
N-acetylcysteine Can Induce Massive Oxidative Stress, Resulting in Cell Death with Apoptotic Features in Human Leukemia Cells



Abstract
:1. Introduction
2. Results
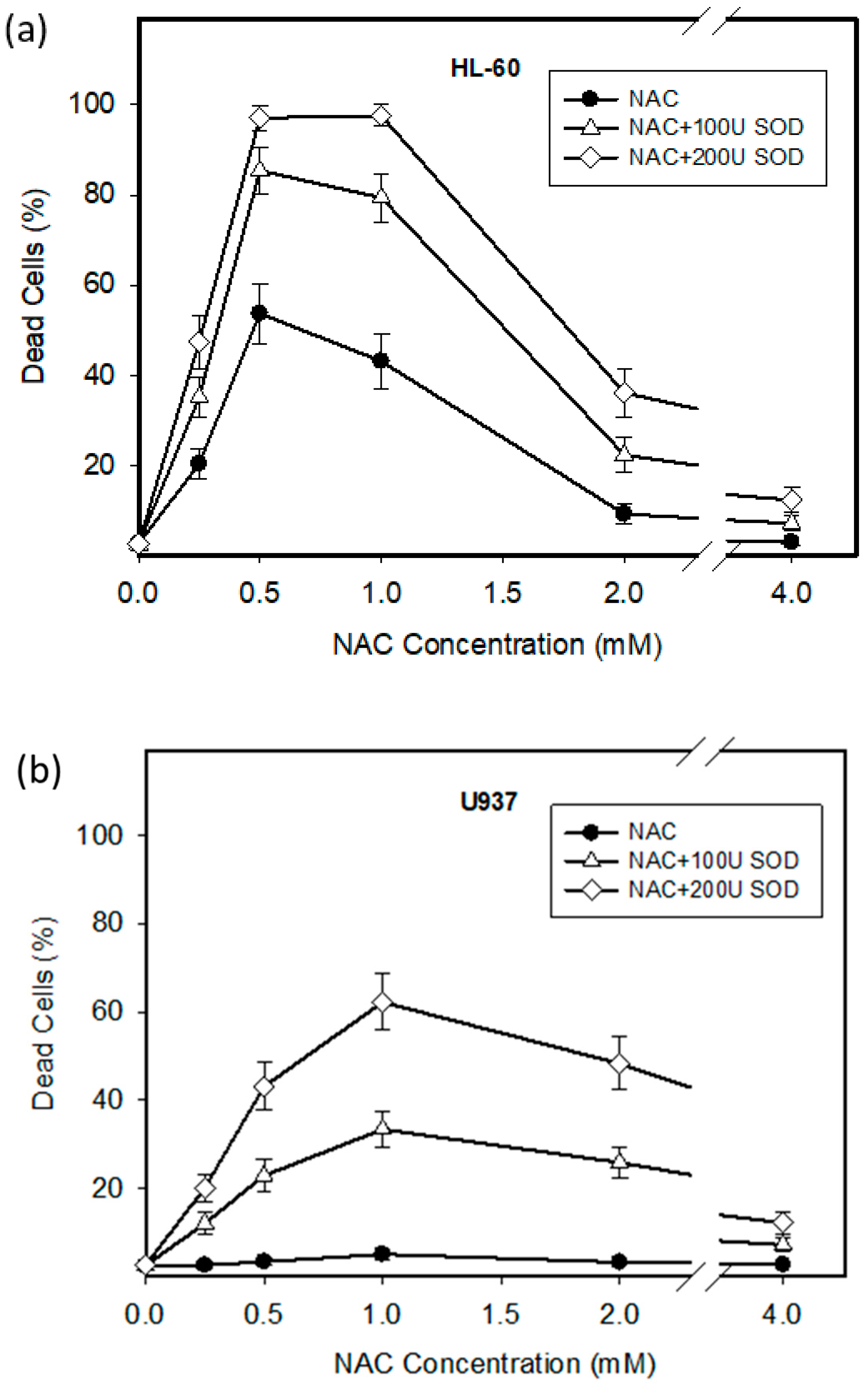
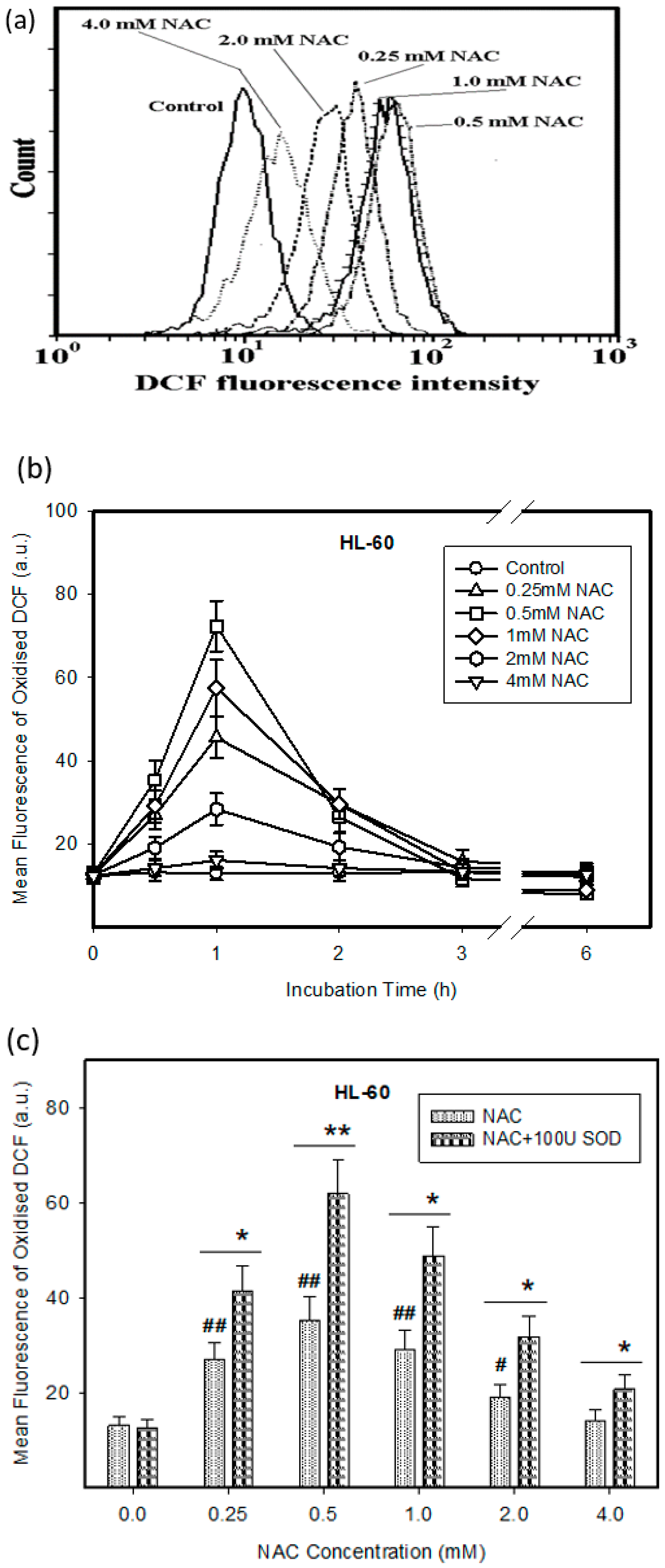
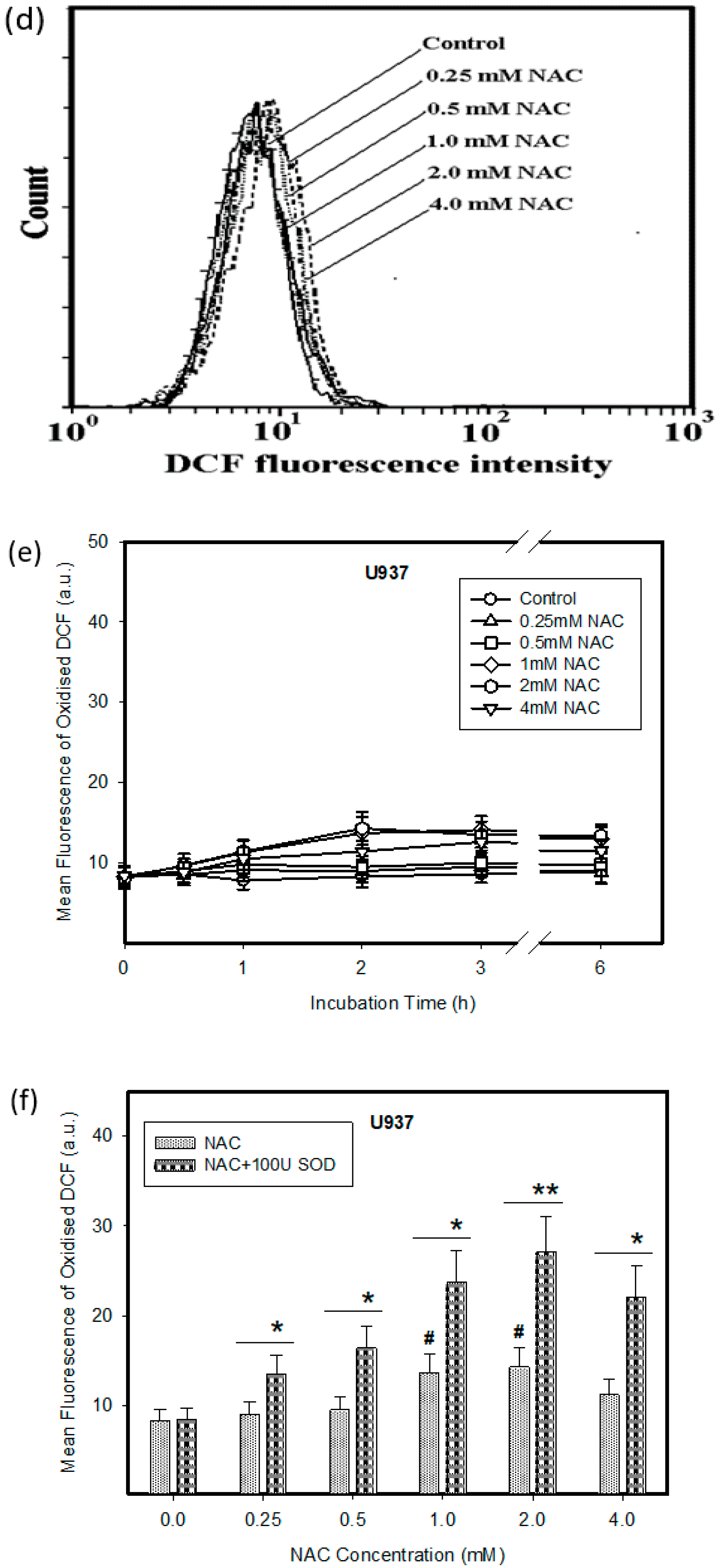
2.1. Cytotoxic Effect of NAC in Human Leukemia Cells and ROS Production
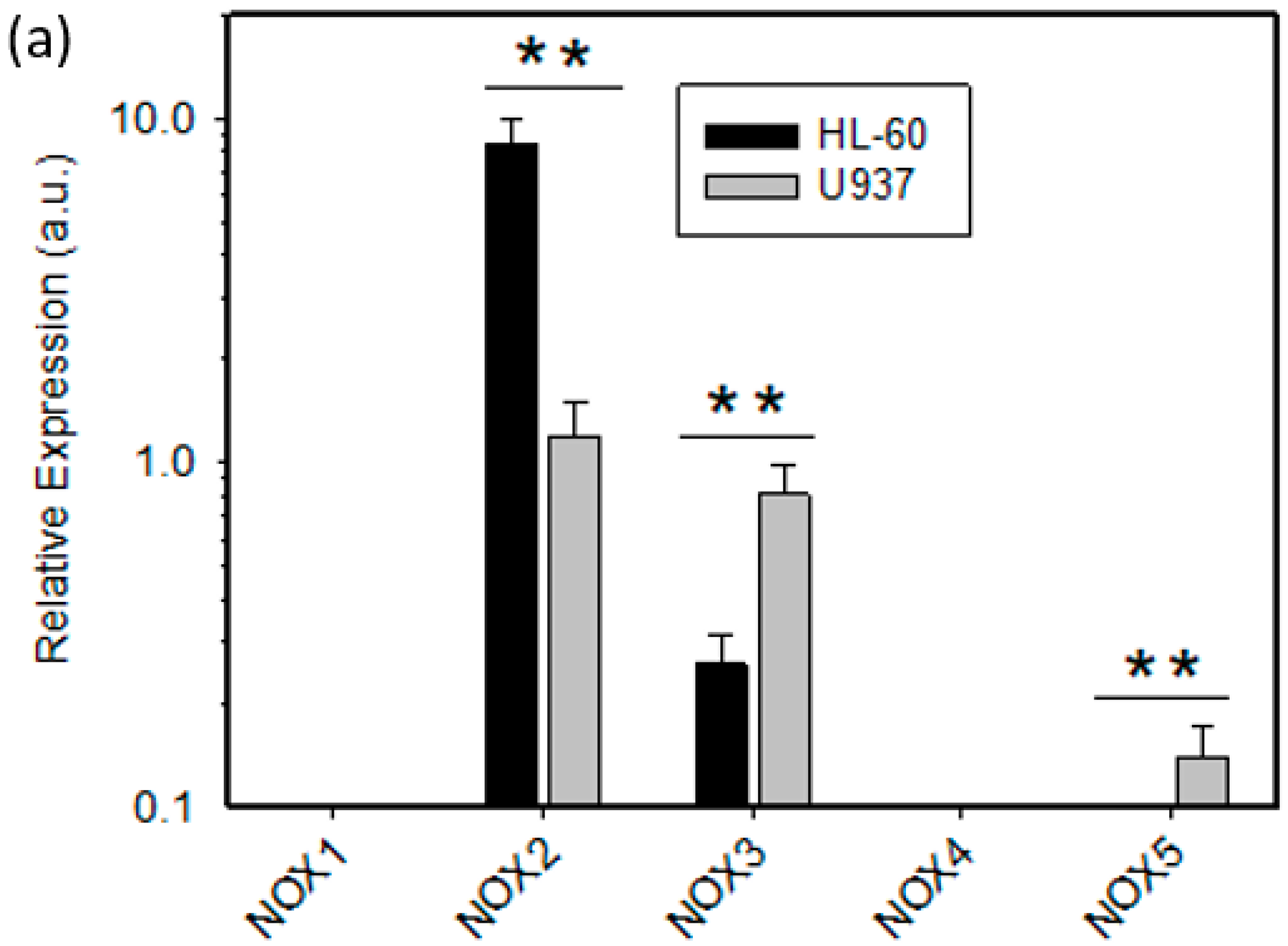
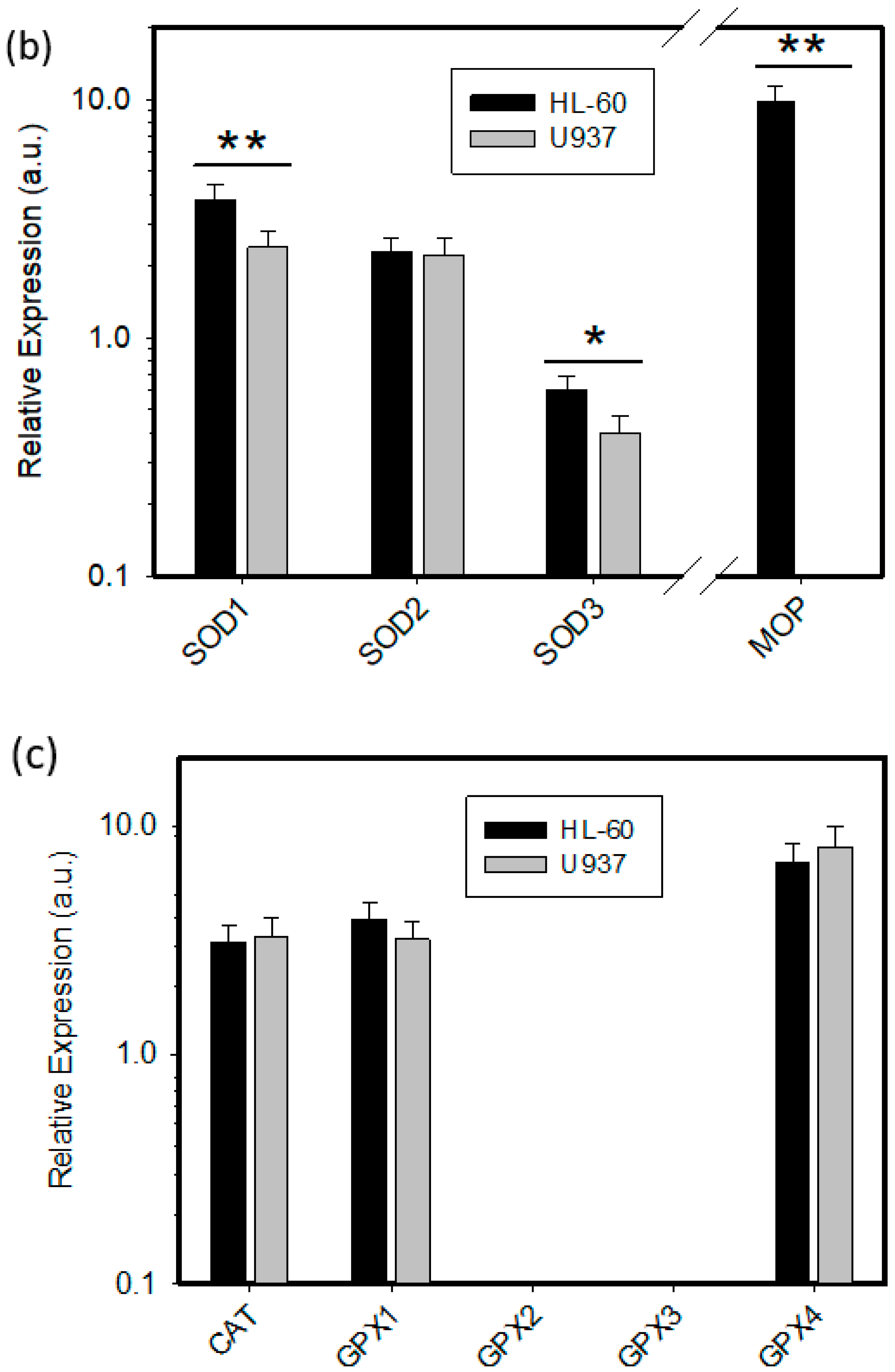
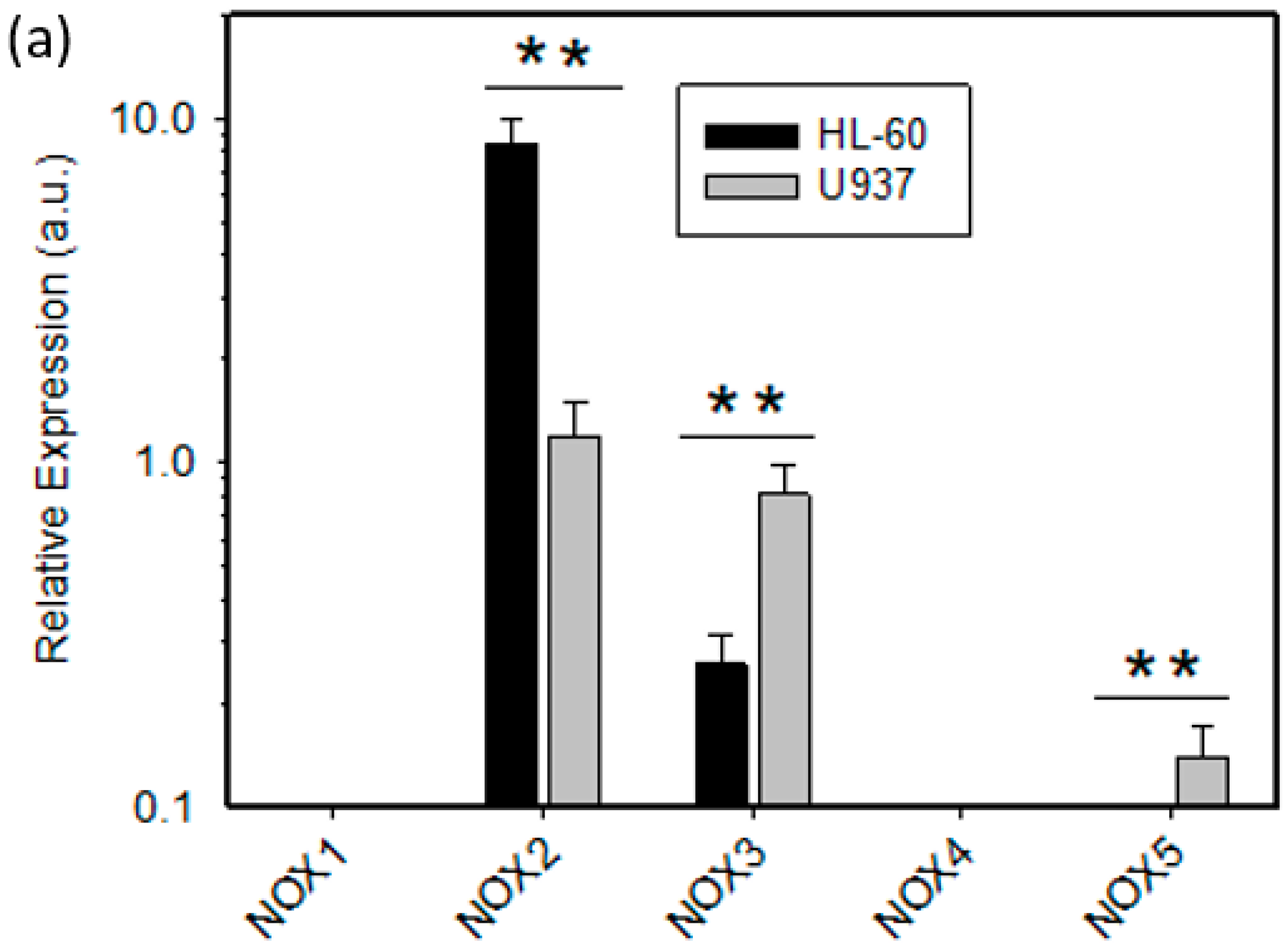
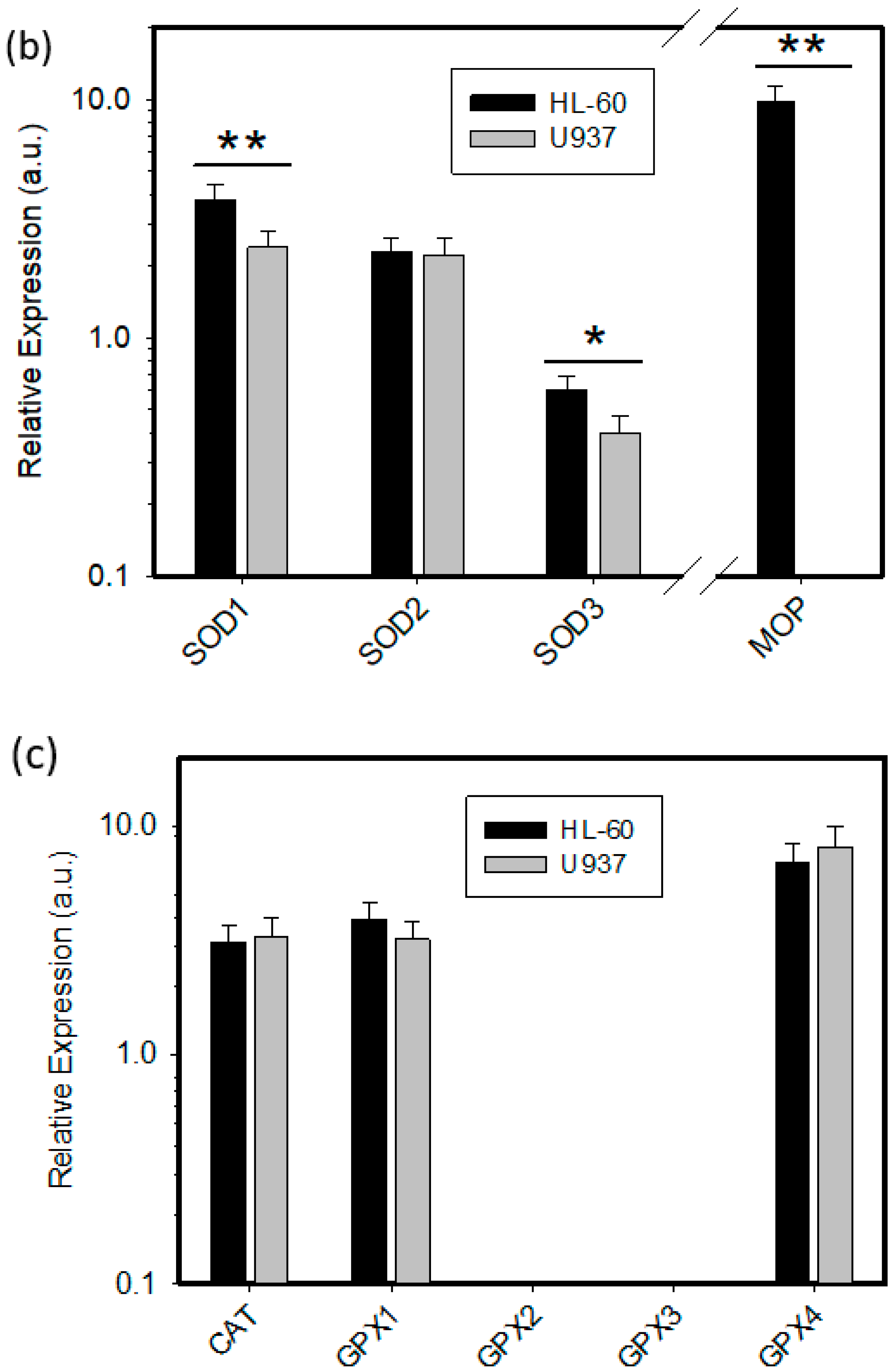
2.2. Expression of ROS Related Genes
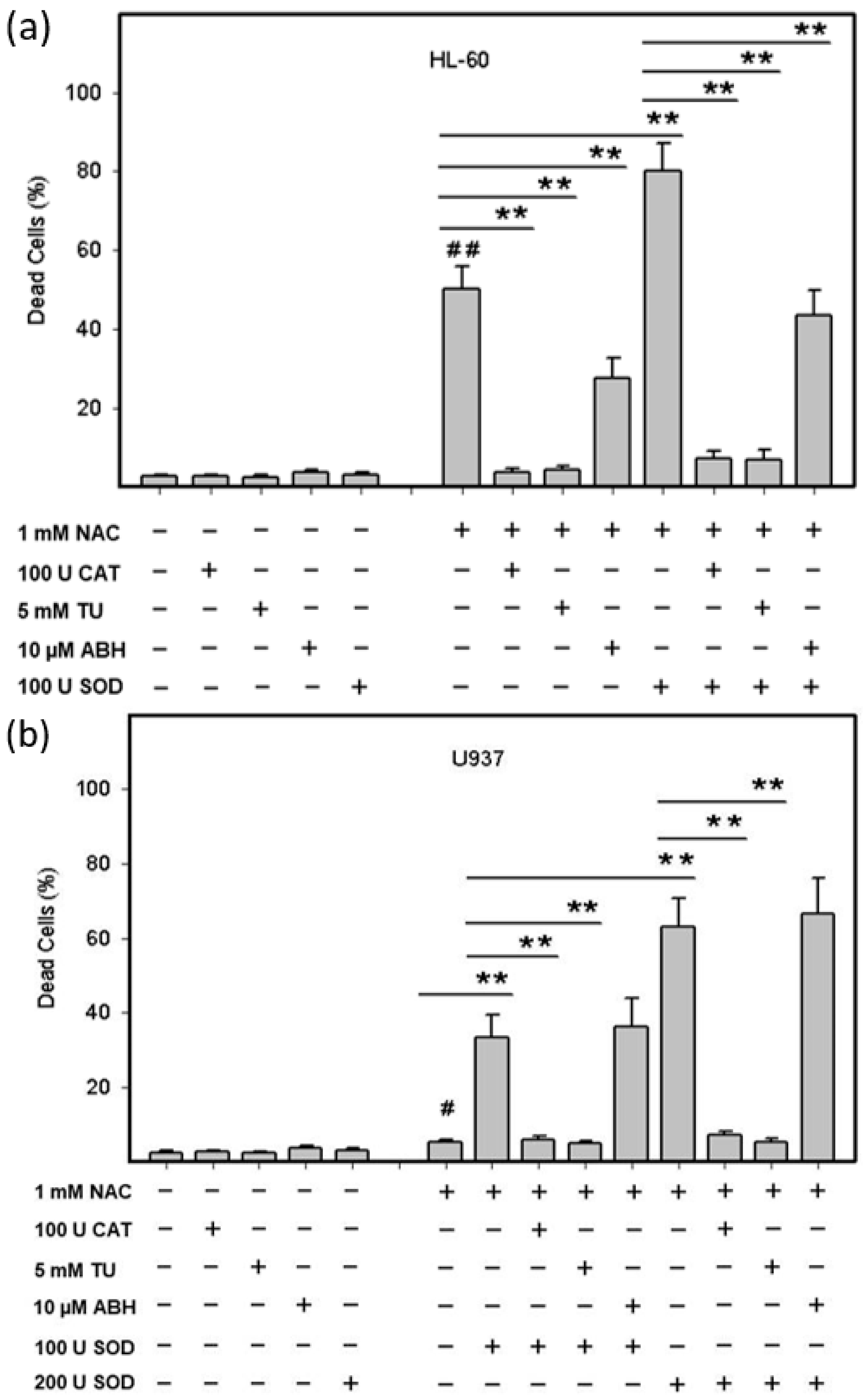
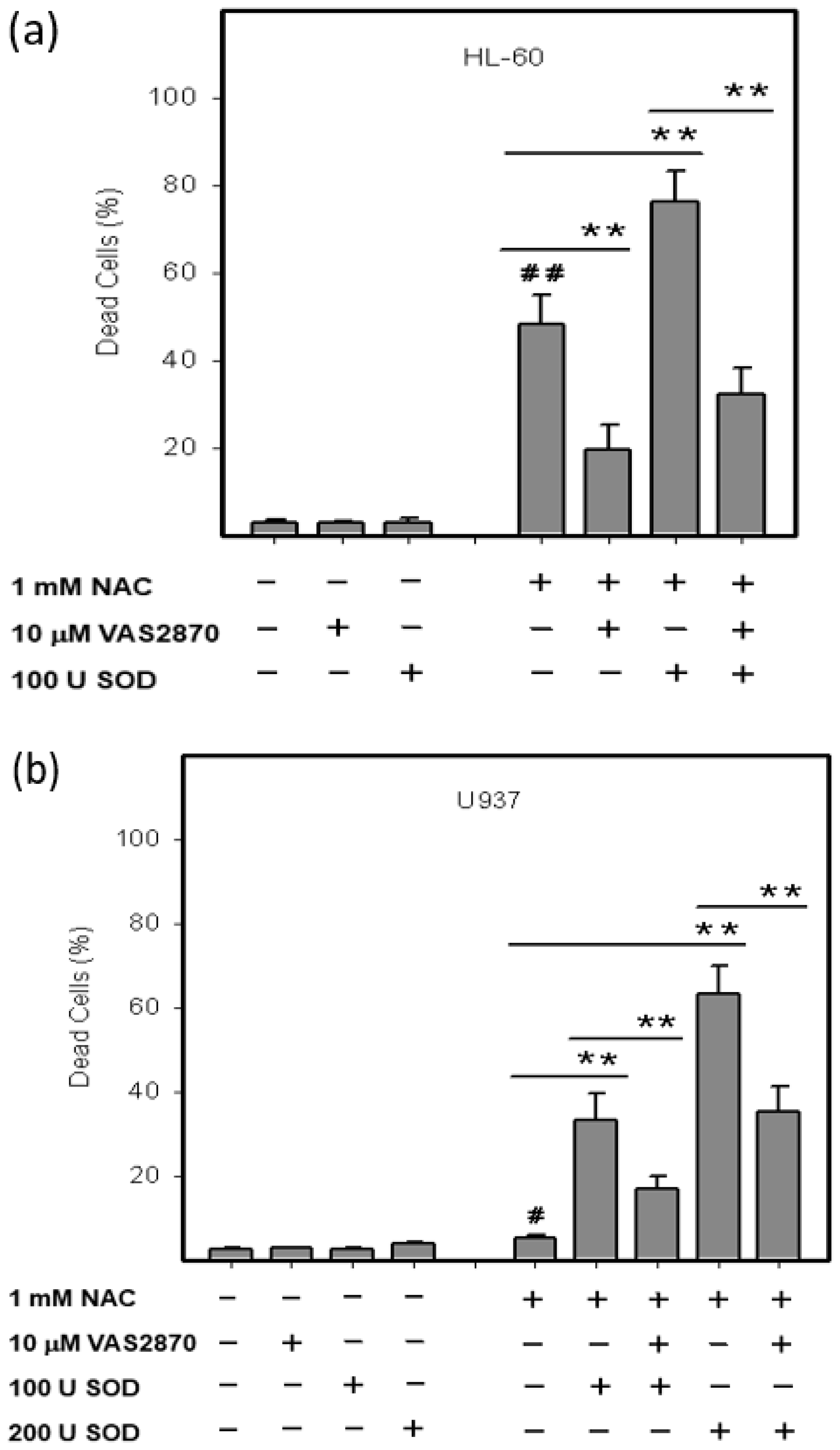
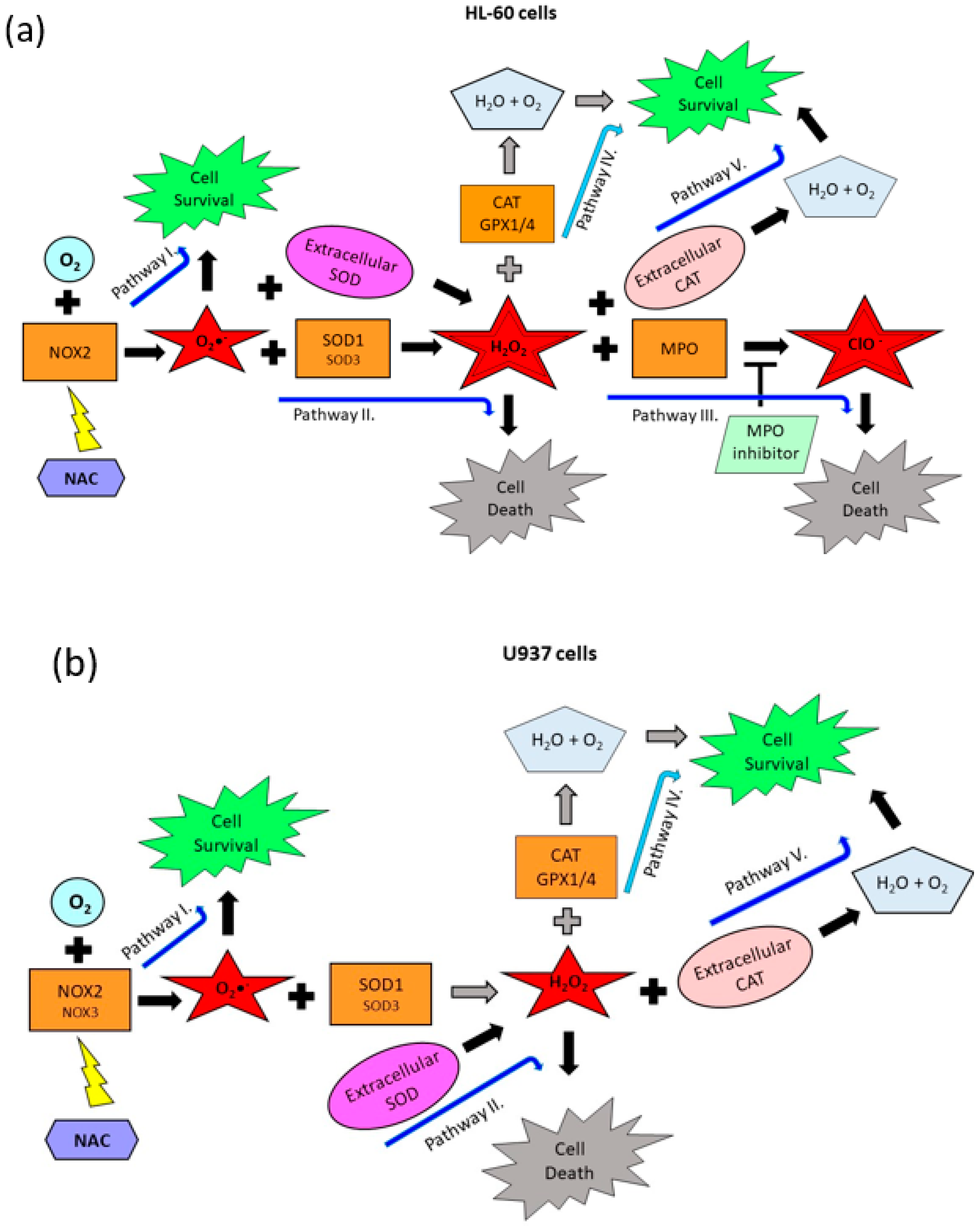
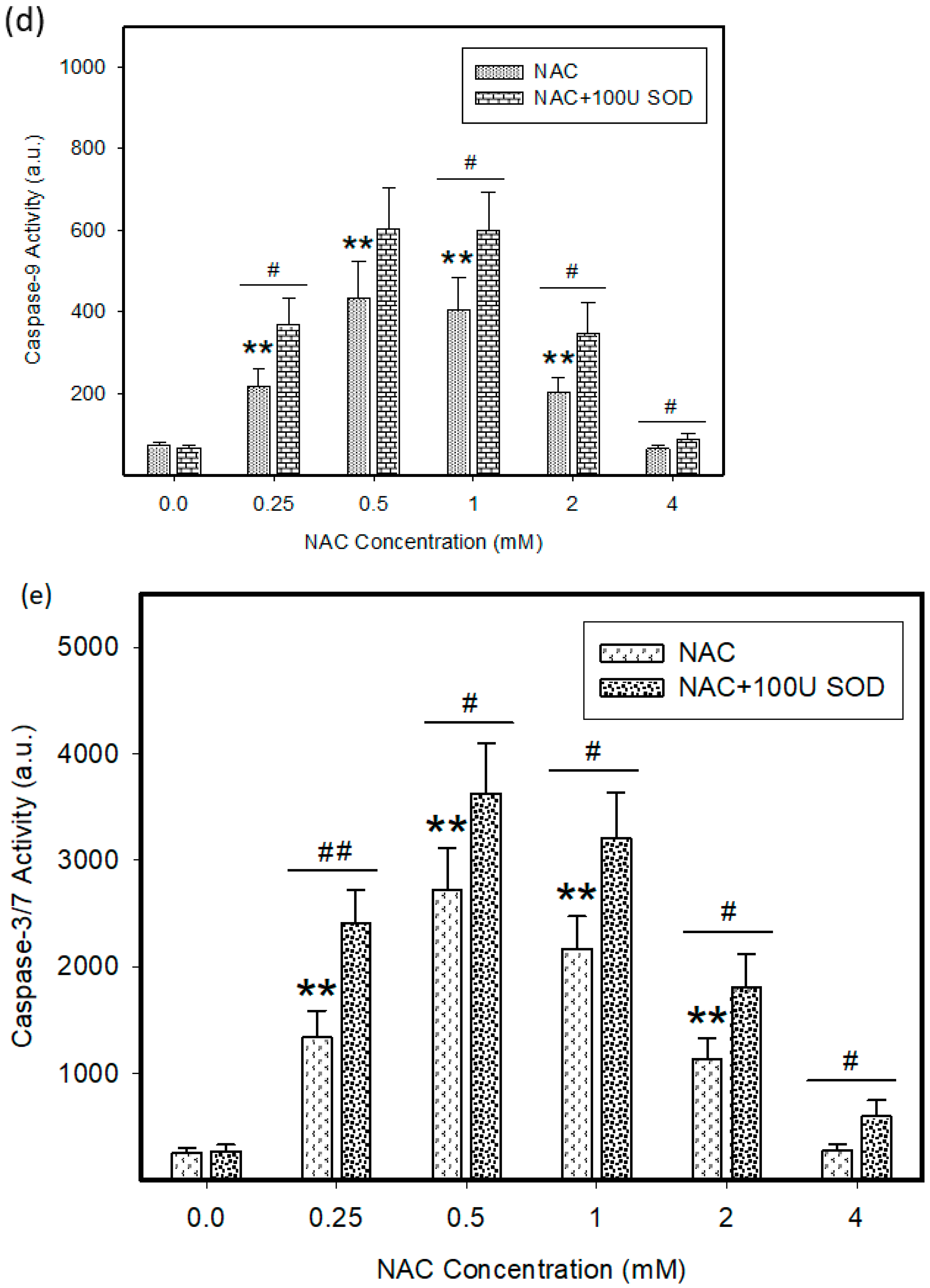
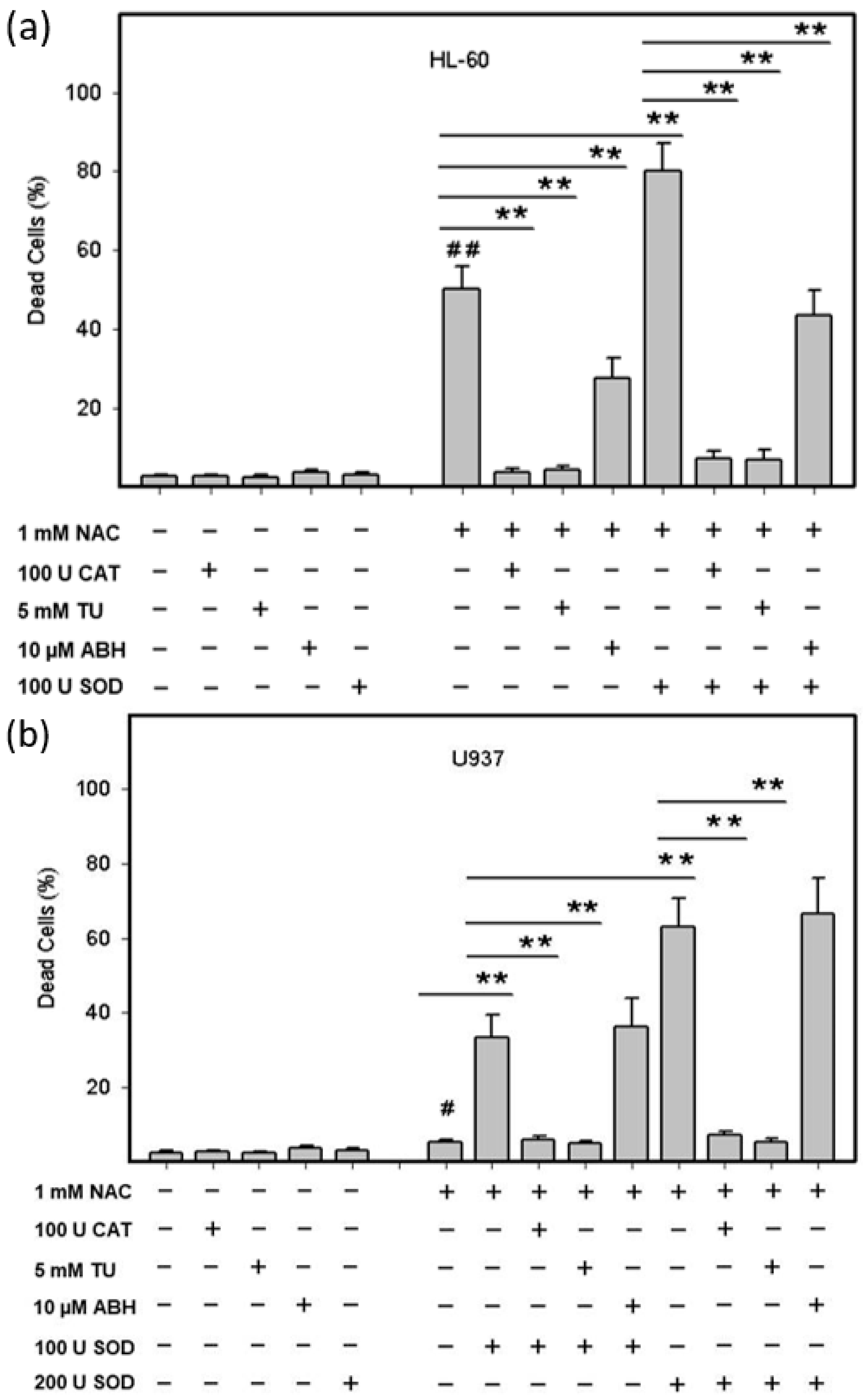
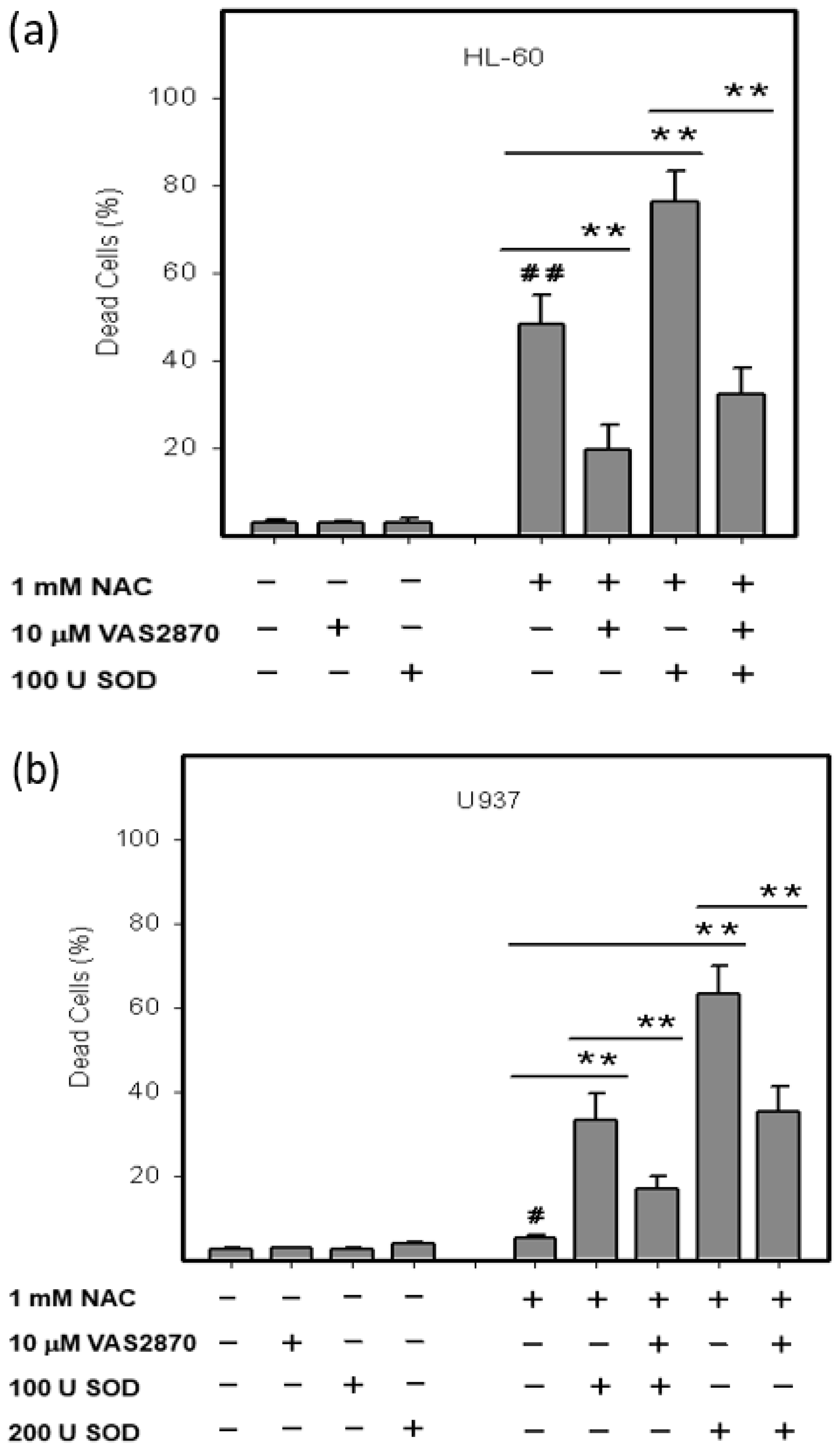
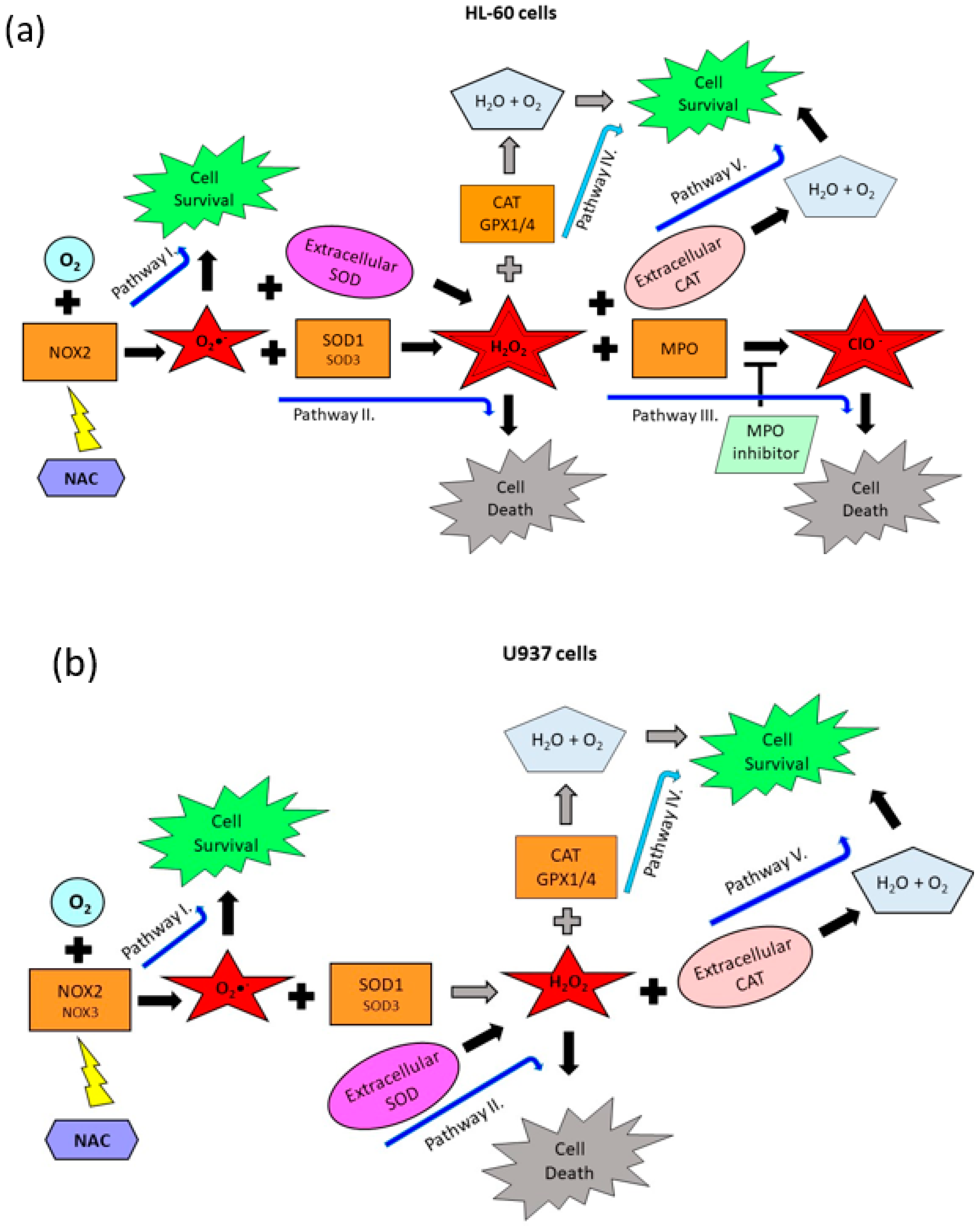
2.3. Origin and Interconversion of ROS and Their Relation to Cell Death
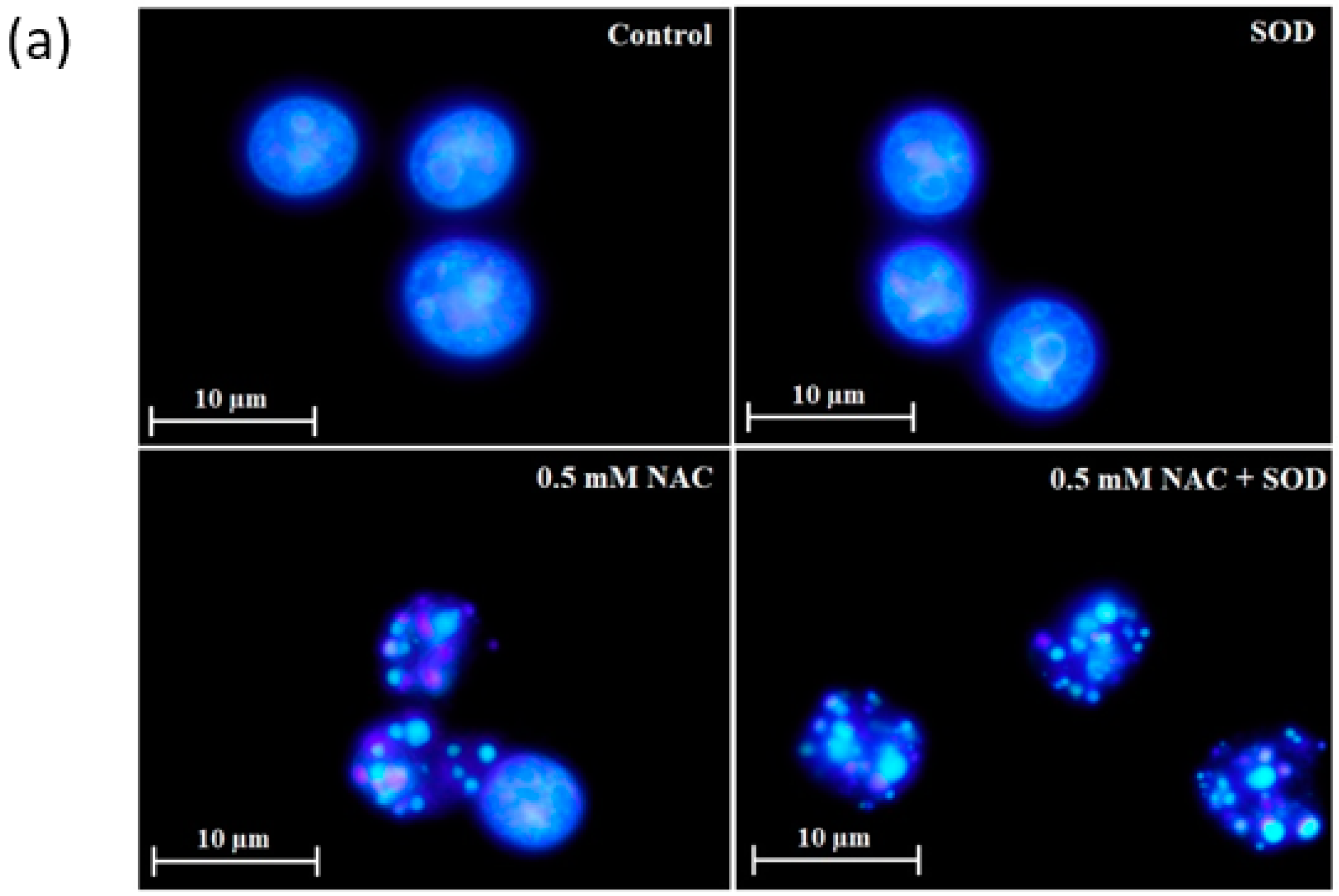
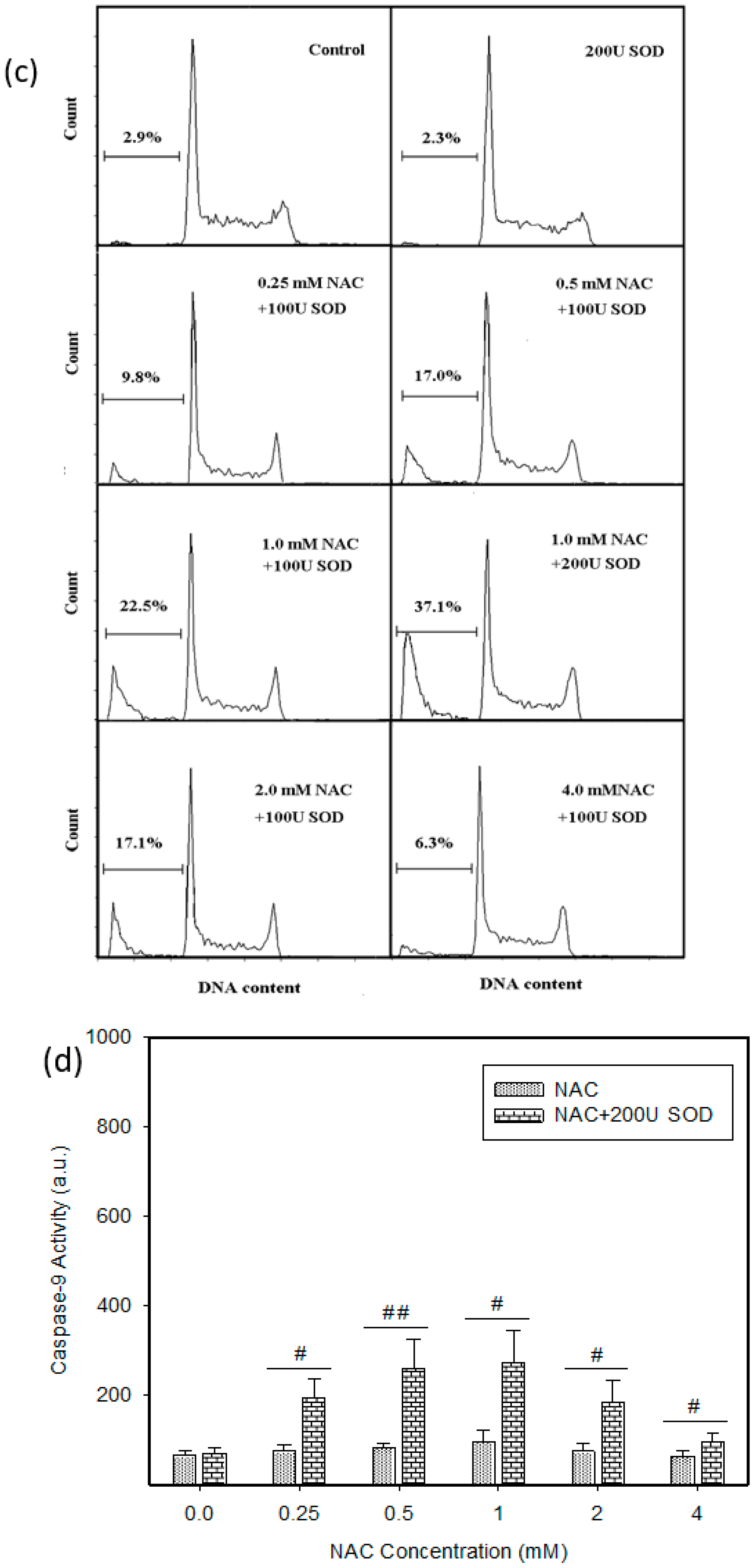
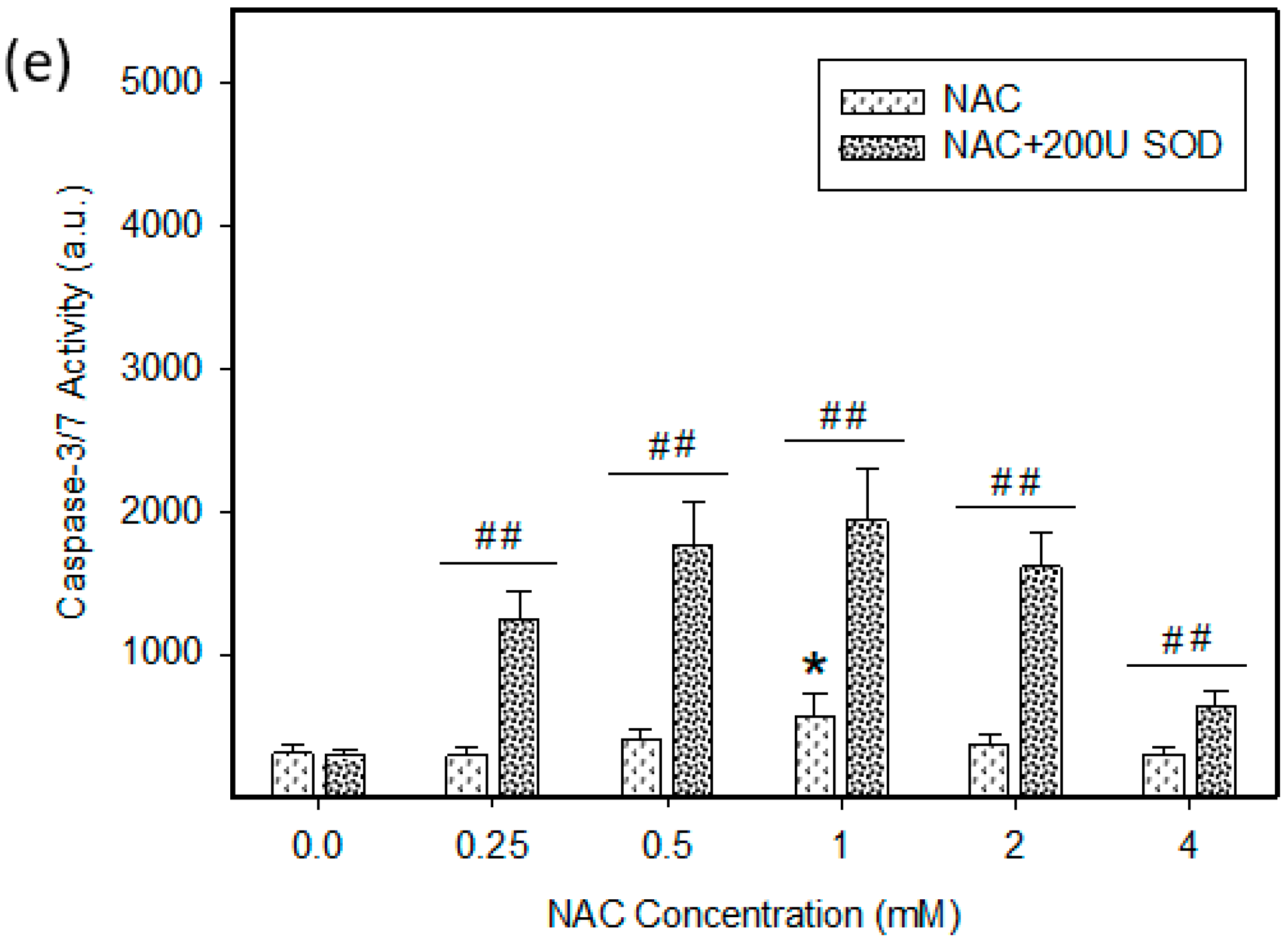
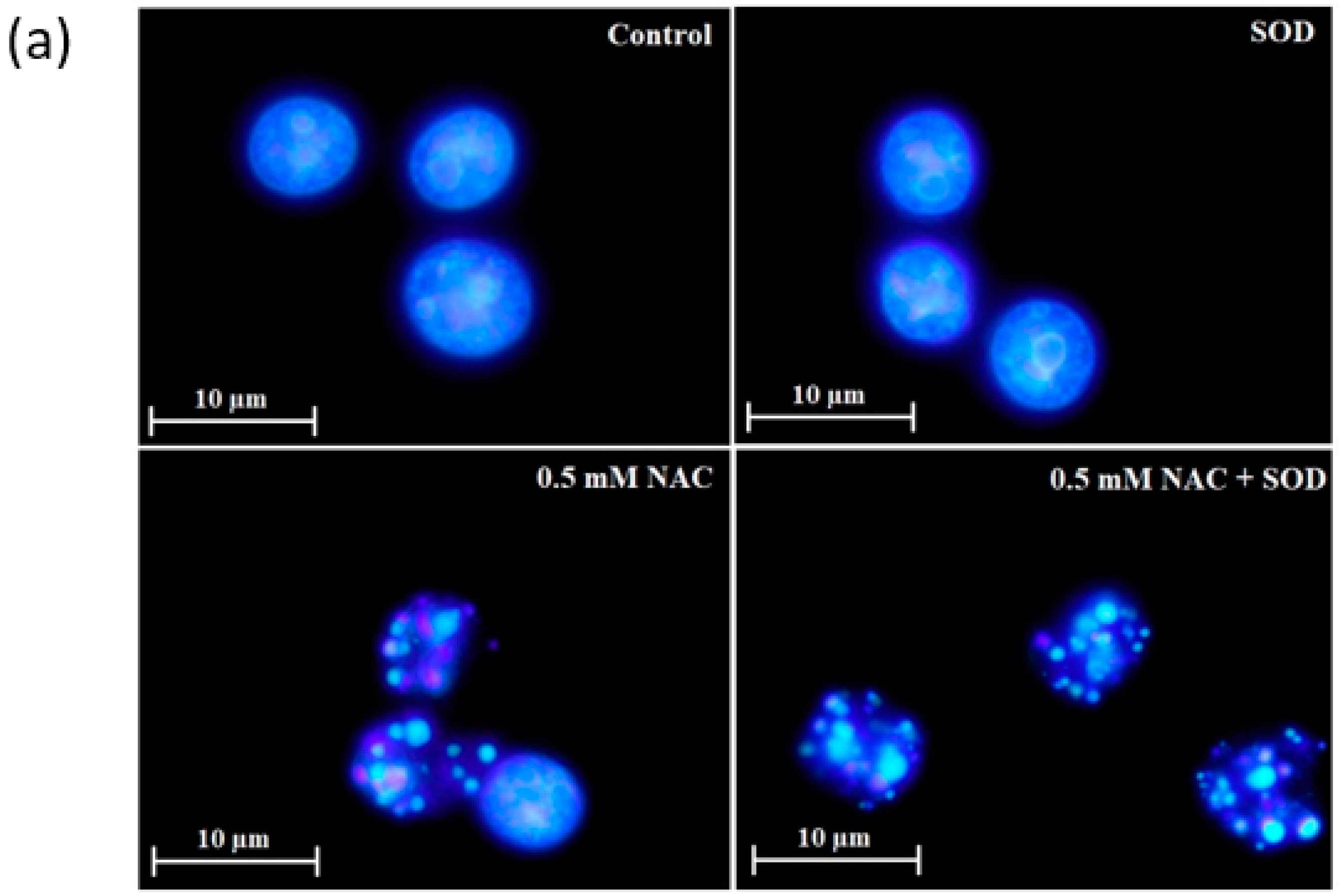
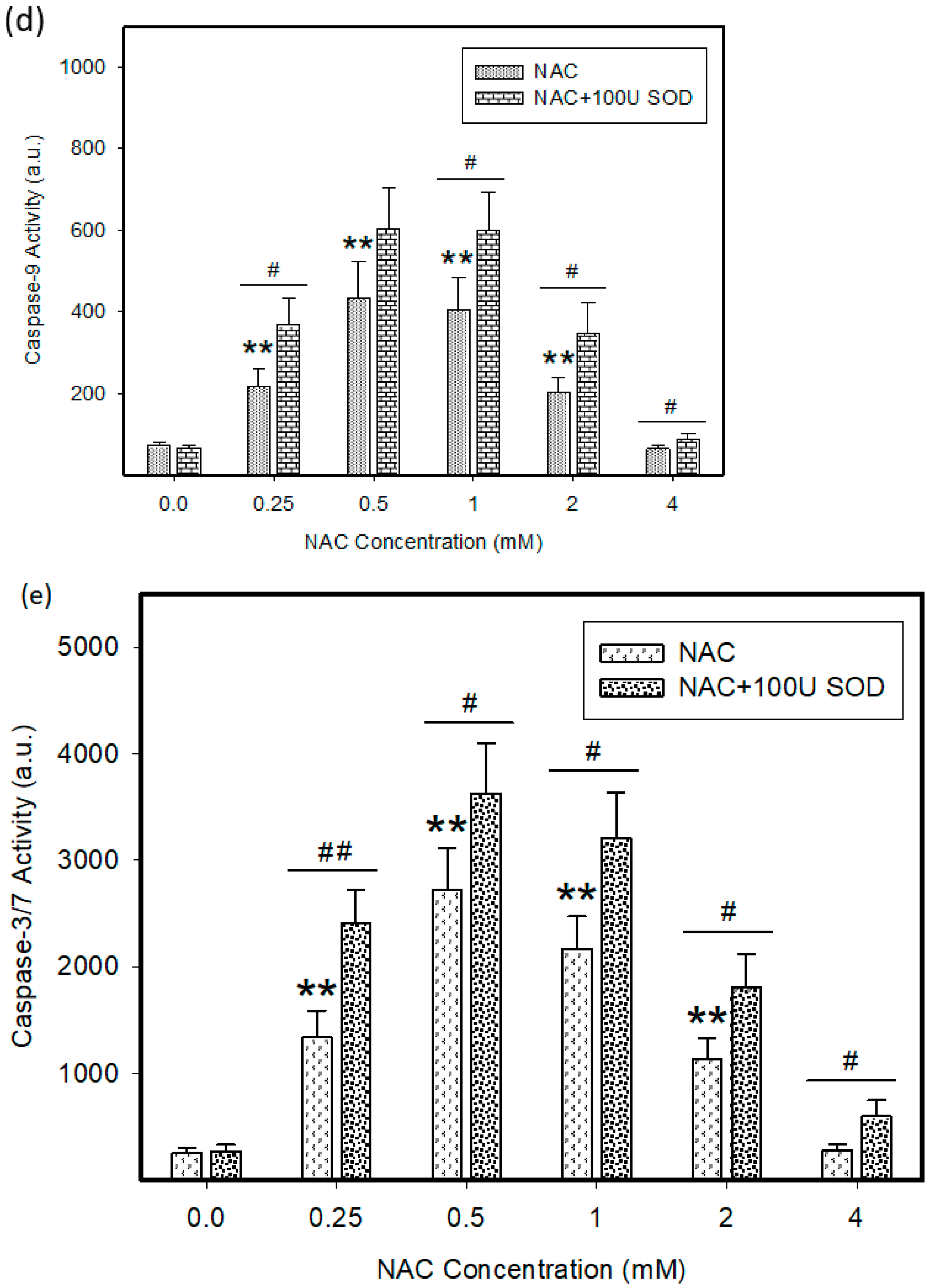
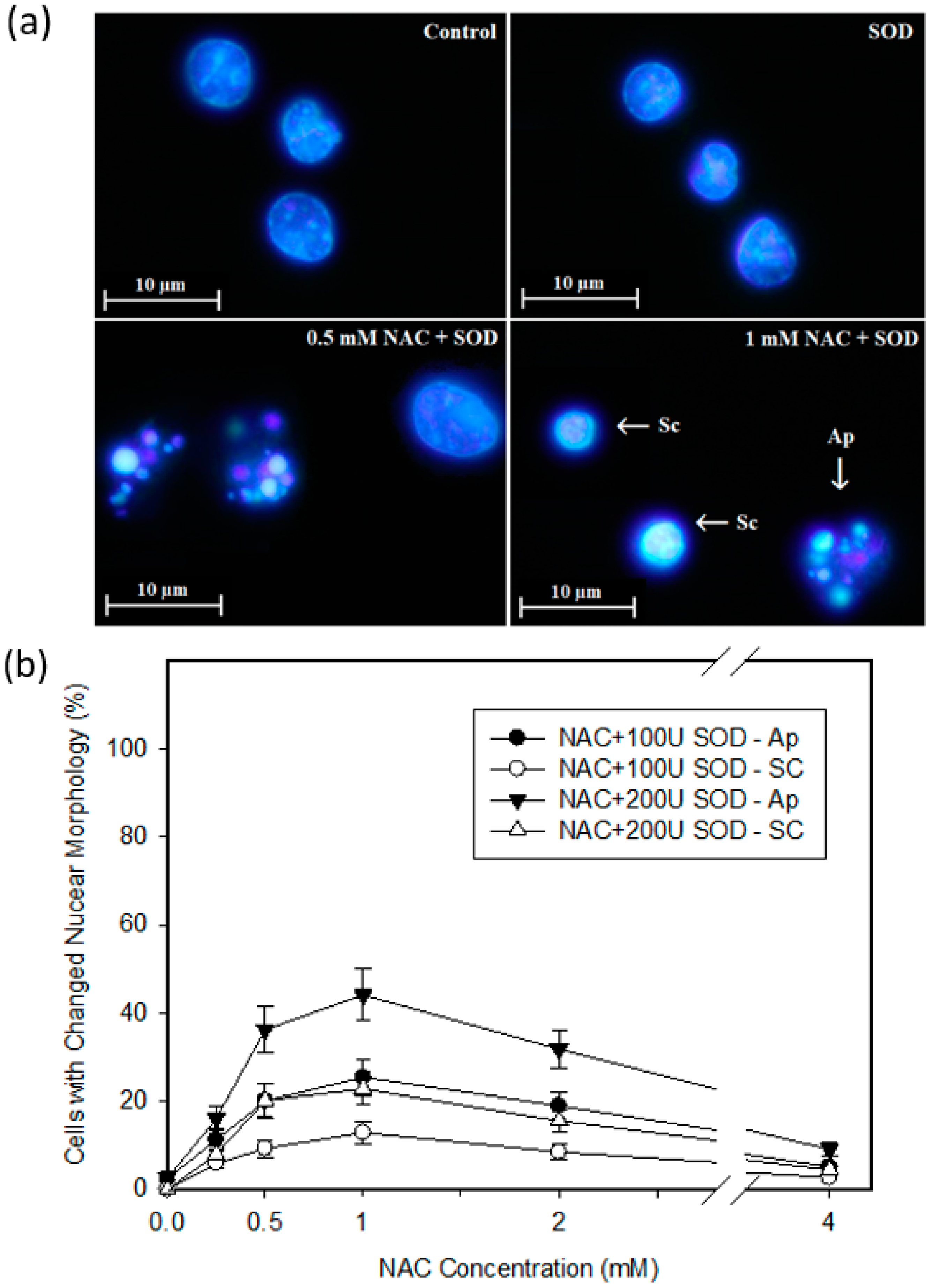
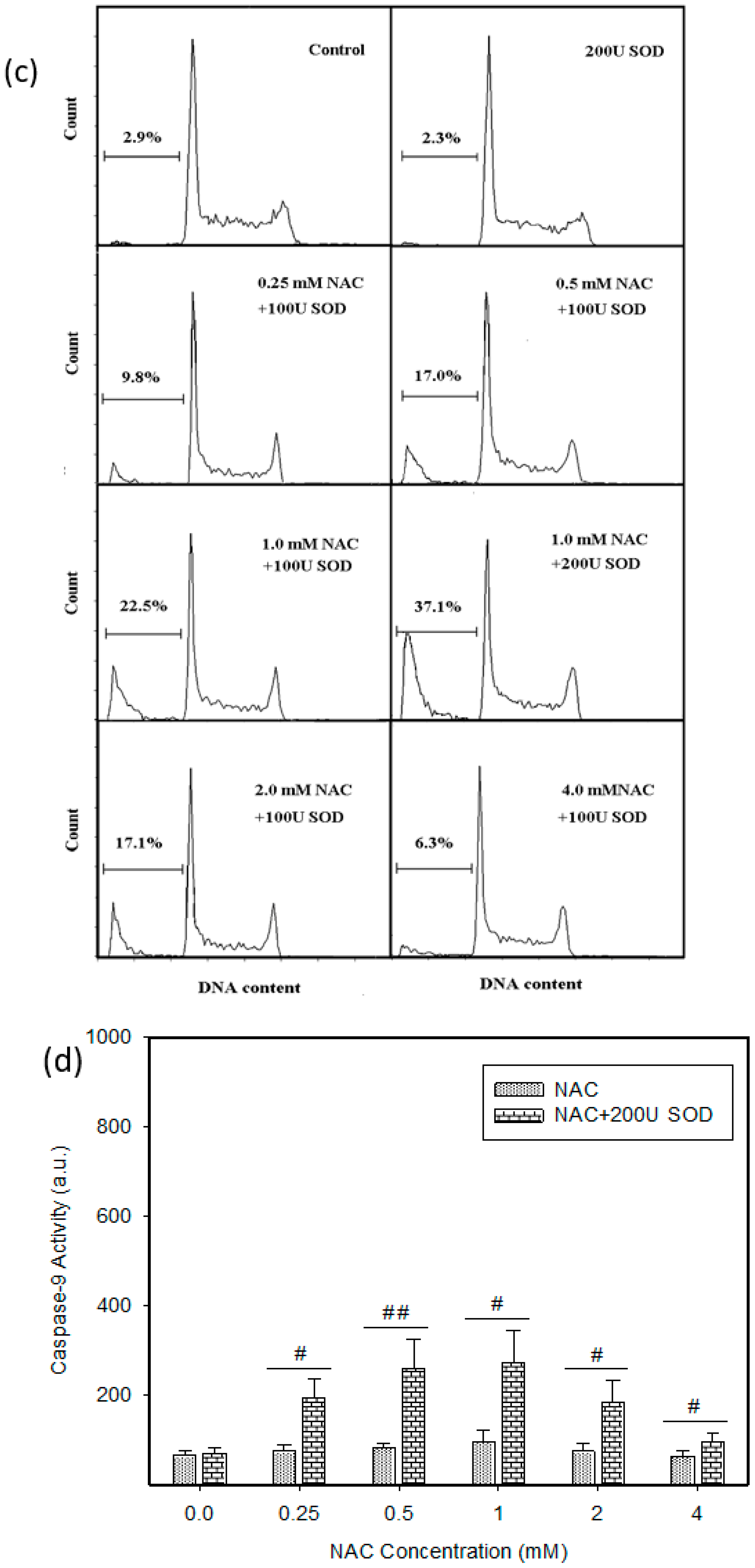
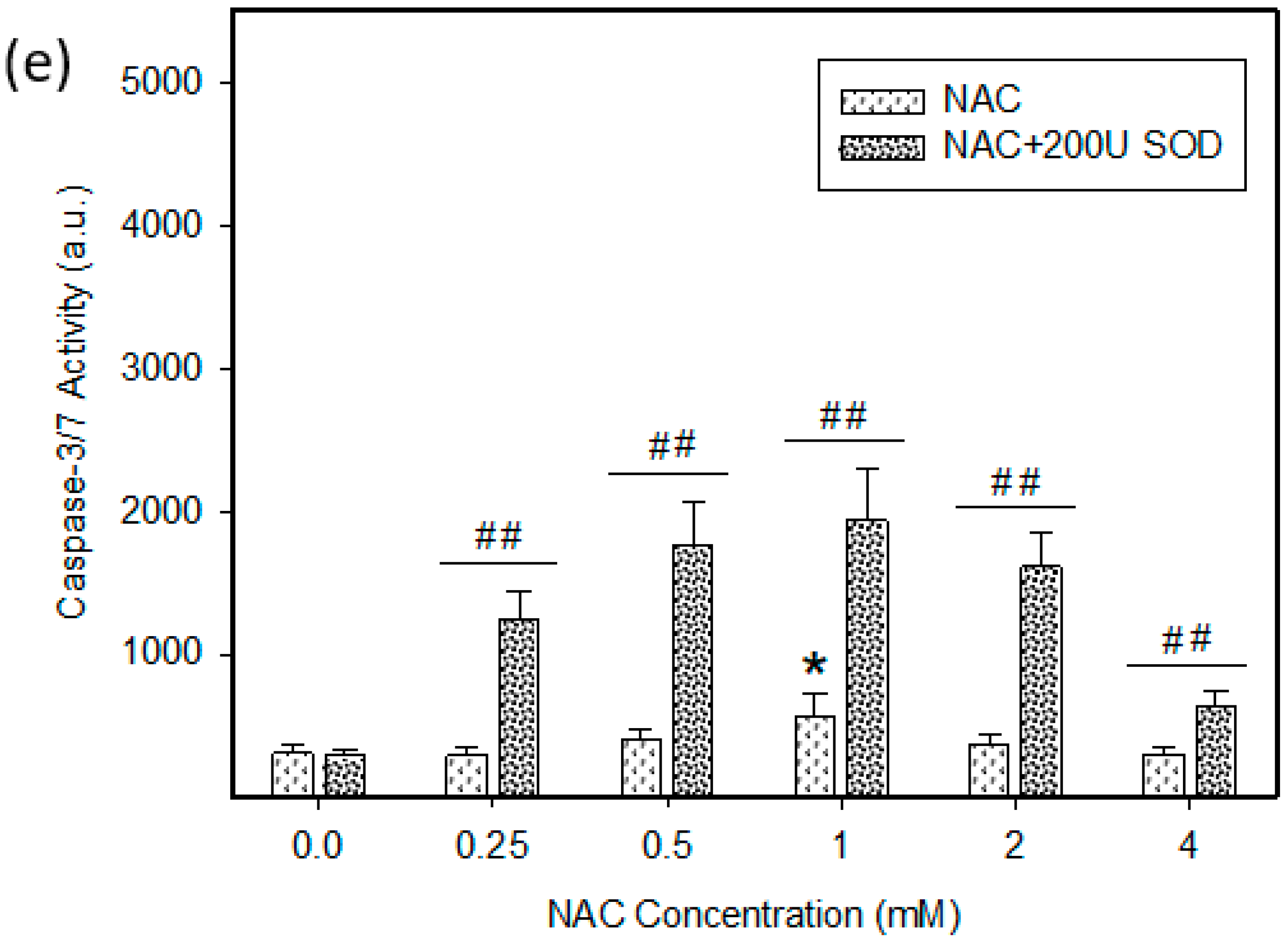
2.4. Mode of Cell Death Induced by NAC
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Chemicals and Cell Treatment
