
Comparative Genome Analysis of Genes Regulating Compound Inflorescences in Tomato

 ,
,
Abstract
:1. Introduction
2. Results
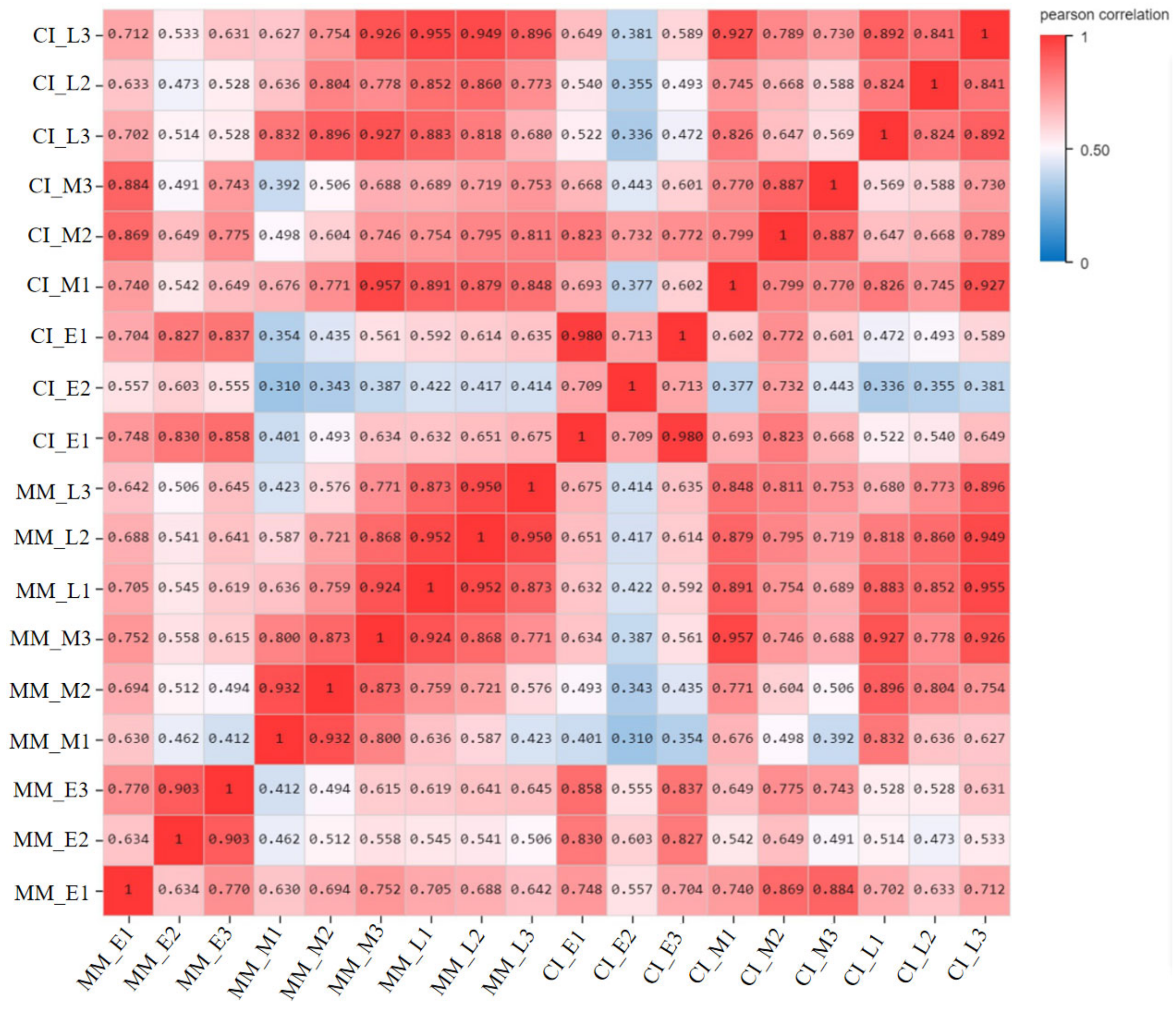
2.1. RNA Sequencing Data and Functional Analysis
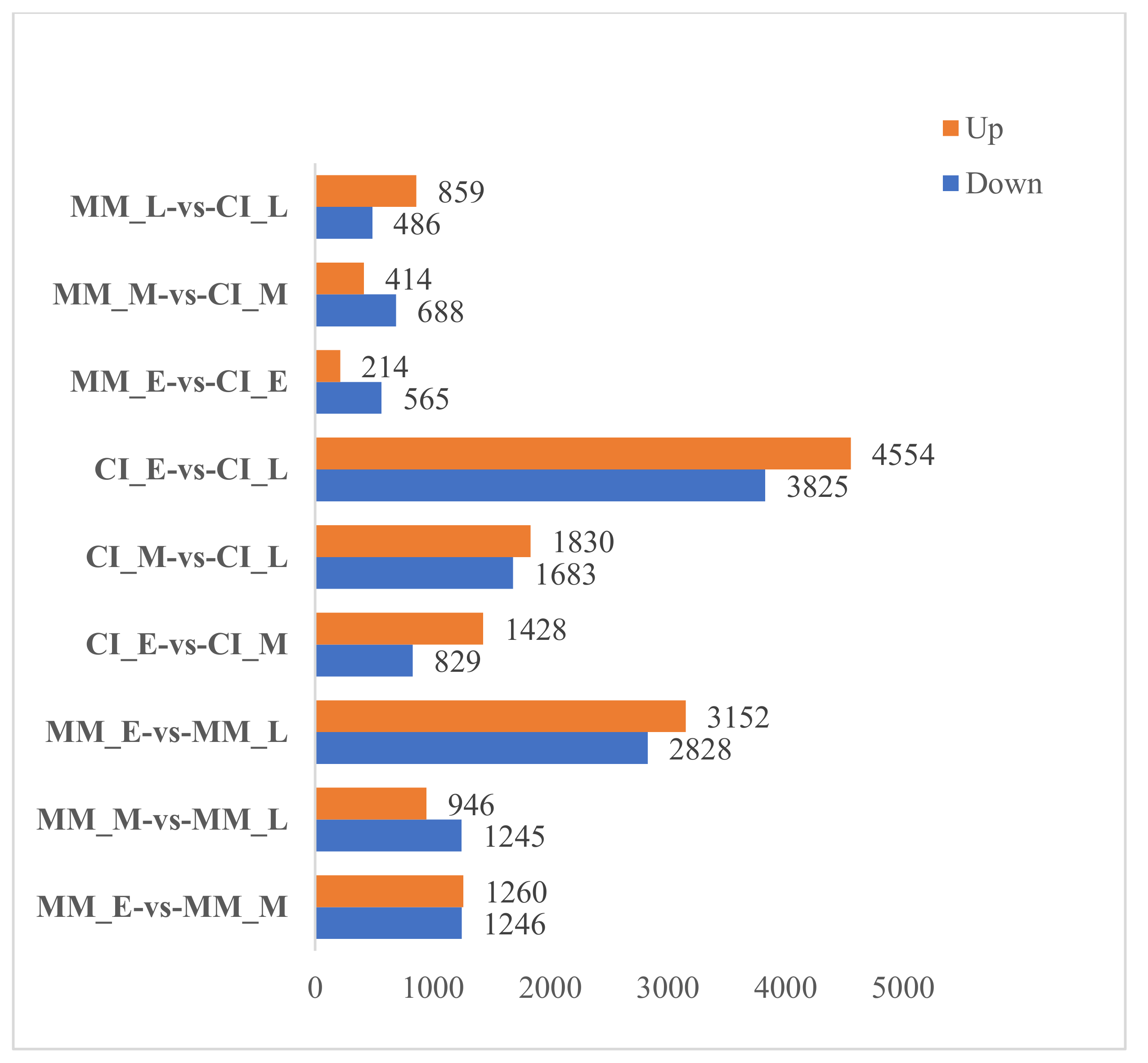
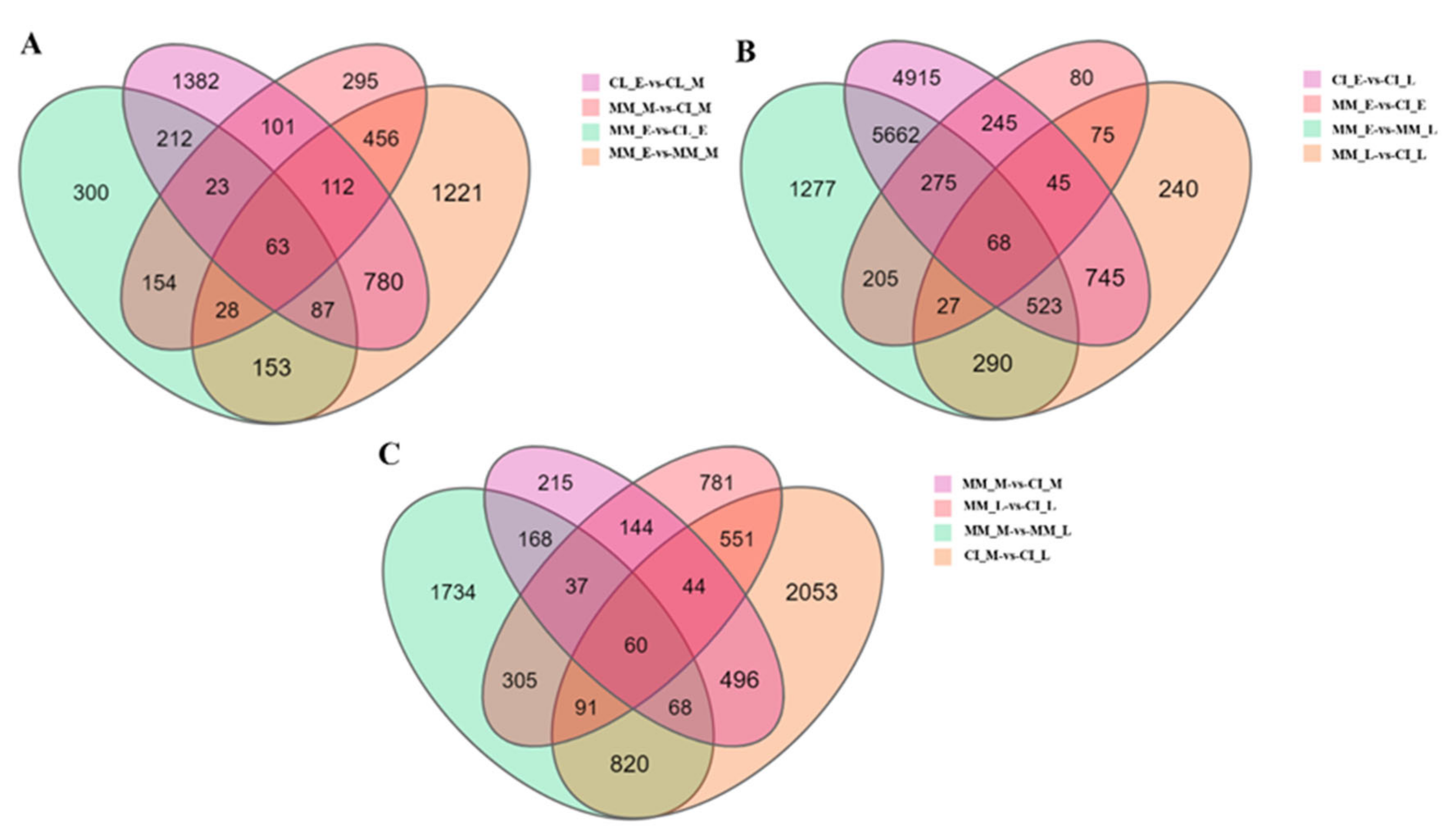
2.2. Expression and Analysis of Differentially Expressed Genes (DEGs) in Tomato Inflorescences
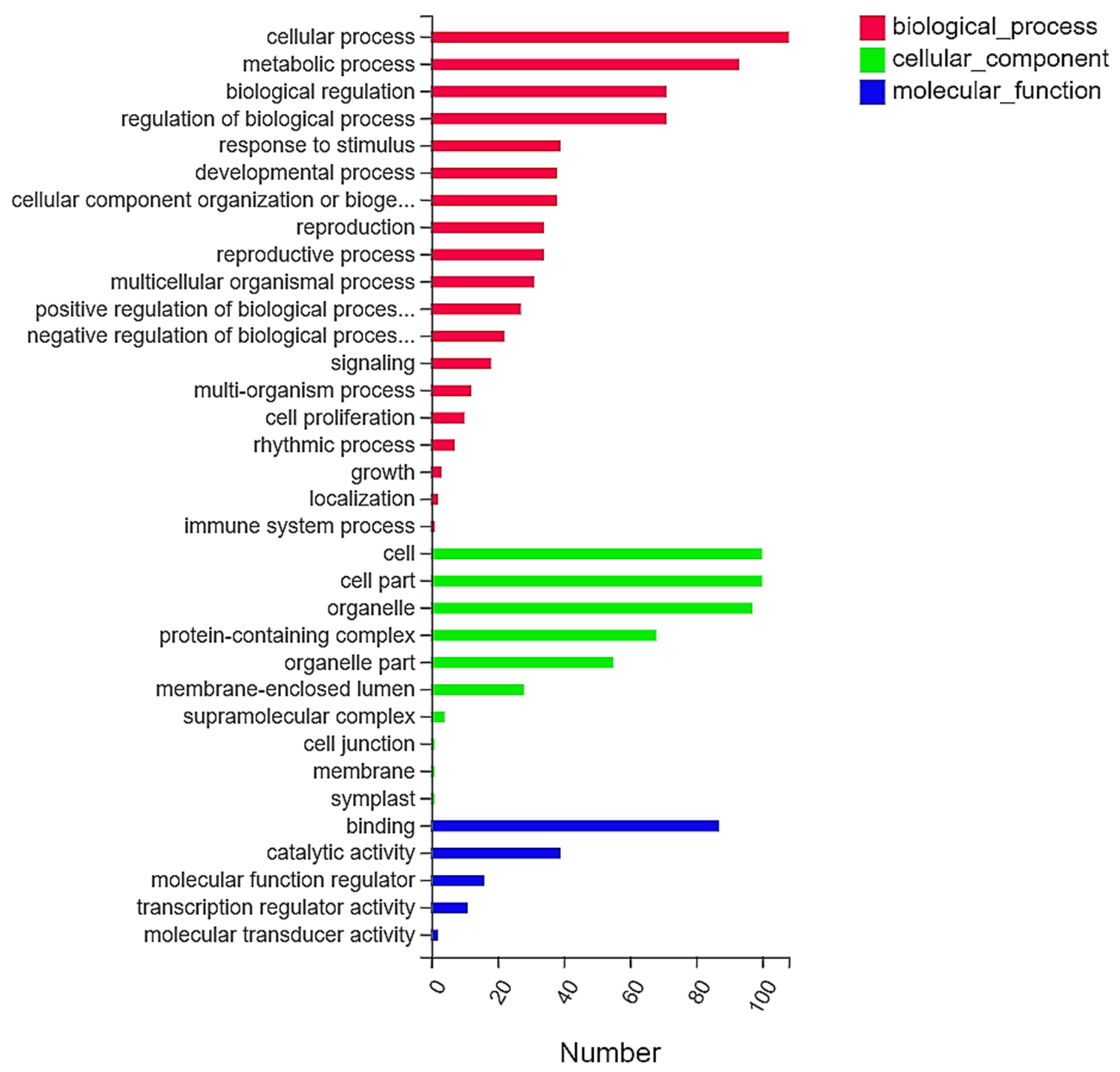
2.3. Gene Ontology (GO) Functional Classification and Analysis of DEGs
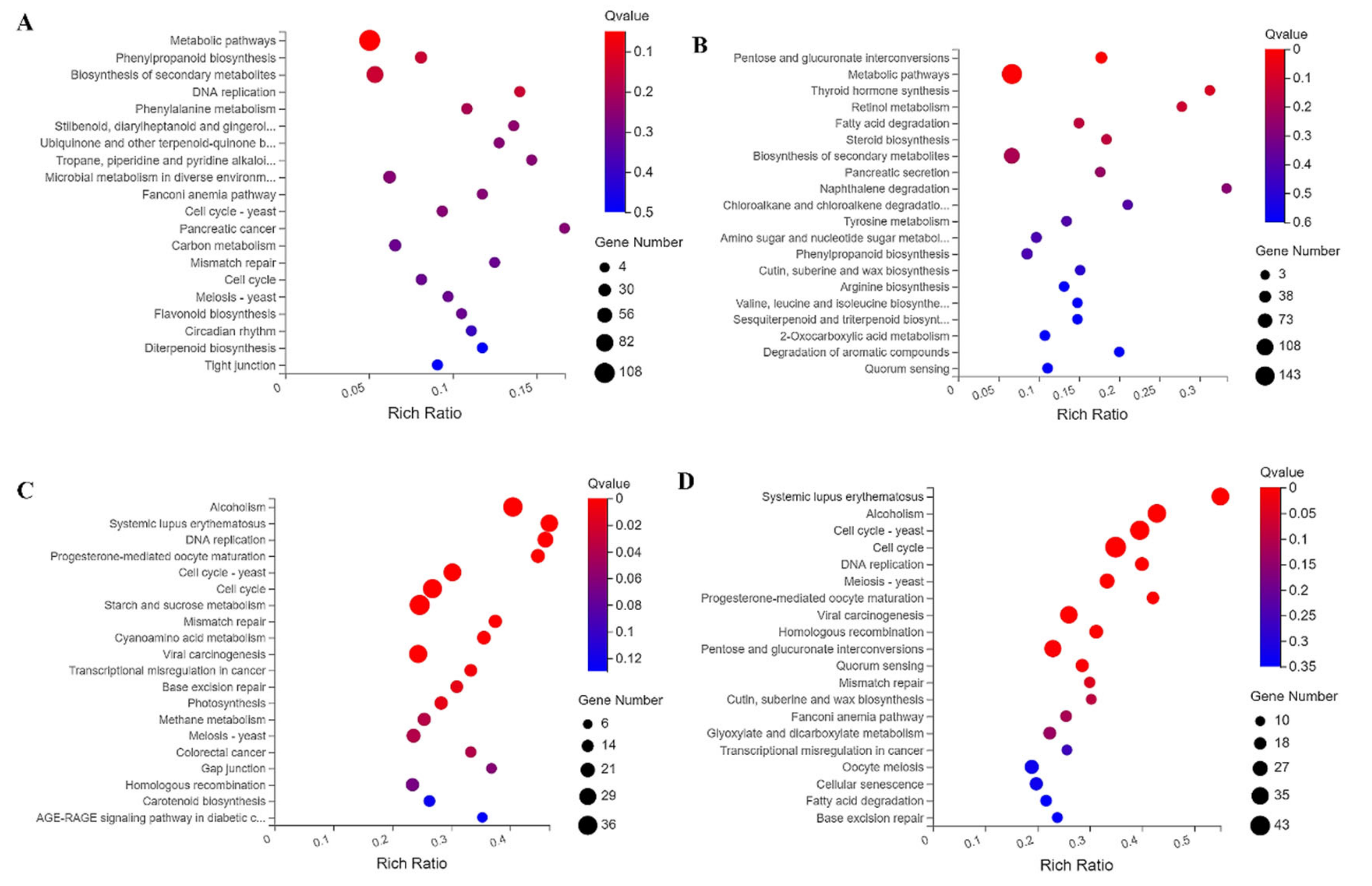
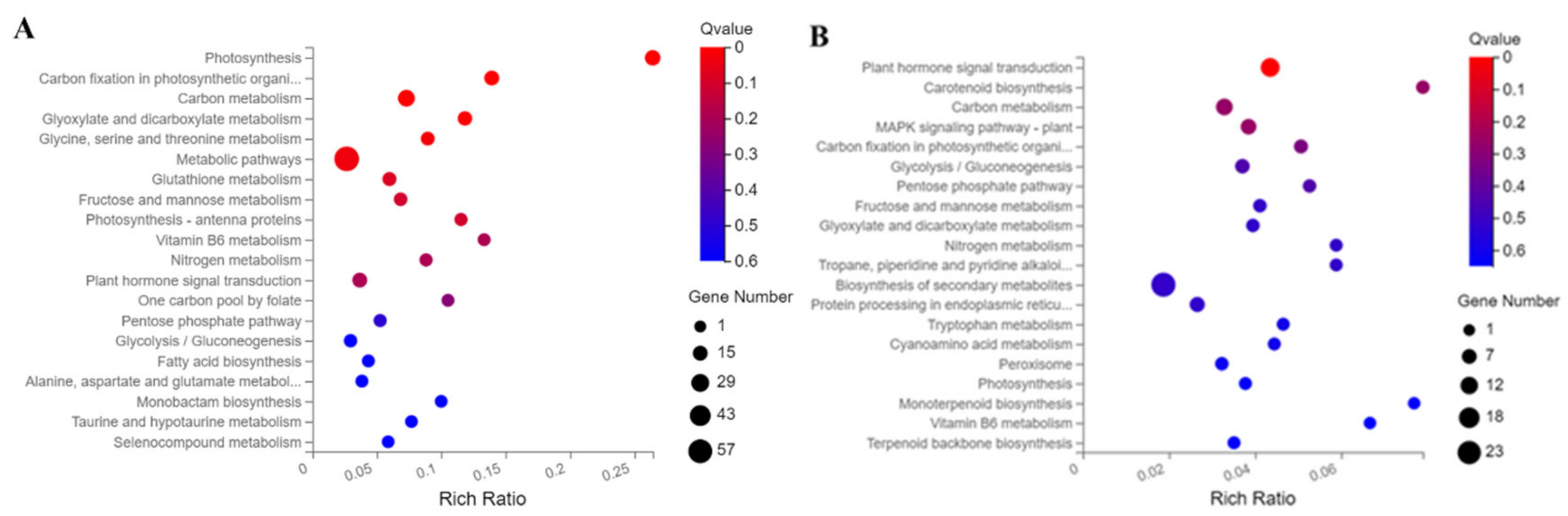
2.4. Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis of DEGs
2.5. Screening and Analysis of Genes Affecting Inflorescence Traits in Tomato
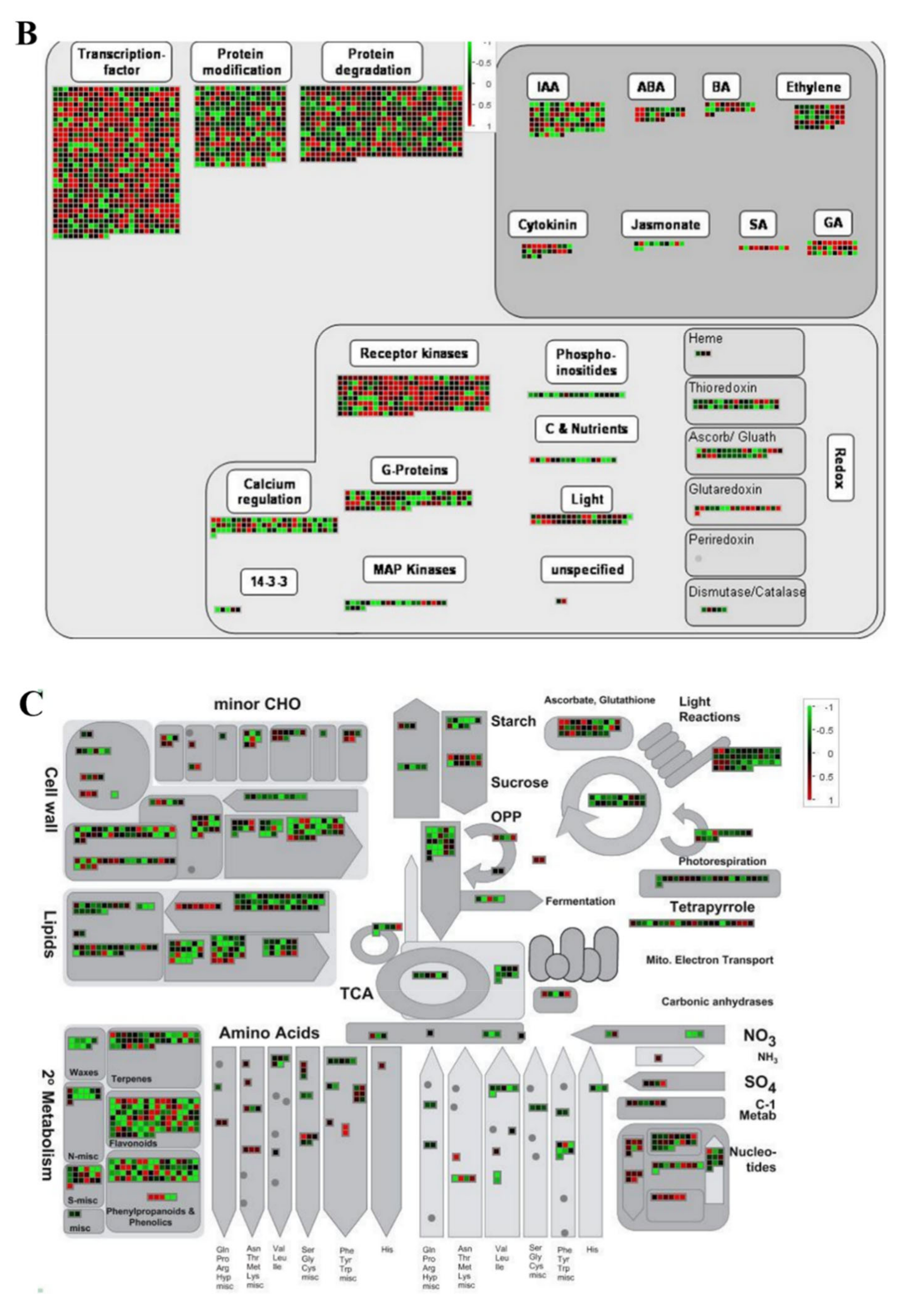
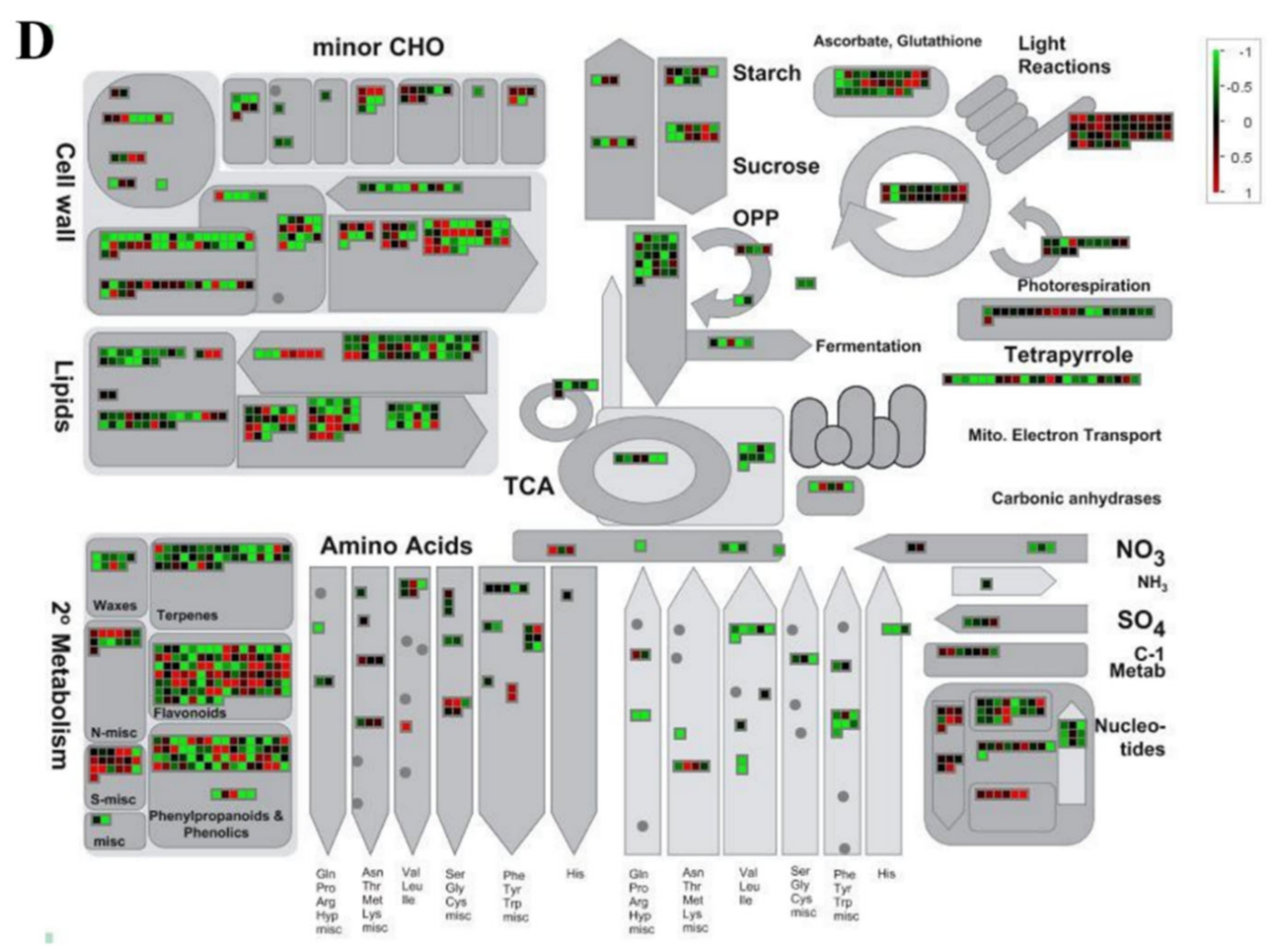
2.6. MapMan Tool-Based Analysis of DEGs
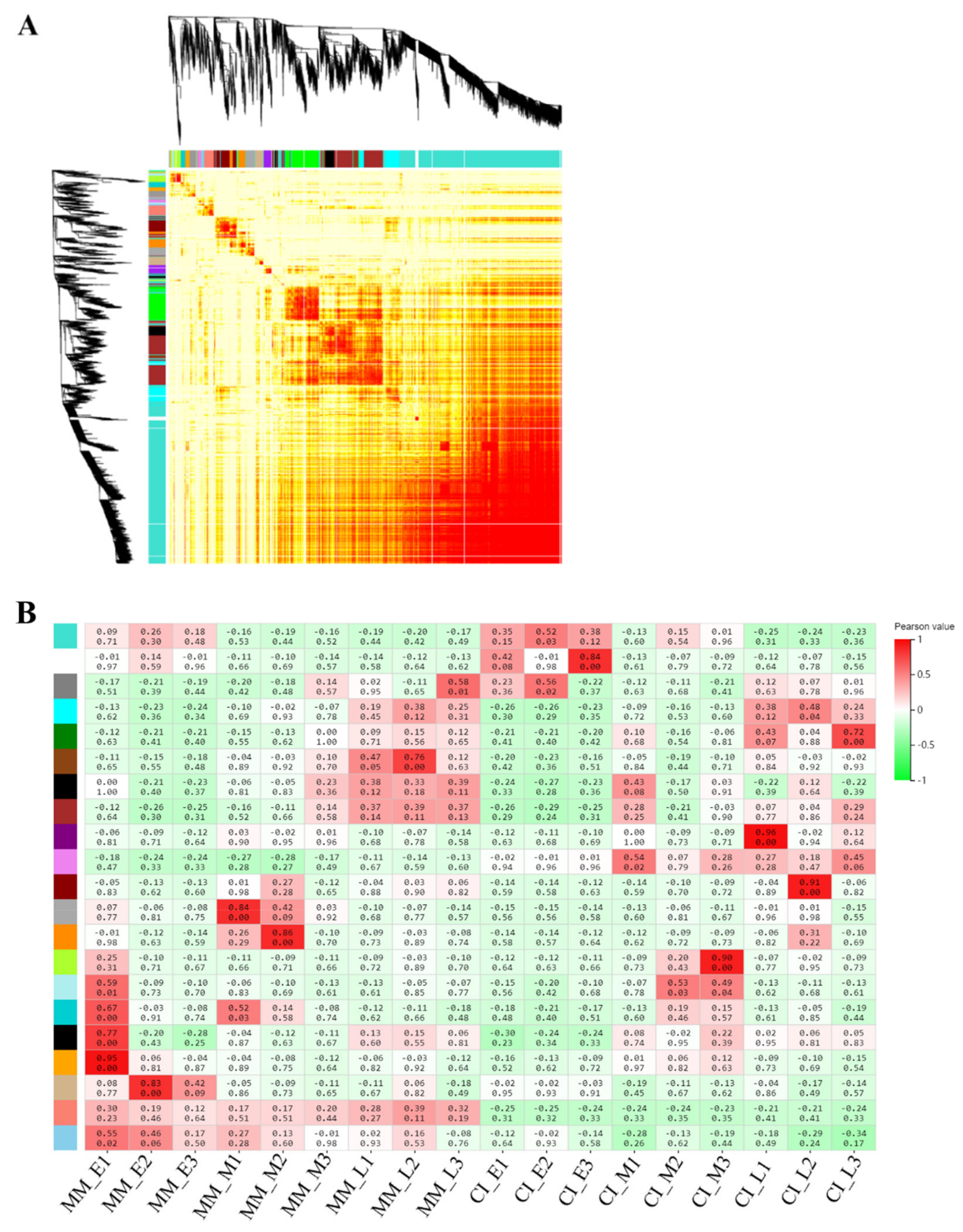
2.7. Weighted Gene Co-Expression Network Analysis (WGCNA) of DEGs
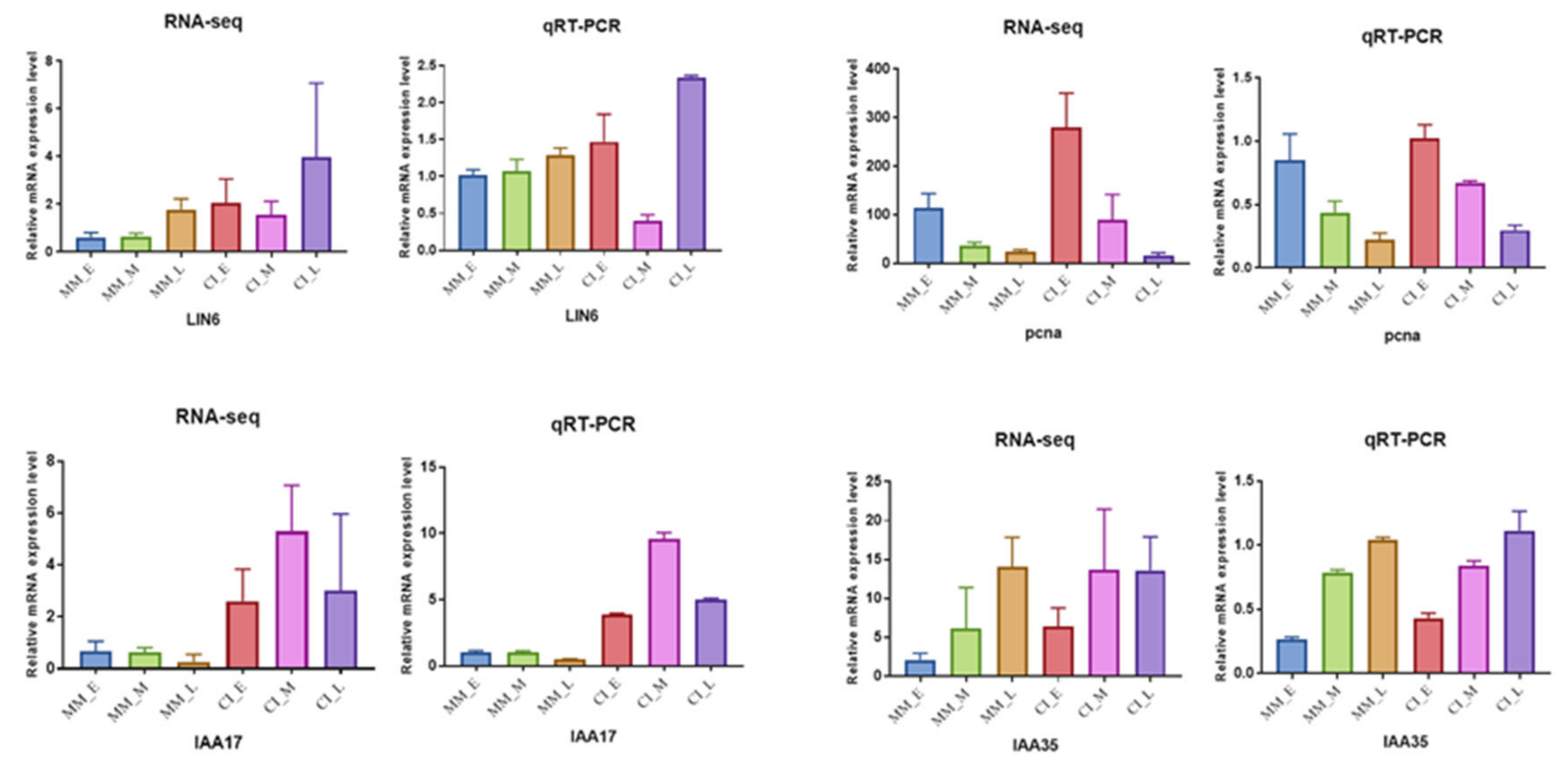
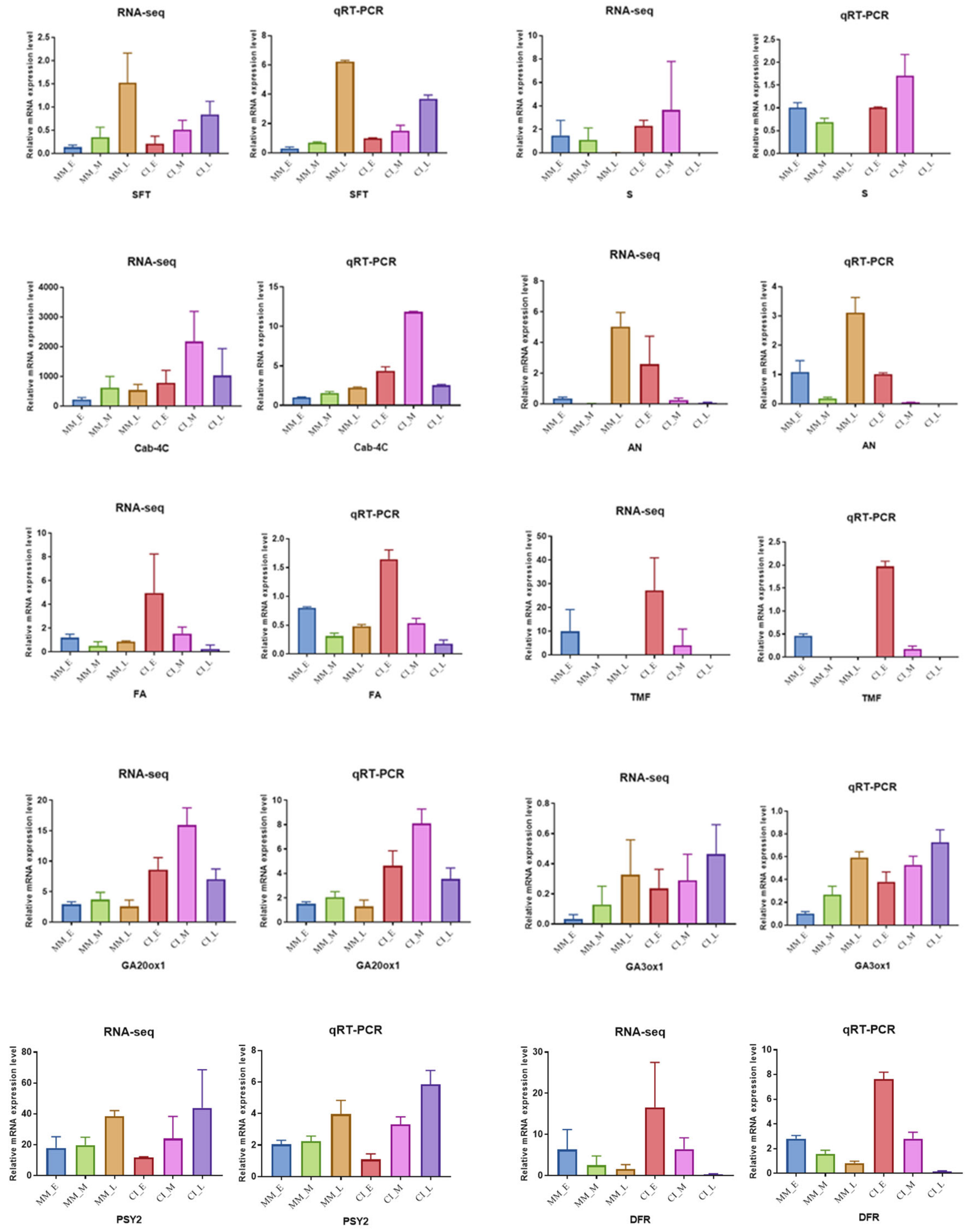
2.8. Verification of DEG Expression Patterns
3. Discussion
4. Materials and Methods
4.1. Test Materials
4.2. RNA Extraction and Transcriptome Sequencing
4.3. RNA-Seq Read Mapping and DEG Identification
4.4. Functional Annotation and DEG Enrichment Analysis
4.5. MapMan Analysis of DEGs
4.6. Weighted Gene Co-Expression Network Analysis (WGCNA)
4.7. qRT-PCR Analysis
4.8. Determination of Endogenous Hormones
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fan, Y.; Zheng, Y.; Teixeira da Silva, J.A.; Yu, X. Comparative transcriptomics and WGCNA reveal candidate genes involved in petaloid stamens in Paeonia lactiflora. J. Hortic. Sci. Biotechnol. 2021, 96, 588–603. [Google Scholar] [CrossRef]
- Huang, X.; Tang, L.; Yu, Y.; Dalrymple, J.; Lippman, Z.B.; Xu, C. Control of flowering and inflorescence architecture in tomato by synergistic interactions between ALOG transcription factors. J. Genet. Genom. 2018, 45, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Giménez, E.; Cara, B.; Capel, J.; Angosto, T. Genetic analysis of reproductive development in tomato. Int. J. Dev. Biol. 2009, 53, 1635–1648. [Google Scholar] [CrossRef]
- Périlleux, C.; Lobet, G.; Tocquin, P. Inflorescence development in tomato: Gene functions within a zigzag model. Front. Plant Sci. 2014, 5, 121. [Google Scholar] [PubMed] [Green Version]
- Quinet, M.; Kinet, J.-M. Transition to Flowering and Morphogenesis of Reproductive Structures in Tomato. Int. J. Plant Dev. Biol. 2007, 1, 64–74. [Google Scholar]
- Huan, Z.; Saneyuki, K. Identification and Validation of New Alleles of FALSIFLORA and COMPOUND INFLORESCENCE Genes Controlling the Number of Branches in Tomato Inflorescence. Int. J. Mol. Sci. 2017, 18, 1572. [Google Scholar]
