
Current Adenosinergic Therapies: What Do Cancer Cells Stand to Gain and Lose?


Abstract
:1. Introduction
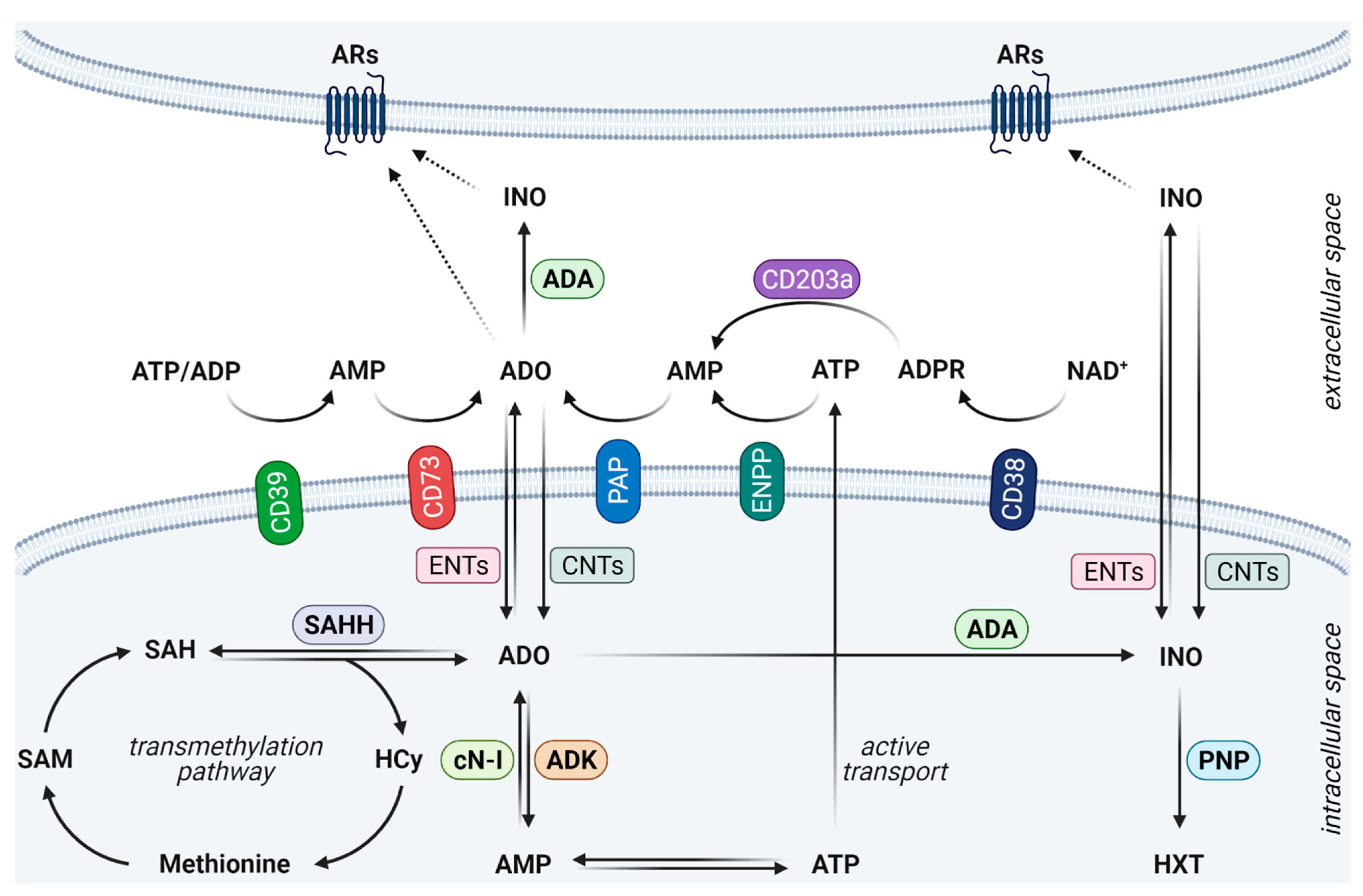
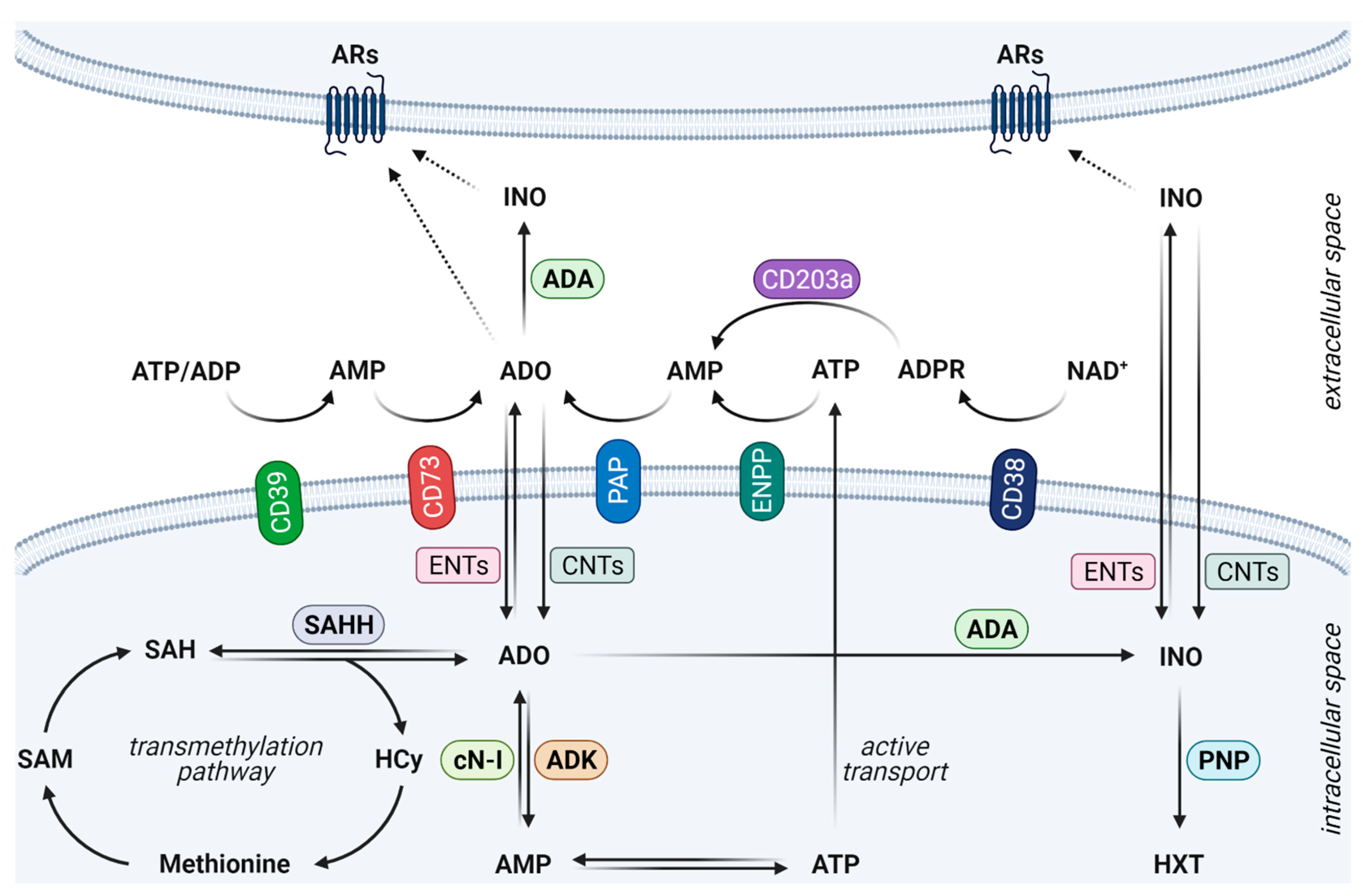
2. Fine-Tuned Orchestration of the Adenosinergic Pathway in Cancer
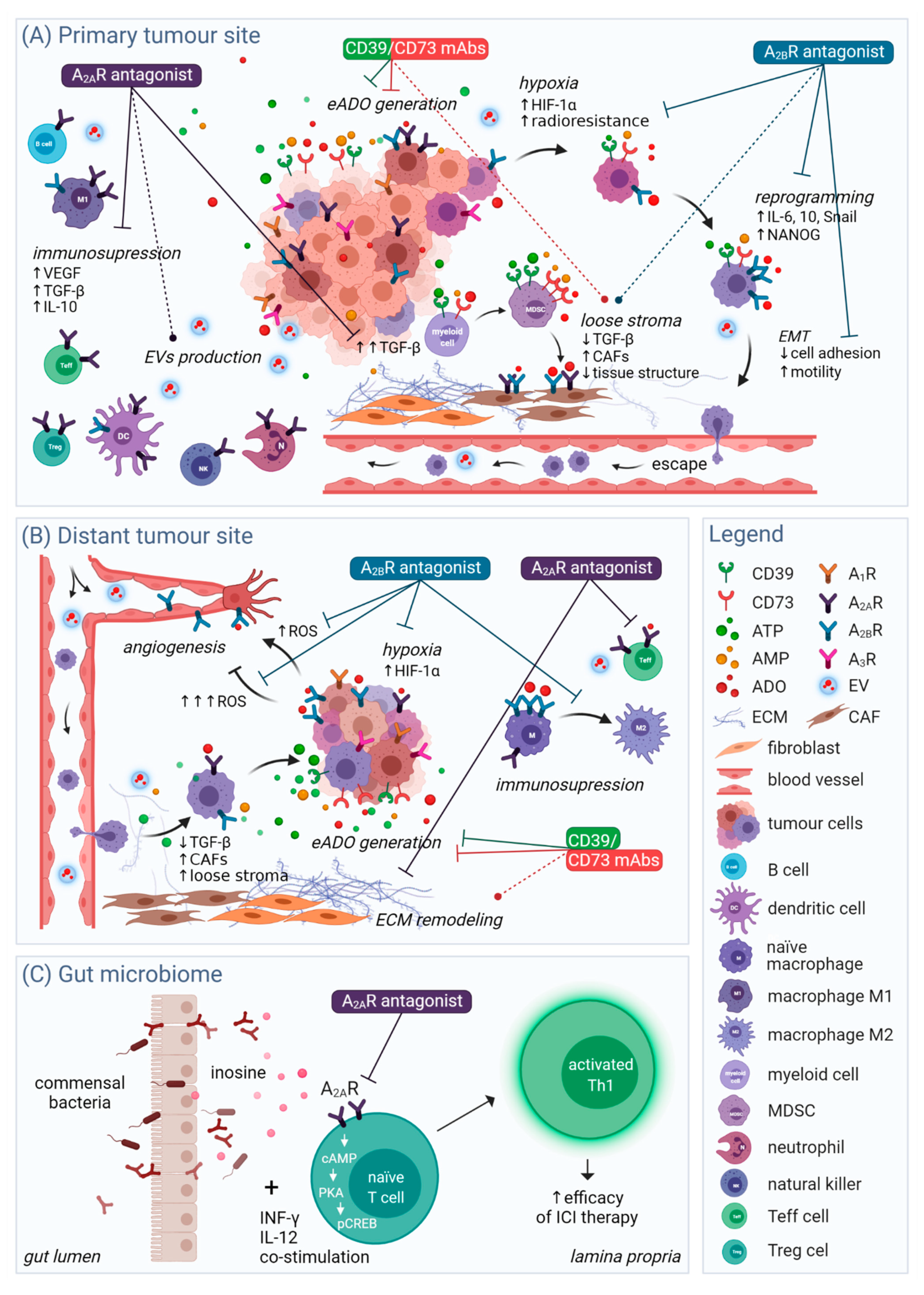
3. Current Therapeutic Focus
4. Targeting ARs on Cancer Cells
4.1. Cancer Cell Proliferation
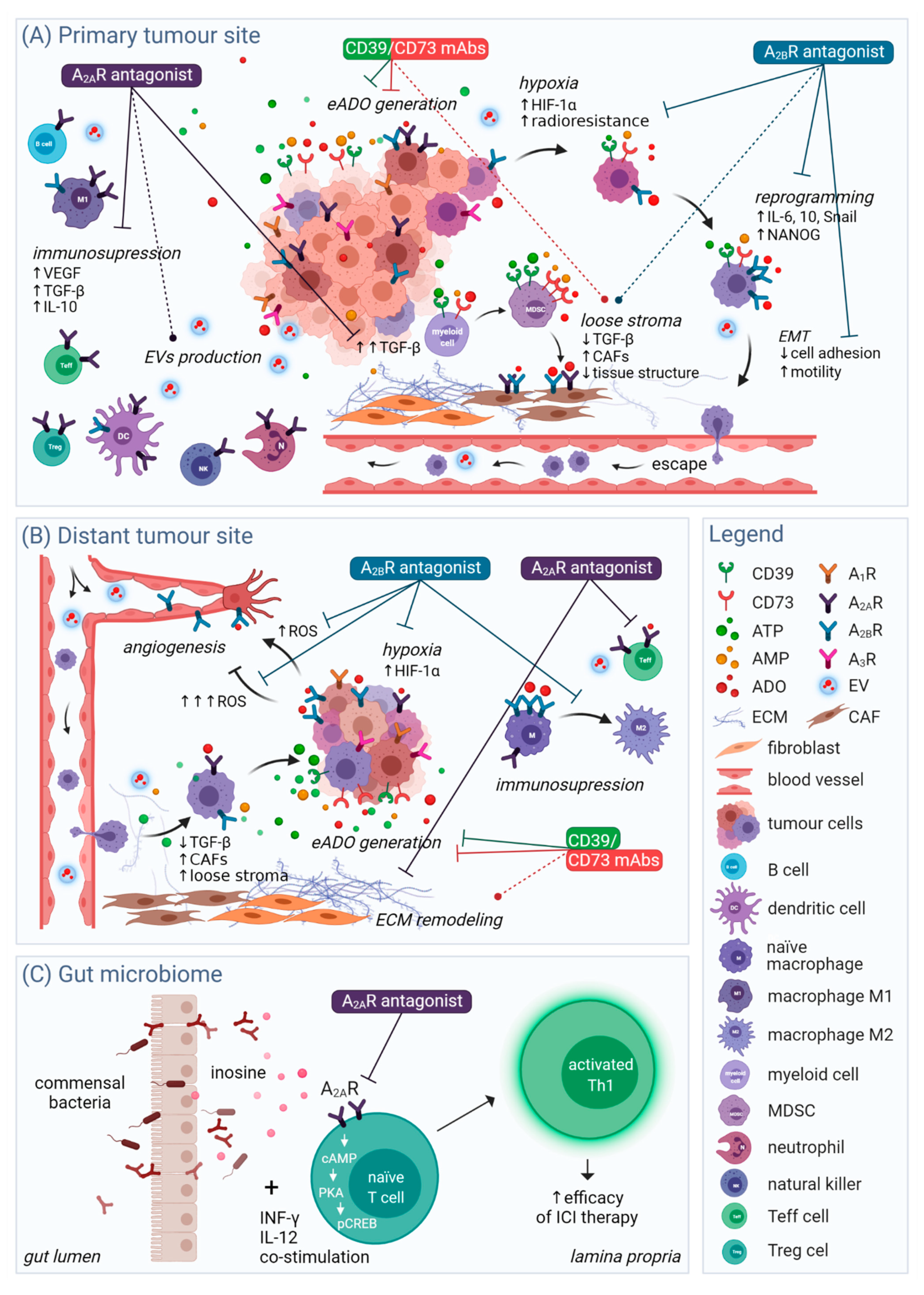
4.2. Hypoxia and Immunomodulation
4.3. Migration and Angiogenesis
4.4. Tumour Cell Stemness and Reprogramming
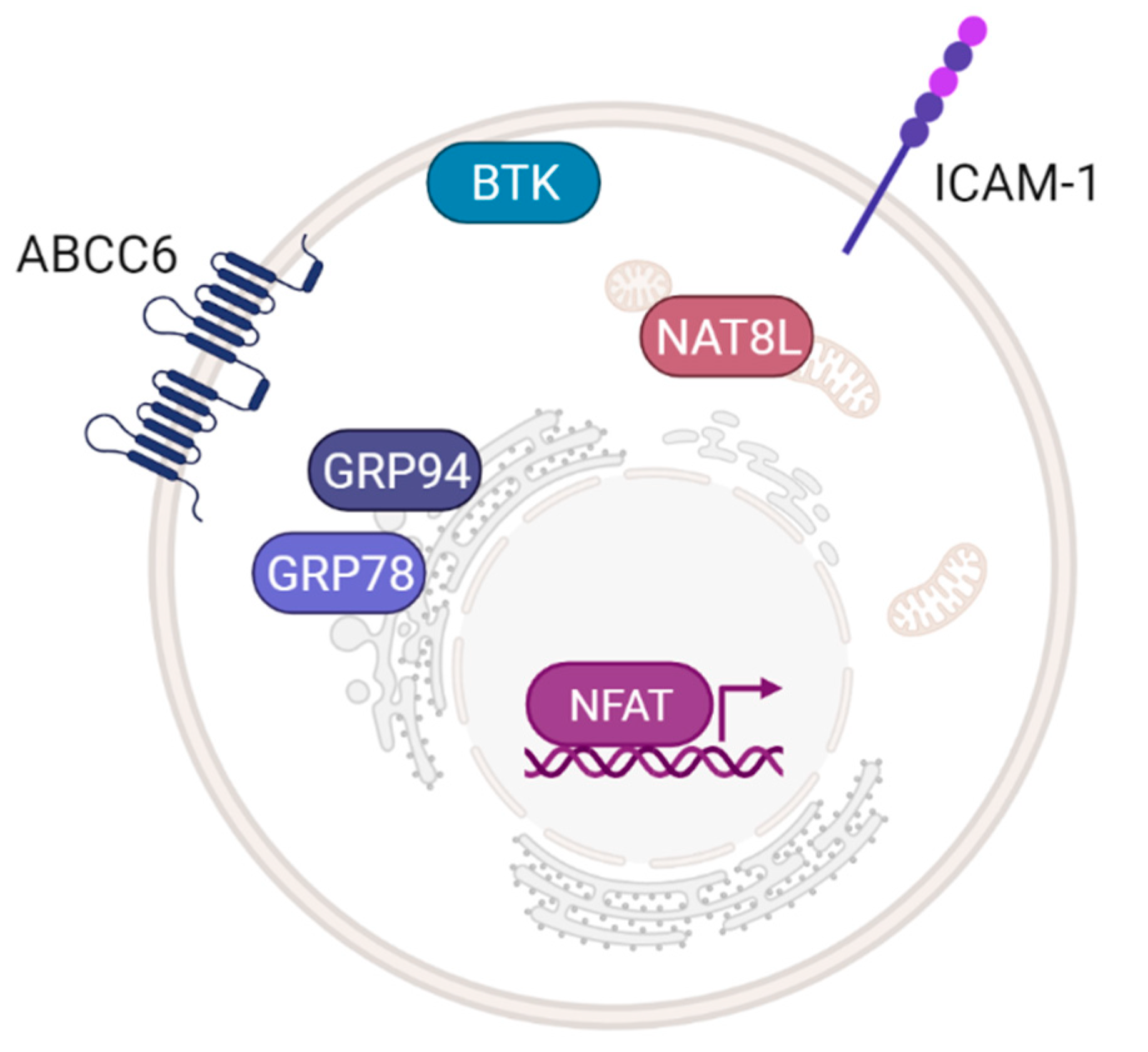
4.5. Extracellular Vesicles
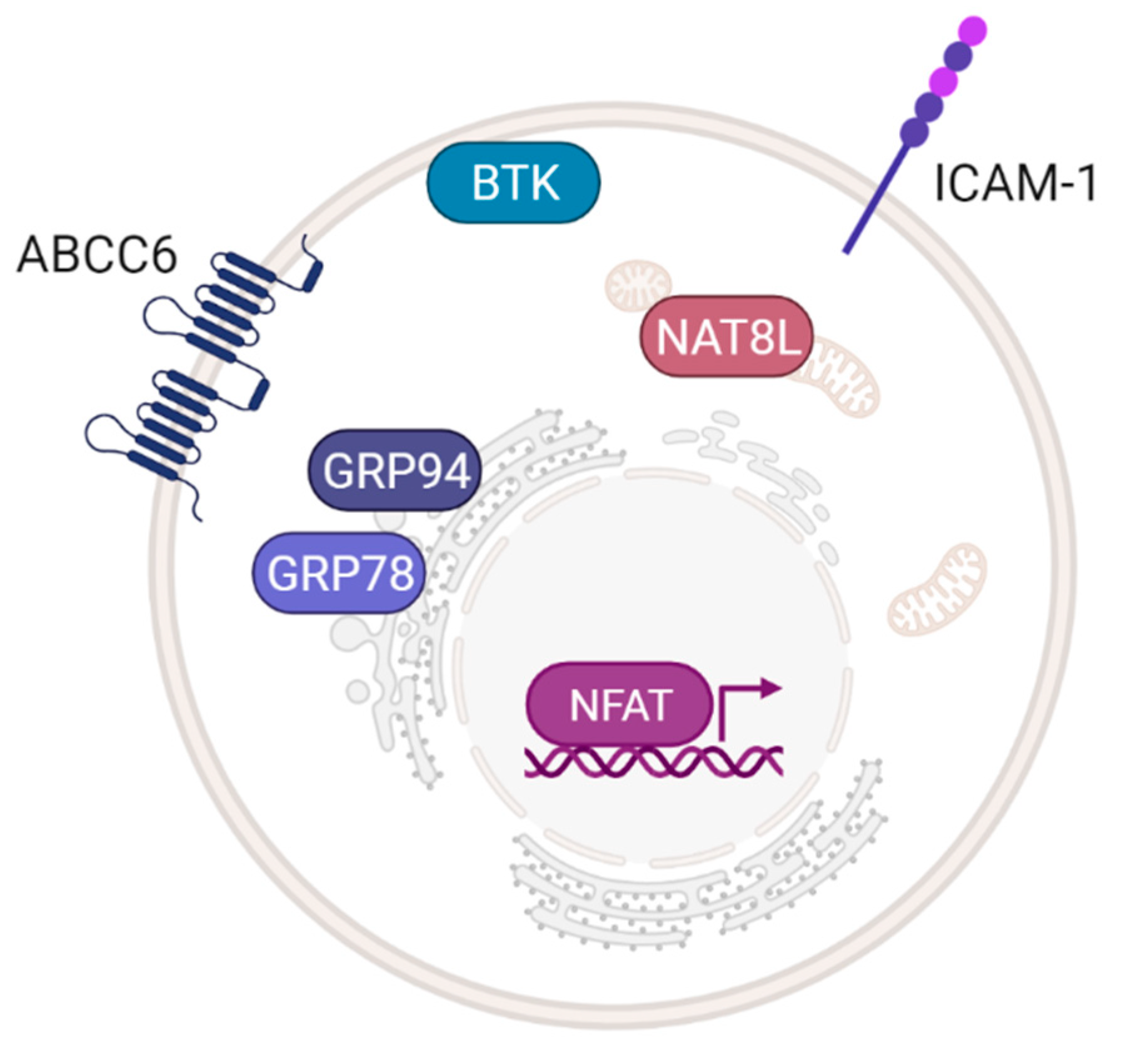
4.6. Prospective Targets of Adenosinergic Therapy
5. Persisting and Potential Limitations of Adenosinergic Therapy
5.1. Structure-Related Limitations
5.2. Context-Related Limitations
6. Perspectives
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4SCAR-T | 4th generation chimeric antigen receptor gene-modified T cells |
| A1R | Adenosine A1 receptor |
| A2AR | Adenosine A2A receptor |
| A2BR | Adenosine A2B receptor |
| A3R | Adenosine A3 receptor |
| ABC | ATP-binding cassette transporter |
| ABCC6 | ATP-binding cassette subfamily C member 6 |
| AC | Adenylyl cyclase |
| ADA | Adenosine deaminase |
| ADK | Adenosine kinase |
| ADK-L | Nuclear isoform of adenosine kinase |
| ADO | Adenosine |
| ADP | Adenosine diphosphate |
| ADPR | Ectonucleotide pyrophosphatase/phosphodiesterase |
| allo HSCT | Allogenic hematopoietic stem cell transplantation |
| AMP | Adenosine monophosphate |
| AR | Adenosine receptor |
| ATP | Adenosine triphosphate |
| BCMA | B cell maturation antigen |
| BTK | Bruton’s tyrosine kinase |
| CAF | Cancer-associated fibroblast |
| cAMP | Cyclic adenosine monophosphate |
| CAR-T | Chimeric antigen receptor T cells |
| cN-I | Cytoplasmic 5′-nucleotidase-I |
| CNT | Concentrative nucleoside transporter |
| CRC | Colorectal cancer |
| CREB | cAMP-response element-binding protein |
| CSC | Cancer stem cells |
| CTC | Circulating tumour cell |
| CTLA4 | Cytotoxic T-lymphocyte-associated protein 4 |
| CXCL1 | C-X-C motif chemokine ligand 1 |
| CXCR4, 5 | C-X-C motif chemokine receptor 4, 5 |
| DA-EPOCH | Dose-adjusted etoposide, prednisone, vincristine sulfate, cyclophosphamide, and doxorubicin hydrochloride |
| DAMP | Damage-associated molecular pattern |
| DC | Dendritic cell |
| eADO | Extracellular adenosine |
| ECL2 | Extracellular loop 2 |
| ECM | Extracellular matrix |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial–mesenchymal transition |
| eNOS | Nitric oxide synthase |
| ENPP | Ectonucleotide pyrophosphatase/phosphodiesterase |
| ENT | Equilibrative nucleoside transporter |
| ePS | Endogenous plastic somatic cell |
| ERK1/2 | Extracellular signal-regulated kinase ½ |
| ETB | Engineered toxin body |
| EV | Extracellular vesicle |
| GPCR | G protein-coupled receptor |
| GRP | Glucose-regulated protein |
| GSK-3β | Glycogen synthase kinase 3β |
| HCC | Hepatocellular carcinoma |
| HCy | Homocysteine |
| HIF-1α | Hypoxia-inducible factor 1α |
| HLA-1 | Major histocompatibility complex class I molecule |
| HNSCC | Head and neck squamous cell carcinomas |
| HXT | Hypoxanthine |
| ICAM-1 | Intercellular adhesion molecule 1 |
| ICI | Immune checkpoint inhibitor |
| IDO | Indoleamine 2,3-dioxygenase |
| IL-1β, 6, 10 | Interleukin-1β, 6, 10 |
| INO | Inosine |
| mAb | Monoclonal antibody |
| MAPK | Mitogen-activated protein kinase |
| MDSC | Myeloid-derived suppressor cell |
| MM | Multiple myeloma |
| NAD+ | Nicotinamide adenine dinucleotide |
| NAT8L | N-acetylaspartate synthetase |
| NF-κB | Nuclear transcription factor-κB |
| NFAT | Nuclear factor of activated T cells |
| NK | Natural killer |
| NSCLC | Non-small cell lung cancer |
| PAP | Prostatic acid phosphatase |
| PD-1 | Programmed cell death protein 1 |
| PDAC | Pancreatic ductal adenocarcinoma |
| PD-L1 | Programmed cell death protein ligand 1 |
| PGVSMC | Rat preglomerular vascular smooth muscle cell |
| PI3K | Phosphoinositide-3-kinase |
| PKA | Protein kinase A |
| PNP | Purine nucleoside phosphorylase |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 |
| RCC | Renal cell carcinoma |
| ROS | Reactive oxygen species |
| RRI | Receptor–receptor interaction |
| SAH | S-adenosylhomocysteine |
| SAHH | S-adenosylhomocysteine hydrolase |
| SAM | S-adenosylmethionine |
| SDF-1 | Stromal cell-derived factor-1 |
| Teff | T effector cell |
| TGF- β | Transforming growth factor β |
| Th1 | T helper cell type 1 |
| TIGIT | T cell immunoreceptor with Ig and ITIM domains |
| TME | Tumour microenvironment |
| TNBC | Triple-negative breast cancer |
| TNF-α | Tumour necrosis factor α |
| TP53 | Tumour protein p53 |
| Treg | T regulatory cell |
| VEGF | Vascular endothelial growth factor |
References
- Moser, G.H.; Schrader, J.; Deussen, A. Turnover of adenosine in plasma of human and dog-blood. Am. J. Physiol. 1989, 256, C799–C806. [Google Scholar] [CrossRef]
- Lofgren, L.; Pehrsson, S.; Hagglund, G.; Tjellstrom, H.; Nylander, S. Accurate measurement of endogenous adenosine in human blood. PLoS ONE 2018, 13, e0205707. [Google Scholar] [CrossRef] [Green Version]
- Fredholm, B.B. Physiological and pathophysiological roles of adenosine. Sleep Biol. Rhythm. 2011, 9, 24–28. [Google Scholar] [CrossRef]
- Andine, P.; Rudolphi, K.A.; Fredholm, B.B.; Hagberg, H. Effect of propentofylline (HWA-285) on extracellular purines and excitatory amino-acids in ca1 of rat hippocampus during transient ischemia. Br. J. Pharmacol. 1990, 100, 814–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedata, F.; Corsi, C.; Melani, A.; Bordoni, F.; Latini, S. Adenosine extracellular brain concentrations and role of A(2A) receptors in ischemia. Ann. N. Y. Acad. Sci. 2001, 939, 74–84. [Google Scholar] [CrossRef]
- Blay, J.; White, T.D.; Hoskin, D.W. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. 1997, 57, 2602–2605. [Google Scholar]
- Boison, D.; Yegutkin, G.G. Adenosine Metabolism: Emerging Concepts for Cancer Therapy. Cancer Cell 2019, 36, 582–596. [Google Scholar] [CrossRef]
- Chiarella, A.M.; Ryu, Y.K.; Manji, G.A.; Rustgi, A.K. Extracellular ATP and Adenosine in Cancer Pathogenesis and Treatment. Trends Cancer 2021, 7, 731–750. [Google Scholar] [CrossRef]
- Williams-Karnesky, R.L.; Sandau, U.S.; Lusardi, T.A.; Lytle, N.K.; Farrell, J.M.; Pritchard, E.M.; Kaplan, D.L.; Boison, D. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J. Clin. Investig. 2013, 123, 3552–3563. [Google Scholar] [CrossRef] [Green Version]
- Boswell-Casteel, R.C.; Hays, F.A. Equilibrative nucleoside transporters A review. Nucleosides Nucleotides Nucleic Acids 2017, 36, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Yegutkin, G.G. Enzymes involved in metabolism of extracellular nucleotides and nucleosides: Functional implications and measurement of activities. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 473–497. [Google Scholar] [CrossRef] [PubMed]
- Drury, A.N.; Szent-Gyorgyi, A. The physiological activity of adenine compounds with especial reference to their action upon the mammalian heart. J. Physiol. 1929, 68, 213–237. [Google Scholar] [CrossRef]
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, S.; Contri, C.; Borea, P.A.; Vincenzi, F.; Varani, K. Adenosine and Inflammation: Here, There and Everywhere. Int. J. Mol. Sci. 2021, 22, 7685. [Google Scholar] [CrossRef]
- Azambuja, J.H.; Ludwig, N.; Braganhol, E.; Whiteside, T.L. Inhibition of the Adenosinergic Pathway in Cancer Rejuvenates Innate and Adaptive Immunity. Int. J. Mol. Sci. 2019, 20, 5698. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Boudreau, A.; Bissell, M.J. Tissue architecture and function: Dynamic reciprocity via extra- and intra-cellular matrices. Cancer Metastasis Rev. 2009, 28, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 873–885. [Google Scholar] [CrossRef] [Green Version]
