
Pseudophosphatases as Regulators of MAPK Signaling

Abstract
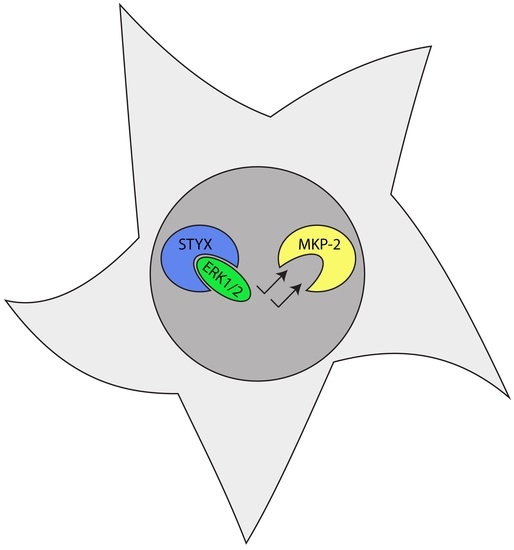
:
1. Introduction
1.1. Mitogen-Activated Protein Kinase (MAPK) Signaling
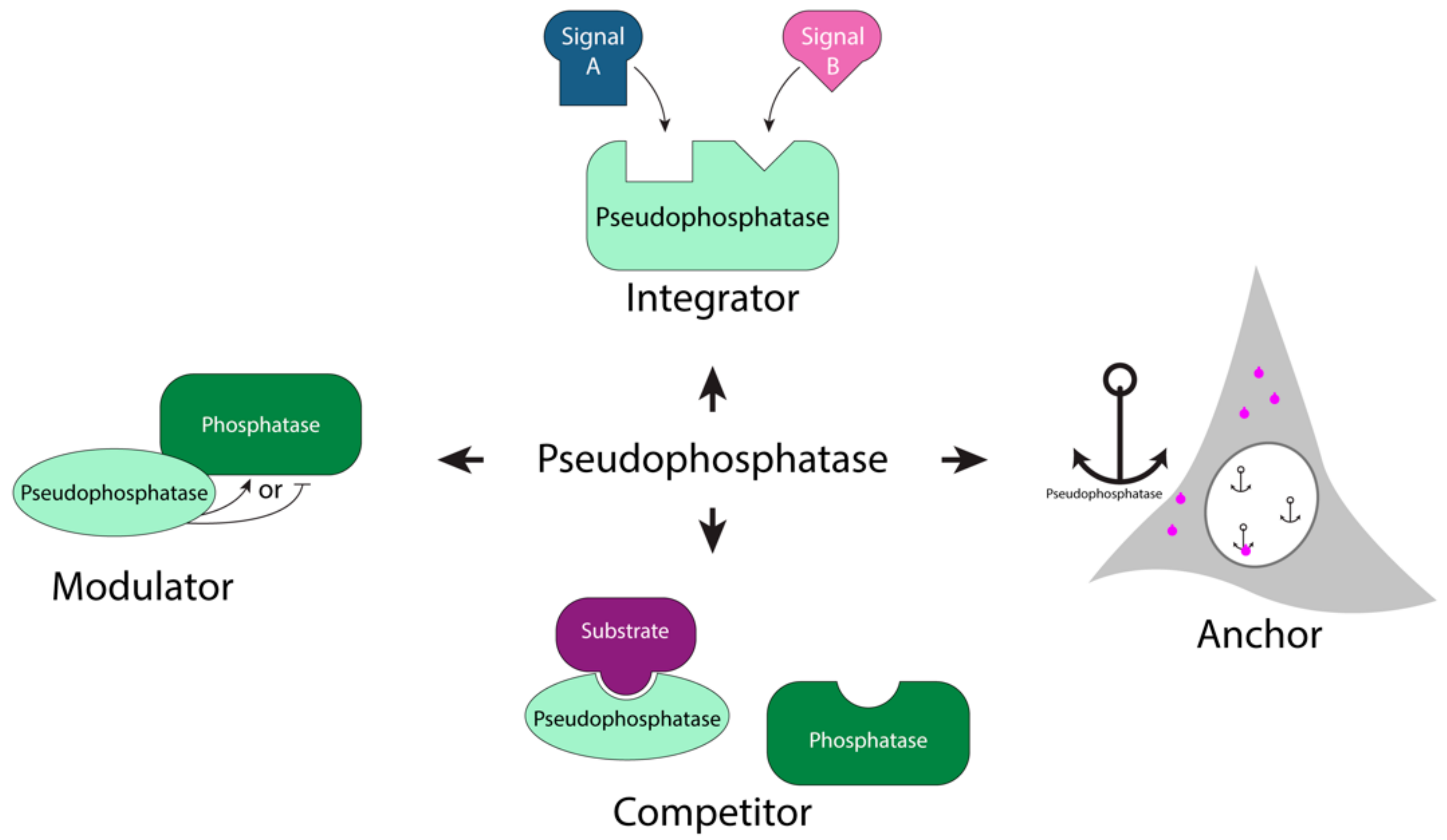
1.2. Pseudophosphatases as Signaling Molecules
2. MAPK Phosphatases (MKPs) Role in MAPK Signaling
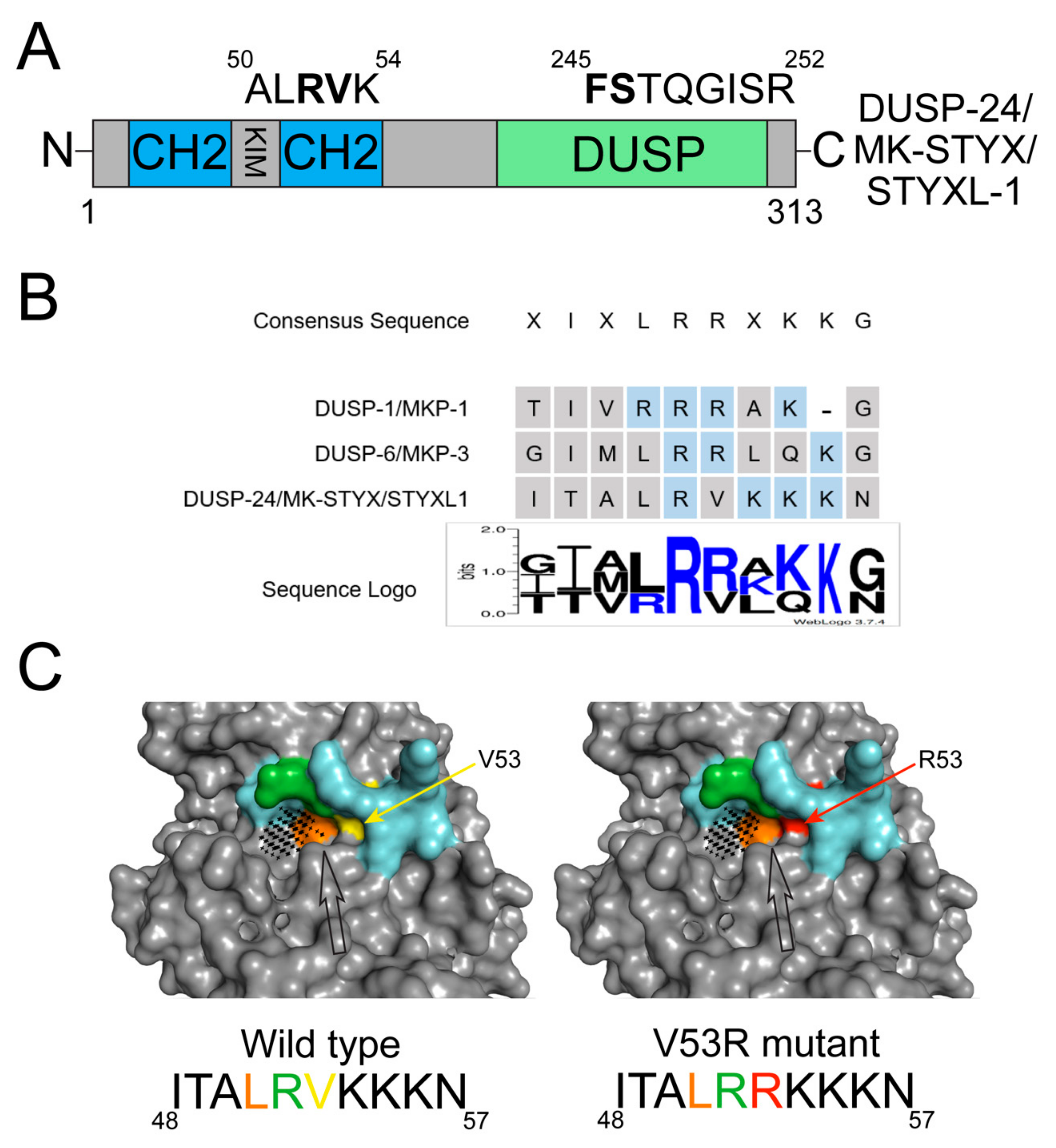
Atypical MKP: MK-STYX
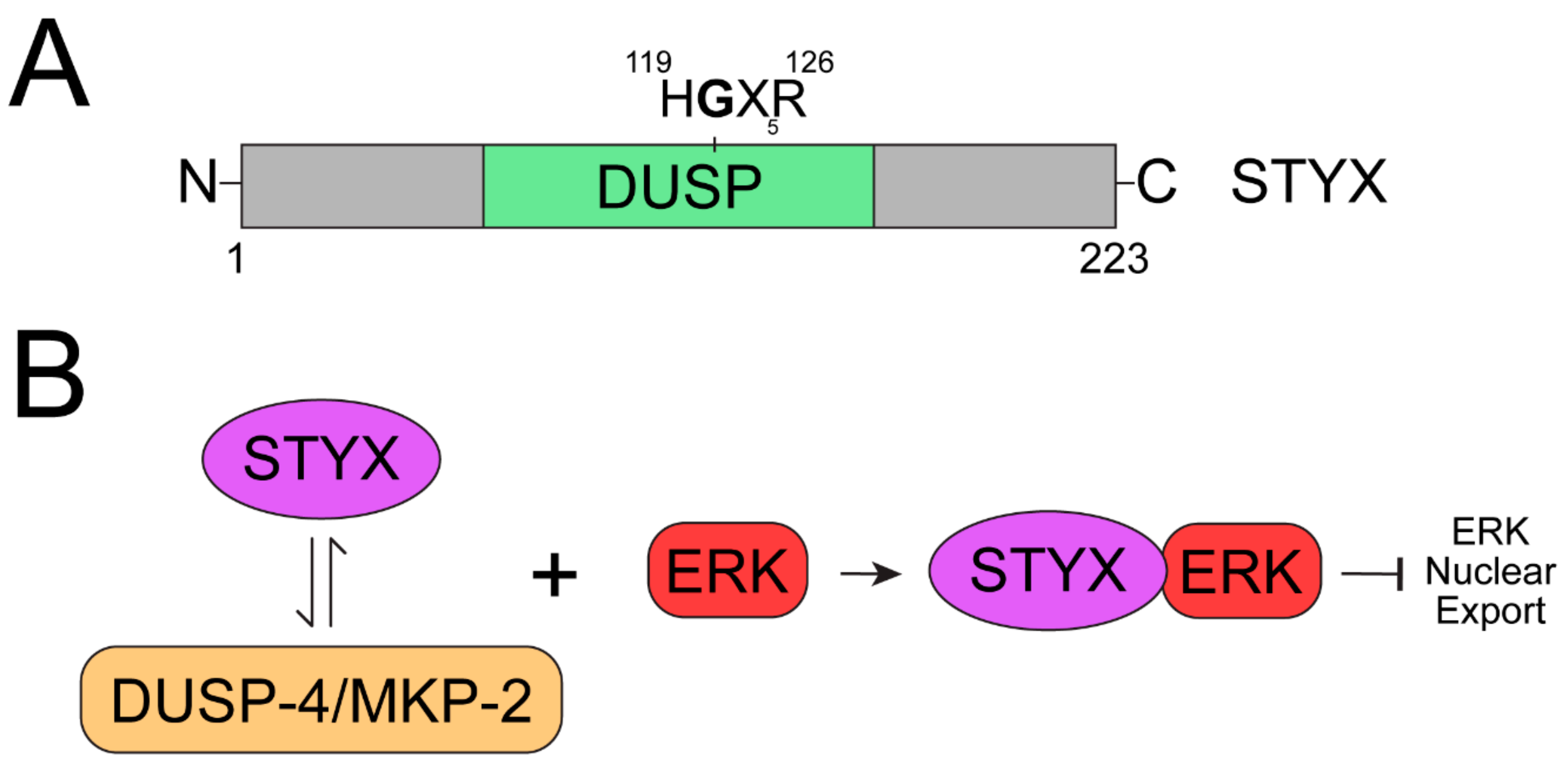
3. STYX
4. TAB1
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ERK | Extracellular signal-regulated kinases 1/2 |
| CH2 | Cell division cycle 25 phosphatase homology 2 |
| DYRK | Dual-specificity tyrosine-phosphorylated and-regulated kinase |
| I-TASSER | Iterative Threading ASSEmbly Refinement |
| FBXW7 | F-box protein WD40 (tryptophan and aspartic acid repeats) domain |
| G3BP-1 | Ras-GTPase-activating protein SH3 domain-binding protein-1 |
| JNK | c-JUN NH2-terminus kinase |
| KIM | Kinase interaction motif |
| MAPK | Mitogen-activated protein kinase |
| MAP2K/MEK | MAPK kinase |
| MAP3K | MAPK kinase kinase |
| MBK-2 | Mini brain kinase 2 |
| MKP | MAP kinase phosphatase |
| MK-STYX | Mitogen-activated protein kinase Phosphoserine/threonine/tyrosine-binding protein |
| MTM | Myotubularins |
| POCASA | Pocket Cavity Search Application |
| PTMs | Post-translational modifications |
| PTPM1 | PTP localized to the mitochondrion 1 |
| PTP | Protein tyrosine phosphatase |
| RTKs | Receptor tyrosine kinases |
| RhoGAP | Rho GTPase-activating protein |
| SCF | SKP/CUL1-F-box |
| STYX | Phosphoserine/threonine/tyrosine-interacting protein |
| TAB 1 | TAK1-binding protein |
| TAK1 | TGF-beta-activated kinase 1 |
References
- Iqbal, J.; Sun, L.; Zaidi, M. Complexity in signal transduction. Ann. N. Y. Acad. Sci. 2010, 1192, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Chauhan, P.; Saha, B.; Kubatzky, K.F. Conceptual Evolution of Cell Signaling. Int. J. Mol. Sci. 2019, 20, 3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, G.; Walther, D. The Roles of Post-translational Modifications in the Context of Protein Interaction Networks. PLoS Comput. Biol. 2015, 11, e1004049. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, In Vivo, and Site-Specific Phosphorylation Dynamics in Signaling Networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Muzio, L.L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- McKay, M.M.; Morrison, D.K. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007, 26, 3113–3121. [Google Scholar] [CrossRef] [Green Version]
- Kharitidi, D.; Manteghi, S.; Pause, A. Pseudophosphatases: Methods of analysis and physiological functions. Methods 2014, 65, 207–218. [Google Scholar] [CrossRef]
- Farooq, A.; Chaturvedi, G.; Mujtaba, S.; Plotnikova, O.; Zeng, L.; Dhalluin, C.; Ashton, R.; Zhou, M.-M. Solution Structure of ERK2 Binding Domain of MAPK Phosphatase MKP-3: Structural Insights into MKP-3 Activation by ERK2. Mol. Cell 2001, 7, 387–399. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Cobb, M.H.; Goldsmith, E.J. How MAP kinases are regulated. J. Biol. Chem. 1995, 270, 14843–14846. [Google Scholar] [CrossRef] [Green Version]