4.3. Measurement of ROS Production
4.4. mRNA Expression Profiling
4.5. Analysis of Cell Cycle and Apoptotic Cells
4.6. Morphological Analysis of Apoptosis
4.7. Measurement of Caspase Enzymatic Activity in Cell Extracts
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Flanagan, R.J.; Meredith, T. Use of N-acetylcysteine in clinical toxicology. Am. J. Med. 1991, 91, S131–S139. [Google Scholar] [CrossRef]
- Kelly, G.S. Clinical applications of N-acetylcysteine. Altern. Med. Rev. J. Clin. Ther. 1998, 3, 114–127. [Google Scholar]
- Millea, P.J. N-acetylcysteine: Multiple clinical applications. Am. Fam. Physician 2009, 80, 265–269. [Google Scholar] [PubMed]
- Rushworth, G.F.; Megson, I.L. Existing and potential therapeutic uses for N-acetylcysteine: The need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol. Ther. 2014, 141, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 4117–4129. [Google Scholar] [CrossRef] [PubMed]
- Aruoma, O.I.; Halliwell, B.; Hoey, B.M.; Butler, J. The antioxidant action of N-acetylcysteine: Its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic. Biol. Med. 1989, 6, 593–597. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Metodiewa, D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 1999, 27, 322–328. [Google Scholar] [CrossRef]