- Stagg, J.; Smyth, M.J. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 2010, 29, 5346–5358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Hasko, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Varani, K. Adenosine as a Multi-Signalling Guardian Angel in Human Diseases: When, Where and How Does it Exert its Protective Effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Allard, D.; Chrobak, P.; Allard, B.; Messaoudi, N.; Stagg, J. Targeting the CD73-adenosine axis in immuno-oncology. Immunol. Lett. 2019, 205, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Long, J.S.; Crighton, D.; O’Prey, J.; MacKay, G.; Zheng, L.; Palmer, T.M.; Gottlieb, E.; Ryanl, K.M. Extracellular Adenosine Sensing-A Metabolic Cell Death Priming Mechanism Downstream of p53. Mol. Cell 2013, 50, 394–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332–1341. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Abdulla, P.; Hoffman, E.; Hamilton, K.E.; Daniels, D.; Schonfeld, C.; Loffler, M.; Reyes, G.; Duszenko, M.; Karhausen, J.; et al. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J. Exp. Med. 2005, 202, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Chambers, E.D.; White, A.; Vang, A.; Wang, Z.K.; Ayala, A.; Weng, T.T.; Blackburn, M.; Choudhary, G.; Rounds, S.; Lu, Q. Blockade of equilibrative nucleoside transporter 1/2 protects against Pseudomonas aeruginosa-induced acute lung injury and NLRP3 inflammasome activation. FASEB J. 2020, 34, 1516–1531. [Google Scholar] [CrossRef] [Green Version]
- Barletta, K.E.; Ley, K.; Mehrad, B. Regulation of Neutrophil Function by Adenosine. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 856–864. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Guo, G.; Huang, L.; Deng, L.B.; Chang, C.S.; Achyut, B.R.; Canning, M.; Xu, N.C.; Arbab, A.S.; Bollag, R.J.; et al. CD73 on cancer-associated fibroblasts enhanced by the A(2B)-mediated feedforward circuit enforces an immune checkpoint. Nat. Commun. 2020, 11, 515. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7, 277ra30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitkovsky, M.V.; Hatfield, S.; Abbott, R.; Belikoff, B.; Lukashev, D.; Ohta, A. Hostile, Hypoxia-A2-Adenosinergic Tumor Biology as the Next Barrier to Overcome for Tumor Immunologists. Cancer Immunol. Res. 2014, 2, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Losenkova, K.; Zuccarini, M.; Karikoski, M.; Laurila, J.; Boison, D.; Jalkanen, S.; Yegutkin, G.G. Compartmentalization of adenosine metabolism in cancer cells and its modulation during acute hypoxia. J. Cell Sci. 2020, 133, jcs241463. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Mittal, D.; Stagg, J.; Smyth, M.J. Targeting Cancer-Derived Adenosine: New Therapeutic Approaches. Cancer Discov. 2014, 4, 879–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.M.; Li, X.Y.; Aguilera, A.R.; Xiao, C.; Jacoberger-Foisac, C.; Nowlan, B.; Robson, S.C.; Beers, C.; Moesta, A.K.; Geetha, N.; et al. Control of Metastases via Myeloid CD39 and NK Cell Effector Function. Cancer Immunol. Res. 2020, 8, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Elsaadi, S.; Misund, K.; Abdollahi, P.; Vandsemb, E.N.; Moen, S.H.; Kusnierczyk, A.; Slupphaug, G.; Standal, T.; Waage, A.; et al. Conversion of ATP to adenosine by CD39 and CD73 in multiple myeloma can be successfully targeted together with adenosine receptor A2A blockade. J. Immunother. Cancer 2020, 8, e000610. [Google Scholar] [CrossRef]
- Harvey, J.B.; Phan, L.H.; Villarreal, O.E.; Bowser, J.L. CD73’s Potential as an Immunotherapy Target in Gastrointestinal Cancers. Front. Immunol. 2020, 11, 508. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.M.; Diao, L.X.; Yang, Y.B.; Yi, X.H.; Rodriguez, L.; Li, Y.L.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masjedi, A.; Ahmadi, A.; Ghani, S.; Malakotikhah, F.; Afjadi, M.N.; Irandoust, M.; Kiani, F.K.; Asl, S.H.; Atyabi, F.; Hassannia, H.; et al. Silencing adenosine A2a receptor enhances dendritic cell-based cancer immunotherapy. Nanomed. Nanotechnol. Biol. Med. 2020, 29, 102240. [Google Scholar] [CrossRef]
- Borodovsky, A.; Barbon, C.M.; Wang, Y.J.; Ye, M.W.; Prickett, L.; Chandra, D.; Shaw, J.; Deng, N.H.; Sachsenmeier, K.; Clarke, J.D.; et al. Small molecule AZD4635 inhibitor of A(2A)R signaling rescues immune cell function including CD103(+) dendritic cells enhancing anti-tumor immunity. J. Immunother. Cancer 2020, 8, e000417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Zhang, Y.; Qu, B.X.; Yang, H.Y.; Hu, S.Q.; Dong, X.W. If small molecules immunotherapy comes, can the prime be far behind? Eur. J. Med. Chem. 2021, 218, 113356. [Google Scholar] [CrossRef] [PubMed]
- Masoumi, E.; Jafarzadeh, L.; Mirzaei, H.R.; Alishah, K.; Fallah-Mehrjardi, K.; Rostamian, H.; Khakpoor-Koosheh, M.; Meshkani, R.; Noorbakhsh, F.; Hadjati, J. Genetic and pharmacological targeting of A2a receptor improves function of anti-mesothelin CAR T cells. J. Exp. Clin. Cancer Res. 2020, 39, 49. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Henderson, M.A.; Giuffrida, L.; Mills, J.K.; Sek, K.; Cross, R.S.; Davenport, A.J.; John, L.B.; Mardiana, S.; Slaney, C.Y.; et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J. Clin. Investig. 2017, 127, 929–941. [Google Scholar] [CrossRef] [Green Version]
- Fong, L.; Hotson, A.; Powderly, J.D.; Sznol, M.; Heist, R.S.; Choueiri, T.K.; George, S.; Hughes, B.G.M.; Hellmann, M.D.; Shepard, D.R.; et al. Adenosine 2A Receptor Blockade as an Immunotherapy for Treatment-Refractory Renal Cell Cancer. Cancer Discov. 2020, 10, 40–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willingham, S.B.; Ho, P.Y.; Hotson, A.; Hill, C.; Piccione, E.C.; Hsieh, J.; Liu, L.; Buggy, J.J.; McCaffery, I.; Miller, R.A. A2AR Antagonism with CPI-444 Induces Antitumor Responses and Augments Efficacy to Anti-PD-(L)1 and Anti-CTLA-4 in Preclinical Models. Cancer Immunol. Res. 2018, 6, 1136–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidders, B.; Zhang, P.; Goodwin, K.; O’Connor, G.; Russell, D.L.; Borodovsky, A.; Armenia, J.; McEwen, R.; Linghu, B.; Bendell, J.C.; et al. Adenosine Signaling Is Prognostic for Cancer Outcome and Has Predictive Utility for Immunotherapeutic Response. Clin. Cancer Res. 2020, 26, 2176–2187. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Ijzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar] [PubMed]
- Fredholm, B.B.; Arslan, G.; Halldner, L.; Kull, B.; Schulte, G.; Wasserman, W. Structure and function of adenosine receptors and their genes. Naunyn-Schmiedebergs Arch. Pharmacol. 2000, 362, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of adenosine receptors: The state of the art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Cordomi, A.; Zelman-Femiak, M.; Brugarolas, M.; Moreno, E.; Aguinaga, D.; Perez-Benito, L.; Cortes, A.; Casado, V.; Mallol, J.; et al. Quaternary structure of a G-protein-coupled receptor heterotetramer in complex with G(i) and G(s). BMC Biol. 2016, 14, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredholm, B.B.; Ijzerman, A.P.; Jacobson, K.A.; Linden, J.; Muller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors—An Update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Townsendnicholson, A.; Baker, E.; Schofield, P.R.; Sutherland, G.R. Localization of the adenosine-A1-receptor subtype gene (ADORA1) to chromosome 1Q32.1. Genomics 1995, 26, 423–425. [Google Scholar] [CrossRef]
- Le, F.; TownsendNicholson, A.; Baker, E.; Sutherland, G.R.; Schofield, P.R. Characterization and chromosomal localization of the human A2a adenosine receptor gene: ADORA2A. Biochem. Biophys. Res. Commun. 1996, 223, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Steingold, J.M.; Hatfield, S.M. Targeting Hypoxia-A2A Adenosinergic Immunosuppression of Antitumor T Cells During Cancer Immunotherapy. Front. Immunol. 2020, 11, 7. [Google Scholar] [CrossRef]
- Yan, L.; Burbiel, J.C.; Maass, A.; Muller, C.E. Adenosine receptor agonists: From basic medicinal chemistry to clinical development. Expert Opin. Emerg. Drugs 2003, 8, 537–576. [Google Scholar] [CrossRef] [PubMed]
- Monitto, C.L.; Levitt, R.C.; Disilvestre, D.; Holroyd, K.J. LOCALIZATION OF THE A(3) ADENOSINE RECEPTOR GENE (ADORA3) TO HUMAN-CHROMOSOME 1P. Genomics 1995, 26, 637–638. [Google Scholar] [CrossRef]
