
Takinib Inhibits Inflammation in Human Rheumatoid Arthritis Synovial Fibroblasts by Targeting the Janus Kinase-Signal Transducer and Activator of Transcription 3 (JAK/STAT3) Pathway

 , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Results
2.1. Inhibition of TAK1 Reduces Proinflammatory Mediators Secreted by RASFs and THP-1 Monocyte-Derived Macrophages
2.2. Takinib Inhibits STAT3 and JNK Phosphorylation in IL-1β, but Not IL-6 Stimulated Human RASFs
2.3. Takinib Inhibits STAT3 Nuclear Translocation and DNA-Binding Activity in Human RASFs
2.4. Takinib Phosphorylates TAK1 and Fails to Inhibit NF-κB or MAPK Signaling in LPS-Stimulated THP-1 Macrophages
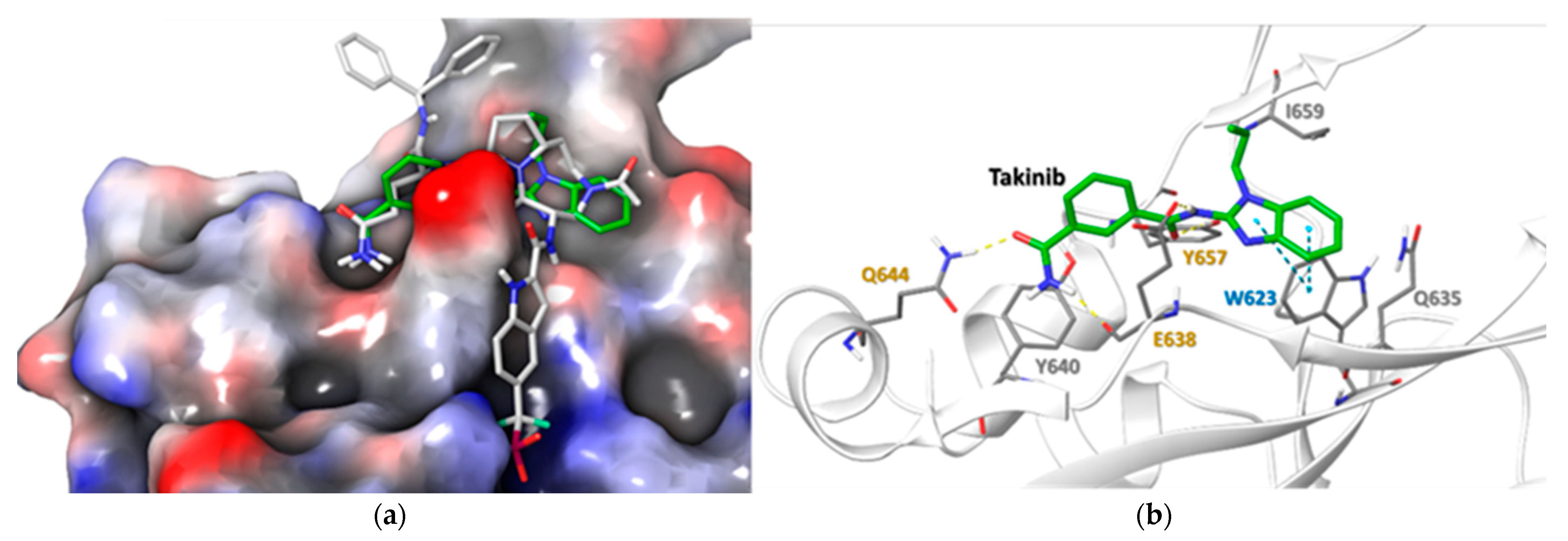
2.5. Docking Simulations Using Takinib and STAT3 Demonstrate a Potential Site of Interaction
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Culturing of Human RASFs and THP1
4.3. Treatment of RASFs and THP-1
4.4. Assay for Cytokine Production
4.5. Western Immunoblotting
4.6. Cell Fractionation
4.7. DNA Binding Assay for STAT3
4.8. Molecular Dynamics (MD) Simulation Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bottini, N.; Firestein, G.S. Duality of fibroblast-like synoviocytes in RA: Passive responders and imprinted aggressors. Nat. Rev. Rheumatol. 2013, 9, 24–33. [Google Scholar] [CrossRef]
- Vandooren, B.; Cantaert, T.; ter Borg, M.; Noordenbos, T.; Kuhlman, R.; Gerlag, D.; Bongartz, T.; Reedquist, K.; Tak, P.P.; Baeten, D. Tumor necrosis factor alpha drives cadherin 11 expression in rheumatoid inflammation. Arthritis Rheum. 2008, 58, 3051–3062. [Google Scholar] [CrossRef] [PubMed]
- Muller-Ladner, U.; Kriegsmann, J.; Franklin, B.N.; Matsumoto, S.; Geiler, T.; Gay, R.E.; Gay, S. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am. J. Pathol. 1996, 149, 1607–1615. [Google Scholar] [PubMed]
- Cronstein, B.N. Interleukin-6—A key mediator of systemic and local symptoms in rheumatoid arthritis. Bull. NYU Hosp. Jt. Dis. 2007, 65 (Suppl. S1), S11–S15. [Google Scholar]
- Houssiau, F.A.; Devogelaer, J.P.; Van Damme, J.; de Deuxchaisnes, C.N.; Van Snick, J. Interlwukin-6 in synovial fluid and serum of patients with rheumatoid arthritis and other inflammatory arthritides. Arthritis Rheum. 1988, 31, 784–788. [Google Scholar] [CrossRef]
- Okamoto, H.; Yamamura, M.; Morita, Y.; Harada, S.; Makino, H.; Ota, Z. The synovial expression and serum levels of interleukin-6, interleukin-11, leukemia inhibitory factor, and oncostatin M in rheumatoid arthritis. Arthritis Rheum. 1997, 40, 1096–1105. [Google Scholar] [CrossRef]
- Jamilloux, Y.; El Jammal, T.; Vuitton, L.; Gerfaud-Valentin, M.; Kerever, S.; Sève, P. JAK inhibitors for the treatment of autoimmune and inflammatory diseases. Autoimmun. Rev. 2019, 18, 102390. [Google Scholar] [CrossRef] [PubMed]
- Rivellese, F.; Lobasso, A.; Barbieri, L.; Liccardo, B.; de Paulis, A.; Rossi, F.W. Novel therapeutic approaches in rheumatoid arthritis: Role of janus kinases inhibitors. Curr. Med. Chem. 2019, 26, 2823–2843. [Google Scholar] [CrossRef]
- Kato, M. New insights into IFN-gamma in rheumatoid arthritis: Role in the era of JAK inhibitors. Immunol. Med. 2020, 43, 72–78. [Google Scholar] [CrossRef]