- de Flora, S.; Cesarone, C.F.; Balansky, R.M.; Albini, A.; D’Agostini, F.; Bennicelli, C.; Bagnasco, M.; Camoirano, A.; Scatolini, L.; Rovida, A.; et al. Chemopreventive properties and mechanisms of N-acetylcysteine. The experimental background. J. Cell. Biochem. 1995, 59, 33–41. [Google Scholar] [CrossRef]
- Mlejnek, P.; Dolezel, P. N-acetylcysteine prevents the geldanamycin cytotoxicity by forming geldanamycin–N-acetylcysteine adduct. Chem.-Biol. Interact. 2014, 220, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Mlejnek, P.; Dolezel, P. Loss of mitochondrial transmembrane potential and glutathione depletion are not sufficient to account for induction of apoptosis by carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone in human leukemia K562 cells. Chem.-Biol. Interact. 2015, 239, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Skoupa, N.; Dolezel, P.; Ruzickova, E.; Mlejnek, P. Apoptosis Induced by the Curcumin Analogue EF-24 Is Neither Mediated by Oxidative Stress-Related Mechanisms nor Affected by Expression of Main Drug Transporters ABCB1 and ABCG2 in Human Leukemia Cells. Int. J. Mol. Sci. 2017, 18, 2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgiou-Siafis, S.K.; Samiotaki, M.; Demopoulos, V.J.; Panayotou, G.; Tsiftsoglou, A.S. Formation of novel N-acetylcysteine-hemin adducts abrogates hemin-induced cytotoxicity and suppresses the NRF2-driven stress response in human pro-erythroid K562 cells. Eur. J. Pharmacol. 2020, 880, 173077. [Google Scholar] [CrossRef] [PubMed]