- Pnueli, L.; Carmel-Goren, L.; Hareven, D.; Gutfinger, T.; Alvarez, J.; Ganal, M.; Zamir, D.; Lifschitz, E. The SELF-PRUNING gene of tomato regulates vegetative to reproductive switching of sympodial meristems and is the ortholog of CEN and TFL1. Development 1998, 125, 1979–1989. [Google Scholar] [CrossRef]
- Sharma, V.; Tripathi, B.N.; Kumar, S. Organ-wise homologies of stipule, leaf and inflorescence between Pisum sativum genetic variants, Delonix regia and Caesalpinia bonduc indicate parallel evolution of morphogenetic regulation. Plant Syst. Evol. 2012, 298, 1167–1175. [Google Scholar] [CrossRef]
- Cheng, X.; Li, G.; Tang, Y.; Wen, J. Dissection of genetic regulation of compound inflorescence development in Medicago truncatula. Development 2018, 145, dev158766. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Zhao, Z.; Chen, J.; Hu, X.; Luo, D. A putative CENTRORADIALIS/TERMINAL FLOWER 1-like gene, Ljcen1, plays a role in phase transition in Lotus japonicus. J. Plant Physiol. 2006, 163, 436–444. [Google Scholar] [CrossRef]
- Ungerer, M.C.; Halldorsdottir, S.S.; Purugganan, M.D.; Mackay, T.F.C. Genotype-environment interactions at quantitative trait loci affecting inflorescence development in Arabidopsis thaliana. Genetics 2003, 165, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Huijser, P.; Klein, J.; Lönnig, W.E.; Meijer, H.; Saedler, H.; Sommer, H. Bracteomania, an inflorescence anomaly, is caused by the loss of function of the MADS-box gene squamosa in Antirrhinum majus. EMBO J. 1992, 11, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.J.; Chuck, G.; Dengler, R.E.; Pelecanda, L.; Riggs, C.D. KNAT1 and ERECTA regulate Inflorescence Architecture in Arabidopsis. Plant Cell 2002, 14, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Venglat, S.P.; Dumonceaux, T.; Rozwadowski, K.; Parnell, L.; Babic, V.; Keller, W.; Martienssen, R.; Selvaraj, G.; Datla, R. The homeobox gene BREVIPEDICELLUS is a key regulator of inflorescence architecture in Arabidopsis. Proc. Natl. Acad. Sci. USA 2002, 99, 4730–4735. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Han, M.S.; Cho, H.W.; Jo, Y.D.; Cho, M.C.; Kim, B.D. Molecular cloning of a pepper gene that is homologous to SELF-PRUNING. Mol. Cells 2006, 22, 89–96. [Google Scholar]
- Kato, K.; Ohta, K.; Komata, Y.; Araki, T.; Kanahama, K.; Kanayama, Y. Morphological and molecular analyses of the tomato floral mutant leafy inflorescence, a new allele of falsiflora. Plant Sci. 2005, 169, 131–138. [Google Scholar] [CrossRef]
- Berbel, A.; Ferrándiz, C.; Hecht, V.; Dalmais, M.; Lund, O.S.; Sussmilch, F.; Taylor, S.A.; Bendahmane, A.; Ellis, T.H.N.; Beltrán, J.P.; et al. VEGETATIVE1 is essential for development of the compound inflorescence in pea. Nat. Commun. 2012, 3, 797. [Google Scholar] [CrossRef] [Green Version]
