
Proteomic Analysis of Marinesco–Sjogren Syndrome Fibroblasts Indicates Pro-Survival Metabolic Adaptation to SIL1 Loss

 , ,
, ,  , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Results
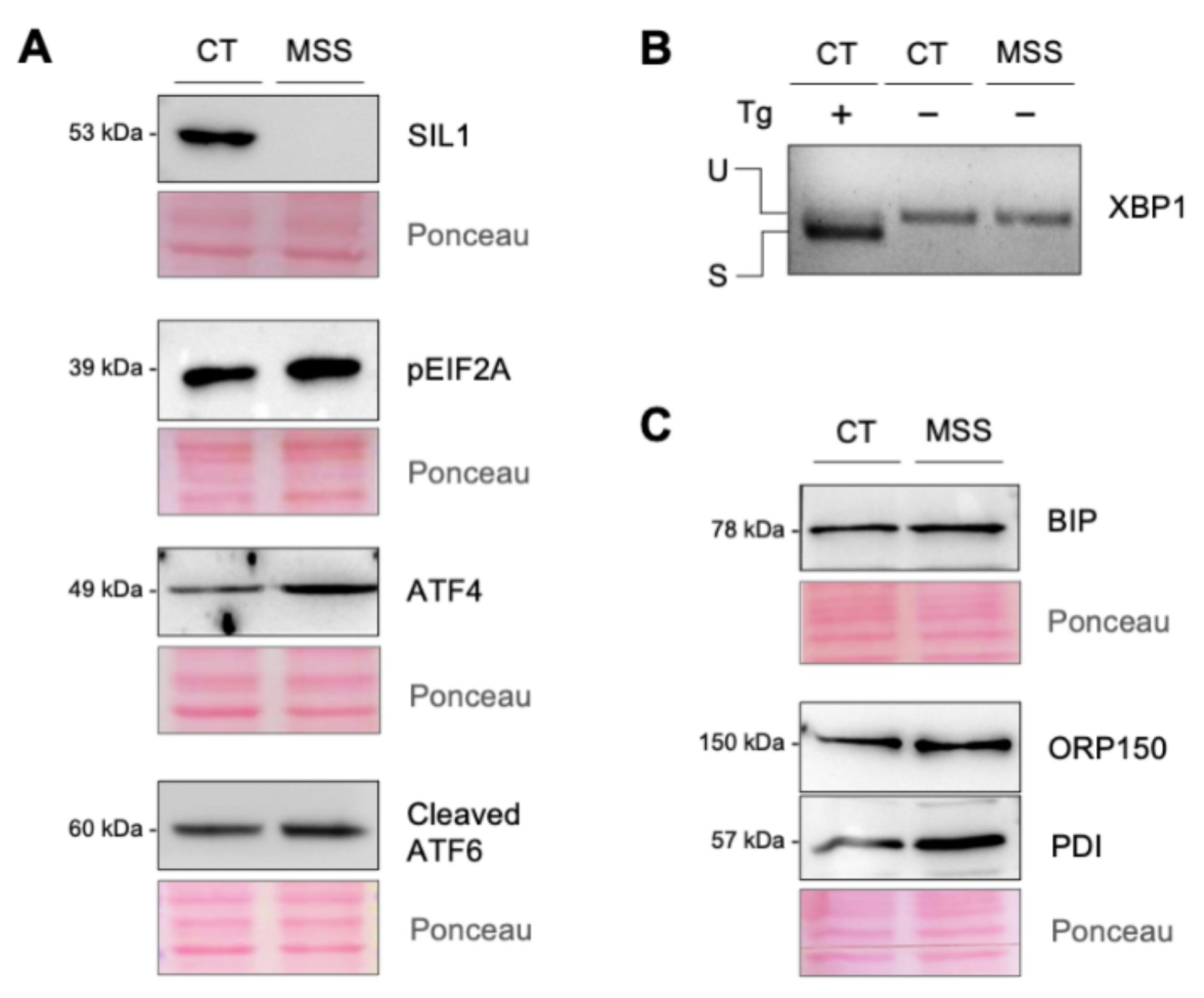
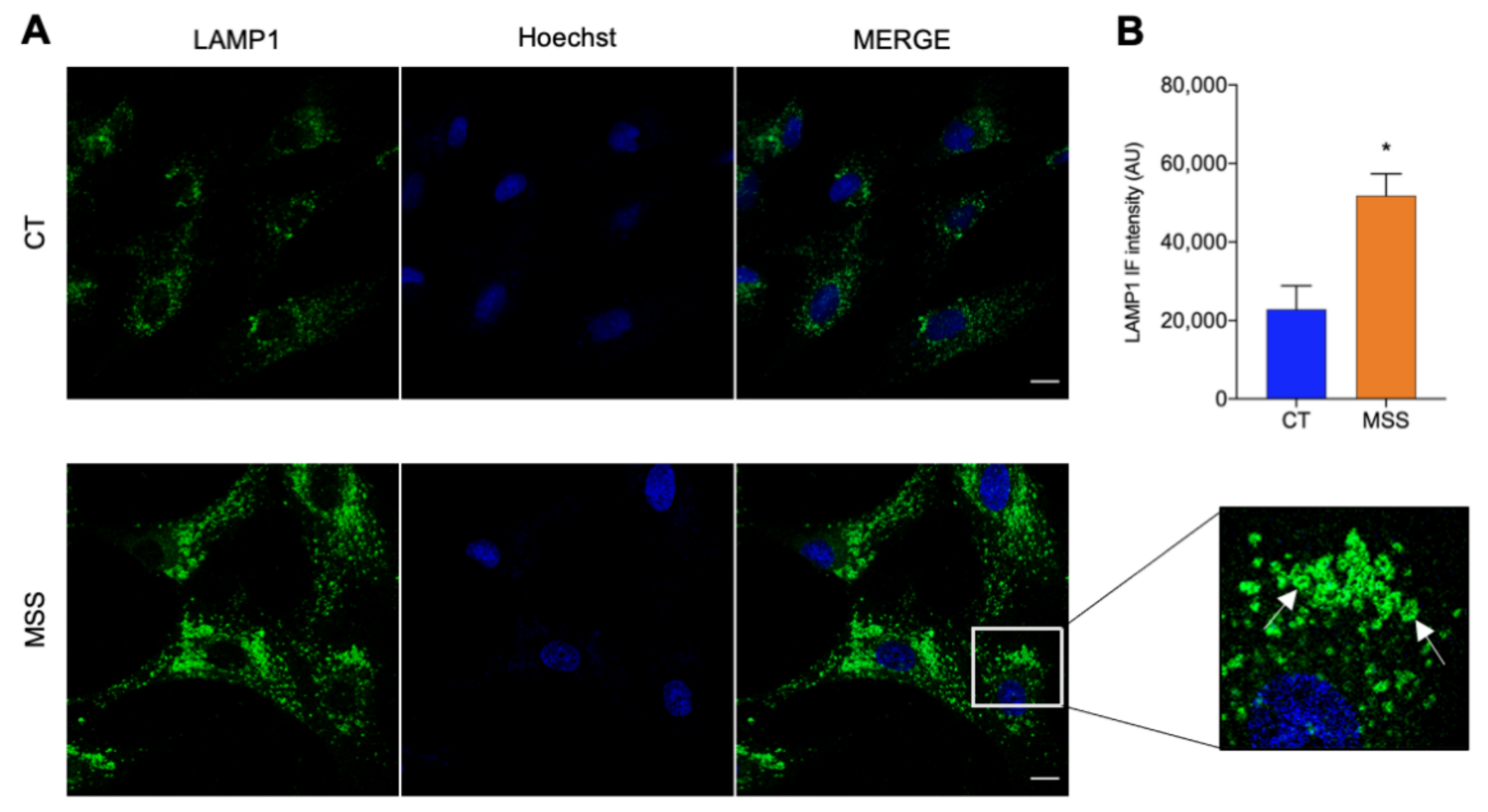
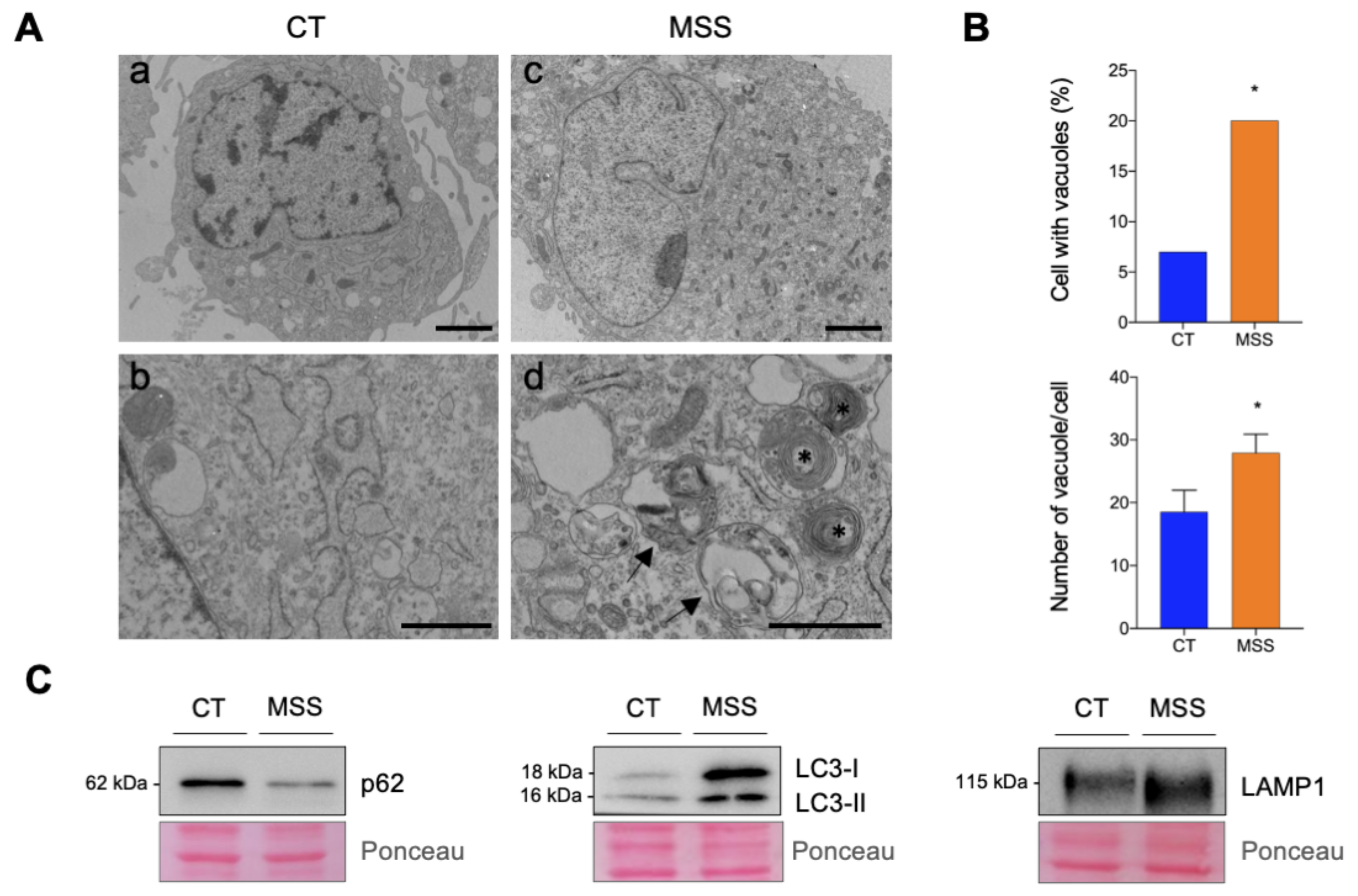
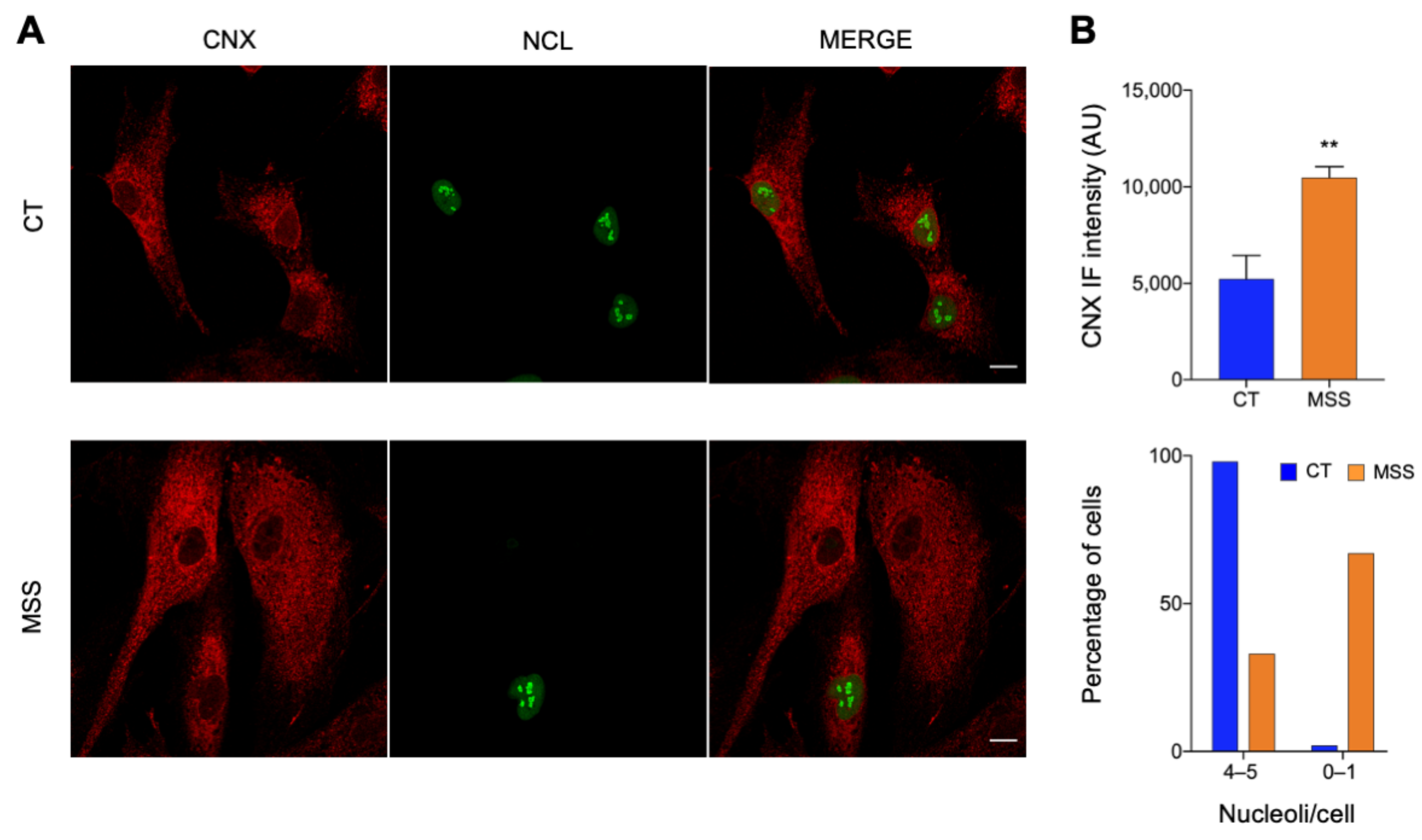
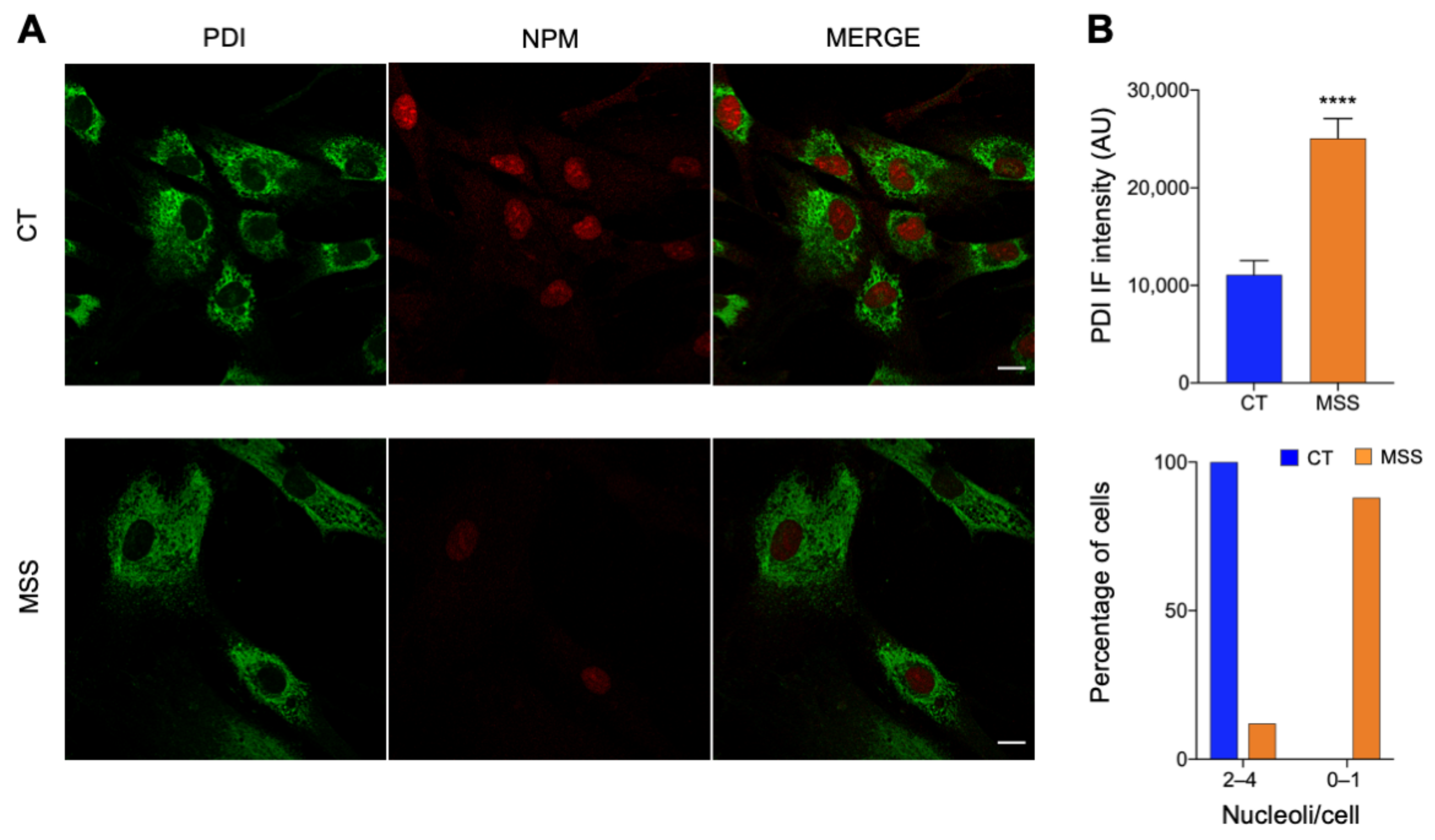
2.1. The Cellular Hallmarks of MSS Are Recapitulated in Cultured Skin Fibroblasts
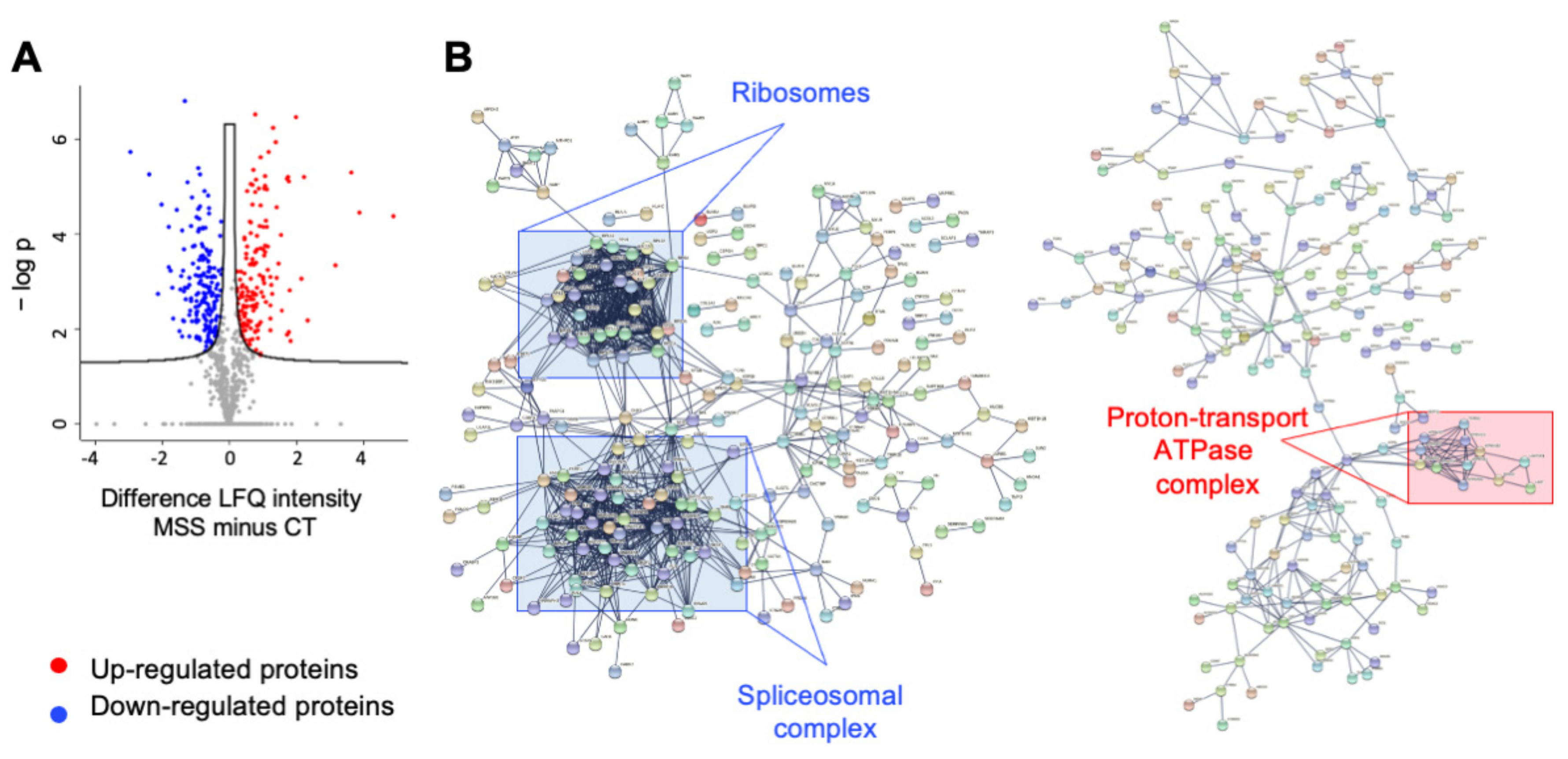
2.2. Proteomics of HF-MSS Identifies 625 Differentially Expressed Proteins
2.3. Ontology Analysis of Differentially Expressed Proteins
2.4. Analysis of Functional Pathway by KEGG
2.5. Analysis of Functional Pathway by IPA
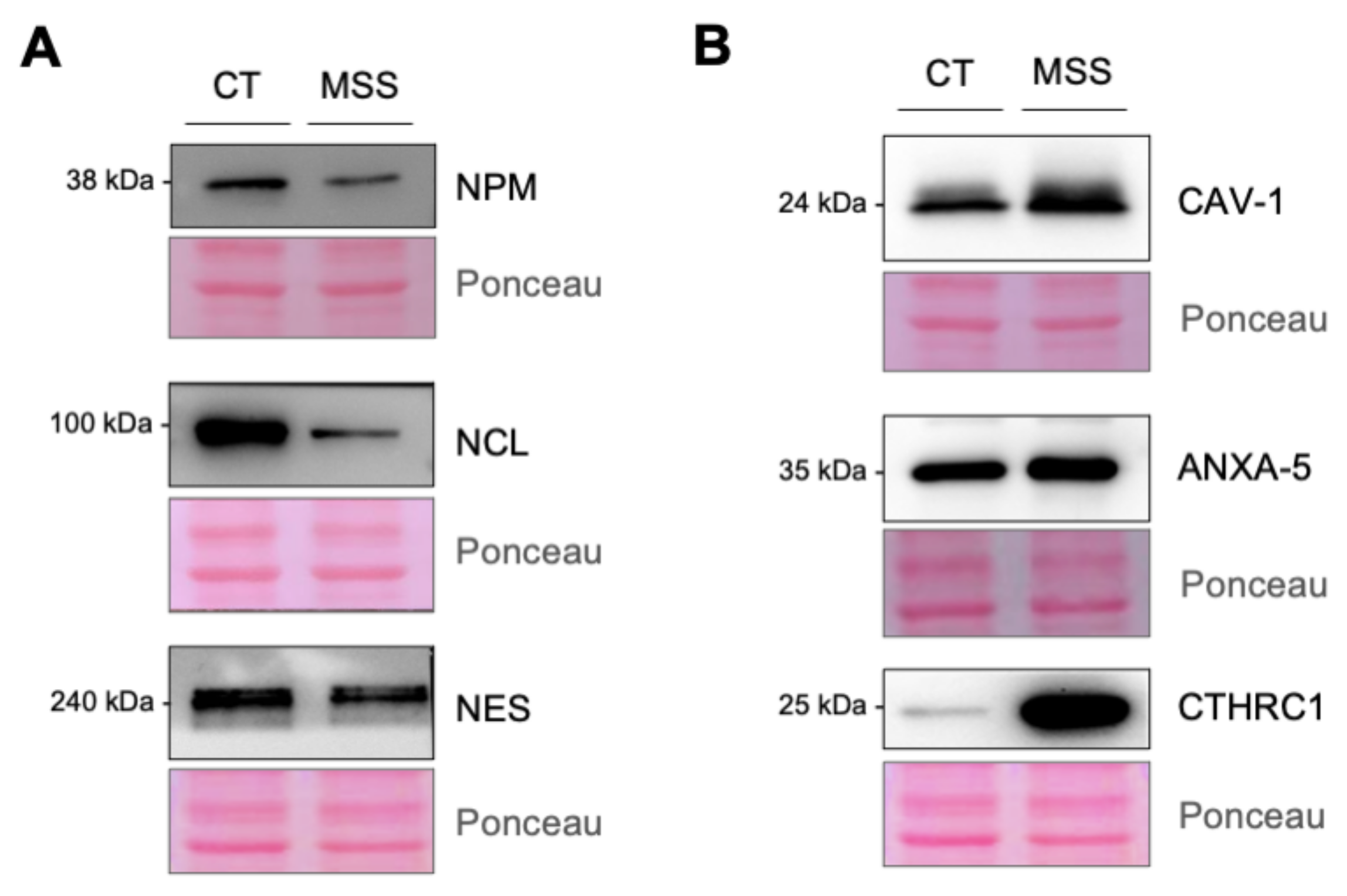
2.6. Validation of the Proteomics
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Fluorescence Immunostaining and Confocal Microscopy
4.4. Western Blot Analysis and PCR
4.5. Proliferation Assay
4.6. Label-Free Proteomics Analysis
4.7. Proteomics Data Processing
4.8. Electron Microscopy (EM)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anttonen, A.K. Marinesco-Sjogren Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Chiesa, R.; Sallese, M. Review: Protein misfolding diseases—The rare case of Marinesco-Sjogren syndrome. Neuropathol. Appl. Neurobiol. 2020, 46, 323–343. [Google Scholar] [CrossRef]
- Ichhaporia, V.P.; Hendershot, L.M. Role of the HSP70 Co-Chaperone SIL1 in Health and Disease. Int. J. Mol. Sci. 2021, 22, 1564. [Google Scholar] [CrossRef]
- Roos, A.; Buchkremer, S.; Kollipara, L.; Labisch, T.; Gatz, C.; Zitzelsberger, M.; Brauers, E.; Nolte, K.; Schroder, J.M.; Kirschner, J.; et al. Myopathy in Marinesco-Sjogren syndrome links endoplasmic reticulum chaperone dysfunction to nuclear envelope pathology. Acta Neuropathol. 2014, 127, 761–777. [Google Scholar] [CrossRef]
- Filezac de L’Etang, A.; Maharjan, N.; Cordeiro Brana, M.; Ruegsegger, C.; Rehmann, R.; Goswami, A.; Roos, A.; Troost, D.; Schneider, B.L.; Weis, J.; et al. Marinesco-Sjogren syndrome protein SIL1 regulates motor neuron subtype-selective ER stress in ALS. Nat. Neurosci. 2015, 18, 227–238. [Google Scholar] [CrossRef]