- Rakshit, S.; Bagchi, J.; Mandal, L.; Paul, K.; Ganguly, D.; Bhattacharjee, S.; Ghosh, M.; Biswas, N.; Chaudhuri, U.; Bandyopadhyay, S. N-acetyl cysteine enhances imatinib-induced apoptosis of Bcr-Abl+ cells by endothelial nitric oxide synthase-mediated production of nitric oxide. Apoptosis 2009, 14, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-S.; Lien, G.-S.; Shen, S.-C.; Yang, L.-Y.; Chen, Y.-C. N-acetyl-L-cysteine enhances fisetin-induced cytotoxicity via induction of ROS-independent apoptosis in human colonic cancer cells. Mol. Carcinog. 2014, 53, E119–E129. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Lou, J.R.; Zhang, X.-X.; Benbrook, D.M.; Hanigan, M.H.; Lind, S.E.; Ding, W.-Q. N-acetylcysteine interacts with copper to generate hydrogen peroxide and selectively induce cancer cell death. Cancer Lett. 2010, 298, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Mlejnek, P.; Dolezel, P.; Maier, V.; Kikalova, K.; Skoupa, N. N-acetylcysteine dual and antagonistic effect on cadmium cytotoxicity in human leukemia cells. Environ. Toxicol. Pharmacol. 2019, 71, 103213. [Google Scholar] [CrossRef] [PubMed]
