Azoxystrobin Impairs Neuronal Migration and Induces ROS Dependent Apoptosis in Cortical Neurons



Abstract
:1. Introduction
2. Results
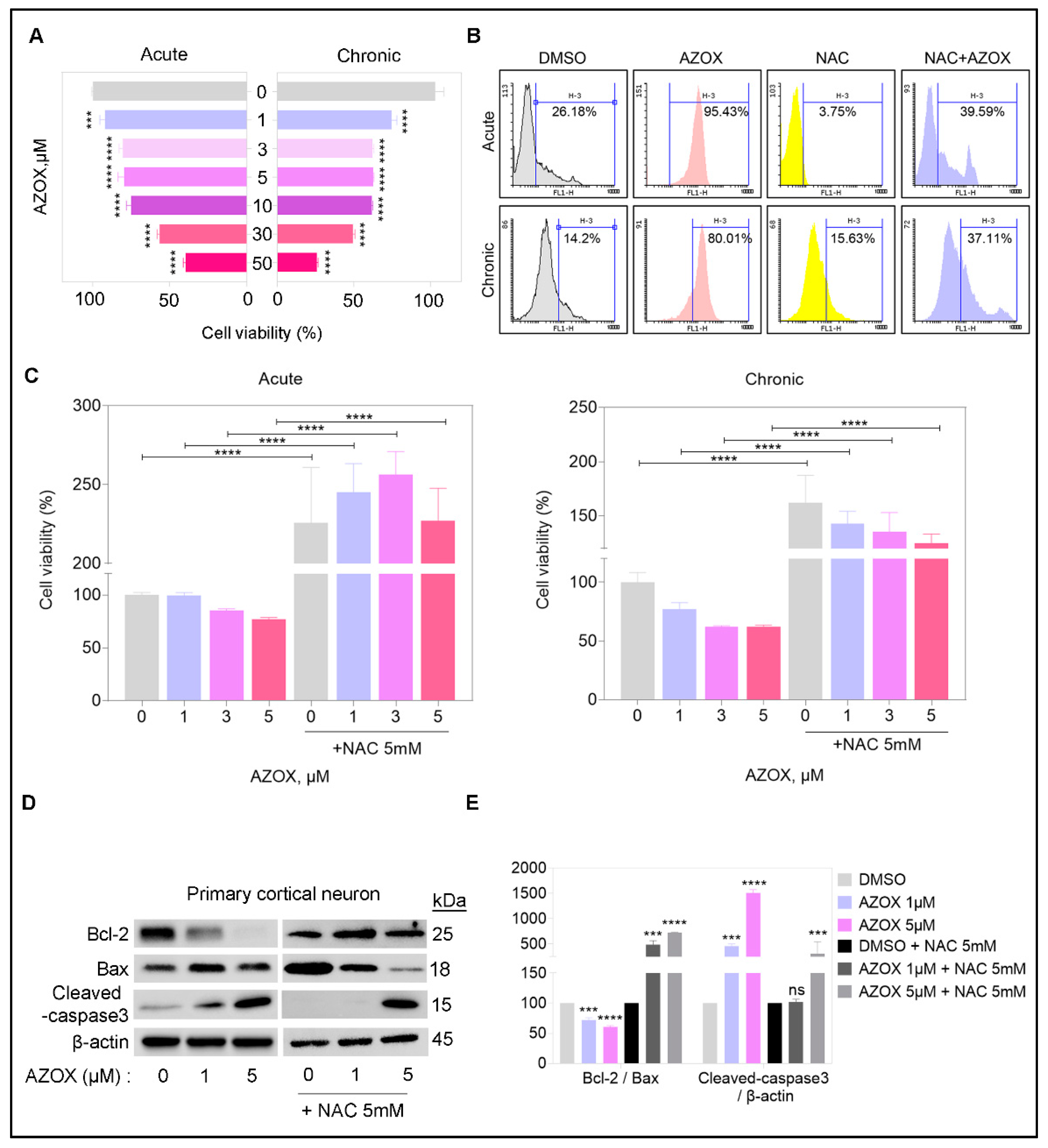
2.1. AZOX Reduced Viability in Primary Cortical Neuron Cultures by Elevating ROS Level
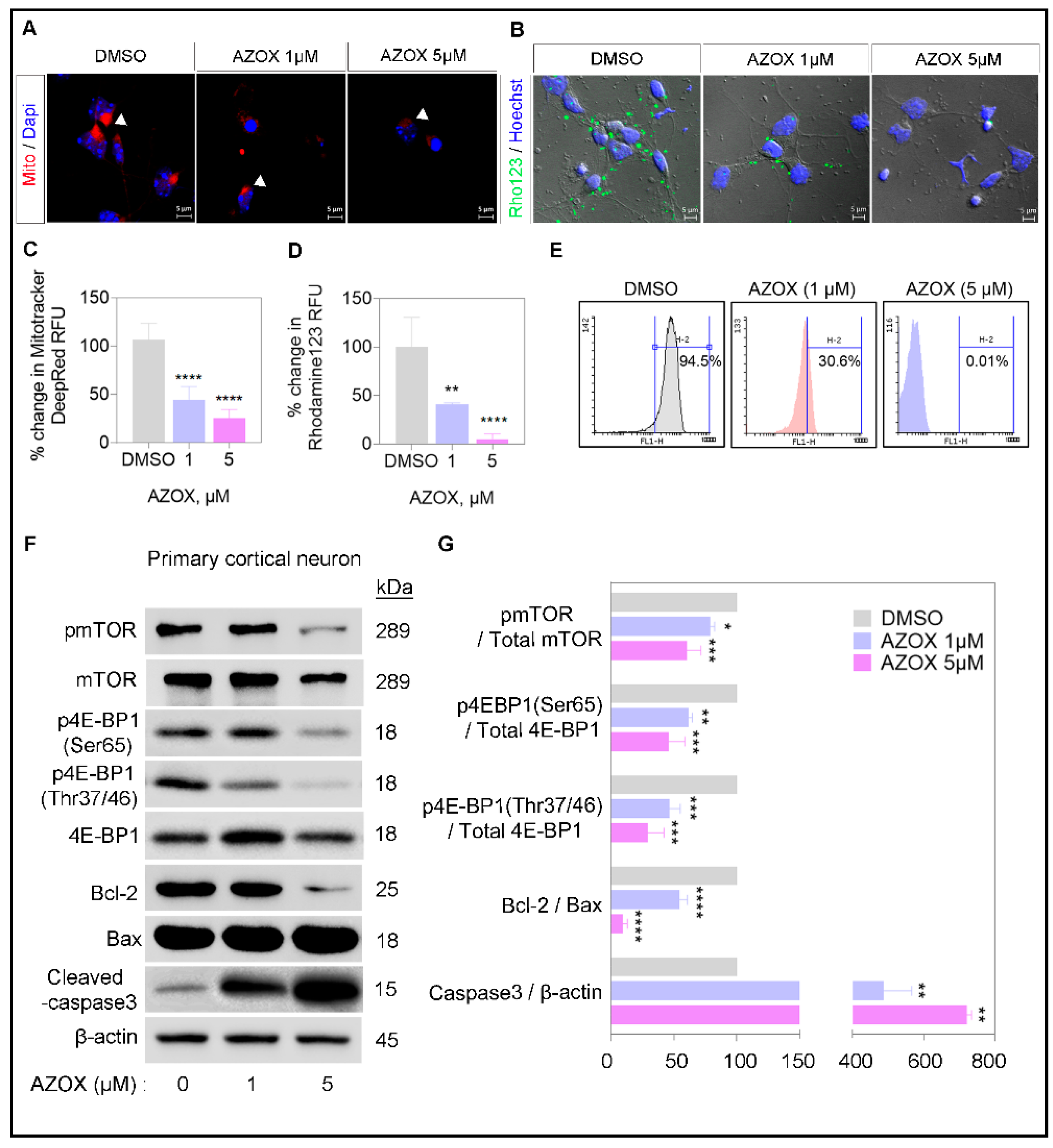
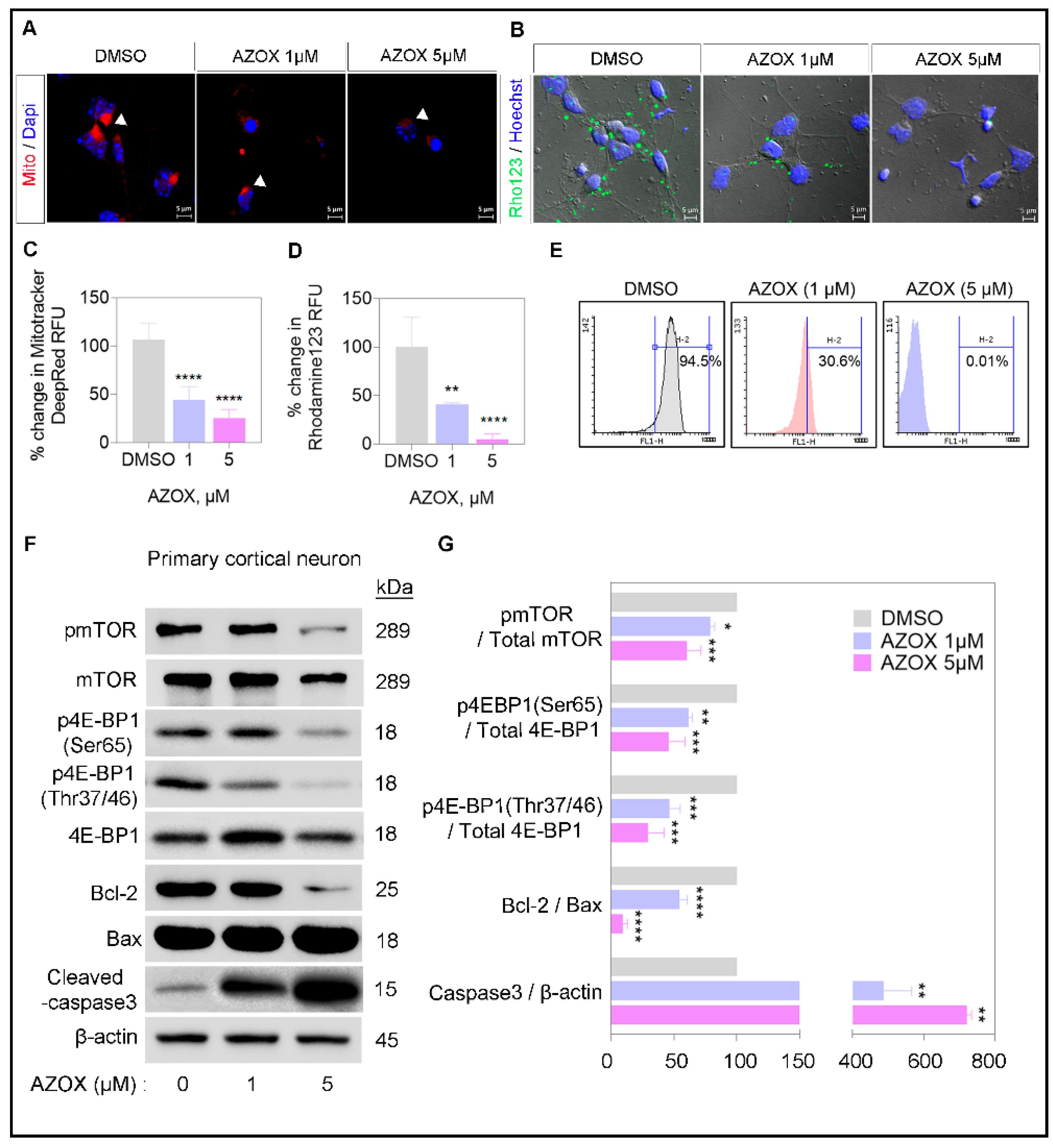
2.2. AZOX Regulated Mitochondrial Mass and Polarization (MMP) and Activated Caspase 3 in Primary Neurons
2.3. AZOX Impaired Radial Migration in Neo-Cortex
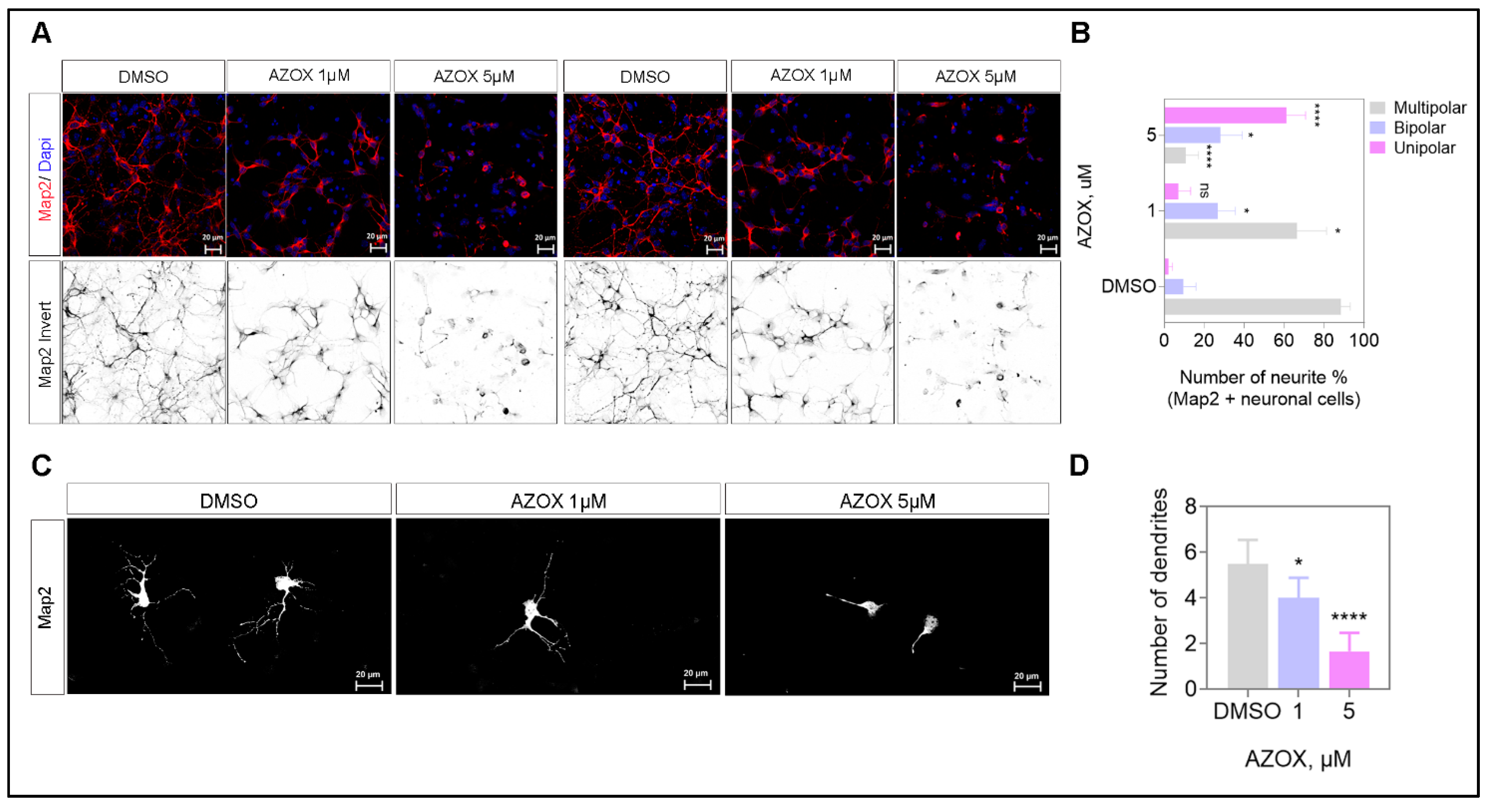
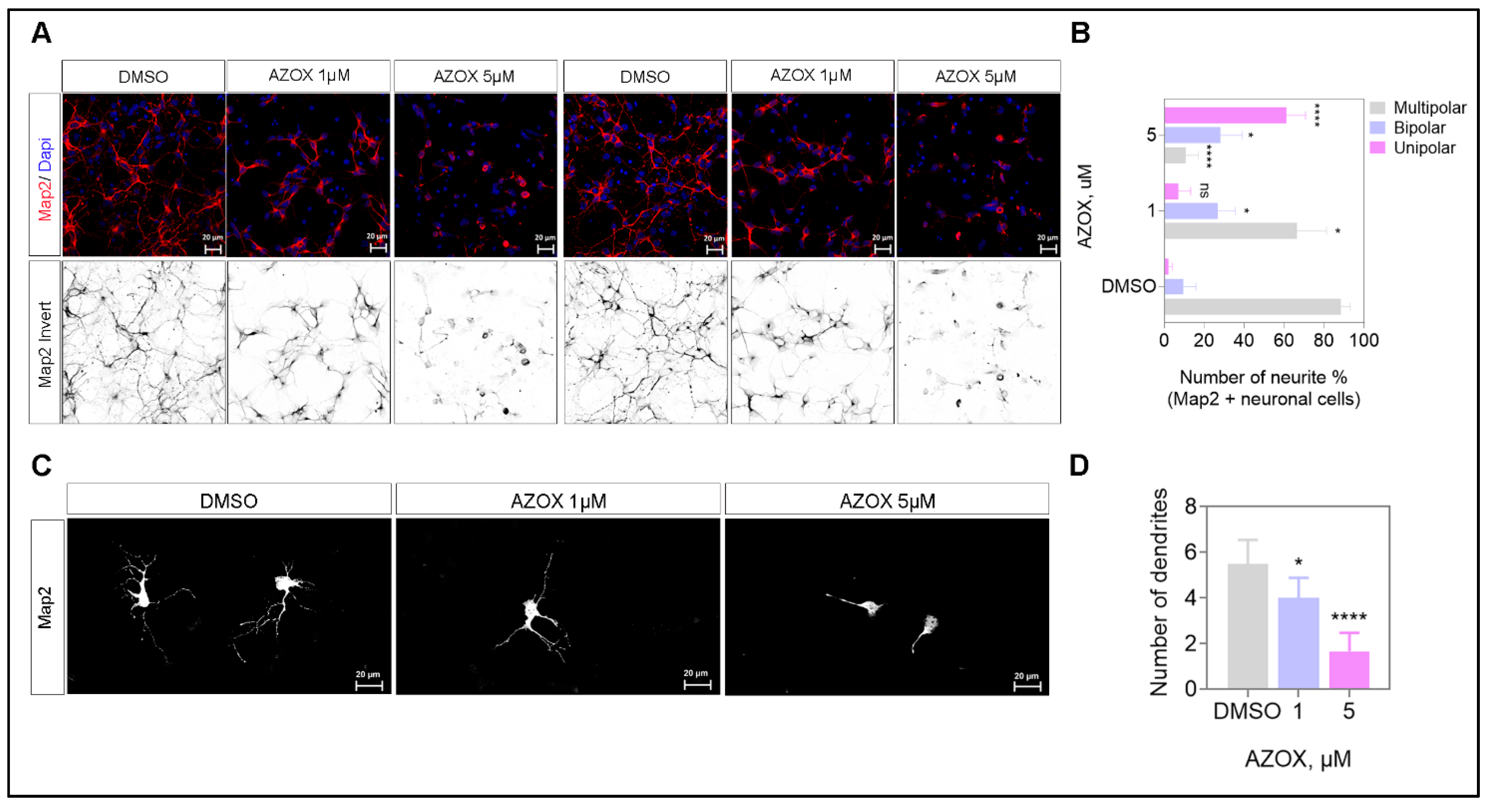
2.4. AZOX Inhibited Neurite Formation, Axons Branching, and Elongation in Primary Neurons
2.5. AZOX Regulated Neurite Outgrowth in mTORC1 Dependent Manner
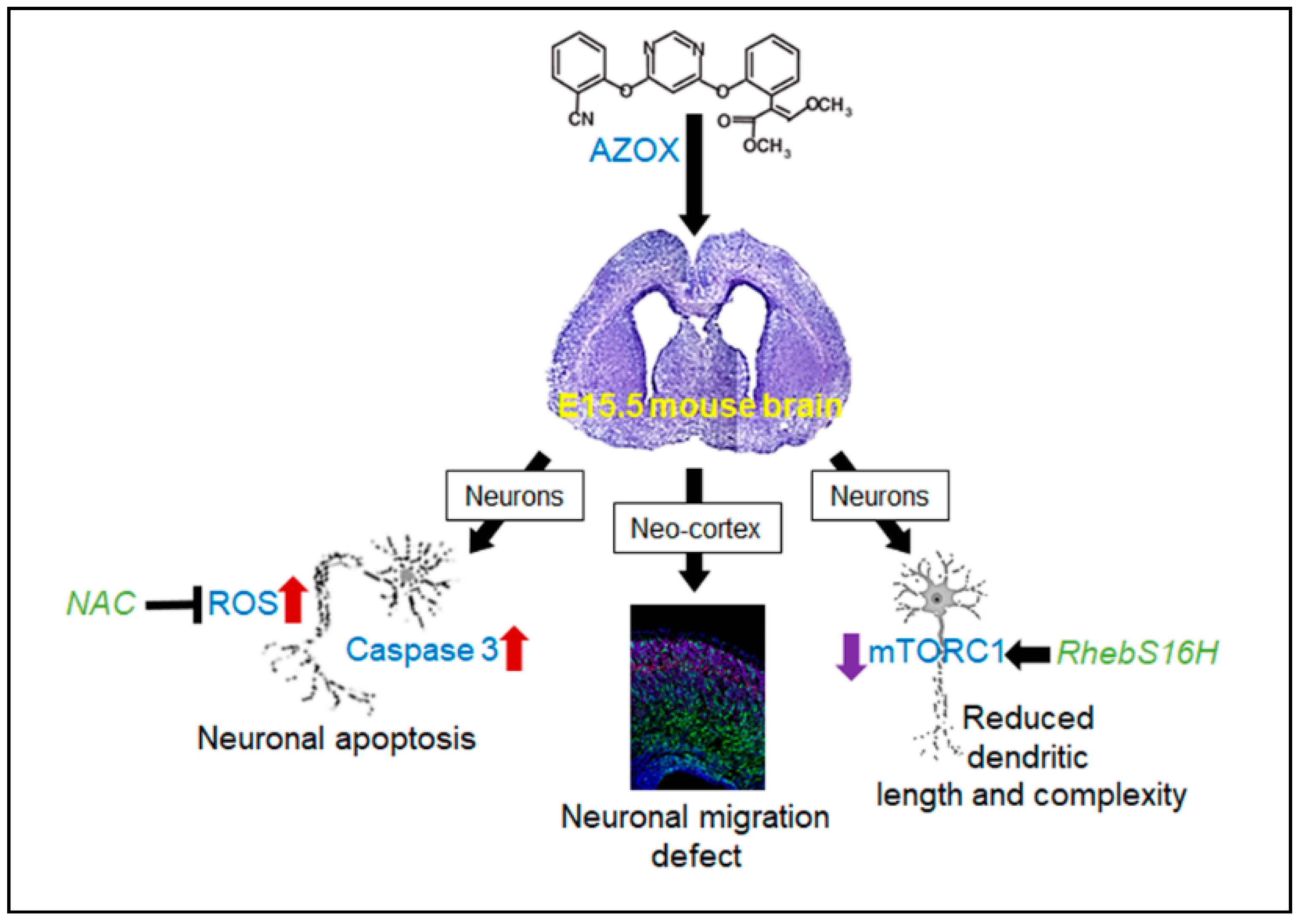
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Isolation of Primary Neurons
4.2. Primary Neuron Cultures
4.3. Azoxystrobin Treatment and Cell Viability Assay
4.4. Identification of ROS Using Oxidized DCFDA and Flowcytometry
4.5. Preparation of Plasmids Constructs and In Utero Electroporation
4.6. Amniotic-Fluid Injection
4.7. SDS-PAGE and Immunoblots
4.8. Immunohistochemistry and Immunocytochemistry
4.9. Neurite Outgrowth Assay
4.10. Mitochondria Staining and Membrane Potential
