
Osteopontin Deficiency Ameliorates Prostatic Fibrosis and Inflammation

 , , , and
, , , and 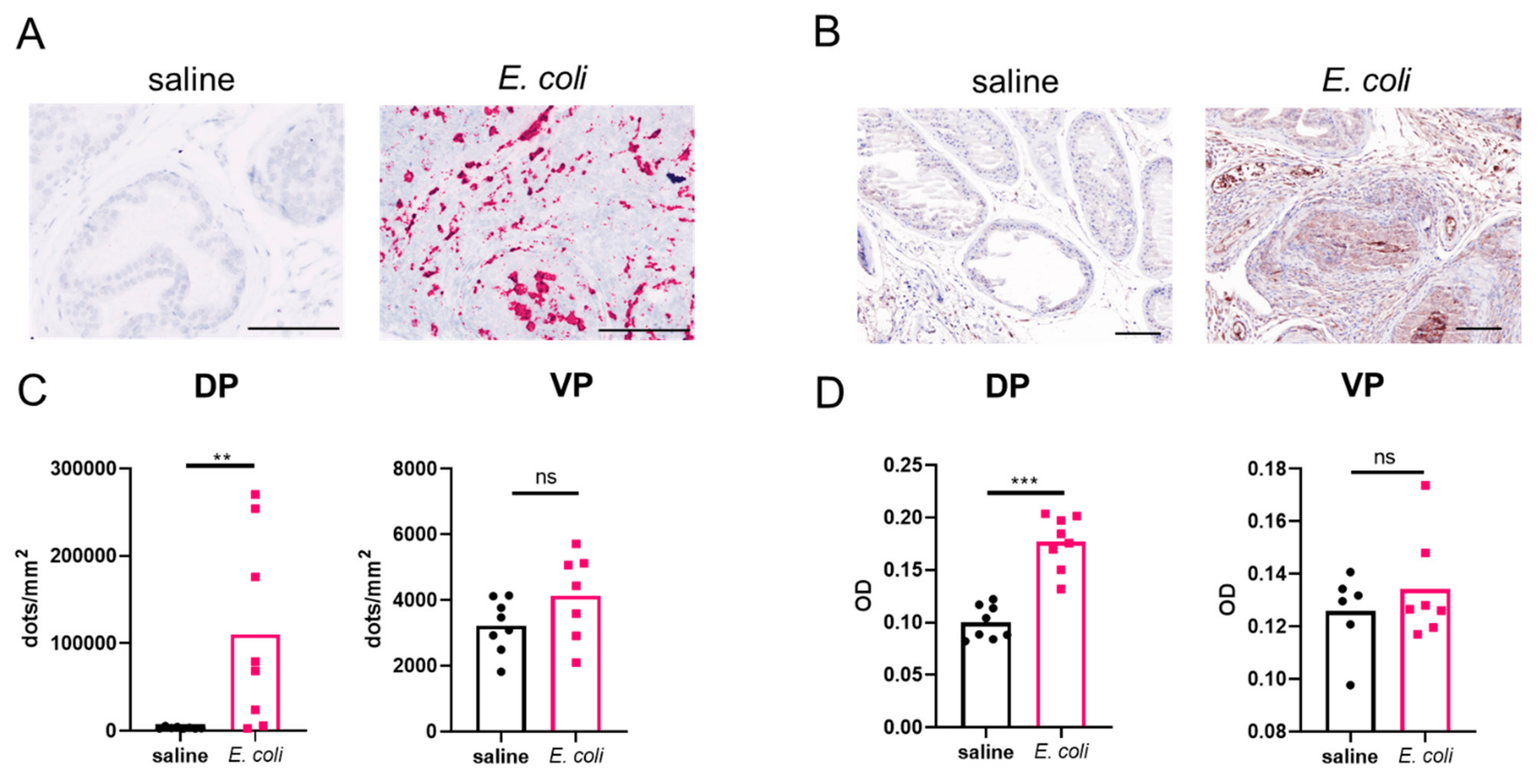{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
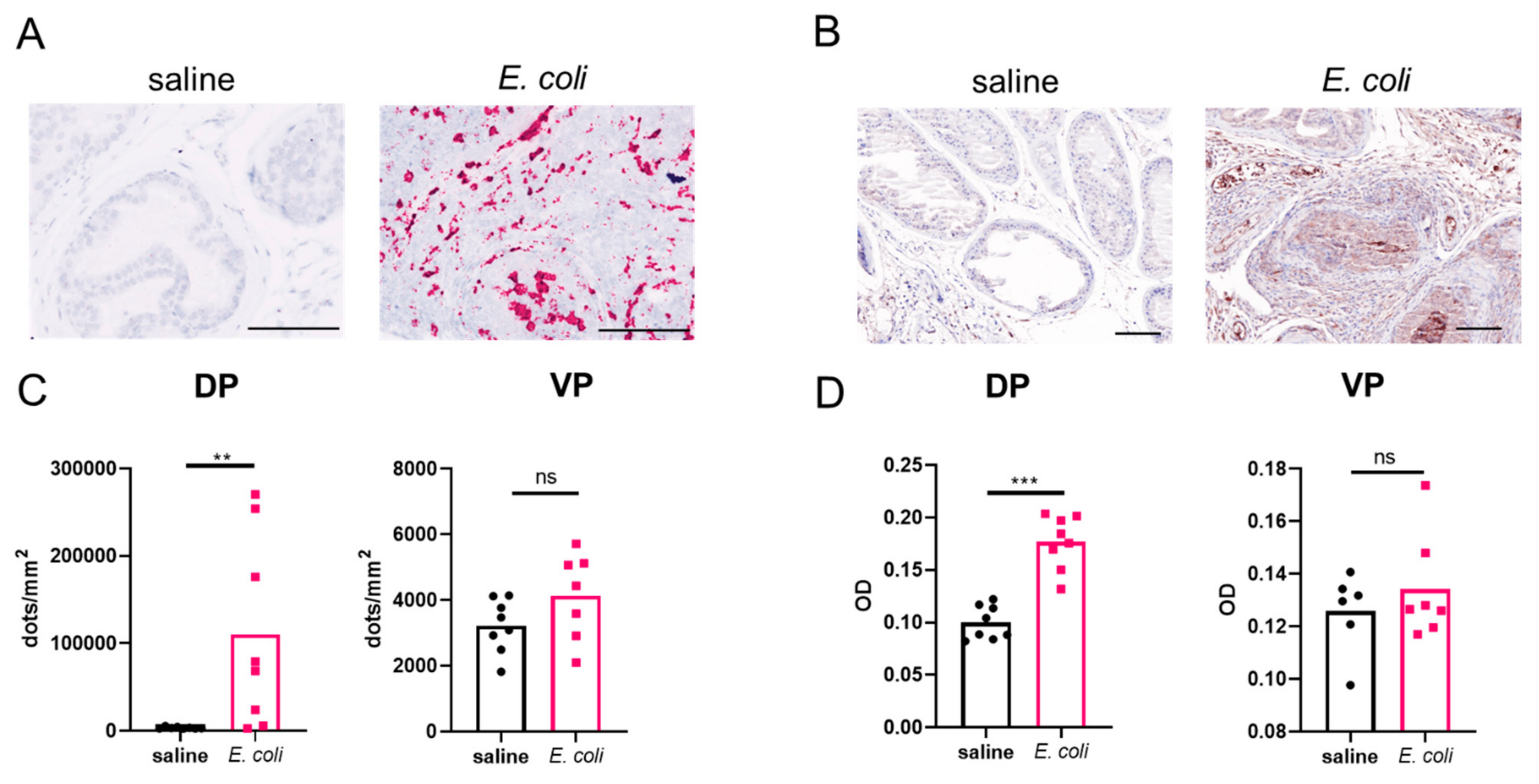
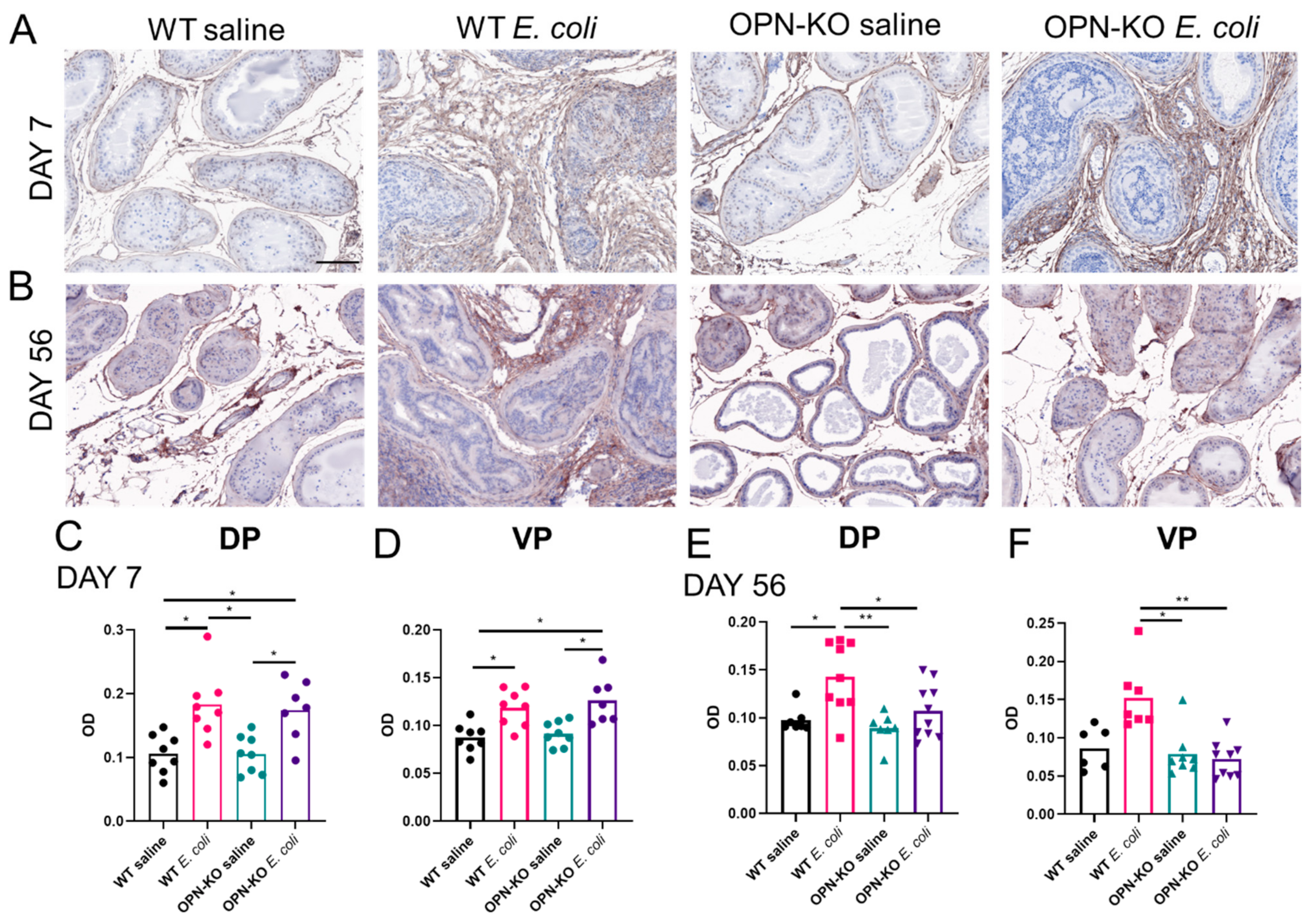
2.1. Prostatic OPN Expression Is Induced in E. coli-Instilled Mice
2.2. Urinary Function Is Improved in OPN-KO Mice
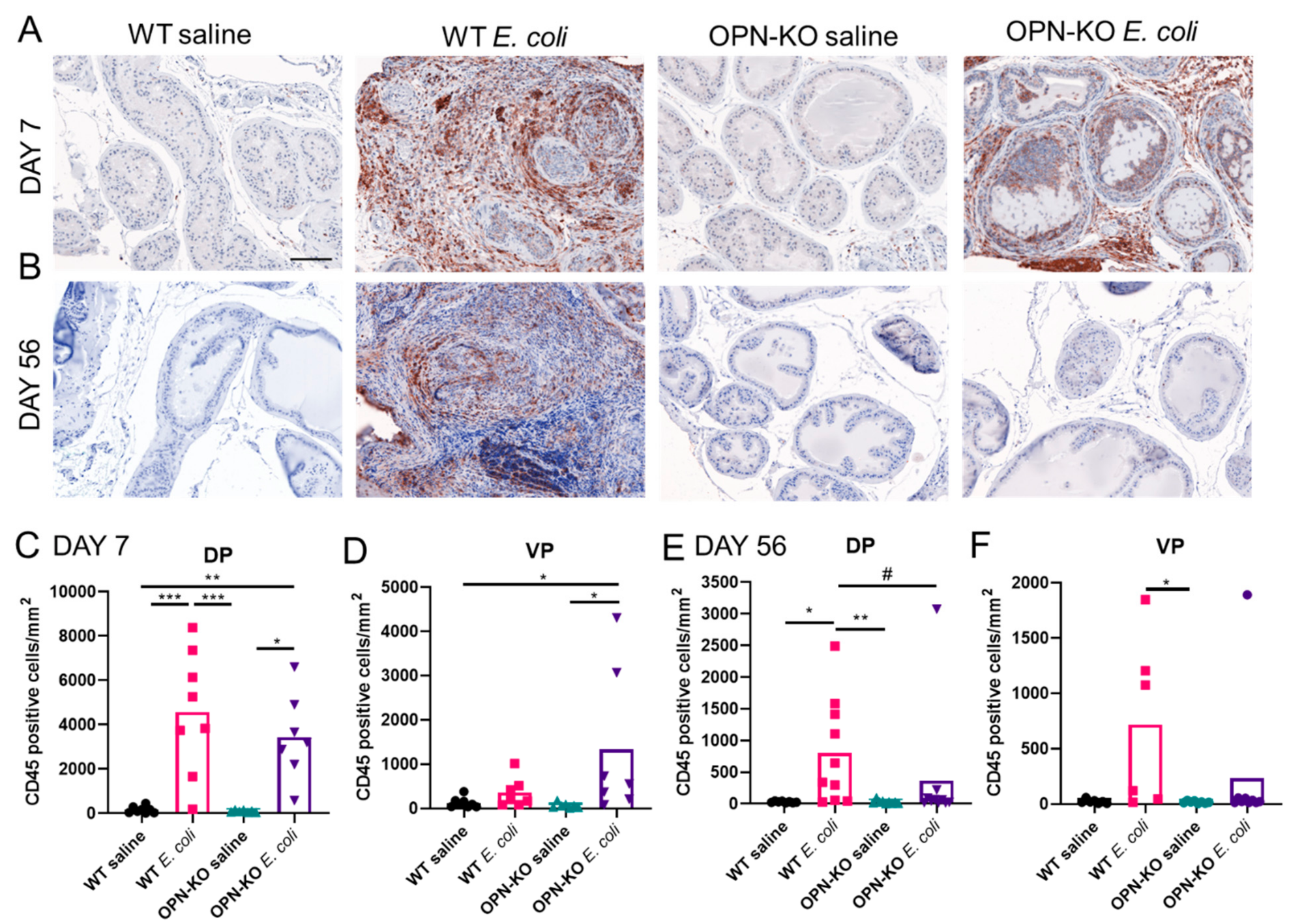
2.3. Bacteria-Induced Inflammation Is Ameliorated in OPN-KO Mice
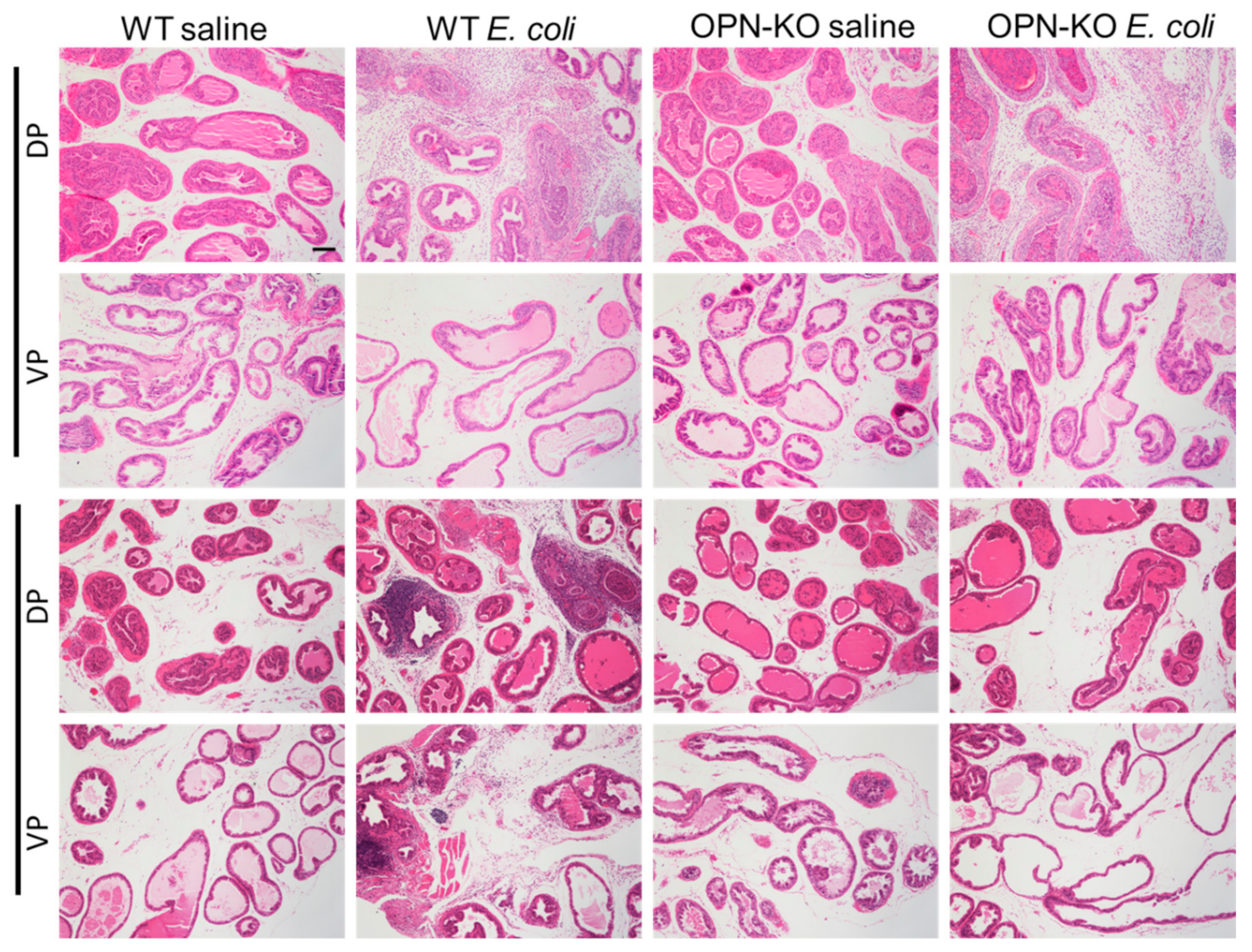
2.4. Chronic Accumulation of Collagen Is Suppressed in OPN-KO Mice
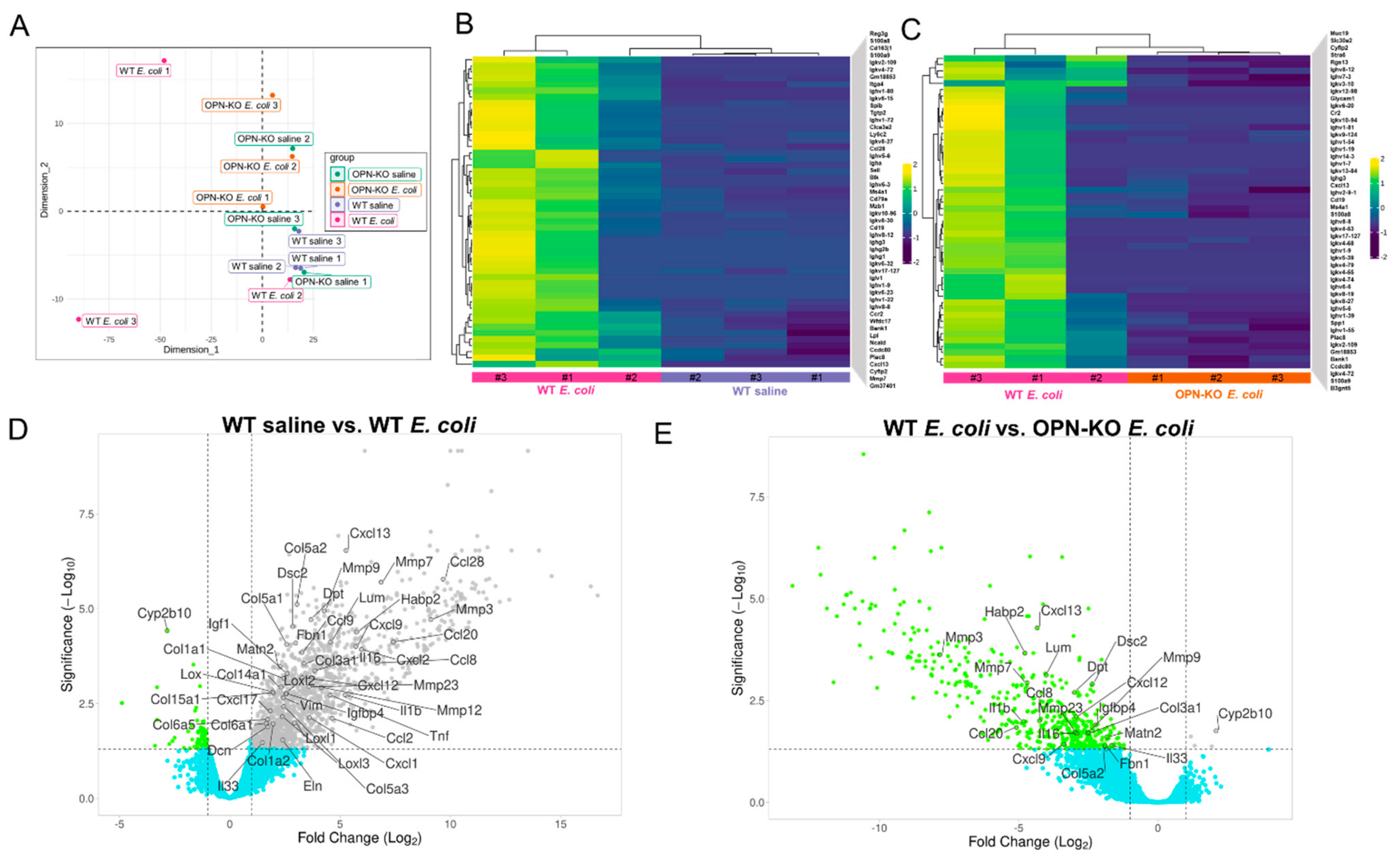
2.5. Identification of Pro-Inflammatory and Pro-Fibrotic Genes Contributing to OPN Action
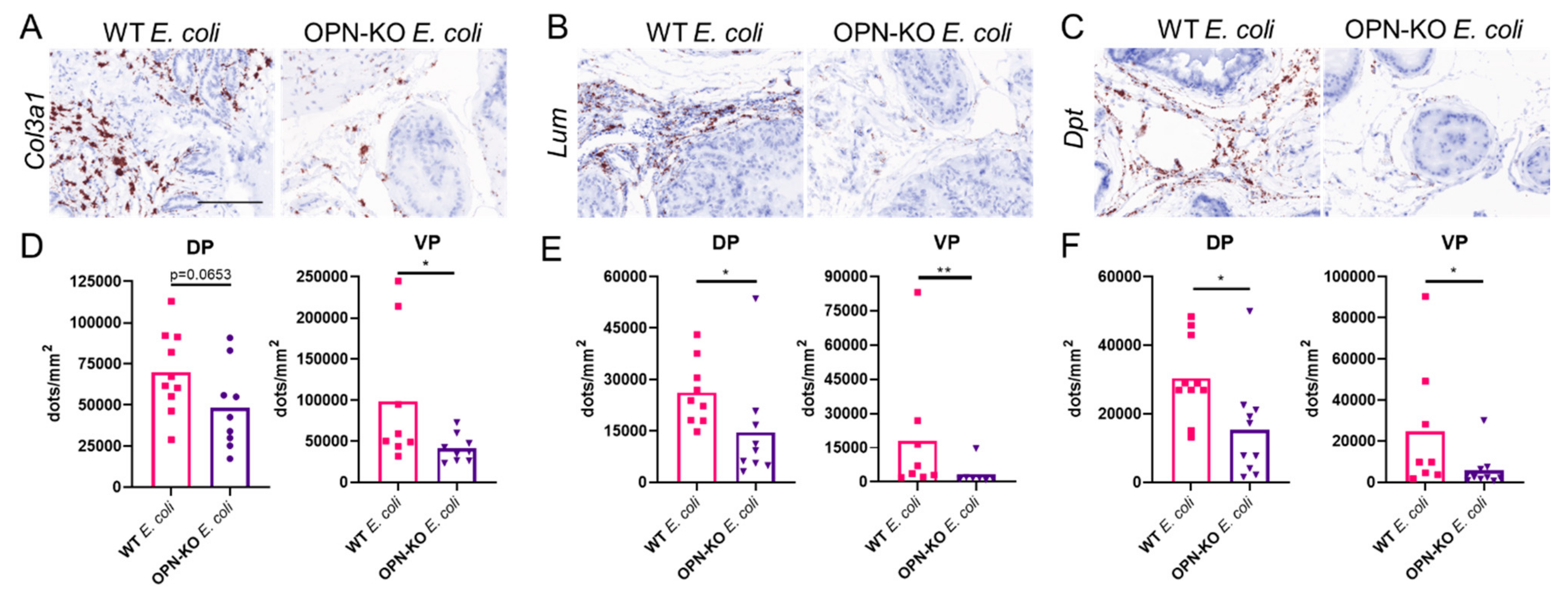
2.6. Validation of Selected Fibrosis Markers
3. Discussion
4. Methods
4.1. Ethics Statement
4.2. Mice
4.3. Bacterial Culture and Transurethral Instillation
4.4. Void Spot Assay
4.5. Picrosirius Red Staining and Polarized Imaging
4.6. Hematoxylin-Eosin (H&E) Staining and Immunohistochemistry (IHC)
4.7. RNAscope
4.8. Image Analysis
4.9. RNA Isolation, Library Preparation, RNA-Seq and Analysis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lepor, H. Pathophysiology of lower urinary tract symptoms in the aging male population. Rev. Urol. 2005, 7 (Suppl. S7), S3–S11. [Google Scholar] [PubMed]
- Lee, S.W.H.; Chan, E.M.C.; Lai, Y.K. The global burden of lower urinary tract symptoms suggestive of benign prostatic hyperplasia: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 7984. [Google Scholar] [CrossRef]
- Parsons, J.K. Benign Prostatic Hyperplasia and Male Lower Urinary Tract Symptoms: Epidemiology and Risk Factors. Curr. Bladder Dysfunct. Rep. 2010, 5, 212–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taub, D.A.; Wei, J.T. The economics of benign prostatic hyperplasia and lower urinary tract symptoms in the United States. Curr. Urol. Rep. 2006, 7, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Vuichoud, C.; Loughlin, K.R. Benign prostatic hyperplasia: Epidemiology, economics and evaluation. Can. J. Urol. 2015, 22, 1–6. [Google Scholar] [PubMed]
- McConnell, J.D.; Roehrborn, C.G.; Bautista, O.M.; Andriole, G.L.; Dixon, C.M.; Kusek, J.W.; Lepor, H.; McVary, K.T.; Nyberg, L.M.; Clarke, H.S.; et al. The Long-Term Effect of Doxazosin, Finasteride, and Combination Therapy on the Clinical Progression of Benign Prostatic Hyperplasia. N. Engl. J. Med. 2003, 349, 2387–2398. [Google Scholar] [CrossRef] [Green Version]
- Theyer, G.; Kramer, G.; Assmann, I.; Sherwood, E.; Preinfalk, W.; Marberger, M.; Zechner, O.; Steiner, G.E. Phenotypic characterization of infiltrating leukocytes in benign prostatic hyperplasia. Lab. Investig. 1992, 66, 96–107. [Google Scholar] [PubMed]
- Kramer, G.; Mitteregger, D.; Marberger, M. Is Benign Prostatic Hyperplasia (BPH) an Immune Inflammatory Disease? Eur. Urol. 2007, 51, 1202–1216. [Google Scholar] [CrossRef] [PubMed]