- Winthrop, K.L.; Park, S.H.; Gul, A.; Cardiel, M.H.; Gomez-Reino, J.J.; Tanaka, Y.; Kwok, K.; Lukic, T.; Mortensen, E.; de Leon, D.P.; et al. Tuberculosis and other opportunistic infections in tofacitinib-treated patients with rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 1133–1138. [Google Scholar] [CrossRef]
- Zhang, Z.; Deng, W.; Wu, Q.; Sun, L. Tuberculosis, hepatitis B and herpes zoster in tofacitinib-treated patients with rheumatoid arthritis. Immunotherapy 2019, 11, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Umar, S.; Riegsecker, S.; Chourasia, M.; Ahmed, S. Regulation of transforming growth factor beta-activated kinase activation by epigallocatechin-3-gallate in rheumatoid arthritis synovial fibroblasts: Suppression of K(63)-linked autoubiquitination of tumor necrosis factor receptor-associated factor 6. Arthritis Rheumatol. 2016, 68, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Fechtner, S.; Fox, D.A.; Ahmed, S. Transforming growth factor beta activated kinase 1: A potential therapeutic target for rheumatic diseases. Rheumatology 2017, 56, 1060–1068. [Google Scholar] [CrossRef][Green Version]
- Singh, A.K.; Fechtner, S.; Chourasia, M.; Sicalo, J.; Ahmed, S. Critical role of IL-1α in IL-1β-induced inflammatory responses: Cooperation with NF-κBp65 in transcriptional regulation. FASEB J. 2019, 33, 2526–2536. [Google Scholar] [CrossRef]
- Jones, D.S.; Jenney, A.P.; Swantek, J.L.; Burke, J.M.; Lauffenburger, D.A.; Sorger, P.K. Profiling drugs for rheumatoid arthritis that inhibit synovial fibroblast activation. Nat. Chem. Biol. 2017, 13, 38–45. [Google Scholar] [CrossRef]
- Kishimoto, K.; Matsumoto, K.; Ninomiya-Tsuji, J. TAK1 mitogen-activated protein kinase kinase kinase is activated by autophosphorylation within its activation loop. J. Biol. Chem. 2000, 275, 7359–7364. [Google Scholar] [CrossRef] [PubMed]
- Singhirunnusorn, P.; Suzuki, S.; Kawasaki, N.; Saiki, I.; Sakurai, H. Critical roles of threonine 187 phosphorylation in cellular stress-induced rapid and transient activation of transforming growth factor-β-activated kinase 1 (TAK1) in a signaling complex containing TAK1-binding protein TAB1 and TAB2. J. Biol. Chem. 2005, 280, 7359–7368. [Google Scholar] [CrossRef]
- Yu, Y.; Ge, N.; Xie, M.; Sun, W.; Burlingame, S.; Pass, A.K.; Nuchtern, J.G.; Zhang, D.; Fu, S.; Schneider, M.D.; et al. Phosphorylation of Thr-178 and Thr-184 in the TAK1 T-loop is required for interleukin (IL)-1-mediated optimal NFκB and AP-1 activation as well as IL-6 gene expression. J. Biol. Chem. 2008, 283, 24497–24505. [Google Scholar] [CrossRef]
- Wu, J.; Powell, F.; Larsen, N.A.; Lai, Z.; Byth, K.F.; Read, J.; Gu, R.F.; Roth, M.; Toader, D.; Saeh, J.C.; et al. Mechanism and in vitro pharmacology of TAK1 inhibition by (5Z)-7-oxozeaenol. ACS Chem. Biol. 2013, 8, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, Z.; Li, Y.; Ji, Q. NG25, an inhibitor of transforming growth factor-β-activated kinase 1, ameliorates neuronal apoptosis in neonatal hypoxic-ischemic rats. Mol. Med. Rep. 2018, 17, 1710–1716. [Google Scholar] [CrossRef]