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abdelraheem, E.M.; Hassan, S.M.; Arief, M.M.; Mohammad, S.G. Validation of quantitative method for azoxystrobin residues in green beans and peas. Food Chem. 2015, 182, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.-K.; Bian, X.-B.; Huang, T.; Wen, B.; Zhao, L.; Mu, H.-X.; Fatima, S.; Fan, B.-M.; Bian, Z.-X.; Huang, L.-F.; et al. Azoxystrobin Induces Apoptosis of Human Esophageal Squamous Cell Carcinoma KYSE-150 Cells through Triggering of the Mitochondrial Pathway. Front. Pharmacol. 2017, 8, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hüttemann, M.; Lee, I.; Samavati, L.; Yu, H.; Doan, J.W. Regulation of mitochondrial oxidative phosphorylation through cell signaling. Biochim. Biophys. Acta Bioenerg. 2007, 1773, 1701–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murali Mahadevan, H.; Hashemiaghdam, A.; Ashrafi, G.; Harbauer, A.B. Mitochondria in Neuronal Health: From Energy Metabolism to Parkinson’s Disease. Adv. Biol. 2021, 5, e2100663. [Google Scholar] [CrossRef]
- De la Rosa, E.J.; de Pablo, F. Cell death in early neural development: Beyond the neurotrophic theory. Trends Neurosci. 2000, 23, 454–458. [Google Scholar] [CrossRef]
- Miodovnik, A. Environmental Neurotoxicants and Developing Brain. Mt. Sinai J. Med. A J. Transl. Pers. Med. 2011, 78, 58–77. [Google Scholar] [CrossRef]