- Bushman, W.A.; Jerde, T.J. The role of prostate inflammation and fibrosis in lower urinary tract symptoms. Am. J. Physiol. Physiol. 2016, 311, F817–F821. [Google Scholar] [CrossRef] [Green Version]
- Kramer, G.; Steiner, G.E.; Handisurya, A.; Stix, U.; Haitel, A.; Knerer, B.; Gessl, A.; Lee, C.; Marberger, M. Increased expression of lymphocyte-derived cytokines in benign hyperplastic prostate tissue, identification of the producing cell types, and effect of differentially expressed cytokines on stromal cell proliferation. Prostate 2002, 52, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Popovics, P.; Schally, A.V.; Salgueiro, L.; Kovacs, K.; Rick, F.G. Antagonists of growth hormone-releasing hormone inhibit proliferation induced by inflammation in prostatic epithelial cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1359–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickel, J.C.; Roehrborn, C.G.; O’Leary, M.P.; Bostwick, D.G.; Somerville, M.C.; Rittmaster, R.S. The Relationship between Prostate Inflammation and Lower Urinary Tract Symptoms: Examination of Baseline Data from the REDUCE Trial. Eur. Urol. 2008, 54, 1379–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, G.L.; Marks, J.M.; Ricke, W.A. Benign Prostatic Hyperplasia and Lower Urinary Tract Symptoms: What Is the Role and Significance of Inflammation? Curr. Urol. Rep. 2019, 20, 54. [Google Scholar] [CrossRef] [PubMed]
- Nickel, J.C.; Roehrborn, C.G.; Castro-Santamaria, R.; Freedland, S.J.; Moreira, D. Chronic Prostate Inflammation is Associated with Severity and Progression of Benign Prostatic Hyperplasia, Lower Urinary Tract Symptoms and Risk of Acute Urinary Retention. J. Urol. 2016, 196, 1493–1498. [Google Scholar] [CrossRef] [PubMed]
- Colinot, D.L.; Garbuz, T.; Bosland, M.C.; Wang, L.; Rice, S.E.; Sullivan, W.J.; Arrizabalaga, G.; Jerde, T.J. The common parasite Toxoplasma gondiiinduces prostatic inflammation and microglandular hyperplasia in a mouse model. Prostate 2017, 77, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- De Marzo, A.M.; Platz, E.A.; Sutcliffe, S.; Xu, J.; Grönberg, H.; Drake, C.G.; Nakai, Y.; Isaacs, W.B.; Nelson, W.G. Inflammation in prostate carcinogenesis. Nat. Rev. Cancer 2007, 7, 256–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Samal, A.G.; Das, B.; Pradhan, B.; Sahu, N.; Mohapatra, D.; Behera, P.K.; Satpathi, P.S.; Mohanty, A.K.; Senapati, S.; et al. Escherichia coli, a common constituent of benign prostate hyperplasia-associated microbiota induces inflammation and DNA damage in prostate epithelial cells. Prostate 2020, 80, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Ruetten, H.; Sandhu, J.K.; Mueller, B.; Wang, P.; Zhang, H.L.; Wegner, K.A.; Cadena, M.T.; Sandhu, S.K.; Abler, L.L.; Zhu, J.; et al. A uropathogenic E. coli UTI89 model of prostatic inflammation and collagen accumulation for use in studying aberrant collagen production in the prostate. Am. J. Physiol. Physiol. 2021, 320, F31–F46. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.; Hutson, P.R.; Bushman, W. Prostatic Inflammation Induces Fibrosis in a Mouse Model of Chronic Bacterial Infection. PLoS ONE 2014, 9, e100770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell-Cohn, A.; Mazur, D.J.; Hall, C.C.; Schaeffer, A.J.; Thumbikat, P. Uropathogenic Escherichia coli-induced fibrosis, leading to lower urinary tract symptoms, is associated with type 2 cytokine signaling. Am. J. Physiol. Physiol. 2019, 316, F682–F692. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Nieves, J.A.; Macoska, J.A. Prostatic fibrosis, lower urinary tract symptoms, and BPH. Nat. Rev. Urol. 2013, 10, 546–550. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Gharaee-Kermani, M.; Kunju, L.; Hollingsworth, J.M.; Adler, J.; Arruda, E.M.; Macoska, J.A. Prostatic Fibrosis is Associated with Lower Urinary Tract Symptoms. J. Urol. 2012, 188, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Bauman, T.; Nicholson, T.M.; Abler, L.L.; Eliceiri, K.; Huang, W.; Vezina, C.; Ricke, W.A. Characterization of Fibrillar Collagens and Extracellular Matrix of Glandular Benign Prostatic Hyperplasia Nodules. PLoS ONE 2014, 9, e109102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macoska, J.A.; Uchtmann, K.S.; Leverson, G.E.; McVary, K.T.; Ricke, W.A. Prostate Transition Zone Fibrosis is Associated with Clinical Progression in the MTOPS Study. J. Urol. 2019, 202, 1240–1247. [Google Scholar] [CrossRef]
- Delella, F.K.; Lacorte, L.M.; Almeida, F.L.A.; Pai, M.D.; Felisbino, S.L. Fibrosis-related gene expression in the prostate is modulated by doxazosin treatment. Life Sci. 2012, 91, 1281–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.K.; Zhao, C.; Choi, B.R.; Chae, H.J.; Kim, D.S.; Park, J.K. Is Transforming Growth Factor-β Signaling Activated in Human Hypertrophied Prostate Treated by 5-Alpha Reductase Inhibitor? Dis. Markers 2013, 35, 679–685. [Google Scholar] [CrossRef] [Green Version]
- Leung, T.-M.; Wang, X.; Kitamura, N.; Fiel, M.I.; Nieto, N. Osteopontin delays resolution of liver fibrosis. Lab. Investig. 2013, 93, 1082–1089. [Google Scholar] [CrossRef]
- Mori, R.; Shaw, T.J.; Martin, P. Molecular mechanisms linking wound inflammation and fibrosis: Knockdown of osteopontin leads to rapid repair and reduced scarring. J. Exp. Med. 2008, 205, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardo, A.; Gibson, K.; Cisneros, J.; Richards, T.J.; Yang, Y.; Becerril, C.; Yousem, S.; Herrera, I.; Ruiz, V.; Selman, M.; et al. Up-Regulation and Profibrotic Role of Osteopontin in Human Idiopathic Pulmonary Fibrosis. PLoS Med. 2005, 2, e251. [Google Scholar] [CrossRef] [Green Version]
- Popovics, P.; Awadallah, W.N.; Kohrt, S.E.; Case, T.C.; Miller, N.L.; Ricke, E.A.; Huang, W.; Grabowska, M.M.; Liu, Q.; Vezina, C.M.; et al. Prostatic osteopontin expression is associated with symptomatic benign prostatic hyperplasia. Prostate 2020, 80, 731–741. [Google Scholar] [CrossRef]