- Allen, K.D.; Sussex, I.M. Falsiflora and anantha control early stages of floral meristem development in tomato (Lycopersicon esculentum Mill.). Planta 1996, 200, 254–264. [Google Scholar] [CrossRef]
- Mero, C.E.; Honma, S. Inheritance of a uniflora mutant in the tomato. J. Hered. 1982, 73, 64–68. [Google Scholar] [CrossRef]
- MacAlister, C.A.; Park, S.J.; Jiang, K.; Marcel, F.; Bendahmane, A.; Izkovich, Y.; Eshed, Y.; Lippman, Z.B. Synchronization of the flowering transition by the tomato TERMINATING FLOWER gene. Nat. Genet. 2012, 44, 1393–1398. [Google Scholar] [CrossRef]
- Thouet, J.; Quinet, M.; Lutts, S.; Kinet, J.-M.; Périlleux, C. Repression of Floral Meristem Fate Is Crucial in Shaping Tomato Inflorescence. PLoS ONE 2012, 7, e31096. [Google Scholar]
- Yuste-Lisbona, F.J.; Quinet, M.; Fernández-Lozano, A.; Pineda, B.; Moreno, V.; Angosto, T.; Lozano, R. Characterization of vegetative inflorescence (mc-vin) mutant provides new insight into the role of MACROCALYX in regulating inflorescence development of tomato. Sci. Rep. 2016, 6, 18796. [Google Scholar] [CrossRef]
- Szymkowiak, E.J.; Irish, E.E. JOINTLESS suppresses sympodial identity in inflorescence meristems of tomato. Planta 2006, 223, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, R.S.; Norton, D.P. Transforming the Balanced Scorecard from Performance Measurement to Strategic Management: Part II. Account. Horiz. 2001, 15, 147–160. [Google Scholar] [CrossRef]
- Koyama, T.; Furutani, M.; Tasaka, M.; Ohme-Takagi, M. TCP Transcription Factors Control the Morphology of Shoot Lateral Organs via Negative Regulation of the Expression of Boundary-Specific Genes in Arabidopsis. Plant Cell 2007, 19, 473–484. [Google Scholar] [CrossRef] [Green Version]
- Kyozuka, J.; Konishi, S.; Nemoto, K.; Izawa, T.; Shimamoto, K. Down-regulation of RFL, the FLO/LFY homolog of rice, accompanied with panicle branch initiation. Proc. Nat. Acad. Sci. USA 1998, 95, 1979–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Yasuno, N.; Sato, Y.; Yoda, M.; Yamazaki, R.; Kimizu, M.; Yoshida, H.; Nagamura, Y.; Kyozuka, J. Inflorescence Meristem Identity in Rice Is Specified by Overlapping Functions of Three AP1/FUL-Like MADS Box Genes and PAP2, a SEPALLATA MADS Box Gene. Plant Cell 2012, 24, 1848–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meeks-Wagner, S.S.R. A Mutation in the Arabidopsis TFL1 Gene Affects Inflorescence Meristem Development. Plant Cell Online 1991, 3, 877–892. [Google Scholar]
- Eliezer, L.; Yuval, E. Universal florigenic signals triggered by FT homologues regulate growth and flowering cycles in perennial day-neutral tomato. Narnia 2006, 57, 3405–3414. [Google Scholar]