- James, S.E.; Burden, H.; Burgess, R.; Xie, Y.; Yang, T.; Massa, S.M.; Longo, F.M.; Lu, Q. Anti-cancer drug induced neurotoxicity and identification of Rho pathway signaling modulators as potential neuroprotectants. NeuroToxicology 2008, 29, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Shelton, J.F.; Geraghty, E.M.; Tancredi, D.J.; Delwiche, L.D.; Schmidt, R.; Ritz, B.; Hansen, R.L.; Hertz-Picciotto, I. Neurodevelopmental Disorders and Prenatal Residential Proximity to Agricultural Pesticides: The CHARGE Study. Environ. Health Perspect. 2014, 122, 1103–1109. [Google Scholar] [CrossRef] [Green Version]
- Ongono, J.S.; Béranger, R.; Baghdadli, A.; Mortamais, M. Pesticides used in Europe and autism spectrum disorder risk: Can novel exposure hypotheses be formulated beyond organophosphates, organochlorines, pyrethroids and carbamates?—A systematic review. Environ. Res. 2020, 187, 109646. [Google Scholar] [CrossRef]
- Pearson, B.L.; Simon, J.M.; McCoy, E.S.; Salazar, G.; Fragola, G.; Zylka, M.J. Identification of chemicals that mimic transcriptional changes associated with autism, brain aging and neurodegeneration. Nat. Commun. 2016, 7, 11173. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, D.W.; Clough, J.M.; Godwin, J.R.; Hall, A.A.; Hamer, M.; Parr-Dobrzanski, B. The strobilurin fungicides. Pest Manag. Sci. 2002, 58, 649–662. [Google Scholar] [CrossRef]