- Kyriakis, J.M.; Avruch, J. Sounding the Alarm: Protein Kinase Cascades Activated by Stress and Inflammation. J. Biol. Chem. 1996, 271, 24313–24316. [Google Scholar] [CrossRef] [Green Version]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK Signal Transduction Pathways Activated by Stress and Inflammation: A 10-Year Update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [Green Version]
- Keshet, Y.; Seger, R. The MAP Kinase Signaling Cascades: A System of Hundreds of Components Regulates a Diverse Array of Physiological Functions. Methods Mol. Biol. 2010, 661, 3–38. [Google Scholar] [CrossRef]
- Caunt, C.J.; Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. FEBS J. 2013, 280, 489–504. [Google Scholar] [CrossRef] [Green Version]
- Hinton, S.D.; Myers, M.P.; Roggero, V.R.; Allison, L.A.; Tonks, N.K. The pseudophosphatase MK-STYX interacts with G3BP and decreases stress granule formation. Biochem. J. 2010, 427, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Wishart, M.J.; Dixon, J.E. Gathering STYX: Phosphatase-like form predicts functions for unique protein-interaction domains. Trends Biochem. Sci. 1998, 23, 301–306. [Google Scholar] [CrossRef]
- Todd, A.E.; Orengo, C.A.; Thornton, J.M. Sequence and Structural Differences between Enzyme and Nonenzyme Homologs. Structure 2002, 10, 1435–1451. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.M.; Mace, P.D.; Eyers, P.A. Live and let die: Insights into pseudoenzyme mechanisms from structure. Curr. Opin. Struct. Biol. 2017, 47, 95–104. [Google Scholar] [CrossRef]
- Hinton, S.D. The role of pseudophosphatases as signaling regulators. Biochim. Biophys. Acta 2018, 1866, 167–174. [Google Scholar] [CrossRef]
- Reiterer, V.; Eyers, P.A.; Farhan, H. Day of the dead: Pseudokinases and pseudophosphatases in physiology and disease. Trends Cell Biol. 2014, 24, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Hinton, S.D. Pseudophosphatase MK-STYX: The atypical member of the MAP kinase phosphatases. FEBS J. 2020, 287, 4221–4231. [Google Scholar] [CrossRef] [PubMed]
- Niemi, N.M.; Lanning, N.J.; Klomp, J.; Tait, S.; Xu, Y.; Dykema, K.J.; Murphy, L.O.; Gaither, L.A.; Xu, H.E.; Furge, K.A.; et al. MK-STYX, a Catalytically Inactive Phosphatase Regulating Mitochondrially Dependent Apoptosis. Mol. Cell. Biol. 2011, 31, 1357–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, M.J.; Denu, J.M.; Williams, J.; Dixon, J.E. A Single Mutation Converts a Novel Phosphotyrosine Binding Domain into a Dual-specificity Phosphatase. J. Biol. Chem. 1995, 270, 26782–26785. [Google Scholar] [CrossRef] [Green Version]
- Reiterer, V.; Pawłowski, K.; DesRochers, G.; Pause, A.; Sharpe, H.J.; Farhan, H. The dead phosphatases society: A review of the emerging roles of pseudophosphatases. FEBS J. 2020, 287, 4198–4220. [Google Scholar] [CrossRef]