- Krieger, M.; Roos, A.; Stendel, C.; Claeys, K.G.; Sonmez, F.M.; Baudis, M.; Bauer, P.; Bornemann, A.; de Goede, C.; Dufke, A.; et al. SIL1 mutations and clinical spectrum in patients with Marinesco-Sjogren syndrome. Brain 2013, 136, 3634–3644. [Google Scholar] [CrossRef] [PubMed]
- Senderek, J.; Krieger, M.; Stendel, C.; Bergmann, C.; Moser, M.; Breitbach-Faller, N.; Rudnik-Schoneborn, S.; Blaschek, A.; Wolf, N.I.; Harting, I.; et al. Mutations in SIL1 cause Marinesco-Sjogren syndrome, a cerebellar ataxia with cataract and myopathy. Nat. Genet. 2005, 37, 1312–1314. [Google Scholar] [CrossRef] [PubMed]
- Anttonen, A.K.; Mahjneh, I.; Hamalainen, R.H.; Lagier-Tourenne, C.; Kopra, O.; Waris, L.; Anttonen, M.; Joensuu, T.; Kalimo, H.; Paetau, A.; et al. The gene disrupted in Marinesco-Sjogren syndrome encodes SIL1, an HSPA5 cochaperone. Nat. Genet. 2005, 37, 1309–1311. [Google Scholar] [CrossRef]
- Chung, K.T.; Shen, Y.; Hendershot, L.M. BAP, a mammalian BiP-associated protein, is a nucleotide exchange factor that regulates the ATPase activity of BiP. J. Biol. Chem. 2002, 277, 47557–47563. [Google Scholar] [CrossRef]
- Zhao, L.; Longo-Guess, C.; Harris, B.S.; Lee, J.W.; Ackerman, S.L. Protein accumulation and neurodegeneration in the woozy mutant mouse is caused by disruption of SIL1, a cochaperone of BiP. Nat. Genet. 2005, 37, 974–979. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Grande, V.; Ornaghi, F.; Comerio, L.; Restelli, E.; Masone, A.; Corbelli, A.; Tolomeo, D.; Capone, V.; Axten, J.M.; Laping, N.J.; et al. PERK inhibition delays neurodegeneration and improves motor function in a mouse model of Marinesco-Sjogren syndrome. Hum. Mol. Genet. 2018, 27, 2477–2489. [Google Scholar] [CrossRef] [PubMed]
- Restelli, E.; Masone, A.; Sallese, M.; Chiesa, R. Neuroprotective modulation of the unfolded protein response in Marinesco-Sjogren syndrome: PERK signaling inhibition and beyond. Neural Regen. Res. 2019, 14, 62–64. [Google Scholar] [CrossRef]
- Capone, V.; Clemente, E.; Restelli, E.; Di Campli, A.; Sperduti, S.; Ornaghi, F.; Pietrangelo, L.; Protasi, F.; Chiesa, R.; Sallese, M. PERK inhibition attenuates the abnormalities of the secretory pathway and the increased apoptotic rate induced by SIL1 knockdown in HeLa cells. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3164–3180. [Google Scholar] [CrossRef] [PubMed]