- Salvatore, C.A.; Jacobson, M.A.; Taylor, H.E.; Linden, J.; Johnson, R.G. Molecular-cloning and characterization of the human-A(3) adenosine receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10365–10369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine receptors as drug targets—What are the challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madi, L.; Ochaion, A.; Rath-Wolfson, L.; Bar-Yehuda, S.; Erlanger, A.; Ohana, G.; Harish, A.; Merimski, O.; Barer, F.; Fishman, P. The A(3) adenosine receptor is highly expressed in tumor versus normal cells: Potential target for tumor growth inhibition. Clin. Cancer Res. 2004, 10, 4472–4479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Wang, Z.Y.; Xue, G.; Qin, X.J.; Wu, J.F.; Zhang, G. ADORA1 is a diagnostic-related biomarker and correlated with immune infiltrates in papillary thyroid carcinoma. J. Cancer 2021, 12, 3997–4010. [Google Scholar] [CrossRef]
- Kamai, T.; Kijima, T.; Tsuzuki, T.; Nukui, A.; Abe, H.; Arai, K.; Yoshida, K.I. Increased expression of adenosine 2A receptors in metastatic renal cell carcinoma is associated with poorer response to anti-vascular endothelial growth factor agents and anti-PD-1/Anti-CTLA4 antibodies and shorter survival. Cancer Immunol. Immunother. 2021, 70, 2009–2021. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Wei, Q.; Yang, L. ADORA1 Promotes Hepatocellular Carcinoma Progression via PI3K/AKT Pathway. OncoTargets Ther. 2020, 13, 12409–12419. [Google Scholar] [CrossRef]
- Pan, S.M.; Liang, S.X.; Wang, X.Y. ADORA1 promotes nasopharyngeal carcinoma cell progression through regulation of PI3K/AKT/GSK-3 beta/beta-catenin signaling. Life Sci. 2021, 278, 119581. [Google Scholar] [CrossRef]
- Ma, H.Y.; Li, Q.Z.; Wang, J.; Pan, J.; Su, Z.D.; Liu, S. Dual Inhibition of Ornithine Decarboxylase and A(1) Adenosine Receptor Efficiently Suppresses Breast Tumor Cells. Front. Oncol. 2021, 11, 636373. [Google Scholar] [CrossRef]
- Shi, L.S.; Wu, Z.Y.; Miao, J.; Du, S.C.; Ai, S.C.; Xu, E.; Feng, M.; Song, J.; Guan, W.X. Adenosine interaction with adenosine receptor A2a promotes gastric cancer metastasis by enhancing PI3K-AKT-mTOR signaling. Mol. Biol. Cell 2019, 30, 2527–2534. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-L.; Shen, M.-N.; Hu, B.; Wang, B.-L.; Yang, W.-J.; Lv, L.-H.; Wang, H.; Zhou, Y.; Jin, A.-L.; Sun, Y.-F.; et al. CD73 promotes hepatocellular carcinoma progression and metastasis via activating PI3K/AKT signaling by inducing Rap1-mediated membrane localization of P110 and predicts poor prognosis. J. Hematol. Oncol. 2019, 12, 37. [Google Scholar] [CrossRef]
- Sitkovsky, M.V. Lessons from the A2A Adenosine Receptor Antagonist-Enabled Tumor Regression and Survival in Patients with Treatment-Refractory Renal Cell Cancer. Cancer Discov. 2020, 10, 16–19. [Google Scholar] [CrossRef]
- Seitz, L.; Jin, L.X.; Leleti, M.; Ashok, D.; Jeffrey, J.; Rieger, A.; Tiessen, R.G.; Arold, G.; Tan, J.B.L.; Powers, J.P.; et al. Safety, tolerability, and pharmacology of AB928, a novel dual adenosine receptor antagonist, in a randomized, phase 1 study in healthy volunteers. Investig. New Drugs 2019, 37, 711–721. [Google Scholar] [CrossRef]
- Harshman, L.C.; Chu, M.; George, S.; Gordon, B.; Hughes, M.; Carthon, B.C.; Fong, L.; Merchan, J.R.; Kwei, L.; Hotson, A.N.; et al. Adenosine receptor blockade with ciforadenant plus/- atezolizumab in advanced metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2020, 38, 129. [Google Scholar] [CrossRef]
- Wilkat, M.; Bast, H.; Drees, R.; Dunser, J.; Mahr, A.; Azoitei, N.; Marienfeld, R.; Frank, F.; Brhel, M.; Ushmorov, A.; et al. Adenosine receptor 2B activity promotes autonomous growth, migration as well as vascularization of head and neck squamous cell carcinoma cells. Int. J. Cancer 2020, 147, 202–217. [Google Scholar] [CrossRef]
- Yi, Y.; Zhou, Y.H.; Chug, X.; Zheng, X.P.; Fei, D.; Lei, J.; Qi, H.Y.; Dai, Y.B. Blockade of Adenosine A2b Receptor Reduces Tumor Growth and Migration in Renal Cell Carcinoma. J. Cancer 2020, 11, 421–431. [Google Scholar] [CrossRef] [Green Version]
- Koussemou, M.; Klotz, K.N. Agonists activate different A(2B) adenosine receptor signaling pathways in MBA-MD-231 breast cancer cells with distinct potencies. Naunyn-Schmiedebergs Arch. Pharmacol. 2019, 392, 1515–1521. [Google Scholar] [CrossRef]
- Koussemou, M.; Lorenz, K.; Klotz, K.-N. The A(2B) adenosine receptor in MDA-MB-231 breast cancer cells diminishes ERK1/2 phosphorylation by activation of MAPK-phosphatase-1. PLoS ONE 2018, 13, e0202914. [Google Scholar] [CrossRef]
- Pottie, E.; Tosh, D.K.; Gao, Z.G.; Jacobson, K.A.; Stove, C.P. Assessment of biased agonism at the A(3) adenosine receptor using beta-arrestin and miniG alpha(i) recruitment assays. Biochem. Pharmacol. 2020, 177, 113934. [Google Scholar] [CrossRef]
- Smith, J.S.; Lefkowitz, R.J.; Rajagopal, S. Biased signalling: From simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243–260. [Google Scholar] [CrossRef]
- Long, J.S.; Schoonen, P.M.; Graczyk, D.; O’Prey, J.; Ryan, K.M. p73 engages A2B receptor signalling to prime cancer cells to chemotherapy-induced death. Oncogene 2015, 34, 5152–5162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.; Ngiow, S.F.; Madore, J.; Reinhardt, J.; Landsberg, J.; Chitsazan, A.; Rautela, J.; Bald, T.; Barkauskas, D.S.; Ahern, E.; et al. Targeting Adenosine in BRAF-Mutant Melanoma Reduces Tumor Growth and Metastasis. Cancer Res. 2017, 77, 4684–4696. [Google Scholar] [CrossRef] [Green Version]
- Fishman, P.; Bar-Yehuda, S.; Barer, F.; Madi, L.; Multani, A.S.; Pathak, S. The A3 adenosine receptor as a new target for cancer therapy and chemoprotection. Exp. Cell Res. 2001, 269, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Merighi, S.; Benini, A.; Mirandola, P.; Gessi, S.; Varani, K.; Leung, E.; Maclennan, S.; Borea, P.A. A(3) adenosine receptor activation inhibits cell proliferation via phosphatidylinositol 3-kinase/Akt-dependent inhibition of the extracellular signal-regulated kinase 1/2 phosphorylation in A375 human melanoma cells. J. Biol. Chem. 2005, 280, 19516–19526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.; Stemmer, S.M.; Zozulya, G.; Ochaion, A.; Patoka, R.; Barer, F.; Bar-Yehuda, S.; Rath-Wolfson, L.; Jacobson, K.A.; Fishman, P. CF102 an A(3) Adenosine Receptor Agonist Mediates Anti-Tumor and Anti-Inflammatory Effects in the Liver. J. Cell. Physiol. 2011, 226, 2438–2447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stemmer, S.M.; Manojlovic, N.S.; Marinca, M.V.; Petrov, P.; Cherciu, N.; Ganea, D.; Ciuleanu, T.E.; Pusca, I.A.; Beg, M.S.; Purcell, W.T.; et al. Namodenoson in Advanced Hepatocellular Carcinoma and Child-Pugh B Cirrhosis: Randomized Placebo-Controlled Clinical Trial. Cancers 2021, 13, 187. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.C.-L.; Zhang, H.; Gilkes, D.M.; Chen, J.; Wei, H.; Chaturvedi, P.; Hubbi, M.E.; Semenza, G.L. Inhibitors of hypoxia-inducible factor 1 block breast cancer metastatic niche formation and lung metastasis. J. Mol. Med. 2012, 90, 803–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Belikoff, B.; Schreiber, T.H.; Sethumadhavan, S.; Abbott, R.; Philbrook, P.; Thayer, M.; Shujia, D.; et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1 alpha-dependent and extracellular adenosine-mediated tumor protection. J. Mol. Med. 2014, 92, 1283–1292. [Google Scholar] [CrossRef]
- Hatfield, S.M.; Sitkovsky, M.V. Antihypoxic oxygenation agents with respiratory hyperoxia to improve cancer immunotherapy. J. Clin. Investig. 2020, 130, 5629–5637. [Google Scholar] [CrossRef]
- Sitkovsky, M.V. T regulatory cells: Hypoxia-adenosinergic suppression and re-direction of the immune response. Trends Immunol. 2009, 30, 102–108. [Google Scholar] [CrossRef]
- Bullen, J.W.; Tchernyshyov, I.; Holewinski, R.J.; DeVine, L.; Wu, F.; Venkatraman, V.; Kass, D.L.; Cole, R.N.; Van Eyk, J.; Semenza, G.L. Protein kinase A-dependent phosphorylation stimulates the transcriptional activity of hypoxia-inducible factor 1. Sci. Signal. 2016, 9, ra56. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.E.; Ko, I.G.; Jin, J.J.; Hwang, L.; Kim, C.J.; Kim, S.H.; Han, J.H.; Jeon, J.W. Polydeoxyribonucleotide Exerts Therapeutic Effect by Increasing VEGF and Inhibiting Inflammatory Cytokines in Ischemic Colitis Rats. BioMed Res. Int. 2020, 2020, 2169083. [Google Scholar] [CrossRef]
- Ernens, I.; Leonard, F.; Vausort, M.; Rolland-Turner, M.; Devaux, Y.; Wagner, D.R. Adenosine up-regulates vascular endothelial growth factor in human macrophages. Biochem. Biophys. Res. Commun. 2010, 392, 351–356. [Google Scholar] [CrossRef]