- Popovics, P.; Cai, R.; Sha, W.; Rick, F.G.; Schally, A.V. Growth hormone-releasing hormone antagonists reduce prostatic enlargement and inflammation in carrageenan-induced chronic prostatitis. Prostate 2018, 78, 970–980. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.X.; Denhardt, D.T. Osteopontin: Role in immune regulation and stress responses. Cytokine Growth Factor Rev. 2008, 19, 333–345. [Google Scholar] [CrossRef] [Green Version]
- Dang, T.; Liou, G.-Y. Macrophage Cytokines Enhance Cell Proliferation of Normal Prostate Epithelial Cells through Activation of ERK and Akt. Sci. Rep. 2018, 8, 7718. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.L.; Jansson, M.; Hwang, E.S.; Werneck, M.B.F.; Glimcher, L.H.; Cantor, H. T-bet-dependent expression of osteopontin contributes to T cell polarization. Proc. Natl. Acad. Sci. USA 2005, 102, 17101–17106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liaw, L.; Birk, D.E.; Ballas, C.B.; Whitsitt, J.S.; Davidson, J.M.; Hogan, B.L. Altered wound healing in mice lacking a functional osteopontin gene (Spp1). J. Clin. Investig. 1998, 101, 1468–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, K.; Hill, W.G.; Li, F.; Shi, B.; Chai, T.C. Early Increased Urinary IL-2 and IL-10 Levels Were Associated with Development of Chronic UTI in a Murine Model. Urology 2020, 141, 188.e1–188.e6. [Google Scholar] [CrossRef]
- Murphy, S.F.; Anker, J.; Mazur, D.J.; Hall, C.; Schaeffer, A.J.; Thumbikat, P. Role of gram-positive bacteria in chronic pelvic pain syndrome (CPPS). Prostate 2019, 79, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Yang, G.; Bushman, W. Prostatic Inflammation Induces Urinary Frequency in Adult Mice. PLoS ONE 2015, 10, e0116827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopinathan, P.A.; Kokila, G.; Jyothi, M.; Ananjan, C.; Pradeep, L.; Nazir, S.H. Study of Collagen Birefringence in Different Grades of Oral Squamous Cell Carcinoma Using Picrosirius Red and Polarized Light Microscopy. Science 2015, 2015, 802980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, J.E.; Dechat, T.; Grin, B.; Helfand, B.; Mendez, M.; Pallari, H.M.; Goldman, R.D. Introducing intermediate filaments: From discovery to disease. J. Clin. Investig. 2009, 119, 1763–1771. [Google Scholar] [CrossRef] [Green Version]
- Ramos, I.; Stamatakis, K.; Oeste, C.L.; Pérez-Sala, D. Vimentin as a Multifaceted Player and Potential Therapeutic Target in Viral Infections. Int. J. Mol. Sci. 2020, 21, 4675. [Google Scholar] [CrossRef] [PubMed]
- Gandaglia, G.; Briganti, A.; Gontero, P.; Mondaini, N.; Novara, G.; Salonia, A.; Sciarra, A.; Montorsi, F. The role of chronic prostatic inflammation in the pathogenesis and progression of benign prostatic hyperplasia (BPH). BJU Int. 2013, 112, 432–441. [Google Scholar] [CrossRef]
- Di Silverio, F.; Gentile, V.; De Matteis, A.; Mariotti, G.; Giuseppe, V.; Luigi, P.A.; Sciarra, A. Distribution of Inflammation, Pre-Malignant Lesions, Incidental Carcinoma in Histologically Confirmed Benign Prostatic Hyperplasia: A Retrospective Analysis. Eur. Urol. 2003, 43, 164–175. [Google Scholar] [CrossRef]