- Quinet, M.; Dubois, C.; Goffin, M.-C.; Chao, J.; Dielen, V.; Batoko, H.; Boutry, M.; Kinet, J.-M. Characterization of tomato (Solanum lycopersicum L.) mutants affected in their flowering time and in the morphogenesis of their reproductive structure. J. Exp. Bot. 2006, 57, 1381–1390. [Google Scholar] [CrossRef]
- Lippman, Z.B.; Cohen, O.; Alvarez, J.P.; Abu-Abied, M.; Pekker, I.; Paran, I.; Eshed, Y.; Zamir, D. The Making of a Compound Inflorescence in Tomato and Related Nightshades. PLoS Biol. 2008, 6, e288. [Google Scholar] [CrossRef]
- Szymkowiak Eugene, J.; Irish Erin, E. Interactions between jointless and Wild-Type Tomato Tissues during Development of the Pedicel Abscission Zone and the Inflorescence Meristem. Plant Cell 1999, 11, 159–175. [Google Scholar] [CrossRef] [Green Version]
- Rickett, H.W. The classifification of inflorescences. Bot. Rev. 1944, 10, 187–231. [Google Scholar] [CrossRef]
- Prusinkiewicz, P.; Erasmus, Y.; Lane, B.; Harder, L.D.; Coen, E. Evolution and development of inflorescences architectures. Science 2007, 316, 1452–1456. [Google Scholar] [CrossRef] [Green Version]
- Danert, S. Die Verzweigung der Solanaceen in reproduktiven Bereich. Abh. Dtsch. Akad. Wiss. Berl. 1958, 6, 1–183. [Google Scholar]
- Child, A. A review of branching patterns in the Solanaceae. In The Biology and Taxonomy of the Solanaceae; Hawkes, J.G., Lester, R.N., Skelding, A.D., Eds.; Academic Press: London, UK, 1979; pp. 345–356. [Google Scholar]
- Halle, F.; Oldeman, R.A.A.; Tomlinson, P.B. Tropical Trees and Forests: An Architectural Analysis; Springer: Berlin/Heidelberg, Germany, 1978. [Google Scholar]
- Ng, M.; Yanofsky, M.F. Three ways to learn the ABCs. Curr. Opin. Plant Biol. 2000, 3, 47–52. [Google Scholar] [CrossRef]
- Gramzow, L.; Theissen, G. A hitchhiker’s guide to the MADS world of plants. Genome Biol. 2010, 11, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Expósito-Rodríguez, M.; Borges, A.A.; Borges-Pérez, A.; Pérez, J.A. Gene structure and spatiotemporal expression profile of tomato genes encoding YUCCA-like flavin monooxygenases: The ToFZY gene family. Plant Physiol. Biochem. 2011, 49, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Pichersky, E.; Bernatzky, R.; Tanksley, S.D.; Breidenbach, R.B.; Kausch, A.P.; Cashmore, A.R. Molecular characterization and genetic mapping of two clusters of genes encoding chlorophyll a/b-binding proteins in Lycopersicon esculentum (tomato). Gene 1985, 40, 247–258. [Google Scholar] [CrossRef]
- Bartley, G.E.; Scolnik, P.A. cDNA cloning, expression during development, and genome mapping of PSY2, a second tomato gene encoding phytoene synthase. J. Biol. Chem. 1993, 268, 25718–25721. [Google Scholar] [CrossRef]
