− mice | 48 h treatment dasatinib (250 nM), quercetin (50 μM) | Reduction in senescent and total cell counts | [144,146] |
| Fisetin | senescent MEFs from Ercc1−/− mice, IMR-90 fibroblasts | 48 h treatment, 1–15 μM | Reduction in the fraction of SA-ß-Gal-positive cells | [341] |
| Curcumin luteolin | senescent MEFs from Ercc1−/− mice | 48 h treatment, 5 μM | Reduction in the fraction of SA-ß-Gal-positive cells | [341] |
| Curcumin analog EF24 | senescent WI-38 and IMR-90 fibroblasts; senescence induced by replication, oncogene and IR | 72 h treatment | Selective killing of senescent cells; EC50 = 0.33–1.74 μM; proteasomal degradation of the Bcl-2 anti-apoptotic protein family proteins; independent of ROS | [342] |
| Rhododendron ferrugineum leaves extract | senescent NHDF; senescence induced by 500 µM H2O2 for 2 h | 48 h treatment, 1% extract | Reduction in SA-ß-Gal-positive cells | [344] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Csekes, E.; Račková, L. Skin Aging, Cellular Senescence and Natural Polyphenols. Int. J. Mol. Sci. 2021, 22, 12641. https://doi.org/10.3390/ijms222312641
Csekes E, Račková L. Skin Aging, Cellular Senescence and Natural Polyphenols. International Journal of Molecular Sciences. 2021; 22(23):12641. https://doi.org/10.3390/ijms222312641
Chicago/Turabian StyleCsekes, Erika, and Lucia Račková. 2021. "Skin Aging, Cellular Senescence and Natural Polyphenols" International Journal of Molecular Sciences 22, no. 23: 12641. https://doi.org/10.3390/ijms222312641
APA StyleCsekes, E., & Račková, L. (2021). Skin Aging, Cellular Senescence and Natural Polyphenols. International Journal of Molecular Sciences, 22(23), 12641. https://doi.org/10.3390/ijms222312641