- Zhang, M.; Luo, C.; Cui, K.; Xiong, T.; Chen, Z. Chronic inflammation promotes proliferation in the prostatic stroma in rats with experimental autoimmune prostatitis: Study for a novel method of inducing benign prostatic hyperplasia in a rat model. World J. Urol. 2020, 38, 2933–2943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lopategi, A.; Ge, X.; Lu, Y.; Kitamura, N.; Urtasun, R.; Leung, T.-M.; Fiel, M.I.; Nieto, N. Osteopontin induces ductular reaction contributing to liver fibrosis. Gut 2014, 63, 1805–1818. [Google Scholar] [CrossRef]
- Hikita, S.T.; Vistica, B.P.; Jones, H.R.; Keswani, J.R.; Watson, M.M.; Ericson, V.R.; Ayoub, G.S.; Gery, I.; Clegg, D.O. Osteopontin Is Proinflammatory in Experimental Autoimmune Uveitis. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4435–4443. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, T.; Matsuda, K.; Uchibori, T.; Sugano, M.; Uehara, T.; Honda, T. Upregulation of osteopontin expression via the interaction of macrophages and fibroblasts under IL-1b stimulation. Cytokine 2018, 110, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.N.; Mori, M.; Mertens, T.C.J.; Siddhuraj, P.; Erjefält, J.S.; Onnerfjord, P.; Hiemstra, P.S.; Egesten, A. Osteopontin Expression in Small Airway Epithelium in Copd is Dependent on Differentiation and Confined to Subsets of Cells. Sci. Rep. 2019, 9, 15566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Gang, Y.; Gu, Y.; Zhao, L.; Chu, J.; Zhou, J.; Cai, X.; Zhang, H.; Xu, L.; Nie, Y.; et al. Osteopontin Contributes to TGF-β1 Mediated Hepatic Stellate Cell Activation. Dig. Dis. Sci. 2012, 57, 2883–2891. [Google Scholar] [CrossRef] [PubMed]
- Irita, J.; Okura, T.; Jotoku, M.; Nagao, T.; Enomoto, D.; Kurata, M.; DeSilva, V.R.; Miyoshi, K.-I.; Matsui, Y.; Uede, T.; et al. Osteopontin deficiency protects against aldosterone-induced inflammation, oxidative stress, and interstitial fibrosis in the kidney. Am. J. Physiol. Physiol. 2011, 301, F833–F844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persy, V.P.; Verhulst, A.; Ysebaert, D.K.; De Greef, K.E.; De Broe, M.E. Reduced postischemic macrophage infiltration and interstitial fibrosis in osteopontin knockout mice. Kidney Int. 2003, 63, 543–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, A.R.; Schnee, J.; Wang, W.; Kim, S.; Fishbein, M.C.; Bruemmer, D.; Law, R.E.; Nicholas, S.; Ross, R.; Hsueh, W.A. Osteopontin modulates angiotensin II- induced fibrosis in the intact murine heart. J. Am. Coll. Cardiol. 2004, 43, 1698–1705. [Google Scholar] [CrossRef] [Green Version]
- Urtasun, R.; Lopategi, A.; George, J.; Leung, T.-M.; Lu, Y.; Wang, X.; Ge, X.; Fiel, M.I.; Nieto, N. Osteopontin, an oxidant stress sensitive cytokine, up-regulates collagen-I via integrin αVβ3 engagement and PI3K/pAkt/NFκB signaling. Hepatology 2012, 55, 594–608. [Google Scholar] [CrossRef] [Green Version]