- Gao, A.-H.; Fu, Y.-Y.; Zhang, K.-Z.; Zhang, M.; Jiang, H.-W.; Fan, L.-X.; Nan, F.-J.; Yuan, C.-G.; Li, J.; Zhou, Y.-B.; et al. Azoxystrobin, a mitochondrial complex III Qo site inhibitor, exerts beneficial metabolic effects in vivo and in vitro. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Garanzini, D.S.; Medici, S.; Moreyra, L.D.; Menone, M.L. Acute exposure to a commercial formulation of Azoxystrobin alters antioxidant enzymes and elicit damage in the aquatic macrophyte Myriophyllum quitense. Physiol. Mol. Biol. Plants 2018, 25, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhu, B.; Wang, G.-X. Azoxystrobin-induced excessive reactive oxygen species (ROS) production and inhibition of photosynthesis in the unicellular green algae Chlorella vulgaris. Environ. Sci. Pollut. Res. 2015, 22, 7766–7775. [Google Scholar] [CrossRef] [PubMed]
- Hartman, E.A.H.; Belden, J.B.; Smith, L.M.; McMurry, S.T. Acute toxicity of three strobilurin fungicide formulations and their active ingredients to tadpoles. Ecotoxicology 2012, 21, 1458–1464. [Google Scholar] [CrossRef]