- Reiterer, V.; Fey, D.; Kolch, W.; Kholodenko, B.N.; Farhan, H. Pseudophosphatase STYX modulates cell-fate decisions and cell migration by spatiotemporal regulation of ERK1/2. Proc. Natl. Acad. Sci. USA 2013, 110, E2934–E2943. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.; Kang, Y.J.; Han, J.; Herschman, H.R.; Stefani, E.; Wang, Y. TAB-1 Modulates Intracellular Localization of p38 MAP Kinase and Downstream Signaling. J. Biol. Chem. 2006, 281, 6087–6095. [Google Scholar] [CrossRef] [Green Version]
- Mattei, A.; Smailys, J.; Hepworth, E.; Hinton, S. The Roles of Pseudophosphatases in Disease. Int. J. Mol. Sci. 2021, 22, 6924. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein tyrosine phosphatases—From housekeeping enzymes to master regulators of signal transduction. FEBS J. 2012, 280, 346–378. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.J.; Dixon, J.E.; Manning, G. Genomics and evolution of protein phosphatases. Sci. Signal. 2017, 10, eaag1796. [Google Scholar] [CrossRef]
- Murphy, J.M.; Farhan, H.; Eyers, P.A. Bio-Zombie: The rise of pseudoenzymes in biology. Biochem. Soc. Trans. 2017, 45, 537–544. [Google Scholar] [CrossRef]
- Tonks, N.K. Pseudophosphatases: Grab and Hold on. Cell 2009, 139, 464–465. [Google Scholar] [CrossRef] [Green Version]
- Bulgari, D.; Jha, A.; Deitcher, D.; Levitan, E.S. Myopic (HD-PTP, PTPN23) selectively regulates synaptic neuropeptide release. Proc. Natl. Acad. Sci. USA 2018, 115, 1617–1622. [Google Scholar] [CrossRef] [Green Version]
- Gahloth, D.; Heaven, G.; Jowitt, T.; Mould, A.P.; Bella, J.; Baldock, C.; Woodman, P.; Tabernero, L. The open architecture of HD-PTP phosphatase provides new insights into the mechanism of regulation of ESCRT function. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Parry, J.M.; Velarde, N.V.; Lefkovith, A.J.; Zegarek, M.H.; Hang, J.S.; Ohm, J.; Klancer, R.; Maruyama, R.; Druzhinina, M.K.; Grant, B.D.; et al. EGG-4 and EGG-5 Link Events of the Oocyte-to-Embryo Transition with Meiotic Progression in C. elegans. Curr. Biol. 2009, 19, 1752–1757. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.C.; Klancer, R.; Singson, A.; Seydoux, G. Regulation of MBK-2/DYRK by CDK-1 and the pseudophosphatases EGG-4 and EGG-5 during the oocyte-to-embryo transition. Cell 2009, 139, 560–572. [Google Scholar] [CrossRef] [Green Version]
- Isrie, M.; Zamani Esteki, M.; Peeters, H.; Voet, T.; Van Houdt, J.; Van Paesschen, W.; Van Esch, H. Homozygous missense mutation in STYXL1 associated with moderate intellectual disability, epilepsy and behavioural complexities. Eur. J. Med. Genet. 2015, 58, 205–210. [Google Scholar] [CrossRef]
- Siligan, C.; Ban, J.; Bachmaier, R.; Spahn, L.; Kreppel, M.; Schaefer, K.L.; Poremba, C.; Aryee, D.N.; Kovar, H. EWS-FLI1 target genes recovered from Ewing’s sarcoma chromatin. Oncogene 2005, 24, 2512–2524. [Google Scholar] [CrossRef] [Green Version]
- Gal, J.; Kuang, L.; Barnett, K.; Zhu, B.Z.; Shissler, S.C.; Korotkov, K.; Hayward, L.; Kasarskis, E.J.; Zhu, H. ALS mutant SOD1 interacts with G3BP1 and affects stress granule dynamics. Acta Neuropathol. 2016, 132, 563–576. [Google Scholar] [CrossRef] [Green Version]
- Robinson, F.L.; Dixon, J.E. Myotubularin phosphatases: Policing 3-phosphoinositides. Trends Cell Biol. 2006, 16, 403–412. [Google Scholar] [CrossRef]