- Kashimada, A.; Hasegawa, S.; Isagai, T.; Uchiyama, T.; Matsuo, M.; Kawai, M.; Goto, M.; Morio, T.; Hayashi, Y.K.; Takagi, M. Targeting the enhanced ER stress response in Marinesco-Sjogren syndrome. J. Neurol. Sci. 2018, 385, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Hentschel, A.; Czech, A.; Munchberg, U.; Freier, E.; Schara-Schmidt, U.; Sickmann, A.; Reimann, J.; Roos, A. Protein signature of human skin fibroblasts allows the study of the molecular etiology of rare neurological diseases. Orphanet J. Rare Dis. 2021, 16, 73. [Google Scholar] [CrossRef] [PubMed]
- Benbrook, D.M.; Long, A. Integration of autophagy, proteasomal degradation, unfolded protein response and apoptosis. Exp. Oncol. 2012, 34, 286–297. [Google Scholar]
- Fregno, I.; Molinari, M. Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 153–163. [Google Scholar] [CrossRef]
- Ichhaporia, V.P.; Kim, J.; Kavdia, K.; Vogel, P.; Horner, L.; Frase, S.; Hendershot, L.M. SIL1, the endoplasmic-reticulum-localized BiP co-chaperone, plays a crucial role in maintaining skeletal muscle proteostasis and physiology. Dis. Models Mech. 2018, 11, dmm033043. [Google Scholar] [CrossRef]
- Piper, R.C.; Katzmann, D.J. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 2007, 23, 519–547. [Google Scholar] [CrossRef]
- Roos, A.; Kollipara, L.; Buchkremer, S.; Labisch, T.; Brauers, E.; Gatz, C.; Lentz, C.; Gerardo-Nava, J.; Weis, J.; Zahedi, R.P. Cellular Signature of SIL1 Depletion: Disease Pathogenesis due to Alterations in Protein Composition Beyond the ER Machinery. Mol. Neurobiol. 2016, 53, 5527–5541. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef]
- Zhao, L.; Rosales, C.; Seburn, K.; Ron, D.; Ackerman, S.L. Alteration of the unfolded protein response modifies neurodegeneration in a mouse model of Marinesco-Sjogren syndrome. Hum. Mol. Genet. 2010, 19, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Anufrieva, K.S.; Shender, V.O.; Arapidi, G.P.; Pavlyukov, M.S.; Shakhparonov, M.I.; Shnaider, P.V.; Butenko, I.O.; Lagarkova, M.A.; Govorun, V.M. Therapy-induced stress response is associated with downregulation of pre-mRNA splicing in cancer cells. Genome Med. 2018, 10, 49. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.M.; Min, S.; Jeoun, U.W.; Sim, M.S.; Jung, G.H.; Hong, S.M.; Jee, B.A.; Woo, H.G.; Lee, C.; Yoon, G. Global spliceosome activity regulates entry into cellular senescence. FASEB J. 2021, 35, e21204. [Google Scholar] [CrossRef]