- Kelner, M.J.; Bagnell, R.; Welch, K.J. Thioureas react with superoxide radicals to yield a sulfhydryl compound. Explanation for protective effect against paraquat. J. Biol. Chem. 1990, 265, 1306–1311. [Google Scholar] [CrossRef]
- Freyhaus, H.T.; Huntgeburth, M.; Wingler, K.; Schnitker, J.; Bäumer, A.T.; Vantler, M.; Bekhite, M.M.; Wartenberg, M.; Sauer, H.; Rosenkranz, S. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc. Res. 2006, 71, 331–341. [Google Scholar] [CrossRef]
- Mlejnek, P. Caspase inhibition and N6-benzyladenosine-induced apoptosis in HL-60 cells. J. Cell. Biochem. 2001, 83, 678–689. [Google Scholar] [CrossRef]
- Mlejnek, P. Caspase-3 Activity and Carbonyl Cyanide m-Chlorophenylhydrazone-induced Apoptosis in HL-60 cells. Altern. Lab. Anim. 2001, 29, 243–249. [Google Scholar] [CrossRef]
- Hayyan, M.; Hashim, M.A.; AlNashef, I.M. Superoxide Ion: Generation and Chemical Implications. Chem. Rev. 2016, 116, 3029–3085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, K.; Krause, K.H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008, 275, 3249–3277. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.; Chen, Y.; Gibson, S.B. Regulation of Autophagy by Reactive Oxygen Species (ROS): Implications for Cancer Progression and Treatment. Antioxid. Redox Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Sedlářová, M.; Balukova, A.; Ovsii, A.; Rác, M.; Křupka, M.; Kasai, S.; Pospíšil, P. Reactive Oxygen Species Imaging in U937 Cells. Front. Physiol. 2020, 11, 552569. [Google Scholar] [CrossRef]