- Chen, H.; Li, L.; Lu, Y.; Shen, Y.; Zhang, M.; Ge, L.; Wang, M.; Yang, J.; Tian, Z.; Tang, X. Azoxystrobin Reduces Oral Carcinogenesis by Suppressing Mitochondrial Complex III Activity and Inducing Apoptosis. Cancer Manag. Res. 2020, 12, 11573–11583. [Google Scholar] [CrossRef]
- Mondal, A.; NeMoyer, R.; Vora, M.; Napoli, L.; Syed, Z.; Langenfeld, E.; Jia, D.; Peng, Y.; Gilleran, J.; Roberge, J.; et al. Bone morphogenetic protein receptor 2 inhibition destabilizes microtubules promoting the activation of lysosomes and cell death of lung cancer cells. Cell Commun. Signal. 2021, 19, 97. [Google Scholar] [CrossRef]
- Garza-Lombó, C.; Schroder, A.; Reyes-Reyes, E.M.; Franco, R. mTOR/AMPK signaling in the brain: Cell metabolism, proteostasis and survival. Curr. Opin. Toxicol. 2018, 8, 102–110. [Google Scholar] [CrossRef]
- De la Cruz López, K.G.; Toledo Guzmán, M.E.; Sánchez, E.O.; Carrancá, A.G. mTORC1 as a Regulator of Mitochondrial Functions and a Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 1373. [Google Scholar] [CrossRef] [Green Version]
- Abe, N.; Borson, S.H.; Gambello, M.J.; Wang, F.; Cavalli, V. Mammalian Target of Rapamycin (mTOR) Activation Increases Axonal Growth Capacity of Injured Peripheral Nerves. J. Biol. Chem. 2010, 285, 28034–28043. [Google Scholar] [CrossRef] [Green Version]