- Shchennikova, A.V.; Dyachenko, E.A.; Efremov, G.I.; Filyushin, M.A.; Kochieva, E.Z. Variability and Phylogeny of the Pepper Phytoene Synthase Paralogs PSY1 and PSY2 in Species of Various Capsicum Complexes. Dokl. Biochem. Biophys. 2020, 495, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Kong, J.; Qin, C.; Yu, S.; Tan, J.; Chen, Y.-r.; Wu, C.; Wang, H.; Shi, Y.; Li, C.; et al. Requirement of CHROMOMETHYLASE3 for somatic inheritance of the spontaneous tomato epimutation Colourless non-ripening. Sci. Rep. 2015, 5, 9192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aya, H.-I.; Ken’ichi, O. Overexpression of GSH1 gene mimics transcriptional response to low temperature during seed vernalization treatment of Arabidopsis. Plant Cell Physiol. 2012, 53, 1195–1203. [Google Scholar]
- Pattison, R.J.; Catalá, C. Evaluating auxin distribution in tomato (Solanum lycopersicum) through an analysis of the PIN and AUX/LAX gene families. Plant J. 2011, 70, 585–598. [Google Scholar] [CrossRef]
- Matsuo, S.; Kikuchi, K.; Fukuda, M.; Honda, I.; Imanishi, S. Roles and regulation of cytokinins in tomato fruit development. J. Exp. Bot. 2012, 63, 5569–5579. [Google Scholar] [CrossRef]
- Okada, K.; Ueda, J.; Komaki, M.K.; Bell, C.J.; Shimura, Y. Requirement of the auxin polar transport system in early stages of Arabidopsis floral bud formation. Plant Cell 1991, 3, 677–684. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, D.; Mandel, T.; Kuhlemeier, C. Auxin regulates the initiation and radial position of plant lateral organs. Plant Cell 2000, 12, 507–518. [Google Scholar] [CrossRef] [Green Version]
- Przemeck, G.K.H.; Mattsson, J.; Hardtke, C.S.; Sung, Z.R.; Berleth, T. Studies on the role of the Aiabidopsis gene MONOPTEROS in vascular development and plant cell axialization. Planta 1996, 200, 229–237. [Google Scholar] [CrossRef]
- Nemhauser, J.L.; Zambryski, P.C.; Roe, J.L. Auxin signaling in Arabidopsis flower development? Curr. Opin. Plant Biol. 1998, 1, 531–535. [Google Scholar] [CrossRef]
- Petrásek, J.; Mravec, J.; Bouchard, R.; Blakeslee, J.J.; Abas, M.; Seifertová, D.; Wisniewska, J.; Tadele, Z.; Kubes, M.; Covanová, M.; et al. PIN proteins perform a rate-limiting function in cellular auxin efflux. Science 2006, 312, 914–918. [Google Scholar] [CrossRef] [Green Version]
- Wisniewska, J.; Xu, J.; Seifertova, D.; BrewerP, B.; Ruzicka, K.; Blilou, I.; Rouquie, D.; Bnekova, E.; Scheres, B.; Friml, J. Polar PIN localization direct auxin flow in plants. Science 2006, 312, 883–885. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.; Jones, M.L.; Banowetz, G.M.; Clark, D.G. Overproduction of cytokinins in petunia flowers transforms with PSAG12-IPT delays corolla senescence and decreases sensitivity to ethylene. Plant Physiol. 2003, 132, 2174–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Cerny, R.E.; Qi, Y.; Bhat, D.; Aydt, C.M.; Hanson, D.D.; Malloy, K.P.; Ness, L.A. Transgenic studies on the involvement of cytokinin and gibberellin in male development. Plant Physiol. 2003, 131, 1270–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, T.E.; Giesler-Lee, J.; Gallie, D.R. Senescence-induced expression of cytokinin reverses pistil abortion during maize flower development. Plant J. 2004, 38, 910–922. [Google Scholar] [CrossRef] [PubMed]