- Prado, F.; Jimeno-Gonzalez, S.; Reyes, J.C. Histone availability as a strategy to control gene expression. RNA Biol. 2017, 14, 281–286. [Google Scholar] [CrossRef]
- Lanz, M.C.; Zatulovskiy, E.; Swaffer, M.P.; Zhang, L.; Zhang, S.; You, D.S.; Marinov, G.; McAlpine, P.; Elias, J.E.; Skotheim, J.M. Increasing cell size remodels the proteome and promotes senescence. bioRxiv 2021. [Google Scholar] [CrossRef]
- Galdieri, L.; Zhang, T.; Rogerson, D.; Vancura, A. Reduced Histone Expression or a Defect in Chromatin Assembly Induces Respiration. Mol. Cell. Biol. 2016, 36, 1064–1077. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Zimmermann, R.; Lang, S. A Little AXER ABC: ATP, BiP, and Calcium Form a Triumvirate Orchestrating Energy Homeostasis of the Endoplasmic Reticulum. Contact 2020, 3, 2515256420926795. [Google Scholar] [CrossRef]
- Yong, J.; Bischof, H.; Burgstaller, S.; Siirin, M.; Murphy, A.; Malli, R.; Kaufman, R.J. Mitochondria supply ATP to the ER through a mechanism antagonized by cytosolic Ca(2). eLife 2019, 8, e49682. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Pendin, D.; Pizzo, P. Mitofusin 2: From functions to disease. Cell Death Dis. 2018, 9, 330. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Hollien, J. Ire1-mediated decay in mammalian cells relies on mRNA sequence, structure, and translational status. Mol. Biol. Cell 2015, 26, 2873–2884. [Google Scholar] [CrossRef]
- Filocamo, M.; Mazzotti, R.; Corsolini, F.; Stroppiano, M.; Stroppiana, G.; Grossi, S.; Lualdi, S.; Tappino, B.; Lanza, F.; Galotto, S.; et al. Cell Line and DNA Biobank From Patients Affected by Genetic Diseases. Open J. Bioresour. 2014, 1, e2. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Falasca, K.; Lanuti, P.; Ucciferri, C.; Pieragostino, D.; Cufaro, M.C.; Bologna, G.; Federici, L.; Miscia, S.; Pontolillo, M.; Auricchio, A.; et al. Circulating extracellular vesicles as new inflammation marker in HIV infection. AIDS 2021, 35, 595–604. [Google Scholar] [CrossRef]
- Brocco, D.; Lanuti, P.; Pieragostino, D.; Cufaro, M.C.; Simeone, P.; Bologna, G.; Di Marino, P.; De Tursi, M.; Grassadonia, A.; Irtelli, L.; et al. Phenotypic and Proteomic Analysis Identifies Hallmarks of Blood Circulating Extracellular Vesicles in NSCLC Responders to Immune Checkpoint Inhibitors. Cancers 2021, 13, 585. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term ID 1 | Term Description 2 | Observed Gene Count 3 | Background Gene Count 4 | False Discovery Rate 5 |