- Hnia, K.; Vaccari, I.; Bolino, A.; Laporte, J. Myotubularin phosphoinositide phosphatases: Cellular functions and disease pathophysiology. Trends Mol. Med. 2012, 18, 317–327. [Google Scholar] [CrossRef]
- Ng, A.A.; Logan, A.M.; Schmidt, E.J.; Robinson, F.L. The CMT4B disease-causing phosphatases Mtmr2 and Mtmr13 localize to the Schwann cell cytoplasm and endomembrane compartments, where they depend upon each other to achieve wild-type levels of protein expression. Hum. Mol. Genet. 2013, 22, 1493–1506. [Google Scholar] [CrossRef] [Green Version]
- Robinson, F.L.; Dixon, J.E.; Kawada, M.; Masuda, T.; Ishizuka, M.; Takeuchi, T. The Phosphoinositide-3-phosphatase MTMR2 Associates with MTMR13, a Membrane-associated Pseudophosphatase Also Mutated in Type 4B Charcot-Marie-Tooth Disease. J. Biol. Chem. 2005, 280, 31699–31707. [Google Scholar] [CrossRef] [Green Version]
- Previtali, S.C.; Quattrini, A.; Bolino, A. Charcot-Marie-Tooth type 4B demyelinating neuropathy: Deciphering the role of MTMR phosphatases. Expert Rev. Mol. Med. 2007, 9, 1–16. [Google Scholar] [CrossRef]
- Dickinson, R.J.; Keyse, S. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J. Cell Sci. 2006, 119, 4607–4615. [Google Scholar] [CrossRef] [Green Version]
- Keyse, S.M.; Ginsburg, M. Amino acid sequence similarity between CL100, a dual-specificity MAP kinase phosphatase and cdc25. Trends Biochem. Sci. 1993, 18, 377–378. [Google Scholar] [CrossRef]
- Bordo, D.; Bork, P. The rhodanese/Cdc25 phosphatase superfamily. EMBO Rep. 2002, 3, 741–746. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-Y.; Tan, T.-H. DUSPs, to MAP kinases and beyond. Cell Biosci. 2012, 2, 24. [Google Scholar] [CrossRef] [Green Version]
- Guan, K.; Broyles, S.S.; Dixon, J.E. A Tyr/Ser protein phosphatase encoded by vaccinia virus. Nature 1991, 350, 359–362. [Google Scholar] [CrossRef]
- Owens, D.M.; Keyse, S.M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar] [CrossRef] [Green Version]
- Tonks, N.K.; Neel, B.G. Combinatorial control of the specificity of protein tyrosine phosphatases. Curr. Opin. Cell Biol. 2001, 13, 182–195. [Google Scholar] [CrossRef]
- Tonks, N.K.; Neel, B. From Form to Function: Signaling by Protein Tyrosine Phosphatases. Cell 1996, 87, 365–368. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.N.; Mortensen, O.H.; Peters, G.H.; Drake, P.G.; Iversen, L.F.; Olsen, O.H.; Jansen, P.G.; Andersen, H.S.; Tonks, N.K.; Møller, N.P.H. Structural and Evolutionary Relationships among Protein Tyrosine Phosphatase Domains. Mol. Cell. Biol. 2001, 21, 7117–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slack, D.N.; Seternes, O.M.; Gabrielsen, M.; Keyse, S.M. Distinct binding determinants for ERK2/p38alpha and JNK map kinases mediate catalytic activation and substrate selectivity of map kinase phosphatase-1. J. Biol. Chem. 2001, 276, 16491–16500. [Google Scholar] [CrossRef] [Green Version]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef]
- Flowers, B.M.; Rusnak, L.E.; Wong, K.; Banks, D.A.; Munyikwa, M.; McFarland, A.G.; Hinton, S.D. The Pseudophosphatase MK-STYX Induces Neurite-Like Outgrowths in PC12 Cells. PLoS ONE 2014, 9, e114535. [Google Scholar] [CrossRef]