- Molinero-Rosales, N.; Jamilena, M.; Zurita, S.; Gómez, P.; Capel, J.; Lozano, R. FALSIFLORA, the tomato orthologue of FLORICAULA and LEAFY, controls flowering time and floral meristem identity. Plant J. 1999, 20, 685–693. [Google Scholar] [CrossRef]
- Siriwardana, N.S.; Lamb, R.S. The poetry of reproduction: The role of LEAFY in Arabidopsis thaliana flower formation. Int. J. Dev. Biol. 2012, 56, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Winter, C.M.; Austin, R.S.; Blanvillain-Baufume, S.; Reback, M.A.; Monniaux, M.; Wu, M.-F.; Sang, Y.; Yamaguchi, A.; Yamaguchi, N.; Parker, J.E. LEAFY target genes reveal floral regulatory logic, cis motifs, and a link to biotic stimulus response. Dev. Cell 2011, 20, 430–443. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Yamaguchi, A.; Abe, M.; Wagner, D.; Komeda, Y. LEAFY controls Arabidopsis pedicel length and orientation by affecting adaxial–abaxial cell fate. Plant J. 2012, 69, 844–856. [Google Scholar] [CrossRef]
- Weigel, D.; Alvarez, J.; Smyth, D.R.; Yanofsky, M.F.; Meyerowitz, E.M. LEAFY controls floral meristem identity in Arabidopsis. Cell 1992, 69, 843–859. [Google Scholar] [CrossRef] [Green Version]
- Schultz, E.A.; Haughn, G.W. LEAFY, a homeotic gene that regulates inflorescence development in Arabidopsis. Plant Cell 1991, 3, 771–781. [Google Scholar] [CrossRef] [Green Version]
- Coen, E.S.; Romero, J.; Doyle, S.; Elliott, R.; Murphy, G.; Carpenter, R. Floricaula: A homeotic gene required for flower development in Antirrhinum majus. Cell 1990, 63, 1311–1322. [Google Scholar] [CrossRef]
- Weigel, D.; Nilsson, O. A developmental switch suffificient for flower initiation in diverse plants. Nature 1995, 377, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Hempel, F.D.; Weigel, D.; Mandel, M.A.; Ditta, G.; Zambryski, P.C.; Feldman, L.J.; Yanofsky, M.F. Floral determination and expression of floral regulatory genes in Arabidopsis. Development 1997, 124, 3845–3853. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Zheng, F.; Wang, J.; Zhang, C.; Xiao, F.; Ye, J.; Li, C.; Ye, Z.; Zhang, J. miR156a-targeted SBP-Box transcription factor SlSPL13 regulates inflorescence morphogenesis by directly activating SFT in tomato. Plant Biotechnol. J. 2020, 18, 1670–1682. [Google Scholar] [CrossRef] [Green Version]