- Kong, T.; Westerman, K.A.; Faigle, M.; Eltzschig, H.K.; Colgan, S.P. HIF-dependent induction of adenosine A2B receptor in hypoxia. FASEB J. 2006, 20, 2242–2250. [Google Scholar] [CrossRef] [PubMed]
- Ngamsri, K.C.; Fabian, F.; Fuhr, A.; Gamper-Tsigaras, J.; Straub, A.; Fecher, D.; Steinke, M.; Walles, H.; Reutershan, J.; Konrad, F.M. Sevoflurane Exerts Protective Effects in Murine Peritonitis-induced Sepsis via Hypoxia-inducible Factor 1 alpha/Adenosine A2B Receptor Signaling. Anesthesiology 2021, 135, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Novitskiy, S.V.; Ryzhov, S.; Zaynagetdinov, R.; Goldstein, A.E.; Huang, Y.H.; Tikhomirov, O.Y.; Blackburn, M.R.; Biaggioni, I.; Carbone, D.P.; Feoktistov, I.; et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 2008, 112, 1822–1831. [Google Scholar] [CrossRef]
- Lan, J.; Lu, H.; Samanta, D.; Salman, S.; Lu, Y.; Semenza, G.L. Hypoxia-inducible factor 1-dependent expression of adenosine receptor 2B promotes breast cancer stem cell enrichment. Proc. Natl. Acad. Sci. USA 2018, 115, E9640–E9648. [Google Scholar] [CrossRef] [Green Version]
- Torres-Pineda, D.B.; Mora-Garcia, M.D.; Garcia-Rocha, R.; Hernandez-Montes, J.; Weiss-Steider, B.; Montesinos-Montesinos, J.J.; Don-Lopez, C.A.; Marin-Aquino, L.A.; Munoz-Godinez, R.; Ibarra, L.R.A.; et al. Corrigendum to “Adenosine augments the production of IL-10 in cervical cancer cells through interaction with the A2B adenosine receptor, resulting in protection against the activity of cytotoxic T cells” [Cytokine 130 (2020) 155082]. Cytokine 2020, 133, 155110. [Google Scholar] [CrossRef] [PubMed]
- Kotanska, M.; Szafarz, M.L.; Mika, K.; Dziubina, A.; Bednarski, M.; Muller, C.E.; Sapa, J.; Kiec-Kononowicz, K. PSB 603-a known selective adenosine A2B receptor antagonist—Has anti-inflammatory activity in mice. Biomed. Pharmacother. 2021, 135. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhang, Y.; Zhang, L.; Song, W.; Shi, L. Hypoxia enhances indoleamine 2,3-dioxygenase production in dendritic cells. Oncotarget 2018, 9, 11572–11580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Fornai, M.; Pellegrini, C.; D’Antongiovanni, V.; Turiello, R.; Morello, S.; Hasko, G.; Blandizzi, C. Adenosine Signaling in the Tumor Microenvironment. In Tumor Microenvironment: Signaling Pathways—Part B (Advances in Experimental Medicine and Biology); Birbrair, A., Ed.; Springer: Cham, Switzerland, 2021; Volume 1270, pp. 145–167. [Google Scholar]
- Rolland-Turner, M.; Goretti, E.; Bousquenaud, M.; Leonard, F.; Nicolas, C.; Zhang, L.; Maskali, F.; Marie, P.-Y.; Devaux, Y.; Wagner, D. Adenosine Stimulates the Migration of Human Endothelial Progenitor Cells. Role ofCXCR4 and MicroRNA-150. PLoS ONE 2013, 8, e54135. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Ou, X.; Song, T.; Zhang, W.; Cong, F.; Zhang, S.; Xiong, Y. Adenosine A(2B) receptor stimulates angiogenesis by inducing VEGF and eNOS in human microvascular endothelial cells. Exp. Biol. Med. 2015, 240, 1472–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryzhov, S.V.; Pickup, M.W.; Chytil, A.; Gorska, A.E.; Zhang, Q.; Owens, P.; Feoktistov, I.; Moses, H.L.; Novitskiy, S.V. Role of TGF-beta Signaling in Generation of CD39(+)CD73(+) Myeloid Cells in Tumors. J. Immunol. 2014, 193, 3155–3164. [Google Scholar] [CrossRef] [Green Version]
- Vasiukov, G.; Novitskaya, T.; Zijlstra, A.; Owens, P.; Ye, F.; Zhao, Z.G.; Moses, H.L.; Blackwell, T.; Feoktistov, I.; Novitskiy, S.V. Myeloid Cell-Derived TGF beta Signaling Regulates ECM Deposition in Mammary Carcinoma via Adenosine-Dependent Mechanisms. Cancer Res. 2020, 80, 2628–2638. [Google Scholar] [CrossRef] [Green Version]
- Howe, A.K. Regulation of actin-based cell migration by cAMP/PKA. Biochim. Et Biophys. Acta-Mol. Cell Res. 2004, 1692, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Angioni, R.; Liboni, C.; Herkenne, S.; Sanchez-Rodriguez, R.; Borile, G.; Marcuzzi, E.; Cali, B.; Muraca, M.; Viola, A. CD73(+) extracellular vesicles inhibit angiogenesis through adenosine A(2B) receptor signalling. J. Extracell. Vesicles 2020, 9, 1757900. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Du, J.; Hourani, S.; Ledent, C.; Li, J.-M. Inactivation of Adenosine A(2A) Receptor Attenuates Basal and Angiotensin II-induced ROS Production by Nox2 in Endothelial Cells. J. Biol. Chem. 2010, 285, 40104–40113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Li, L.D.; Zhou, C.S.; Chen, X.; Cao, Y.Q. Adenosine A2A receptor activation reduces brain metastasis via SDF-1/CXCR4 axis and protecting blood-brain barrier. Mol. Carcinog. 2020, 59, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Bours, M.J.L.; Swennen, E.L.R.; Di Virgilio, F.; Cronstein, B.N.; Dagnelie, P.C. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 2006, 112, 358–404. [Google Scholar] [CrossRef] [PubMed]
- Popielarski, M.; Ponamarczuk, H.; Stasiak, M.; Gdula, A.; Bednarek, R.; Wolska, N.; Swiatkowska, M. P2Y(12) receptor antagonists and AR receptor agonists regulates Protein Disulfide Isomerase secretion from platelets and endothelial cells. Biochem. Biophys. Res. Commun. 2020, 526, 756–763. [Google Scholar] [CrossRef]
- Bowser, J.L.; Broaddus, R.R. CD73s protection of epithelial integrity: Thinking beyond the barrier. Tissue Barriers 2016, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntantie, E.; Gonyo, P.; Lorimer, E.L.; Hauser, A.D.; Schuld, N.; McAllister, D.; Kalyanaraman, B.; Dwinell, M.B.; Auchampach, J.A.; Williams, C.L. An Adenosine-Mediated Signaling Pathway Suppresses Prenylation of the GTPase Rap1B and Promotes Cell Scattering. Sci. Signal. 2013, 6, ra39. [Google Scholar] [CrossRef] [Green Version]
- Hinz, S.; Jung, D.; Hauert, D.; Bachmann, H.S. Molecular and Pharmacological Characterization of the Interaction between Human Geranylgeranyltransferase Type I and Ras-Related Protein Rap1B. Int. J. Mol. Sci. 2021, 22, 2501. [Google Scholar] [CrossRef] [PubMed]
- Lupia, M.; Angiolini, F.; Bertalot, G.; Freddi, S.; Sachsenmeier, K.F.; Chisci, E.; Kutryb-Zajac, B.; Confalonieri, S.; Smolenski, R.T.; Giovannoni, R.; et al. CD73 Regulates Stemness and Epithelial-Mesenchymal Transition in Ovarian Cancer-Initiating Cells. Stem Cell Rep. 2018, 10, 1412–1425. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.-L.; Hu, B.; Tang, W.-G.; Xie, S.-H.; Ren, N.; Guo, L.; Lu, R.-Q. CD73 sustained cancer-stem-cell traits by promoting SOX9 expression and stability in hepatocellular carcinoma. J. Hematol. Oncol. 2020, 13, 11. [Google Scholar] [CrossRef] [Green Version]
- Tsiampali, J.; Neumann, S.; Giesen, B.; Koch, K.; Maciaczyk, D.; Janiak, C.; Hanggi, D.; Maciaczyk, J. Enzymatic Activity of CD73 Modulates Invasion of Gliomas via Epithelial-Mesenchymal Transition-Like Reprogramming. Pharmaceuticals 2020, 13, 378. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, U.; Ukena, D.; Lohse, M.J. Xanthine derivatives as antagonists at A1 and A2 adenosine receptors. Naunyn-Schmiedebergs Arch. Pharmacol. 1985, 330, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.; Vargas, Y.; Uribe, D.; Jaramillo, C.; Gleisner, A.; Salazar-Onfray, F.; Lopez, M.N.; Melo, R.; Oyarzun, C.; San Martin, R.; et al. Adenosine A(3) receptor elicits chemoresistance mediated by multiple resistance-associated protein-1 in human glioblastoma stem-like cells. Oncotarget 2016, 7, 67373–67386. [Google Scholar] [CrossRef] [Green Version]
- Gessi, S.; Sacchetto, V.; Fogli, E.; Merighi, S.; Varani, K.; Baraldi, P.G.; Tabrizi, M.A.; Leung, E.; Maclennan, S.; Borea, P.A. Modulation of metalloproteinase-9 in U87MG glioblastoma cells by A(3) adenosine receptors. Biochem. Pharmacol. 2010, 79, 1483–1495. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.; Erices, J.I.; Sanchez, F.; Ehrenfeld, P.; Turchi, L.; Virolle, T.; Uribe, D.; Niechi, I.; Spichiger, C.; Rocha, J.D.; et al. Extracellular adenosine promotes cell migration/invasion of Glioblastoma Stem-like Cells through A(3) Adenosine Receptor activation under hypoxia. Cancer Lett. 2019, 446, 112–122. [Google Scholar] [CrossRef]