- Yoo, K.; Thornhill, B.; Forbes, M.; Coleman, C.; Marcinko, E.; Liaw, L.; Chevalier, R. Osteopontin regulates renal apoptosis and interstitial fibrosis in neonatal chronic unilateral ureteral obstruction. Kidney Int. 2006, 70, 1735–1741. [Google Scholar] [CrossRef] [Green Version]
- Clemente, N.; Raineri, D.; Cappellano, G.; Boggio, E.; Favero, F.; Soluri, M.F.; Dianzani, C.; Comi, C.; Dianzani, U.; Chiocchetti, A. Osteopontin Bridging Innate and Adaptive Immunity in Autoimmune Diseases. J. Immunol. Res. 2016, 2016, 7675437. [Google Scholar] [CrossRef]
- Psallidas, I.; Stathopoulos, G.; Maniatis, N.A.; Magkouta, S.; Moschos, C.; Karabela, S.P.; Kollintza, A.; Simoes, D.C.M.; Kardara, M.; Vassiliou, S.; et al. Secreted phosphoprotein-1 directly provokes vascular leakage to foster malignant pleural effusion. Oncogene 2012, 32, 528–535. [Google Scholar] [CrossRef]
- Zhang, Y.; Du, W.; Chen, Z.; Xiang, C. Upregulation of PD-L1 by SPP1 mediates macrophage polarization and facilitates immune escape in lung adenocarcinoma. Exp. Cell Res. 2017, 359, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Ashkar, S.; Weber, G.F.; Panoutsakopoulou, V.; Sanchirico, M.E.; Jansson, M.; Zawaideh, S.; Rittling, S.R.; Denhardt, D.T.; Glimcher, M.J.; Cantor, H. Eta-1 (Osteopontin): An Early Component of Type-1 (Cell-Mediated) Immunity. Science 2000, 287, 860–864. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Jia, G.; Guttman, A.; DePianto, D.J.; Morshead, K.B.; Sun, K.-H.; Ramamoorthi, N.; Heiden, J.A.V.; Modrusan, Z.; Wolters, P.J.; et al. Osteopontin Links Myeloid Activation and Disease Progression in Systemic Sclerosis. Cell Rep. Med. 2020, 1, 100140. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular Matrix Degradation and Remodeling in Development and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef] [PubMed]
- Giannandrea, M.; Parks, W.C. Diverse functions of matrix metalloproteinases during fibrosis. Dis. Model. Mech. 2014, 7, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Park, P.W.; Wilson, C.L.; Parks, W.C. Matrilysin Shedding of Syndecan-1 Regulates Chemokine Mobilization and Transepithelial Efflux of Neutrophils in Acute Lung Injury. Cell 2002, 111, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, S.; Petroll, W.M.; Hassell, J.R.; Jester, J.; Lass, J.H.; Paul, J.; Birk, D.E. Corneal opacity in lumican-null mice: Defects in collagen fibril structure and packing in the posterior stroma. Investig. Opthalmol. Vis. Sci. 2000, 41, 3365–3373. [Google Scholar]
- Krishnan, A.; Li, X.; Kao, W.-Y.; Viker, K.; Butters, K.; Masuoka, H.; Knudsen, B.; Gores, G.; Charlton, M. Lumican, an extracellular matrix proteoglycan, is a novel requisite for hepatic fibrosis. Lab. Investig. 2012, 92, 1712–1725. [Google Scholar] [CrossRef]