- Dookie, M.; Ali, O.; Ramsubhag, A.; Jayaraman, J. Flowering gene regulation in tomato plants treated with brown seaweed extracts. Sci. Hortic. 2020, 276, 109715. [Google Scholar] [CrossRef]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, S.; Hu, B.; Zhou, Q.; Yu, Q.; Zhang, Z. Comparative analysis of the silk gland transcriptomes between the domestic and wild silkworms. BMC Genom. 2015, 16, 60. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Liu, W.; Zhao, Z.; Yang, H.; Bao, Y.; Zhang, D.; Wang, Z.; Jiang, J.; Xu, Y.; Zhang, H.; et al. Transcriptome profiling reveals the response process of tomato carrying Cf-19 and Cladosporium fulvum interaction. BMC Plant Biol. 2019, 19, 572. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M. Pathway databases and higher order function. Adv. Protein Chem. 2000, 54, 381–408. [Google Scholar] [PubMed]
- Thimm, O.; Blaesing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wu, X.; Ding, C.; Zhang, G.; Li, M.; Lv, L.; Li, Y.; Sun, D.; Zhao, J. Weighted gene co-expression network analysis identifies specific modules and hub genes related to Parkinson’s disease. Neuroreport 2021, 32, 1073–1081. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Raw Reads (M) | Total Clean Reads (M) | Total Clean Bases (Gb) | Clean Reads Q 20 (%) | Clean Reads Q 30 (%) | Clean Reads Ratio (%) |
|---|---|---|---|---|---|---|
| MM_E1 | 45.57 | 44.22 | 6.63 | 97.9 | 94.07 | 97.04 |
| MM_E2 | 47.33 | 45.43 | 6.81 | 97.89 | 94.08 | 95.98 |
| MM_E3 | 45.57 | 43.92 | 6.59 | 97.97 | 94.34 | 96.37 |
| MM_M1 | 44.78 | 43.54 | 6.53 | 97.85 | 93.97 | 97.24 |
| MM_M2 | 45.57 | 44.16 | 6.62 | 97.81 | 93.87 | 96.89 |
| MM_M3 | 45.57 | 44.4 | 6.66 | 97.97 | 94.28 | 97.43 |
| MM_L1 | 45.57 | 43.95 | 6.59 | 97.87 | 94.03 | 96.44 |
| MM_L2 | 45.57 | 44.32 | 6.65 | 97.91 | 94.13 | 97.24 |
| MM_L3 | 45.57 | 44.41 | 6.66 | 97.68 | 93.5 | 97.44 |
| CI_E1 | 45.57 | 43.95 | 6.59 | 97.27 | 92.66 | 96.43 |
| CI_E2 | 47.33 | 45.02 | 6.75 | 97.45 | 93.17 | 95.13 |
| CI_E3 | 45.57 | 44.35 | 6.65 | 97.3 | 92.75 | 97.31 |
| CI_M1 | 45.57 | 44.09 | 6.61 | 97.41 | 93.03 | 96.75 |
| CI_M2 | 45.57 | 44.25 | 6.64 | 97.5 | 93.28 | 97.09 |
| CI_M3 | 45.57 | 44.32 | 6.65 | 97.34 | 92.82 | 97.25 |
| CI_L1 | 45.57 | 44.23 | 6.63 | 97.39 | 92.99 | 97.06 |
| CI_L2 | 45.57 | 44.34 | 6.65 | 97.28 | 92.68 | 97.28 |
| CI_L3 | 45.57 | 44.03 | 6.6 | 97.33 | 92.82 | 96.6 |
| Pathway | Gene ID | Gene Symbol | Log2 Fold-Change | |
|---|---|---|---|---|
| MM_E vs. CI_E | MM_M vs. CI_M | |||
| Metabolic pathways | 101260940 | FZY4 | 5.8 | 2.65 |
| Metabolic pathways | 101251255 | ACO2 | 1.58 | 0.56 |
| Metabolic pathways | 108491835 | Cab-3C | 1.86 | 1.68 |
| Metabolic pathways | 543502 | LIN6 | 1.69 | 1.12 |
| Metabolic pathways | 544150 | DFR | 1.35 | 1.14 |
| Metabolic pathways | 544149 | CHI3 | 1.25 | 1.26 |
| Metabolic pathways | 543959 | TPX2 | 1.07 | 1.08 |
| Metabolic pathways | 101255734 | AnthOMT | 2.13 | 1.61 |
| Metabolic pathways | 543964 | PSY2 | 6.66 | 3.36 |