- Liu, T.Z.; Wang, X.; Bai, Y.F.; Liao, H.Z.; Qiu, S.C.; Yang, Y.Q.; Yan, X.H.; Chen, J.; Guo, H.B.; Zhang, S.Z. The HIF-2alpha dependent induction of PAP and adenosine synthesis regulates glioblastoma stem cell function through the A2B adenosine receptor. Int. J. Biochem. Cell Biol. 2014, 49, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Giacomelli, C.; Daniele, S.; Romei, C.; Tavanti, L.; Neri, T.; Piano, I.; Celi, A.; Martini, C.; Trincavelli, M.L. The A(2B) Adenosine Receptor Modulates the Epithelial-Mesenchymal Transition through the Balance of cAMP/PKA and MAPK/ERK Pathway Activation in Human Epithelial Lung Cells. Front. Pharmacol. 2018, 9, 54. [Google Scholar] [CrossRef] [Green Version]
- Daniele, S.; Zappelli, E.; Natali, L.; Martini, C.; Trincavelli, M.L. Modulation of A(1) and A(2B) adenosine receptor activity: A new strategy to sensitise glioblastoma stem cells to chemotherapy. Cell Death Dis. 2014, 5, e1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafari, S.M.; Joshaghani, H.R.; Panjehpour, M.; Aghaei, M.; Balajam, N.Z. Apoptosis and cell cycle regulatory effects of adenosine by modulation of GLI-1 and ERK1/2 pathways in CD44(+) and CD24(−) breast cancer stem cells. Cell Prolif. 2017, 50, e12345. [Google Scholar] [CrossRef]
- Jafari, S.M.; Joshaghani, H.R.; Panjehpour, M.; Aghaei, M. A2B adenosine receptor agonist induces cell cycle arrest and apoptosis in breast cancer stem cells via ERK1/2 phosphorylation. Cell. Oncol. 2018, 41, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Jafari, S.M.; Panjehpour, M.; Aghaei, M.; Joshaghani, H.R.; Enderami, S.E. A3 Adenosine Receptor Agonist Inhibited Survival of Breast Cancer Stem Cells via GLI-1 and ERK1/2 Pathway. J. Cell. Biochem. 2017, 118, 2909–2920. [Google Scholar] [CrossRef]
- Pan, D.; Roy, S.; Gascard, P.; Zhao, J.; Chen-Tanyolac, C.; Tlsty, T.D. SOX2, OCT3/4 and NANOG expression and cellular plasticity in rare human somatic cells requires CD73. Cell. Signal. 2016, 28, 1923–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Gascard, P.; Dumont, N.; Zhao, J.; Pan, D.; Petrie, S.; Margeta, M.; Tlsty, T.D. Rare somatic cells from human breast tissue exhibit extensive lineage plasticity. Proc. Natl. Acad. Sci. USA 2013, 110, 4598–4603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitabatake, K.; Kaji, T.; Tsukimoto, M. Involvement of CD73 and A2B Receptor in Radiation-Induced DNA Damage Response and Cell Migration in Human Glioblastoma A172 Cells. Biol. Pharm. Bull. 2021, 44, 197–210. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kitabatake, K.; Abe, R.; Tsukimoto, M. Involvement of A2B Receptor in DNA Damage Response and Radiosensitizing Effect of A2B Receptor Antagonists on Mouse B16 Melanoma. Biol. Pharm. Bull. 2020, 43, 516–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitabatake, K.; Yoshida, E.; Kaji, T.; Tsukimoto, M. Involvement of adenosine A2B receptor in radiation-induced translocation of epidermal growth factor receptor and DNA damage response leading to for radioresistance in human lung cancer cells. Biochim. Biophys. Acta—Gen. Subj. 2020, 1864, 14. [Google Scholar] [CrossRef]
- Moeller, B.J.; Cao, Y.T.; Li, C.Y.; Dewhirst, M.W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: Role of reoxygenation, free radicals, and stress granules. Cancer Cell 2004, 5, 429–441. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Lee, J.S.; Kim, D.C.; Ko, Y.S.; Lee, G.W.; Kim, H.J. Increased Extracellular Adenosine in Radiotherapy-Resistant Breast Cancer Cells Enhances Tumor Progression through A2AR-Akt-beta-Catenin Signaling. Cancers 2021, 13, 2105. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hutter, J.-C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Di Iorio, P.; Ciccarelli, R. Adenine-Based Purines and Related Metabolizing Enzymes: Evidence for Their Impact on Tumor Extracellular Vesicle Activities. Cells 2021, 10, 188. [Google Scholar] [CrossRef]
- Srinivasan, S.; Vannberg, F.O.; Dixon, J.B. Lymphatic transport of exosomes as a rapid route of information dissemination to the lymph node. Sci. Rep. 2016, 6, 24436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, A.; Al-Taei, S.; Webber, J.; Mason, M.D.; Tabi, Z. Cancer Exosomes Express CD39 and CD73, Which Suppress T Cells through Adenosine Production. J. Immunol. 2011, 187, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, H.; Hirayama, A.; Kudo, R.; Hasebe, M.; Yoshioka, Y.; Matsuzaki, J.; Yamamoto, Y.; Sugimoto, M.; Soga, T.; Ochiya, T. Adenosine leakage from perforin-burst extracellular vesicles inhibits perforin secretion by cytotoxic T-lymphocytes. PLoS ONE 2020, 15, e0231430. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Yerneni, S.S.; Azambuja, J.H.; Gillespie, D.G.; Menshikova, E.V.; Jackson, E.K.; Whiteside, T.L. Tumor-derived exosomes promote angiogenesis via adenosine A(2B) receptor signaling. Angiogenesis 2020, 23, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.A.; Ratnasothy, K.; Tsang, J.Y.S.; Boardman, D.; Warley, A.; Lechler, R.; Lombardi, G. CD73 expression on extracellular vesicles derived from CD4(+)CD25(+)Foxp3(+) T cells contributes to their regulatory function. Eur. J. Immunol. 2013, 43, 2430–2440. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Li, R.; Yang, Y.; Shi, C.; Shen, Y.; Lu, C.; Chen, Y.; Zhou, W.; Lin, A.; Yu, L.; et al. Specific Decrease in B-Cell-Derived Extracellular Vesicles Enhances Post-Chemotherapeutic CD8(+) T Cell Responses. Immunity 2019, 50, 738–750. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiva, A.; Guzman-Gutierrez, E.; Contreras-Duarte, S.; Fuenzalida, B.; Cantin, C.; Carvajal, L.; Salsoso, R.; Gutierrez, J.; Pardo, F.; Sobrevia, L. Adenosine receptors: Modulators of lipid availability that are controlled by lipid levels. Mol. Asp. Med. 2017, 55, 26–44. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, N.; Azambuja, J.H.; Rao, A.; Gillespie, D.G.; Jackson, E.K.; Whiteside, T.L. Adenosine receptors regulate exosome production. Purinergic Signal. 2020, 16, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Man, S.; Lu, Y.; Yin, L.; Cheng, X.; Ma, L. Potential and promising anticancer drugs from adenosine and its analogs. Drug Discov. Today 2021, 26, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, R.W.; Yuvaraj, S.; Kil, L.P. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat. Rev. Cancer 2014, 14, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Jeske, S.S.; Brand, M.; Ziebart, A.; Laban, S.; Doescher, J.; Greve, J.; Jackson, E.K.; Hoffmann, T.K.; Brunner, C.; Schuler, P.J. Adenosine-producing regulatory B cells in head and neck cancer. Cancer Immunol. Immunother. 2020, 69, 1205–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.H.; Kokabee, L.; Kokabee, M.; Conklin, D.S. Bruton’s Tyrosine Kinase and Its Isoforms in Cancer. Front. Cell Dev. Biol. 2021, 9, 668996. [Google Scholar] [CrossRef]
- de Gorter, D.J.J.; Beuling, E.A.; Kersseboom, R.; Middendorp, S.; van Gils, J.M.; Hendriks, R.W.; Pals, S.T.; Spaargaren, M. Bruton’s tyrosine kinase and phospholipase C gamma 2 mediate chemokine-controlled B cell migration and homing. Immunity 2007, 26, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Lou, T.-F.; Sethuraman, D.; Dospoy, P.; Srivastva, P.; Kim, H.S.; Kim, J.; Ma, X.; Chen, P.-H.; Huffman, K.E.; Frink, R.E.; et al. Cancer-Specific Production of N-Acetylaspartate via NAT8L Overexpression in Non-Small Cell Lung Cancer and Its Potential as a Circulating Biomarker. Cancer Prev. Res. 2016, 9, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zand, B.; Previs, R.A.; Zacharias, N.M.; Rupaimoole, R.; Mitamura, T.; Nagaraja, A.S.; Guindani, M.; Dalton, H.J.; Yang, L.; Baddour, J.; et al. Role of Increased n-acetylaspartate Levels in Cancer. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, M.; Yang, Z.L.; Li, X.F.; Zhang, Z.X.; Yin, A.K. Screening of Methylation Gene Sites as Prognostic Signature in Lung Adenocarcinoma. Yonsei Med. J. 2020, 61, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Menga, A.; Favia, M.; Spera, I.; Vegliante, M.C.; Gissi, R.; De Grassi, A.; Laera, L.; Campanella, A.; Gerbino, A.; Carra, G.; et al. N-acetylaspartate release by glutaminolytic ovarian cancer cells sustains protumoral macrophages. EMBO Rep. 2021, 22, e51981. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.H.; Wu, X.R.; Lan, N.; Zheng, X.B.; Zhou, C.; Hu, T.; Chen, Y.F.; Cai, Z.R.; Chen, Z.X.; Lan, P.; et al. CD73 promotes colitis-associated tumorigenesis in mice. Oncol. Lett. 2020, 20, 1221–1230. [Google Scholar] [CrossRef]