- Silva, C.G.; Peyre, E.; Adhikari, M.H.; Tielens, S.; Tanco, S.; Van Damme, P.; Magno, L.; Krusy, N.; Agirman, G.; Magiera, M.M.; et al. Cell-Intrinsic Control of Interneuron Migration Drives Cortical Morphogenesis. Cell 2018, 172, 1063–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LiCausi, F.; Hartman, N.W. Role of mTOR Complexes in Neurogenesis. Int. J. Mol. Sci. 2018, 19, 1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, M.; Gravel, S.-P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 Controls Mitochondrial Activity and Biogenesis through 4E-BP-Dependent Translational Regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahimi-Balaei, M.; Bergen, H.; Kong, J.; Marzban, H. Neuronal Migration During Development of the Cerebellum. Front. Cell. Neurosci. 2018, 12, 484. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.-H.; Wu, N.; Yuan, X.-B. Toward a Better Understanding of Neuronal Migration Deficits in Autism Spectrum Disorders. Front. Cell Dev. Biol. 2019, 7, 205. [Google Scholar] [CrossRef]
- Tarkowski, B.; Kuchcinska, K.; Blazejczyk, M.; Jaworski, J.; Bartosz, T.; Kinga, K.; Magdalena, B.; Jacek, J. Pathological mTOR mutations impact cortical development. Hum. Mol. Genet. 2019, 28, 2107–2119. [Google Scholar] [CrossRef]