- Mohammadzadeh, N.; Melleby, A.O.; Palmero, S.; Sjaastad, I.; Chakravarti, S.; Engebretsen, K.V.; Christensen, G.; Lunde, I.G.; Tønnessen, T. Moderate Loss of the Extracellular Matrix Proteoglycan Lumican Attenuates Cardiac Fibrosis in Mice Subjected to Pressure Overload. Cardiology 2020, 145, 187–198. [Google Scholar] [CrossRef]
- Romanic, A.; Adachi, E.; Kadler, K.; Hojima, Y.; Prockop, D. Copolymerization of pNcollagen III and collagen I. pNcollagen III decreases the rate of incorporation of collagen I into fibrils, the amount of collagen I incorporated, and the diameter of the fibrils formed. J. Biol. Chem. 1991, 266, 12703–12709. [Google Scholar] [CrossRef]
- Kottmann, R.M.; Sharp, J.A.; Owens, K.; Salzman, P.; Xiao, G.-Q.; Phipps, R.P.; Sime, P.J.; Brown, E.B.; Perry, S.W. Second harmonic generation microscopy reveals altered collagen microstructure in usual interstitial pneumonia vs. healthy lung. Respir. Res. 2015, 16, 61. [Google Scholar] [CrossRef] [Green Version]
- Kato, A.; Okamoto, O.; Wu, W.; Matsuo, N.; Kumai, J.; Yamada, Y.; Katagiri, F.; Nomizu, M.; Fujiwara, S. Identification of fibronectin binding sites in dermatopontin and their biological function. J. Dermatol. Sci. 2014, 76, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, O.; Fujiwara, S.; Abe, M.; Sato, Y. Dermatopontin interacts with transforming growth factor beta and enhances its biological activity. Biochem. J. 1999, 337, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, R.; Crawford, H.C.; Haro, H.; Matrisian, L.M.; Havrda, M.C.; Liaw, L. Osteopontin, a Novel Substrate for Matrix Metalloproteinase-3 (Stromelysin-1) and Matrix Metalloproteinase-7 (Matrilysin). J. Biol. Chem. 2001, 276, 28261–28267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, C.M.; Dolgonos, L.; Zemans, R.L.; Young, S.K.; Robertson, J.; Briones, N.; Suzuki, T.; Campbell, M.N.; Gauldie, J.; Radisky, D.C.; et al. Matrix Metalloproteinase 3 Is a Mediator of Pulmonary Fibrosis. Am. J. Pathol. 2011, 179, 1733–1745. [Google Scholar] [CrossRef]
- Lin, H.-L.; Zhang, H.; Kenaan, C.; Hollenberg, P.F. Roles of Residues F206 and V367 in Human CYP2B6: Effects of Mutations on Androgen Hydroxylation, Mechanism-Based Inactivation, and Reversible Inhibition. Drug Metab. Dispos. 2016, 44, 1771–1779. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, J.; Fujimura, T.; Takahashi, S.; Urano, T.; Ogushi, T.; Horie-Inoue, K.; Ouchi, Y.; Kitamura, T.; Muramatsu, M.; Blumberg, B.; et al. Cytochrome P450 2B6 is a growth-inhibitory and prognostic factor for prostate cancer. Prostate 2007, 67, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- De Jong, L.M.; Jiskoot, W.; Swen, J.J.; Manson, M.L. Distinct Effects of Inflammation on Cytochrome P450 Regulation and Drug Metabolism: Lessons from Experimental Models and a Potential Role for Pharmacogenetics. Genes 2020, 11, 1509. [Google Scholar] [CrossRef]