- Borutaite, V.; Brown, G.C. Caspases are reversibly inactivated by hydrogen peroxide. FEBS Lett. 2001, 500, 114–118. [Google Scholar] [CrossRef]
- Phillips, H.; Terryberry, J. Counting actively metabolizing tissue cultured cells. Exp. Cell Res. 1957, 13, 341–347. [Google Scholar] [CrossRef]
- LeBel, C.P.; Ischiropoulos, H.; Bondy, S.C. Evaluation of the probe 2’,7’-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem. Res. Toxicol. 1992, 5, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Dyskova, T.; Fillerova, R.; Novosad, T.; Kudelka, M.; Zurkova, M.; Gajdos, P.; Kolek, V.; Kriegova, E. Correlation Network Analysis Reveals Relationships between MicroRNAs, Transcription FactorT-bet, and Deregulated Cytokine/Chemokine-Receptor Network in Pulmonary Sarcoidosis. Mediat. Inflamm. 2015, 2015, 121378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrackova, A.; Horak, P.; Radvansky, M.; Fillerova, R.; Kraiczova, V.S.; Kudelka, M.; Mrazek, F.; Skacelova, M.; Smrzova, A.; Kriegova, E. Revealed heterogeneity in rheumatoid arthritis based on multivariate innate signature analysis. Clin. Exp. Rheumatol 2019, 38, 289–298. [Google Scholar] [PubMed]