|---|---|---|---|---|
| GO:0016071 | mRNA metabolic process | 83 | 667 | 2.5 × 10−21 |
| GO:0000398 | mRNA splicing, via spliceosome | 45 | 284 | 6.16 × 10−15 |
| GO:0006397 | mRNA processing | 54 | 456 | 2.70 × 10−13 |
| GO:1903311 | regulation of mRNA metabolic process | 36 | 238 | 1.89 × 10−11 |
| GO:0043488 | regulation of mRNA stability | 21 | 113 | 4.54 × 10−8 |
| GO:0050684 | regulation of mRNA processing | 18 | 114 | 4.33 × 10−6 |
| GO:0048024 | regulation of mRNA splicing, via spliceosome | 15 | 77 | 4.38 × 10−6 |
| GO:0006413 | translational initiation | 30 | 142 | 1.17 × 10−12 |
| GO:0006412 | translation | 44 | 362 | 3.99 × 10−11 |
| GO:0006417 | regulation of translation | 38 | 327 | 3.73 × 10−9 |
| GO:0002181 | cytoplasmic translation | 15 | 57 | 1.90 × 10−7 |
| GO:0006446 | regulation of translational initiation | 8 | 71 | 0.0246 |
| GO:0006614 | SRP-dependent cotranslational protein targeting to membrane | 23 | 92 | 6.11 × 10−11 |
| GO:0016192 | vesicle-mediated transport | 139 | 1699 | 6.02 × 10−21 |
| GO:0060627 | regulation of vesicle-mediated transport | 30 | 480 | 0.0077 |
| GO:0006892 | post-Golgi vesicle-mediated transport | 9 | 83 | 0.0197 |
| GO:0055114 | oxidation-reduction process | 94 | 932 | 4.04 × 10−9 |
| GO:0015980 | energy derivation by oxidation of organic compounds | 17 | 217 | 0.0112 |
| GO:0044281 | small molecule metabolic process | 134 | 1779 | 1.21 × 10−17 |
| GO:0062012 | regulation of small molecule metabolic process | 21 | 332 | 0.0272 |
| Term ID 1 | Term Description 2 | Observed Gene Count 3 | Background Gene Count 4 | False Discovery Rate 5 |
|---|---|---|---|---|
| GO:0003723 | RNA binding | 86 | 850 | 2.57 × 10−17 |
| GO:0003729 | mRNA binding | 36 | 198 | 2.76 × 10−13 |
| GO:0003727 | single-stranded RNA binding | 13 | 80 | 0.00011 |
| GO:0003725 | double-stranded RNA binding | 11 | 70 | 0.00058 |
| GO:0019843 | rRNA binding | 10 | 60 | 0.00079 |
| GO:0008135 | translation factor activity, RNA binding | 11 | 84 | 0.0021 |
| GO:0000049 | tRNA binding | 8 | 56 | 0.0079 |
| GO:0061980 | regulatory RNA binding | 5 | 34 | 0.0424 |
| GO:0016491 | oxidoreductase activity | 75 | 716 | 8.70 × 10−16 |
| GO:0016860 | intramolecular oxidoreductase activity | 12 | 47 | 5.01 × 10−6 |