- Tee, A.R.; Sampson, J.R.; Pal, D.K.; Bateman, J.M. The role of mTOR signalling in neurogenesis, insights from tuberous sclerosis complex. Semin. Cell Dev. Biol. 2016, 52, 12–20. [Google Scholar] [CrossRef]
- Hansen, S.M.; Berezin, V.; Bock, E. Signaling mechanisms of neurite outgrowth induced by the cell adhesion molecules NCAM and N-Cadherin. Cell. Mol. Life Sci. 2008, 65, 3809–3821. [Google Scholar] [CrossRef]
- Andrews, M.G.; Subramanian, L.; Kriegstein, A.R. mTOR signaling regulates the morphology and migration of outer radial glia in developing human cortex. eLife 2020, 9, e58737. [Google Scholar] [CrossRef]
- Yu, S.B.; Pekkurnaz, G. Mechanisms Orchestrating Mitochondrial Dynamics for Energy Homeostasis. J. Mol. Biol. 2018, 430, 3922–3941. [Google Scholar] [CrossRef]
- Feng, Y.; Huang, Y.; Zhan, H.; Bhatt, P.; Chen, S. An Overview of Strobilurin Fungicide Degradation:Current Status and Future Perspective. Front. Microbiol. 2020, 11, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Organization for Economic Co-Operation and Development (OECD). Case Study on the Use of Integrated Approaches to Testing and Assessment for Mitochondrial Complex-Iiimediated Neurotoxicity of Azoxystrobin—Read-Across to Other Strobilurins. 2020, p. 9. Available online: https://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV/JM/MONO(2020)23&docLanguage=en (accessed on 15 November 2021).