- Kriegova, E.; Arakelyan, A.; Fillerova, R.; Zatloukal, J.; Mrazek, F.; Navratilova, Z.; Kolek, V.; Du Bois, R.M.; Petrek, M. PSMB2 and RPL32 are suitable denominators to normalize gene expression profiles in bronchoalveolar cells. BMC Mol. Biol. 2008, 9, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Bruno, S.; del Bino, G.; Gorczyca, W.; Hotz, M.A.; Lassota, P.; Traganos, F. Features of apoptotic cells measured by flow cytometry. Cytometry 1992, 13, 795–808. [Google Scholar] [CrossRef]
- Mlejnek, P.; Kuglík, P. Induction of apoptosis in HL-60 cells by N(6)-benzyladenosine. J. Cell. Biochem. 2000, 77, 6–17. [Google Scholar] [CrossRef]
- Frydrych, I.; Mlejnek, P. Serine protease inhibitors N-α-Tosyl-L-Lysinyl-Chloromethylketone (TLCK) and N-Tosyl-L-Phenylalaninyl-Chloromethylketone (TPCK) are potent inhibitors of activated caspase proteases. J. Cell. Biochem. 2008, 103, 1646–1656. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| NOX1 | AAGGATCCTCCGGTTTTACC | TTTGGATGGGTGCATAACAA |
| NOX2 | GGCTTCCTCAGCTACAACATCT | GTGCACAGCAAAGTGATTGG |
| SOD1 | TCATCAATTTCGAGCAGAAGG | GCAGGCCTTCAGTCAGTCC |
| SOD2 | GCACTAGCAGCATGTTGAGC | CCGTAGTCGTAGGGCAGGT |
| SOD3 | GGTGCAGCTCTCTTTTCAGG | AACACAGTAGCGCCAGCAT |
| CAT | TCATCAGGGATCCCATATTGTT | CCTTCAGATGTGTCTGAGGATTT |