| GO:0000166 | nucleotide binding | 134 | 2097 | 2.45 × 10−12 |
| GO:0017076 | purine nucleotide binding | 108 | 1865 | 1.36 × 10−7 |
| GO:0030554 | adenyl nucleotide binding | 79 | 1524 | 0.00039 |
| GO:0008092 | cytoskeletal protein binding | 71 | 882 | 4.82 × 10−10 |
| GO:0046961 | proton-transporting ATPase activity, rotational mechanism | 9 | 21 | 4.97 × 10−6 |
| GO:0008553 | proton-exporting ATPase activity, phosphorylative mechanism | 9 | 23 | 8.55 × 10−6 |
| GO:0015078 | proton transmembrane transporter activity | 12 | 120 | 0.0082 |
| GO:0005178 | integrin binding | 19 | 122 | 2.30 × 10−6 |
| GO:0050839 | cell adhesion molecule binding | 24 | 200 | 3.68 × 10−6 |
| GO:0098634 | cell-matrix adhesion mediator activity | 3 | 7 | 0.0229 |
| Term ID 1 | Term Description 2 | Observed Gene Count 3 | Background Gene Count 4 | False Discovery Rate 5 |
|---|---|---|---|---|
| GO:0043226 | organelle | 566 | 12432 | 1.37 × 10−50 |
| GO:0043229 | intracellular organelle | 560 | 12193 | 3.30 × 10−50 |
| GO:0043227 | membrane-bounded organelle | 516 | 11244 | 1.07 × 10−37 |
| GO:0005783 | endoplasmic reticulum | 120 | 1796 | 9.40 × 10−13 |
| GO:0005788 | endoplasmic reticulum lumen | 30 | 299 | 1.86 × 10−6 |
| GO:0005793 | endoplasmic reticulum-Golgi intermediate compartment | 13 | 113 | 0.00095 |
| GO:0031982 | vesicle | 173 | 2318 | 4.41 × 10−24 |
| GO:0031410 | cytoplasmic vesicle | 166 | 2226 | 6.49 × 10−23 |
| GO:0099503 | secretory vesicle | 96 | 948 | 1.62 × 10−20 |
| GO:0030139 | endocytic vesicle | 35 | 275 | 8.75 × 10−10 |
| GO:0045335 | phagocytic vesicle | 23 | 122 | 2.25 × 10−9 |
| GO:0042470 | melanosome | 35 | 105 | 1.55 × 10−20 |
| GO:1990904 | ribonucleoprotein complex | 89 | 770 | 2.33 × 10−22 |
| GO:0030532 | small nuclear ribonucleoprotein complex | 11 | 67 | 0.00021 |
| GO:0005773 | vacuole | 74 | 682 | 4.05 × 10−17 |
| GO:0005775 | vacuolar lumen | 30 | 172 | 2.47 × 10−11 |
| GO:0005774 | vacuolar membrane | 31 | 311 | 1.36 × 10−6 |
| GO:0005764 | lysosome | 67 | 582 | 1.24 × 10−16 |
| GO:0015629 | actin cytoskeleton | 42 | 432 | 1.75 × 10−8 |
| GO:0032432 | actin filament bundle | 12 | 57 | 1.38 × 10−5 |
| Term ID 1 | Term Description 2 | Observed Gene Count 3 | Background Gene Count 4 | False Discovery Rate 5 |
|---|---|---|---|---|