- Dahal, A.; Hinton, S.D. Antagonistic roles for STYX pseudophosphatases in neurite outgrowth. Biochem. Soc. Trans. 2017, 45, 381–387. [Google Scholar] [CrossRef]
- Peti, W.; Page, R. Molecular basis of MAP kinase regulation. Protein Sci. 2013, 22, 1698–1710. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Shuvo, M.H.; Gulfam, M.; Bhattacharya, D. DeepRefiner: High-accuracy protein structure refinement by deep network calibration. Nucleic Acids Res. 2021, 49, w147–w152. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.
- Yu, J.; Zhou, Y.; Tanaka, I.; Yao, M. Roll: A new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics 2010, 26, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Ittisoponpisan, S.; Islam, S.A.; Khanna, T.; Alhuzimi, E.; David, A.; Sternberg, M.J.E. Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J. Mol. Biol. 2019, 431, 2197–2212. [Google Scholar] [CrossRef]
- Bell, A.; Christian, L.; Hecht, D.; Huisinga, K.; Rakus, J.; Bell, E. Teaching virtual protein-centric CUREs and UREs using computational tools. Biochem. Mol. Biol. Educ. 2020, 48, 646–647. [Google Scholar] [CrossRef]
- Niemi, N.M.; Sacoman, J.L.; Westrate, L.M.; Gaither, L.A.; Lanning, N.J.; Martin, K.; MacKeigan, J.P. The Pseudophosphatase MK-STYX Physically and Genetically Interacts with the Mitochondrial Phosphatase PTPMT1. PLoS ONE 2014, 9, e93896. [Google Scholar] [CrossRef] [Green Version]
- Barr, J.E.; Munyikwa, M.R.; Frazier, E.A.; Hinton, S.D. The pseudophosphatase MK-STYX inhibits stress granule assembly independently of Ser149 phosphorylation of G3BP-1. FEBS J. 2013, 280, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Banks, D.A.; Dahal, A.; McFarland, A.G.; Flowers, B.M.; Stephens, C.A.; Swack, B.; Gugssa, A.; Anderson, W.A.; Hinton, S.D. MK-STYX Alters the Morphology of Primary Neurons, and Outgrowths in MK-STYX Overexpressing PC-12 Cells Develop a Neuronal Phenotype. Front. Mol. Biosci. 2017, 4, 76. [Google Scholar] [CrossRef] [Green Version]
- Vaudry, D.; Stork, P.J.S.; Lazarovici, P.; Eiden, L.E. Signaling Pathways for PC12 Cell Differentiation: Making the Right Connections. Science 2002, 296, 1648–1649. [Google Scholar] [CrossRef]
- Reiterer, V.; Pawłowski, K.; Farhan, H. STYX: A versatile pseudophosphatase. Biochem. Soc. Trans. 2017, 45, 449–456. [Google Scholar] [CrossRef]
- Reiterer, V.; Figueras-Puig, C.; Le Guerroue, F.; Confalonieri, S.; Vecchi, M.; Jalapothu, D.; Kanse, S.M.; Deshaies, R.J.; Di Fiore, P.P.; Behrends, C.; et al. The pseudophosphatase STYX targets the F-box of FBXW7 and inhibits SCFFBXW7 function. EMBO J. 2017, 36, 260–273. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.J.; Welcker, M.; Clurman, B.E. Tumor Suppression by the Fbw7 Ubiquitin Ligase: Mechanisms and Opportunities. Cancer Cell 2014, 26, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ye, X.; Liu, Y.; Wei, W.; Wang, Z. Aberrant regulation of FBW7 in cancer. Oncotarget 2014, 5, 2000–2015. [Google Scholar] [CrossRef] [Green Version]