- Abel, B.; Tosh, D.K.; Durell, S.R.; Murakami, M.; Vahedi, S.; Jacobson, K.A.; Ambudkar, S.V. Evidence for the Interaction of A(3) Adenosine Receptor Agonists at the Drug-Binding Site(s) of Human P-glycoprotein (ABCB1). Mol. Pharmacol. 2019, 96, 180–192. [Google Scholar] [CrossRef]
- Mlejnek, P.; Dolezel, P.; Kosztyu, P. P-glycoprotein mediates resistance to A3 adenosine receptor agonist 2-chloro-N-6-(3-iodobenzyl)-adenosine-5′-n-methyluronamide in human leukemia cells. J. Cell. Physiol. 2012, 227, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Ostuni, A.; Carmosino, M.; Miglionico, R.; Abruzzese, V.; Martinelli, F.; Russo, D.; Laurenzana, I.; Petillo, A.; Bisaccia, F. Inhibition of ABCC6 Transporter Modifies Cytoskeleton and Reduces Motility of HepG2 Cells via Purinergic Pathway. Cells 2020, 9, 1410. [Google Scholar] [CrossRef] [PubMed]
- Miglionico, R.; Armentano, M.F.; Carmosino, M.; Salvia, A.M.; Cuviello, F.; Bisaccia, F.; Ostuni, A. Dysregulation of gene expression in ABCC6 knockdown HepG2 cells. Cell. Mol. Biol. Lett. 2014, 19, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Shali, S.; Yu, J.; Zhang, X.; Wang, X.; Jin, Y.; Su, M.; Liao, X.; Yu, J.; Zhi, X.; Zhou, P. Ecto-5-nucleotidase (CD73) is a potential target of hepatocellular carcinoma. J. Cell. Physiol. 2019, 234, 10248–10259. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.-w.; Wang, H.-p.; Lin, F.; Wang, X.; Long, M.; Zhang, H.-z.; Dong, K. CD73 promotes proliferation and migration of human cervical cancer cells independent of its enzyme activity. BMC Cancer 2017, 17, 135. [Google Scholar] [CrossRef] [Green Version]
- Lappas, C.M.; Rieger, J.M.; Linden, J. A(2A) adenosine receptor induction inhibits IFN-gamma production in murine CD4(+) T cells. J. Immunol. 2005, 174, 1073–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himer, L.; Csoka, B.; Selmeczy, Z.; Koscso, B.; Pocza, T.; Pacher, P.; Nemeth, Z.H.; Deitch, E.A.; Vizi, E.S.; Cronstein, B.N.; et al. Adenosine A(2A) receptor activation protects CD4(+) T lymphocytes against activation-induced cell death. FASEB J. 2010, 24, 2631–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shou, J.; Jing, J.; Xie, J.; You, L.; Jing, Z.; Yao, J.; Han, W.; Pan, H. Nuclear factor of activated T cells in cancer development and treatment. Cancer Lett. 2015, 361, 174–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokrani, M.B.; Klibi, J.; Bluteau, D.; Bismuth, G.; Mami-Chouaib, F. Smad and NFAT Pathways Cooperate To Induce CD103 Expression in Human CD8 T Lymphocytes. J. Immunol. 2014, 192, 2471–2479. [Google Scholar] [CrossRef] [Green Version]
- Kjaergaard, J.; Hatfield, S.; Jones, G.; Ohta, A.; Sitkovsky, M. A(2A) Adenosine Receptor Gene Deletion or Synthetic A2A Antagonist Liberate Tumor-Reactive CD8(+) T Cells from Tumor-Induced Immunosuppression. J. Immunol. 2018, 201, 782–791. [Google Scholar] [CrossRef] [Green Version]
- Nie, J.; Liu, A.; Tan, Q.; Zhao, K.; Hu, K.; Li, Y.; Yan, B.; Zhou, L. AICAR activates ER stress-dependent apoptosis in gallbladder cancer cells. Biochem. Biophys. Res. Commun. 2017, 482, 246–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.-F.; Wei, B.-L.; Guo, Y.-T.; Ye, Y.-Q.; Li, G.-P.; Pu, Z.-J.; Feng, J.-L. Apoptosis induced by adenosine involves endoplasmic reticulum stress in EC109 cells. Int. J. Mol. Med. 2012, 30, 797–804. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.-F.; Guo, Y.-T.; Zhang, Q.-H.; Xiang, M.-Q.; Deng, W.; Ye, Y.-Q.; Pu, Z.-J.; Feng, J.-L.; Huang, G.-Y. Enhanced Antitumor Effects of Adenoviral-Mediated siRNA against GRP78 Gene on Adenosine-Induced Apoptosis in Human Hepatoma HepG2 Cells. Int. J. Mol. Sci. 2014, 15, 525–544. [Google Scholar] [CrossRef]
- Tosh, D.K.; Brackett, C.M.; Jung, Y.H.; Gao, Z.G.; Banerjee, M.; Blagg, B.S.J.; Jacobson, K.A. Biological Evaluation of 5′-(N-Ethylcarboxamido)adenosine Analogues as Grp94-Selective Inhibitors. ACS Med. Chem. Lett. 2021, 12, 373–379. [Google Scholar] [CrossRef]
- Kumari, N.; Reabroi, S.; North, B.J. Unraveling the Molecular Nexus between GPCRs, ERS, and EMT. Mediat. Inflamm. 2021, 2021, 6655417. [Google Scholar] [CrossRef] [PubMed]
- Hassanian, S.M.; Dinarvand, P.; Rezaie, A.R. Adenosine Regulates the Proinflammatory Signaling Function of Thrombin in Endothelial Cells. J. Cell. Physiol. 2014, 229, 1292–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.K.; Iwagaki, H.; Hamano, R.; Wake, H.; Kanke, T.; Liu, K.; Yoshino, T.; Tanaka, N.; Nishibori, M. Effects of adenosine on adhesion molecule expression and cytokine production in human PBMC depend on the receptor subtype activated. Br. J. Pharmacol. 2007, 150, 816–822. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wang, Y.; Yan, S.; Yang, Q.; Zhou, Y.; Zeng, X.; Liu, Z.; An, X.; Toque, H.A.; Dong, Z.; et al. Regulation of endothelial intracellular adenosine via adenosine kinase epigenetically modulates vascular inflammation. Nat. Commun. 2017, 8, 943. [Google Scholar] [CrossRef]
- Schuster, E.; Taftaf, R.; Reduzzi, C.; Albert, M.K.; Romero-Calvo, I.; Liu, H. Better together: Circulating tumor cell clustering in metastatic cancer. Trends Cancer 2021, 7, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112. [Google Scholar] [CrossRef] [Green Version]
- Vecchio, E.A.; Baltos, J.A.; Nguyen, A.T.N.; Christopoulos, A.; White, P.J.; May, L.T. New paradigms in adenosine receptor pharmacology: Allostery, oligomerization and biased agonism. Br. J. Pharmacol. 2018, 175, 4036–4046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidolin, D.; Marcoli, M.; Tortorella, C.; Maura, G.; Agnati, L.F. Receptor-Receptor Interactions as a Widespread Phenomenon: Novel Targets for Drug Development? Front. Endocrinol. 2019, 10, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Jiang, H.; Zhang, H.; Smith, R.G. Modification of ghrelin receptor signaling by somatostatin receptor-5 regulates insulin release. Proc. Natl. Acad. Sci. USA 2012, 109, 19003–19008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonas, K.C.; Hanyaloglu, A.C. Impact of G protein-coupled receptor heteromers in endocrine systems. Mol. Cell. Endocrinol. 2017, 449, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervetto, C.; Venturini, A.; Passalacqua, M.; Guidolin, D.; Genedani, S.; Fuxe, K.; Borroto-Esquela, D.O.; Cortelli, P.; Woods, A.; Maura, G.; et al. A2A-D2 receptor-receptor interaction modulates gliotransmitter release from striatal astrocyte processes. J. Neurochem. 2017, 140, 268–279. [Google Scholar] [CrossRef]
- Tonazzini, I.; Trincavelli, M.L.; Montali, M.; Martini, C. Regulation of A(1) adenosine receptor functioning induced by P2Y(1) purinergic receptor activation in human astroglial cells. J. Neurosci. Res. 2008, 86, 2857–2866. [Google Scholar] [CrossRef] [PubMed]
- Tonazzini, I.; Trincavelli, M.L.; Storm-Mathisen, J.; Martini, C.; Bergersen, L.H. Co-localization and functional cross-talk between A(1) and P2Y(1) purine receptors in rat hippocampus. Eur. J. Neurosci. 2007, 26, 890–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conde, S.V.; Obeso, A.; Monteiro, E.C.; Gonzalez, C. The A(2B)-D-2 Receptor Interaction that Controls Carotid Body Catecholamines Release Locates Between the Last Two Steps of Hypoxic Transduction Cascade. Arter. Chemorecept. 2009, 648, 161–168. [Google Scholar] [CrossRef]
- Conde, S.V.; Gonzalez, C.; Batuca, J.R.; Monteiro, E.C.; Obeso, A. An antagonistic interaction between A(2B) adenosine and D-2 dopamine receptors modulates the function of rat carotid body chemoreceptor cells. J. Neurochem. 2008, 107, 1369–1381. [Google Scholar] [CrossRef]
- Moreno, E.; Andradas, C.; Medrano, M.; Caffarel, M.M.; Perez-Gomez, E.; Blasco-Benito, S.; Gomez-Canas, M.; Ruth Pazos, M.; Irving, A.J.; Lluis, C.; et al. Targeting CB2-GPR55 Receptor Heteromers Modulates Cancer Cell Signaling. J. Biol. Chem. 2014, 289, 21960–21972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomsen, A.R.B.; Plouffe, B.; Cahill, T.J.; Shukla, A.K.; Tarrasch, J.T.; Dosey, A.M.; Kahsai, A.W.; Strachan, R.T.; Pani, B.; Mahoney, J.P.; et al. GPCR-G Protein-beta-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell 2016, 166, 907–919. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; van Westen, G.J.P.; Heitman, L.H.; Ijzerman, A.P. G protein-coupled receptors expressed and studied in yeast. The adenosine receptor as a prime example. Biochem. Pharmacol. 2021, 187, 114370. [Google Scholar] [CrossRef]