| hsa03040 | spliceosome | 26 | 130 | 3.14 × 10−10 |
| hsa03010 | ribosome | 21 | 130 | 3.48 × 10−7 |
| hsa03013 | RNA transport | 14 | 159 | 0.0074 |
| hsa04145 | phagosome | 26 | 145 | 1.35 × 10−9 |
| hsa04142 | lysosome | 24 | 123 | 1.39 × 10−9 |
| hsa01100 | metabolic pathways | 99 | 1250 | 1.52 × 10−13 |
| hsa00020 | citrate cycle (TCA cycle) | 9 | 30 | 7.00 × 10−5 |
| hsa01212 | fatty acid metabolism | 10 | 48 | 0.00024 |
| hsa01230 | biosynthesis of amino acids | 11 | 72 | 0.00075 |
| hsa00010 | glycolysis/Gluconeogenesis | 10 | 68 | 0.0017 |
| hsa00030 | pentose phosphate pathway | 5 | 30 | 0.0259 |
| Ingenuity Canonical Pathway 1 | −log (p-Value) 2 | z-Score 3 |
|---|---|---|
| phagosome maturation | 14.6 | |
| EIF2 signaling | 14 | −2.357 |
| caveolar-mediated endocytosis signaling | 7.97 | |
| fatty acid β-oxidation I | 7.88 | 2.53 |
| TCA cycle II (Eukaryotic) | 6.89 | 2.121 |
| isoleucine degradation I | 6.83 | 2.236 |
| mitochondrial dysfunction | 6.78 | |
| valine degradation I | 4.99 | 2.236 |
| tryptophan degradation III (Eukaryotic) | 4.49 | 1.342 |
| spliceosomal cycle | 3.51 | −2.646 |
| tRNA charging | 2.43 | −1.342 |
| Disease or Functions Annotation 1 | p-Value | z-Score 2 | Predicted Activation State 3 |
|---|---|---|---|
| fatty acid metabolism | 4.57 × 10−12 | 2.017 | Increased |
| cellular homeostasis | 5.29 × 10−11 | 2.129 | Increased |
| cell death of tumor cell lines | 8.41 × 10−24 | 2.15 | Increased |
| endocytosis by eukaryotic cells | 5.24 × 10−11 | 2.311 | Increased |
| metabolism of glycosphingolipid | 0.00000134 | 2.387 | Increased |
| cell death of tumor cells | 1.13 × 10−13 | 2.416 | Increased |
| sensitivity of cells | 0.000000386 | 2.56 | Increased |
| phagocytosis | 3.93 × 10−8 | 2.708 | Increased |
| phagocytosis of cells | 0.000000438 | 2.714 | Increased |
| degradation of extracellular matrix | 7.31 × 10−8 | 2.905 | Increased |
| cell death of cancer cells | 1.17 × 10−10 | 2.995 | Increased |
| cell death of osteosarcoma cells | 1.76 × 10−12 | 3.545 | Increased |
| homologous recombination | 9.48 × 10−9 | −3.637 | Decreased |
| accumulation of lipid | 2.49 × 10−9 | −3.636 | Decreased |
| homologous recombination of cells | 5.85 × 10−9 | −3.499 | Decreased |
| glucose metabolism disorder | 0.00000102 | −3.279 | Decreased |
| DNA recombination | 6.51 × 10−9 | −3.235 | Decreased |