- Anderson, P.; Kedersha, N. Visibly stressed: The role of eIF2, TIA-1, and stress granules in protein translation. Cell Stress Chaperones 2002, 7, 213–221. [Google Scholar] [CrossRef]
- Conner, S.H.; Kular, G.; Peggie, M.; Shepherd, S.; Schüttelkopf, A.W.; Cohen, P.; Van Aalten, D.M.F. TAK1-binding protein 1 is a pseudophosphatase. Biochem. J. 2006, 399, 427–434. [Google Scholar] [CrossRef]
- Wolf, A.; Beuerlein, K.; Eckart, C.; Weiser, H.; Dickkopf, B.; Müller, H.; Sakurai, H.; Kracht, M. Identification and Functional Characterization of Novel Phosphorylation Sites in TAK1-Binding Protein (TAB) 1. PLoS ONE 2011, 6, e29256. [Google Scholar] [CrossRef]
- Hirata, Y.; Takahashi, M.; Morishita, T.; Noguchi, T.; Matsuzawa, A. Post-Translational Modifications of the TAK1-TAB Complex. Int. J. Mol. Sci. 2017, 18, 205. [Google Scholar] [CrossRef]
- Campbell, D.; Cohen, P.; Gaestel, M.; Arthur, J.S.C.; Burness, K.; Ronkina, N.; Shim, J.-H.; Mendoza, H.; Johnson, G.; Davis, R.; et al. Roles for TAB1 in regulating the IL-1-dependent phosphorylation of the TAB3 regulatory subunit and activity of the TAK1 complex. Biochem. J. 2008, 409, 711–722. [Google Scholar] [CrossRef]
- Cheung, P.C.; Campbell, D.G.; Nebreda, A.R.; Cohen, P. Feedback control of the protein kinase TAK1 by SAPK2a/p38alpha. EMBO J. 2003, 22, 5793–5805. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Regunath, K.; Jacq, X.; Prives, C. Cisplatin causes cell death via TAB1 regulation of p53/MDM2/MDMX circuitry. Genes Dev. 2013, 27, 1739–1751. [Google Scholar] [CrossRef] [Green Version]
- Eyers, P.A.; Murphy, J.M. The evolving world of pseudoenzymes: Proteins, prejudice and zombies. BMC Biol. 2016, 14, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Fan, G.; Hao, Y.; Hammell, M.; Wilkinson, J.E.; Tonks, N.K. Suppression of protein tyrosine phosphatase N23 predisposes to breast tumorigenesis via activation of FYN kinase. Genes Dev. 2017, 31, 1939–1957. [Google Scholar] [CrossRef] [PubMed]
- Brognard, J.; Sierecki, E.; Gao, T.; Newton, A.C. PHLPP and a Second Isoform, PHLPP2, Differentially Attenuate the Amplitude of Akt Signaling by Regulating Distinct Akt Isoforms. Mol. Cell 2007, 25, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, G.; Funato, Y.; Chen, Y.S.; Zhang, Z.; Illes, K.; Miki, H.; Gehring, K. PRL3 pseudophosphatase activity is necessary and sufficient to promote metastatic growth. J. Biol. Chem. 2020, 295, 11682–11692. [Google Scholar] [CrossRef]
- Tomar, V.S.; Baral, T.K.; Nagavelu, K.; Somasundaram, K. Serine/threonine/tyrosine-interacting-like protein 1 (STYXL1), a pseudo phosphatase, promotes oncogenesis in glioma. Biochem. Biophys. Res. Commun. 2019, 515, 241–247. [Google Scholar] [CrossRef]
- Wu, J.-Z.; Jiang, N.; Lin, J.-M.; Liu, X. STYXL1 promotes malignant progression of hepatocellular carcinoma via downregulating CELF2 through the PI3K/Akt pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2977–2985. [Google Scholar]
- Winter, J.M.; Curry, N.L.; Gildea, D.M.; Williams, K.A.; Lee, M.; Hu, Y.; Crawford, N.P.S. Modifier locus mapping of a transgenic F2 mouse population identifies CCDC115 as a novel aggressive prostate cancer modifier gene in humans. BMC Genom. 2018, 19, 450. [Google Scholar] [CrossRef] [Green Version]