- Totzke, J.; Gurbani, D.; Raphemot, R.; Hughes, P.F.; Bodoor, K.; Carlson, D.A.; Loiselle, D.R.; Bera, A.K.; Eibschutz, L.S.; Perkins, M.M.; et al. Takinib, a selective TAK1 inhibitor, broadens the therapeutic efficacy of TNFα inhibition for cancer and autoimmune disease. Cell Chem. Biol. 2017, 24, 1029–1039. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, H.; Xu, R.; Zhao, Y.; Chinnaswamy, K.; McEachern, D.; Chen, J.; Yang, C.Y.; Liu, Z.; Wang, M.; et al. A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell 2019, 36, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2020-3; Schrödinger, LLC: New York, NY, USA, 2021.
- Shao, H.; Xu, X.; Jing, N.; Tweardy, D.J. Unique structural determinants for Stat3 recruitment and activation by the granulocyte colony-stimulating factor receptor at phosphotyrosine ligands 704 and 744. J. Immunol. 2006, 176, 2933–2941. [Google Scholar] [CrossRef] [PubMed]
- Yu-Jung, C.; Kuan-Wei, C.; Ching-Jen, C.; Ming-Hsing, L.; Yuh-Ju, S.; Jia-Lin, L.; Ing-Ming, C.; Linyi, C. SH2B1β interacts with STAT3 and enhances fibroblast growth factor 1-induced gene expression during neuronal differentiation. Mol. Cell. Biol. 2014, 34, 1003–1019. [Google Scholar] [CrossRef][Green Version]
- Pilati, C.; Amessou, M.; Bihl, M.P.; Balabaud, C.; Nhieu, J.T.; Paradis, V.; Nault, J.C.; Izard, T.; Bioulac-Sage, P.; Couchy, G.; et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J. Exp. Med. 2011, 208, 1359–1366. [Google Scholar] [CrossRef]
- Brambilla, L.; Genini, D.; Laurini, E.; Merulla, J.; Perez, L.; Fermeglia, M.; Carbone, G.M.; Pricl, S.; Catapano, C.V. Hitting the right spot: Mechanism of action of OPB-31121, a novel and potent inhibitor of the signal transducer and activator of transcription 3 (STAT3). Mol. Oncol. 2015, 9, 1194–1206. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Scarneo, S.A.; Eibschutz, L.S.; Bendele, P.J.; Yang, K.W.; Totzke, J.; Hughes, P.; Fox, D.A.; Haystead, T.A.J. Pharmacological inhibition of TAK1, with the selective inhibitor takinib, alleviates clinical manifestation of arthritis in CIA mice. Arthritis Res. Ther. 2019, 21, 292. [Google Scholar] [CrossRef]
- Scarneo, S.A.; Mansourati, A.; Eibschutz, L.S.; Totzke, J.; Roques, J.R.; Loiselle, D.; Carlson, D.; Hughes, P.; Haystead, T.A.J. Genetic and pharmacological validation of TAK1 inhibition in macrophages as a therapeutic strategy to effectively inhibit TNF secretion. Sci. Rep. 2018, 8, 17058. [Google Scholar] [CrossRef]
- Sakurai, H.; Miyoshi, H.; Toriumi, W.; Sugita, T. Functional interactions of transforming growth factor beta-activated kinase 1 with IκB kinases to stimulate NF-κB activation. J. Biol. Chem. 1999, 274, 10641–10648. [Google Scholar] [CrossRef]
- Ninomiya-Tsuji, J.; Kishimoto, K.; Hiyama, A.; Inoue, J.-I.; Cao, Z.; Matsumoto, K. The kinase TAK1 can activate the NIK-IκB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 1999, 398, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Dinarello, C.A. Treating rheumatological diseases and co-morbidities with interleukin-1 blocking therapies. Rheumatology 2015, 54, 2134–2144. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Polzer, K.; Joosten, L.; Gasser, J.; Distler, J.H.; Ruiz, G.; Baum, W.; Redlich, K.; Bobacz, K.; Smolen, J.S.; Berg, W.V.D.; et al. Interleukin-1 is essential for systemic inflammatory bone loss. Ann. Rheum. Dis. 2010, 69, 284–290. [Google Scholar] [CrossRef]