- Li, J.; Yousefi, K.; Ding, W.; Singh, J.; Shehadeh, L.A. Osteopontin RNA aptamer can prevent and reverse pressure overload-induced heart failure. Cardiovasc. Res. 2017, 113, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Bandopadhyay, M.; Bulbule, A.; Butti, R.; Chakraborty, G.; Ghorpade, P.; Ghosh, P.; Gorain, M.; Kale, S.; Kumar, D.; Kumar, S.; et al. Osteopontin as a therapeutic target for cancer. Expert Opin. Ther. Targets 2014, 18, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Mysorekar, I.U.; Hultgren, S.J. Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc. Natl. Acad. Sci. USA 2006, 103, 14170–14175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulvey, M.; Schilling, J.D.; Hultgren, S.J. Establishment of a Persistent Escherichia coli Reservoir during the Acute Phase of a Bladder Infection. Infect. Immun. 2001, 69, 4572–4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keil, K.P.; Abler, L.L.; Altmann, H.M.; Bushman, W.; Marker, P.C.; Li, L.; Ricke, W.; Bjorling, D.E.; Vezina, C.M. Influence of animal husbandry practices on void spot assay outcomes in C57BL/6J male mice. Neurourol. Urodyn. 2014, 35, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Wegner, K.; Abler, L.L.; Oakes, S.R.; Mehta, G.S.; Ritter, K.E.; Hill, W.; Zwaans, B.M.M.; Lamb, L.E.; Wang, Z.; Bjorling, D.E.; et al. Void spot assay procedural optimization and software for rapid and objective quantification of rodent voiding function, including overlapping urine spots. Am. J. Physiol. Physiol. 2018, 315, F1067–F1080. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Lei, R.; Ding, S.-W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiner, A.; Yekutieli, D.; Benjamini, Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 2003, 19, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Goedhart, J.; Luijsterburg, M.S. VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci. Rep. 2020, 10, 20560. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popovics, P.; Jain, A.; Skalitzky, K.O.; Schroeder, E.; Ruetten, H.; Cadena, M.; Uchtmann, K.S.; Vezina, C.M.; Ricke, W.A. Osteopontin Deficiency Ameliorates Prostatic Fibrosis and Inflammation. Int. J. Mol. Sci. 2021, 22, 12461. https://doi.org/10.3390/ijms222212461
Popovics P, Jain A, Skalitzky KO, Schroeder E, Ruetten H, Cadena M, Uchtmann KS, Vezina CM, Ricke WA. Osteopontin Deficiency Ameliorates Prostatic Fibrosis and Inflammation. International Journal of Molecular Sciences. 2021; 22(22):12461. https://doi.org/10.3390/ijms222212461
Chicago/Turabian StylePopovics, Petra, Asha Jain, Kegan O. Skalitzky, Elise Schroeder, Hannah Ruetten, Mark Cadena, Kristen S. Uchtmann, Chad M. Vezina, and William A. Ricke. 2021. "Osteopontin Deficiency Ameliorates Prostatic Fibrosis and Inflammation" International Journal of Molecular Sciences 22, no. 22: 12461. https://doi.org/10.3390/ijms222212461
APA StylePopovics, P., Jain, A., Skalitzky, K. O., Schroeder, E., Ruetten, H., Cadena, M., Uchtmann, K. S., Vezina, C. M., & Ricke, W. A. (2021). Osteopontin Deficiency Ameliorates Prostatic Fibrosis and Inflammation. International Journal of Molecular Sciences, 22(22), 12461. https://doi.org/10.3390/ijms222212461