- Wang, X.S.; Jespers, W.; Bongers, B.J.; Jansen, M.; Stangenberger, C.M.; Dilweg, M.A.; Gutierrez-de-Teran, H.; Ijzerman, A.P.; Heitman, L.H.; van Westen, G.J.P. Characterization of cancer-related somatic mutations in the adenosine A2B receptor. Eur. J. Pharmacol. 2020, 880, 173126. [Google Scholar] [CrossRef]
- Vecchio, E.A.; Tan, C.Y.R.; Gregory, K.J.; Christopoulos, A.; White, P.J.; May, L.T. Ligand-Independent Adenosine A(2B) Receptor Constitutive Activity as a Promoter of Prostate Cancer Cell Proliferation. J. Pharmacol. Exp. Ther. 2016, 357, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeill, S.M.; Baltos, J.A.; White, P.J.; May, L.T. Biased agonism at adenosine receptors. Cell. Signal. 2021, 82, 109954. [Google Scholar] [CrossRef] [PubMed]
- Storme, J.; Cannaert, A.; Van Craenenbroeck, K.; Stove, C.P. Molecular dissection of the human A(3) adenosine receptor coupling with beta-arrestin2. Biochem. Pharmacol. 2018, 148, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.H.; Ahn, S.; Kim, H.J.; Lee, M.; Kim, J.; Jin, S.H.; Lee, E.; Kim, G.; Cheong, J.H.; Jacobson, K.A.; et al. Polypharmacology of N-6-(3-lodobenzyl)adenosine-5′-N-methyluronamide (IB-MECA) and Related A(3) Adenosine Receptor Ligands: Peroxisome Proliferator Activated Receptor (PPAR) gamma Partial Agonist and PPAR delta Antagonist Activity Suggests Their Antidiabetic Potential. J. Med. Chem. 2017, 60, 7459–7475. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.; Johnson, L.A.A.; Jacobson, P.A.; Kachler, S.; Kirstein, M.N.; Lamba, J.; Klotz, K.-N. Cytotoxic purine nucleoside analogues bind to A(1), A(2A), and A(3) adenosine receptors. Naunyn-Schmiedebergs Arch. Pharmacol. 2012, 385, 519–525. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.X.; Zou, Y.; Li, L.; Lu, F.P.; Xu, H.T.; Ren, P.X.; Bai, F.; Niedermann, G.; Zhu, X.K. BAY 60-6583 Enhances the Antitumor Function of Chimeric Antigen Receptor-Modified T Cells Independent of the Adenosine A2b Receptor. Front. Pharmacol. 2021, 12, 274. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.; Lebon, G. Human Adenosine A(2A) Receptor: Molecular Mechanism of Ligand Binding and Activation. Front. Pharmacol. 2017, 8, 898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seibt, B.F.; Schiedel, A.C.; Thimm, D.; Hinz, S.; Sherbiny, F.F.; Mueller, C.E. The second extracellular loop of GPCRs determines subtype-selectivity and controls efficacy as evidenced by loop exchange study at A(2) adenosine receptors. Biochem. Pharmacol. 2013, 85, 1317–1329. [Google Scholar] [CrossRef]
- De Filippo, E.; Hinz, S.; Pellizzari, V.; Deganutti, G.; El-Tayeb, A.; Navarro, G.; Franco, R.; Moro, S.; Schiedel, A.C.; Mueller, C.E. A(2A) and A(2B) adenosine receptors: The extracellular loop 2 determines high (A(2A)) or low affinity (A(2B)) for adenosine. Biochem. Pharmacol. 2020, 172, 113718. [Google Scholar] [CrossRef]
- Bowser, J.L.; Blackburn, M.R.; Shipley, G.L.; Molina, J.G.; Dunner, K., Jr.; Broaddus, R.R. Loss of CD73-mediated actin polymerization promotes endometrial tumor progression. J. Clin. Investig. 2016, 126, 220–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurnit, K.C.; Draisey, A.; Kazen, R.C.; Chung, C.; Phan, L.H.; Harvey, J.B.; Feng, J.P.; Xie, S.S.; Broaddus, R.R.; Bowser, J.L. Loss of CD73 shifts transforming growth factor-beta 1 (TGF-beta 1) from tumor suppressor to promoter in endometrial cancer. Cancer Lett. 2021, 505, 75–86. [Google Scholar] [CrossRef]
- Dziedzic, K.; Wegrzyn, P.; Galezowski, M.; Bonkowska, M.; Grycuk, K.; Satala, G.; Wiatrowska, K.; Wiklik, K.; Brzozka, K.; Nowak, M. Release of adenosine-induced immunosuppression: Comprehensive characterization of dual A(2A)/A(2B) receptor antagonist. Int. Immunopharmacol. 2021, 96, 107645. [Google Scholar] [CrossRef]
- Moriyama, K.; Sitkovsky, M.V. Adenosine A2A Receptor Is Involved in Cell Surface Expression of A2B Receptor. J. Biol. Chem. 2010, 285, 39271–39288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arruga, F.; Serra, S.; Vitale, N.; Guerra, G.; Papait, A.; Gyau, B.B.; Tito, F.; Efremov, D.; Vaisitti, T.; Deaglio, S. Targeting the A2A adenosine receptor counteracts immunosuppression in vivo in a mouse model of chronic lymphocytic leukemia. Haematologica 2021, 106, 1343–1353. [Google Scholar] [CrossRef] [Green Version]
- Ott, M.; Tomaszowski, K.H.; Marisetty, A.; Kong, L.Y.; Wei, J.; Duna, M.; Blumberg, K.; Ji, X.R.; Jacobs, C.; Fuller, G.N.; et al. Profiling of patients with glioma reveals the dominant immunosuppressive axis is refractory to immune function restoration. JCI Insight 2020, 5, e134386. [Google Scholar] [CrossRef] [PubMed]
- Vasiukov, G.; Menshikh, A.; Owens, P.; Novitskaya, T.; Hurley, P.; Blackwell, T.; Feoktistov, I.; Novitskiy, S.V. Adenosine/TGF beta axis in regulation of mammary fibroblast functions. PLoS ONE 2021, 16, e0252424. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Mirandola, P.; Milani, D.; Varani, K.; Gessi, S.; Klotz, K.N.; Leung, E.; Baraldi, P.G.; Borea, P.A. Adenosine receptors as mediators of both cell proliferation and cell death of cultured human melanoma cells. J. Investig. Dermatol. 2002, 119, 923–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, G.; Sitkovsky, M.V. Differential requirement for A2a and A3 adenosine receptors for the protective effect of inosine in vivo. Blood 2003, 102, 4472–4478. [Google Scholar] [CrossRef] [PubMed]
- Welihinda, A.A.; Kaur, M.; Greene, K.; Zhai, Y.; Amento, E.P. The adenosine metabolite inosine is a functional agonist of the adenosine A(2A) receptor with a unique signaling bias. Cell. Signal. 2016, 28, 552–560. [Google Scholar] [CrossRef] [Green Version]
- Serrano-del Valle, A.; Naval, J.; Anel, A.; Marzo, I. Novel Forms of Immunomodulation for Cancer Therapy. Trends Cancer 2020, 6, 518–532. [Google Scholar] [CrossRef] [PubMed]
- Mager, L.F.; Burkhard, R.; Pett, N.; Cooke, N.C.A.; Brown, K.; Ramay, H.; Paik, S.; Stagg, J.; Groves, R.A.; Gallo, M.; et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020, 369, 1481–1489. [Google Scholar] [CrossRef]
- Luo, B.H.; Zhang, Y.B.; Zhang, C.Q.; Liu, X.Q.; Shi, C.H. Intestinal microbiota: A potential target for enhancing the antitumor efficacy and reducing the toxicity of immune checkpoint inhibitors. Cancer Lett. 2021, 509, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Reitman, M.L. Adenosine-Related Mechanisms in Non-Adenosine Receptor Drugs. Cells 2020, 9, 956. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, L.; Sek, K.; Henderson, M.A.; Lai, J.Y.; Chen, A.X.Y.; Meyran, D.; Todd, K.L.; Petley, E.V.; Mardiana, S.; Molck, C.; et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat. Commun. 2021, 12, 3236. [Google Scholar] [CrossRef] [PubMed]
- Kipniss, N.H.; Dingal, P.C.D.P.; Abbott, T.R.; Gao, Y.; Wang, H.; Dominguez, A.A.; Labanieh, L.; Qi, L.S. Engineering cell sensing and responses using a GPCR-coupled CRISPR-Cas system. Nat. Commun. 2017, 8, 2212. [Google Scholar] [CrossRef] [Green Version]
- Vargason, A.M.; Anselmo, A.C.; Mitragotri, S. The evolution of commercial drug delivery technologies. Nat. Biomed. Eng. 2021, 5, 951–967. [Google Scholar] [CrossRef] [PubMed]
- Esfahani, K.; Elkrief, A.; Calabrese, C.; Lapointe, R.; Hudson, M.; Routy, B.; Miller, W.H.; Calabrese, L. Moving towards personalized treatments of immune-related adverse events. Nat. Rev. Clin. Oncol. 2020, 17, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Newton, H.S.; Chimote, A.A.; Arnold, M.J.; Wise-Draper, T.M.; Conforti, L. Targeted knockdown of the adenosine A(2A) receptor by lipid NPs rescues the chemotaxis of head and neck cancer memory T cells. Mol. Ther.-Methods Clin. Dev. 2021, 21, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi-Chaleshtari, M.; Kiaie, S.H.; Irandoust, M.; Karami, H.; Afjadi, M.N.; Ghani, S.; Vanda, N.A.; Sede, M.J.G.; Ahmadi, A.; Masjedi, A.; et al. Concomitant blockade of A2AR and CTLA-4 by siRNA-loaded polyethylene glycol-chitosan-alginate nanoparticles synergistically enhances antitumor T-cell responses. J. Cell. Physiol. 2020, 235, 10068–10080. [Google Scholar] [CrossRef] [PubMed]