| Metabolic pathways | 543819 | PLDa1 | 3.57 | 8.38 |
| Metabolic pathways | 101252616 | PMK2 | 5.19 | 4.14 |
| Metabolic pathways | 101265056 | CMT3 | 1.63 | 5.4 |
| Metabolic pathways | 543536 | GSH1 | 5.03 | 4.94 |
| Metabolic pathways | 544258 | GABA-TP3 | 2.66 | 2 |
| Plant hormone signal transduction | 101055578 | CYP735A2-TP3 | 4.83 | 3.56 |
| Plant hormone signal transduction | 543553 | GA20ox1 | 1.55 | 1.61 |
| Plant hormone signal transduction | 543712 | EREB | 2.86 | 4.84 |
| Plant hormone signal transduction | 543887 | FUL2 | 0.87 | 1.38 |
| Plant hormone signal transduction | 101251833 | GH3-4 | 1.25 | 2.46 |
| Plant hormone signal transduction | 543503 | GA3ox1 | 2.92 | 1.02 |
| Plant hormone signal transduction | 543544 | IAA17 | 1.89 | 2.85 |
| Plant hormone signal transduction | 100191111 | LOC100191111 | 1.55 | 1.99 |
| Plant hormone signal transduction | 100736541 | SLAX5 | 1.21 | 1.73 |
| Plant hormone signal transduction | 101055555 | IAA35 | 1.65 | 1.04 |
| Plant hormone signal transduction | 101251833 | SIGH3.4 | 1.25 | 2.47 |
| Plant hormone signal transduction | 101267832 | ERF68 | 1.89 | 1.1 |
| Plant hormone signal transduction | 543644 | TKR | 0.2 | 1.15 |
| Cell cycle | 101250197 | cdc20-1 | 0.55 | 1 |
| Cell cycle | 544248 | pcna | 1.23 | 1.06 |
| Cell cycle | 543730 | cycB1.1 | 1.02 | 1.16 |
| Circadian rhythm | 101246029 | SFT | 0.6 | 0.39 |
| 543584 | Ls | 0.6 | 5.85 | |
| 101253723 | TMF | 1.32 | 7.29 | |
| 543707 | MADSMC | −1.22 | −1.89 | |
| 543884 | MADS1 | 0.44 | 1.24 | |
| 100301925 | AN | 2.87 | 4.12 | |
| 543630 | FA | 2 | 1.38 | |
| 543703 | BLIND | 2.81 | 3.57 | |
| 543770 | TM29 | −1.62 | −0.18 | |
| 100240705 | S | 0.43 | 1.57 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Yang, H.; Tan, Y.; Zhao, T.; Xu, X.; Li, J.; Jiang, J. Comparative Genome Analysis of Genes Regulating Compound Inflorescences in Tomato. Int. J. Mol. Sci. 2021, 22, 12548. https://doi.org/10.3390/ijms222212548
Yang Y, Yang H, Tan Y, Zhao T, Xu X, Li J, Jiang J. Comparative Genome Analysis of Genes Regulating Compound Inflorescences in Tomato. International Journal of Molecular Sciences. 2021; 22(22):12548. https://doi.org/10.3390/ijms222212548
Chicago/Turabian StyleYang, Yahui, Huanhuan Yang, Yinxiao Tan, Tingting Zhao, Xiangyang Xu, Jingfu Li, and Jingbin Jiang. 2021. "Comparative Genome Analysis of Genes Regulating Compound Inflorescences in Tomato" International Journal of Molecular Sciences 22, no. 22: 12548. https://doi.org/10.3390/ijms222212548
APA StyleYang, Y., Yang, H., Tan, Y., Zhao, T., Xu, X., Li, J., & Jiang, J. (2021). Comparative Genome Analysis of Genes Regulating Compound Inflorescences in Tomato. International Journal of Molecular Sciences, 22(22), 12548. https://doi.org/10.3390/ijms222212548