- Wishart, M.J.; Dixon, J.E. The archetype STYX/dead-phosphatase complexes with a spermatid mRNA-binding protein and is essential for normal sperm production. Proc. Natl. Acad. Sci. USA 2002, 99, 2112–2117. [Google Scholar] [CrossRef] [Green Version]
- He, D.; Ma, Z.; Fang, C.; Ding, J.; Yang, W.; Chen, P.; Huang, L.; Wang, C.; Yu, Y.; Yang, L.; et al. Pseudophosphatase STYX promotes tumor growth and metastasis by inhibiting FBXW7 function in colorectal cancer. Cancer Lett. 2019, 454, 53–65. [Google Scholar] [CrossRef]
- Liu, L.; Jiang, H.; Wang, X.; Wang, X.; Zou, L. STYX/FBXW7 axis participates in the development of endometrial cancer cell via Notch-mTOR signaling pathway. Biosci. Rep. 2020, 40, BSR20200057. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.D.; Fjeld, C.C.; Denu, J.M. Probing the function of conserved residues in the serine/threonine phosphatase PP2Calpha. Biochemistry 2003, 42, 8513–8521. [Google Scholar] [CrossRef]
- Lei, X.; Han, N.; Xiao, X.; Jin, Q.; He, B.; Wang, J. Enterovirus 71 3C inhibits cytokine expression through cleavage of the TAK1/TAB1/TAB2/TAB3 complex. J. Virol. 2014, 88, 9830–9841. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pseudophosphatase | Critical Mutation(s) | Implications in MAPK Signaling | Implications in Other Pathways | Links to Diseases |
|---|---|---|---|---|
| MK-STYX (STYXL1/DUSP-24) | Contains the active site sequence FSX5R instead of HCX5R which results in the loss of catalytic activity. The KIM of MK-STYX lacks consecutive arginines (R53V), which may be the reason that MK-STYX does not bind MAPK/ERK. | Does not function like the other MKPs in that MK-STYX does not impact MAPK/ERK signaling. | The overexpression of MK-STYX decreases stress granule formation through interaction with G3BP1 and alters the localization of HDAC6. MK-STYX modulates the activity of PTPM1 in order to induce stress-activated mitochondrial-dependent apoptosis. In PC12 cells, the overexpression of MK-STYX induces neurite formation by affecting cofilin and decreasing RhoA activation. | MK-STYX has a potential oncogenic role in Ewing’s sarcoma family tumors (ESFT) linked to the EWS-FLI1-driven overexpression of MK-STYX in these tumors [38]. Potential oncogenic role in glioblastoma (GBM) as MK-STYX was found to be upregulated and promoted aggressive phenotypes in gliomas [86]. Increased expression of MK-STYX implicated in the proliferation of hepatocellular carcinoma (HCC) by inhibiting apoptosis [87]. Increased expression of MK-STYX noted in breast cancer and prostate cancer [88]. |
| STYX | The essential active site cysteine (C) is replaced by a glycine (G) and results in the loss of catalytic activity. | Competes with MKP-2 to serve as a spatiotemporal regulator of ERK1/2 and reduce downstream MAPK activation. Downregulation of STYX inhibits Golgi polarization in an ERK-dependent manner. | STYX associates with CRHSP-24 (calcium-regulated heat-stable protein of 24 kDa) to serve as a critical regulator of spermatogenesis in mice and the deletion of STYX results in male sterility [89]. STYX regulates ubiquitination by interacting with F-box proteins and inhibiting the associated SCF complex. | The ability of STYX to bind and inhibit the F-box protein FBXW7, along with imbalances in the relative expression of these two proteins, has been implicated in breast cancer, colorectal cancer, and endometrial cancer [72,90,91]. |