- Zwerina, J.; Hayer, S.; Tohidast-Akrad, M.; Bergmeister, H.; Redlich, K.; Feige, U.; Dunstan, C.; Kollias, G.; Smolen, J.; Schett, G. Single and combined inhibition of tumor necrosis factor, interleukin-1, and RANKL pathways in tumor necrosis factor-induced arthritis: Effects on synovial inflammation, bone erosion, and cartilage destruction. Arthritis Rheum. 2004, 50, 277–290. [Google Scholar] [CrossRef]
- Zwerina, J.; Redlich, K.; Polzer, K.; Joosten, L.; Krönke, G.; Distler, J.; Hess, A.; Pundt, N.; Pap, T.; Hoffmann, O.; et al. TNF-induced structural joint damage is mediated by IL-1. Proc. Natl. Acad. Sci. USA 2007, 104, 11742–11747. [Google Scholar] [CrossRef]
- Zhu, H.; Jian, Z.; Zhong, Y.; Ye, Y.; Zhang, Y.; Hu, X.; Pu, B.; Gu, L.; Xiong, X. Janus kinase inhibition ameliorates ischemic stroke injury and neuroinflammation through reducing NLRP3 inflammasome activation via JAK2/STAT3 pathway inhibition. Front. Immunol. 2021, 12, 714943. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.L.; Duan, W.; Su, C.Y.; Mao, F.Y.; Lv, Y.P.; Teng, Y.S.; Yu, P.W.; Zhuang, Y.; Zhao, Y.L. Interleukin 6 induces M2 macrophage differentiation by STAT3 activation that correlates with gastric cancer progression. Cancer Immunol. Immunother. 2017, 66, 1597–1608. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | Takinib Interaction | Function |
|---|---|---|
| E638 | H-Bond | Amide H-Bond works with Q644 to stabilize ligand [24] |
| Q644 | H-Bond | Stabilizes side chain H-bonds [25] |
| Y640 | Hydrophobic | Modulates Tyr705 phosphorylation, pocket hydrophobicity [26] |
| Y657 | H-bond, Hydrophobic | Works with E638 & Y640 to modulate pocket hydrophobicity [22,24,26] |
| I659 | Hydrophobic | Ligand stabilization, pocket hydrophobicity [24,26] |
| W623 | Hydrophobic, π-π | Induces ligand conformational fit, stabilizes ligand in pocket [24,27] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panipinto, P.M.; Singh, A.K.; Shaikh, F.S.; Siegel, R.J.; Chourasia, M.; Ahmed, S. Takinib Inhibits Inflammation in Human Rheumatoid Arthritis Synovial Fibroblasts by Targeting the Janus Kinase-Signal Transducer and Activator of Transcription 3 (JAK/STAT3) Pathway. Int. J. Mol. Sci. 2021, 22, 12580. https://doi.org/10.3390/ijms222212580
Panipinto PM, Singh AK, Shaikh FS, Siegel RJ, Chourasia M, Ahmed S. Takinib Inhibits Inflammation in Human Rheumatoid Arthritis Synovial Fibroblasts by Targeting the Janus Kinase-Signal Transducer and Activator of Transcription 3 (JAK/STAT3) Pathway. International Journal of Molecular Sciences. 2021; 22(22):12580. https://doi.org/10.3390/ijms222212580
Chicago/Turabian StylePanipinto, Paul M., Anil K. Singh, Farheen S. Shaikh, Ruby J. Siegel, Mukesh Chourasia, and Salahuddin Ahmed. 2021. "Takinib Inhibits Inflammation in Human Rheumatoid Arthritis Synovial Fibroblasts by Targeting the Janus Kinase-Signal Transducer and Activator of Transcription 3 (JAK/STAT3) Pathway" International Journal of Molecular Sciences 22, no. 22: 12580. https://doi.org/10.3390/ijms222212580
APA StylePanipinto, P. M., Singh, A. K., Shaikh, F. S., Siegel, R. J., Chourasia, M., & Ahmed, S. (2021). Takinib Inhibits Inflammation in Human Rheumatoid Arthritis Synovial Fibroblasts by Targeting the Janus Kinase-Signal Transducer and Activator of Transcription 3 (JAK/STAT3) Pathway. International Journal of Molecular Sciences, 22(22), 12580. https://doi.org/10.3390/ijms222212580