- Pineux, F.; Federico, S.; Klotz, K.N.; Kachler, S.; Michiels, C.; Sturlese, M.; Prato, M.; Spalluto, G.; Moro, S.; Bonifazi, D. Targeting G Protein-Coupled Receptors with Magnetic Carbon Nanotubes: The Case of the A(3)Adenosine Receptor. Chemmedchem 2020, 15, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Reis, R.I.; Moraes, I. Probing Membrane Protein Assembly into Nanodiscs by In Situ Dynamic Light Scattering: A(2A) Receptor as a Case Study. Biology 2020, 9, 400. [Google Scholar] [CrossRef] [PubMed]
- Psaraki, A.; Ntari, L.; Karakostas, C.; Korrou-Karava, D.; Roubelakis, M.G. Extracellular vesicles derived from Mesenchymal Stem/Stromal Cells: The regenerative impact in liver diseases. Hepatology 2021, Accepted. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, D. Exosomes in cancer development, metastasis, and immunity. Biochim. Biophys. Acta-Rev. Cancer 2019, 1871, 455–468. [Google Scholar] [CrossRef]
- Cheng, J.; Chen, M.Z.; Wang, S.Y.; Liang, T.J.; Chen, H.; Chen, C.J.; Feng, Z.W.; Xie, X.Q. Binding Characterization of Agonists and Antagonists by MCCS: A Case Study from Adenosine A(2A) Receptor. ACS Chem. Neurosci. 2021, 12, 1606–1620. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Hou, X.; Lee, J.H.; Nayak, A.; Alexander, V.; Sharma, P.K.; Chang, H.; Phan, K.; Gao, Z.-G.; Jacobson, K.A.; et al. Subtle Chemical Changes Cross the Boundary between Agonist and Antagonist: New A3 Adenosine Receptor Homology Models and Structural Network Analysis Can Predict This Boundary. J. Med. Chem. 2021, 64, 12525–12536. [Google Scholar] [CrossRef]
- Martynowycz, M.W.; Shiriaeva, A.; Ge, X.; Hattne, J.; Nannenga, B.L.; Cherezov, V.; Gonen, T. MicroED structure of the human adenosine receptor determined from a single nanocrystal in LCP. Proc. Natl. Acad. Sci. USA 2021, 118, e2106041118. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; de Graaf, C.; Swain, N.A.; Tate, C.G. Impact of GPCR Structures on Drug Discovery. Cell 2020, 181, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Voronova, V.; Peskov, K.; Kosinsky, Y.; Helmlinger, G.; Chu, L.L.; Borodovsky, A.; Woessner, R.; Sachsenmeier, K.; Shao, W.L.; Kumar, R.; et al. Evaluation of Combination Strategies for the A(2A)R Inhibitor AZD4635 Across Tumor Microenvironment Conditions via a Systems Pharmacology Model. Front. Immunol. 2021, 12, 16. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| NCT Number | Target | Type of Agent | Agent | Combination Therapy | Condition | Phases |
|---|---|---|---|---|---|---|
| NCT04280328 | A2AR | Antagonist | Ciforadenant (CPI-444) | Daratumumab (CD38) | Relapsed or refractory MM | I |
| NCT04381832 | A2AR/A2BR | Dual antagonist | AB928 (etrumadenant) | Zimberelimab (PD-1) ± enzalutamide (androgen receptor), docetaxel or AB680 (CD73) ± zimberelimab (PD-1) | Metastatic castrate resistant prostate cancer | I/II |
| NCT04660812 | A2AR/A2BR | Dual antagonist | AB928 (etrumadenant) | Zimberelimab (PD-1) ± mFOLFOX6, bevacizumab (VEGF), regorafenib (kinases inhibitor) | Metastatic CRC | I/II |
| NCT04017130 | CD38 | ETB targeting CD38 | TAK-169 | - | Relapsed or refractory MM | I |
| NCT04083898 | CD38 | IgG1 anti-CD38 mAb | Isatuximab | Bendamustine, prednisone | Relapsed or refractory MM | I/II |
| NCT04352205 | CD38 | IgG1 anti-CD38 mAb | Daratumumab | Bortezomib, dexamethasone ± thalidomide or lenalidomide | MM with renal failure | II |
| NCT04430530 | CD38 | CAR-T | 4SCAR-T specific to CD22/CD123 /CD38/CD10/CD20 | - | CD19 negative B-cell malignancies | I/II |
| NCT04270409 | CD38 | IgG1 anti-CD38 mAb | Isatuximab | Lenalidomide, dexamethasone | Smoldering MM | III |
| NCT03841565 | CD38 | IgG1 anti-CD38 mAb | Daratumumab | Pomalidomide, dexamethasone | Relapsed MM | II |
| NCT04251065 | CD38 | IgG1 anti-CD38 mAb | Daratumumab | Gemcitabine, cisplatin, dexamethasone | Relapsed or refractory T-cell lymphoma | II |
| NCT04230304 | CD38 | IgG1 anti-CD38 mAb | Daratumumab | Ibrutinib (BTK inhibitor) | Relapsed or refractory chronic lymphocytic leukaemia | II |
| NCT04566328 | CD38 | IgG1 anti-CD38 mAb with hyaluronidase | Daratumumab and hyaluronidase-fihj | Lenalidomide, dexamethasone ± bortezomib | MM | III |
| NCT04316442 | CD38, tubulin polymerization | Antibody-drug conjugate of anti-CD38 mAb and duostatin 5.2 | STI-6129 | - | Relapsed or refractory systemic AL amyloidosis | I |
| NCT04407442 | CD38 | IgG1 anti-CD38 mAb | Daratumumab | Azacitidine, dexamethasone | Relapsed or refractory MM | II |
| NCT04150692 | CD38 | IgG1 anti-CD38 mAb with hyaluronidase | Daratumumab and hyaluronidase-fihj | - | Relapsed or refractory MM | II |
| NCT04824794 | CD38 | IgG1 anti-CD38 mAb | GEN3014 (HexaBody-CD38) | - | Relapsed or refractory MM | I/II |
| NCT04758767 | CD38 | IgG1 anti-CD38 mAb | CID-103 | - | Relapsed or refractory MM | I |
| NCT04139304 | CD38 | IgG1 anti-CD38 mAb | Daratumumab | DA-EPOCH | Plasmablastic lymphoma | I |
| NCT04802031 | CD38 | IgG1 anti-CD38 mAb | Isatuximab | - | Relapsed or refractory MM | II |
| NCT04892264 | CD38 | IgG1 anti-CD38 mAb | Daratumumab | Belantamab (BCMA), mafodotin (microtubule inhibitor), lenalidomide | Untreated, relapsed or refractory MM | I/II |
| NCT04763616 | CD38 | IgG1 anti-CD38 mAb | Isatuximab | Cemiplimab (PD-1) | Relapsed or refractory NK/T-cell lymphoid malignancy | II |
| NCT05011097 | CD38, CD3 | Anti-CD38 and anti-CD3 bispecific antibody | Y150 | - | Relapsed or refractory MM | I |
| NCT04751877 | CD38 | IgG1 anti-CD38 mAb | Isatuximab | Lenalidomide and dexamethasone ± bortezomib | MM | III |
| NCT04336098 | CD39 | Anti-CD39 mAb | SRF617 | ±gemcitabine + paclitaxel or pembrolizumab (PD-1) | Advanced solid tumours | I |
| NCT04306900 | CD39 | Anti-CD39 mAb | TTX-030 | mFOLFOX6, docetaxel, nab-paclitaxel, gemcitabine and/or budigalimab (PD-1) or pembrolizumab (PD-1) | Advanced solid tumours | I |
| NCT04672434 | CD73 | anti-CD73 mAb | Sym024 | ±Sym021 (PD-1) | Advanced solid tumours | I |
| NCT04668300 | CD73 | IgG1 anti-CD73 mAb | Oleclumab | Durvalumab (PD-L1) | Recurrent, refractory, or metastatic sarcoma | II |
| NCT04262375 † | CD73 | IgG1 anti-CD73 mAb | Oleclumab | Durvalumab (PD-L1) | Advanced NSCLC or RCC | II |
| NCT04262388 † | CD73 | IgG1 anti-CD73 mAb | Oleclumab | Durvalumab (PD-L1) | PDAC, NSCLC and HNSCC | II |
| NCT04776018 * | SUMOylation | Small molecule inhibitor | TAK-981 (subasumstat) | Mezagitamab (CD38) ± daratumumab and hyaluronidase-fihj (CD38) | Relapsed or refractory MM | I/II |
| NCT05060432 * | TIGIT | IgG1 anti-TIGIT mAb | EOS-448 | Pembrolizumab (PD-1) or inupadenant (A2AR) | Advanced solid tumours | I/II |
| NCT04205240 * | - | allo HSCT | - | Cyclophosphamide, fludarabine, melphalan; mycophenolate mofetil, tacrolimus (immunotherapy); daratumumab (CD38) | Relapsed MM | II |
| 1 | Based on current knowledge, intracellular ADO triggers epigenetic reprogramming independently of ARs. Low levels of intracellular ADO can boost DNA methylation, whereas its accumulation blocks epigenetic changes [9]. Could there be a direct feed-forward loop between intracellular ADO and AR expression? |
| 2 | When the therapy targets ADO-rich tumours and blocks A2AR on immune cells by A2AR antagonist for instance, what happens to the excessive eADO in the niche? Could continuously generated ADO backfire as a result? Will the ADO metabolites engage other pro-tumoral molecular processes? What pathways will be heightened? |
| 3 | How to better understand the inconsistencies of adenosinergic pathways in different tumour models? |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotulová, J.; Hajdúch, M.; Džubák, P. Current Adenosinergic Therapies: What Do Cancer Cells Stand to Gain and Lose? Int. J. Mol. Sci. 2021, 22, 12569. https://doi.org/10.3390/ijms222212569
Kotulová J, Hajdúch M, Džubák P. Current Adenosinergic Therapies: What Do Cancer Cells Stand to Gain and Lose? International Journal of Molecular Sciences. 2021; 22(22):12569. https://doi.org/10.3390/ijms222212569
Chicago/Turabian StyleKotulová, Jana, Marián Hajdúch, and Petr Džubák. 2021. "Current Adenosinergic Therapies: What Do Cancer Cells Stand to Gain and Lose?" International Journal of Molecular Sciences 22, no. 22: 12569. https://doi.org/10.3390/ijms222212569
APA StyleKotulová, J., Hajdúch, M., & Džubák, P. (2021). Current Adenosinergic Therapies: What Do Cancer Cells Stand to Gain and Lose? International Journal of Molecular Sciences, 22(22), 12569. https://doi.org/10.3390/ijms222212569