| TAB1 (MAP3K7IP1) | The N-terminal of TAB1 lacks the three catalytic residues of PP2C (protein phosphatase 2C): -Asp282 (substituted with Glu356 in TAB1), -His62 (substituted with Tyr71 in TAB1), and -Arg33 (missing in TAB1) [76,92]. Four of the active site aspartic acid residues that co-ordinate the metal ions required for catalytic function in PP2C have been substituted: -Asp60 (substituted with Asn69 in TAB1), -Asp239 (substituted with Glu290 in TAB1), -Asp 282 (substituted with Glu356) this is also a catalytic residue, and -Asn283 (substituted with Asp357 in TAB1) [76,92]. | Downstream regulator of the MAP3K TAK1 by activating and changing the localization of p38 MAPK in order to recruit p38 MAPK to the TAK1 complex in order to activate TAK1. | Blocks inhibition of p53 by inhibiting the negative regulator MDM2. | TAB1 as part of the TAK1 complex is linked with the outcome of viral infection with enterovirus 71 (EV71), the pathogen responsible for hand, foot, and mouth disease via inhibition of NF-κB activation [93]. TAB1 is implicated as a potential tumor suppressor and lower levels of TAB1 are associated with cancerous ovarian tumors [81]. Implicated in rheumatoid arthritis and other pro-inflammatory diseases [77,78,79]. |
| EGG4/EGG5 | Catalytic cysteine (C) residue replaced by aspartic acid (D) in the active site motif [35,36]. | Does not regulate MAPK signaling. However, EGG4/5 do regulate dual-specificity tyrosine-regulated kinase (DYRK) signaling. EGG4/5 regulates the oocyte-to-zygote transition in Caenorhabditis elegans via competition with mini brain kinase 2 (MBK-2). | EGG4/5 regulates the localization of EGG3 and CHS-1 during meiotic progression and EGG4/5 localize to the cortex in developing oocytes independently of MBK-2. | Loss of function of both EGG4/5 results in maternal-effect lethality in the nematode C. elegans. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hepworth, E.M.W.; Hinton, S.D. Pseudophosphatases as Regulators of MAPK Signaling. Int. J. Mol. Sci. 2021, 22, 12595. https://doi.org/10.3390/ijms222212595
Hepworth EMW, Hinton SD. Pseudophosphatases as Regulators of MAPK Signaling. International Journal of Molecular Sciences. 2021; 22(22):12595. https://doi.org/10.3390/ijms222212595
Chicago/Turabian StyleHepworth, Emma Marie Wilber, and Shantá D. Hinton. 2021. "Pseudophosphatases as Regulators of MAPK Signaling" International Journal of Molecular Sciences 22, no. 22: 12595. https://doi.org/10.3390/ijms222212595
APA StyleHepworth, E. M. W., & Hinton, S. D. (2021). Pseudophosphatases as Regulators of MAPK Signaling. International Journal of Molecular Sciences, 22(22), 12595. https://doi.org/10.3390/ijms222212595