| GPX1 | CAACCAGTTTGGGCATCAG | TCTCGAAGAGCATGAAGTTGG |
| GPX4 | TTCCCGTGTAACCAGTTCG | CGGCGAACTCTTTGATCTCT |
| MPO | CGTCAACTGCGAGACCAG | GTCATTGGGCGGGATCTT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mlejnek, P.; Dolezel, P.; Kriegova, E.; Pastvova, N. N-acetylcysteine Can Induce Massive Oxidative Stress, Resulting in Cell Death with Apoptotic Features in Human Leukemia Cells. Int. J. Mol. Sci. 2021, 22, 12635. https://doi.org/10.3390/ijms222312635
Mlejnek P, Dolezel P, Kriegova E, Pastvova N. N-acetylcysteine Can Induce Massive Oxidative Stress, Resulting in Cell Death with Apoptotic Features in Human Leukemia Cells. International Journal of Molecular Sciences. 2021; 22(23):12635. https://doi.org/10.3390/ijms222312635
Chicago/Turabian StyleMlejnek, Petr, Petr Dolezel, Eva Kriegova, and Nikola Pastvova. 2021. "N-acetylcysteine Can Induce Massive Oxidative Stress, Resulting in Cell Death with Apoptotic Features in Human Leukemia Cells" International Journal of Molecular Sciences 22, no. 23: 12635. https://doi.org/10.3390/ijms222312635
APA StyleMlejnek, P., Dolezel, P., Kriegova, E., & Pastvova, N. (2021). N-acetylcysteine Can Induce Massive Oxidative Stress, Resulting in Cell Death with Apoptotic Features in Human Leukemia Cells. International Journal of Molecular Sciences, 22(23), 12635. https://doi.org/10.3390/ijms222312635