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T. Signal Transduction by Mitochondrial Oxidants. J. Biol. Chem. 2012, 287, 4434–4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokhtari, V.; Afsharian, P.; Shahhoseini, M.; Kalantar, S.M.; Moini, A. A Review on Various Uses of N-Acetyl Cysteine. Cell J 2016, 19, 11–17. [Google Scholar]
- Cloëtta, D.; Thomanetz, V.; Baranek, C.; Lustenberger, R.M.; Lin, S.; Oliveri, F.; Atanasoski, S.; Rüegg, M.A. Inactivation of mTORC1 in the Developing Brain Causes Microcephaly and Affects Gliogenesis. J. Neurosci. 2013, 33, 7799–7810. [Google Scholar] [CrossRef] [Green Version]
- Saito, T. In vivo electroporation in the embryonic mouse central nervous system. Nat. Protoc. 2006, 1, 1552–1558. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Antibody | Species | Applications | Dilutions | Manufacturer | Cat. No. |
|---|---|---|---|---|---|---|
| 1 | Phospho-mTOR (S2448) | Rabbit | WB | 1:1000 | Cell Signaling Technology | 5536 |
| 2 | mTOR | Rabbit | WB | 1:1000 | Cell Signaling Technology | 2983 |
| 3 | Phospho-4E-BP1 (Ser65) | Rabbit | WB | 1:1000 | Cell Signaling Technology | 9451S |
| 4 | Phospho-4E-BP1 (Thr37/46) | Rabbit | WB | 1:1000 | Cell Signaling Technology | 2855S |
| 5 | 4E-BP1 | Rabbit | WB | 1:1000 | Cell Signaling Technology | 9452S |
| 6 | Phospho-S6 | Rabbit | WB, ICC | 1:1000 | Cell Signaling Technology | 5364 |
| 7 | S6 | Mouse | WB | 1:1000 | Cell Signaling Technology | 2317S |
| 8 | Bcl-2 | Rabbit | WB | 1:1000 | Cell Signaling Technology | 2876 |
| 9 | Bax | Rabbit | WB | 1:1000 | Cell Signaling Technology | 2772 |
| 10 | Caspase3 | Rabbit | WB | 1:1000 | Abcam | ab4051 |
| 11 | Map2 | Rabbit | ICC | 1:500 | Abcam | ab32454 |
| 12 | beta-3-tubulin (Tuj1) | Mouse | ICC | 1:100 | Cell Signaling Technology | 4466 |
| 13 | GFP | Chicken | ICC, IHC-IF | 1:1000 | Abcam | ab13970 |
| 14 | Ctip2 | Rat | IHC-IF | 1:500 | Abcam | ab18465 |
| 15 | Satb2 | Mouse | IHC-IF | 1:500 | Abcam | ab51502 |
| 16 | β-actin | Rabbit | WB | 1:1000 | Cell Signaling Technology | 4967 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, J.; Bishayee, K.; Huh, S.-O. Azoxystrobin Impairs Neuronal Migration and Induces ROS Dependent Apoptosis in Cortical Neurons. Int. J. Mol. Sci. 2021, 22, 12495. https://doi.org/10.3390/ijms222212495
Kang J, Bishayee K, Huh S-O. Azoxystrobin Impairs Neuronal Migration and Induces ROS Dependent Apoptosis in Cortical Neurons. International Journal of Molecular Sciences. 2021; 22(22):12495. https://doi.org/10.3390/ijms222212495
Chicago/Turabian StyleKang, Jieun, Kausik Bishayee, and Sung-Oh Huh. 2021. "Azoxystrobin Impairs Neuronal Migration and Induces ROS Dependent Apoptosis in Cortical Neurons" International Journal of Molecular Sciences 22, no. 22: 12495. https://doi.org/10.3390/ijms222212495
APA StyleKang, J., Bishayee, K., & Huh, S.-O. (2021). Azoxystrobin Impairs Neuronal Migration and Induces ROS Dependent Apoptosis in Cortical Neurons. International Journal of Molecular Sciences, 22(22), 12495. https://doi.org/10.3390/ijms222212495