| motor dysfunction or movement disorder | 1.01 × 10−18 | −2.934 | Decreased |
| movement Disorders | 9.11 × 10−19 | −2.666 | Decreased |
| lysosomal storage disease | 2.68 × 10−10 | −2.591 | Decreased |
| repair of DNA | 0.000000166 | −2.545 | Decreased |
| cell death of breast cancer cell lines | 9.12 × 10−8 | −2.374 | Decreased |
| astrocytosis | 0.00000147 | −2.219 | Decreased |
| degeneration of central nervous system | 8.66 × 10−9 | −2.218 | Decreased |
| neurodegeneration | 0.000000465 | −2.185 | Decreased |
| degeneration of nervous system | 0.000000827 | −2.128 | Decreased |
| amyloidosis | 7.3 × 10−13 | −2.08 | Decreased |
| degeneration of brain | 1.46 × 10−8 | −2.032 | Decreased |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potenza, F.; Cufaro, M.C.; Di Biase, L.; Panella, V.; Di Campli, A.; Ruggieri, A.G.; Dufrusine, B.; Restelli, E.; Pietrangelo, L.; Protasi, F.; et al. Proteomic Analysis of Marinesco–Sjogren Syndrome Fibroblasts Indicates Pro-Survival Metabolic Adaptation to SIL1 Loss. Int. J. Mol. Sci. 2021, 22, 12449. https://doi.org/10.3390/ijms222212449
Potenza F, Cufaro MC, Di Biase L, Panella V, Di Campli A, Ruggieri AG, Dufrusine B, Restelli E, Pietrangelo L, Protasi F, et al. Proteomic Analysis of Marinesco–Sjogren Syndrome Fibroblasts Indicates Pro-Survival Metabolic Adaptation to SIL1 Loss. International Journal of Molecular Sciences. 2021; 22(22):12449. https://doi.org/10.3390/ijms222212449
Chicago/Turabian StylePotenza, Francesca, Maria Concetta Cufaro, Linda Di Biase, Valeria Panella, Antonella Di Campli, Anna Giulia Ruggieri, Beatrice Dufrusine, Elena Restelli, Laura Pietrangelo, Feliciano Protasi, and et al. 2021. "Proteomic Analysis of Marinesco–Sjogren Syndrome Fibroblasts Indicates Pro-Survival Metabolic Adaptation to SIL1 Loss" International Journal of Molecular Sciences 22, no. 22: 12449. https://doi.org/10.3390/ijms222212449
APA StylePotenza, F., Cufaro, M. C., Di Biase, L., Panella, V., Di Campli, A., Ruggieri, A. G., Dufrusine, B., Restelli, E., Pietrangelo, L., Protasi, F., Pieragostino, D., De Laurenzi, V., Federici, L., Chiesa, R., & Sallese, M. (2021). Proteomic Analysis of Marinesco–Sjogren Syndrome Fibroblasts Indicates Pro-Survival Metabolic Adaptation to SIL1 Loss. International Journal of Molecular Sciences, 22(22), 12449. https://doi.org/10.3390/ijms222212449