
Aryl Hydrocarbon Receptor (AhR) Activation by 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) Dose-Dependently Shifts the Gut Microbiome Consistent with the Progression of Non-Alcoholic Fatty Liver Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
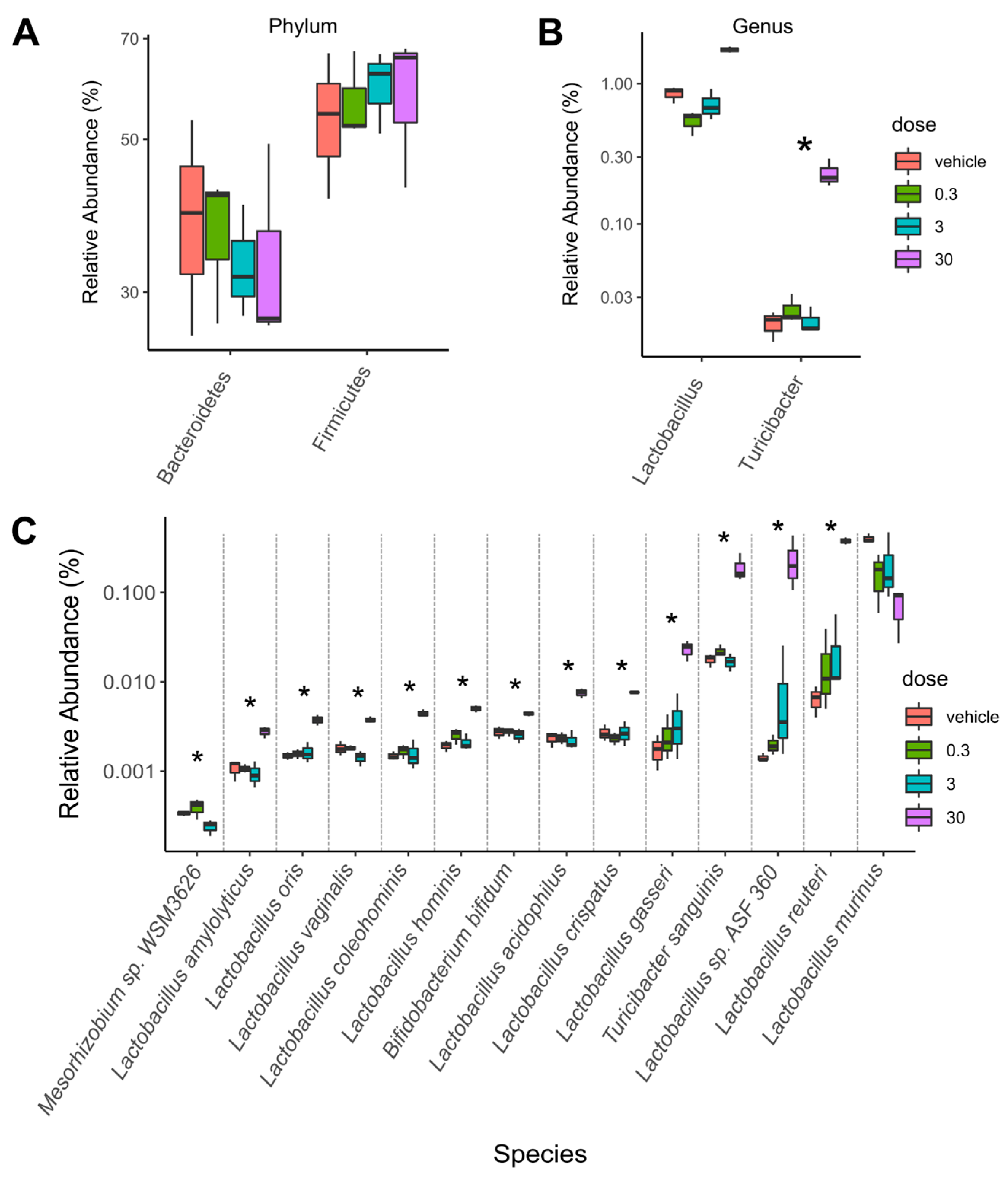
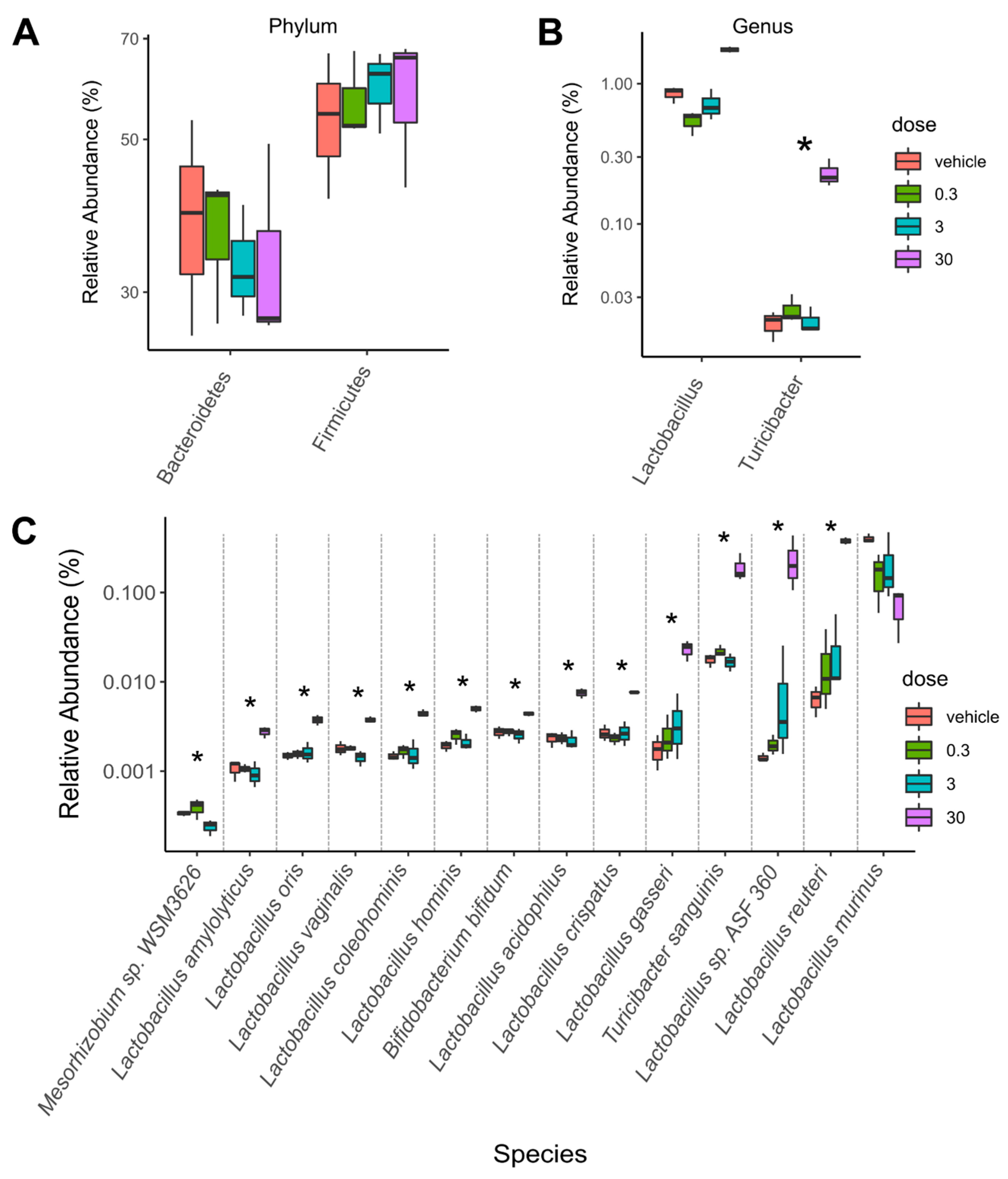
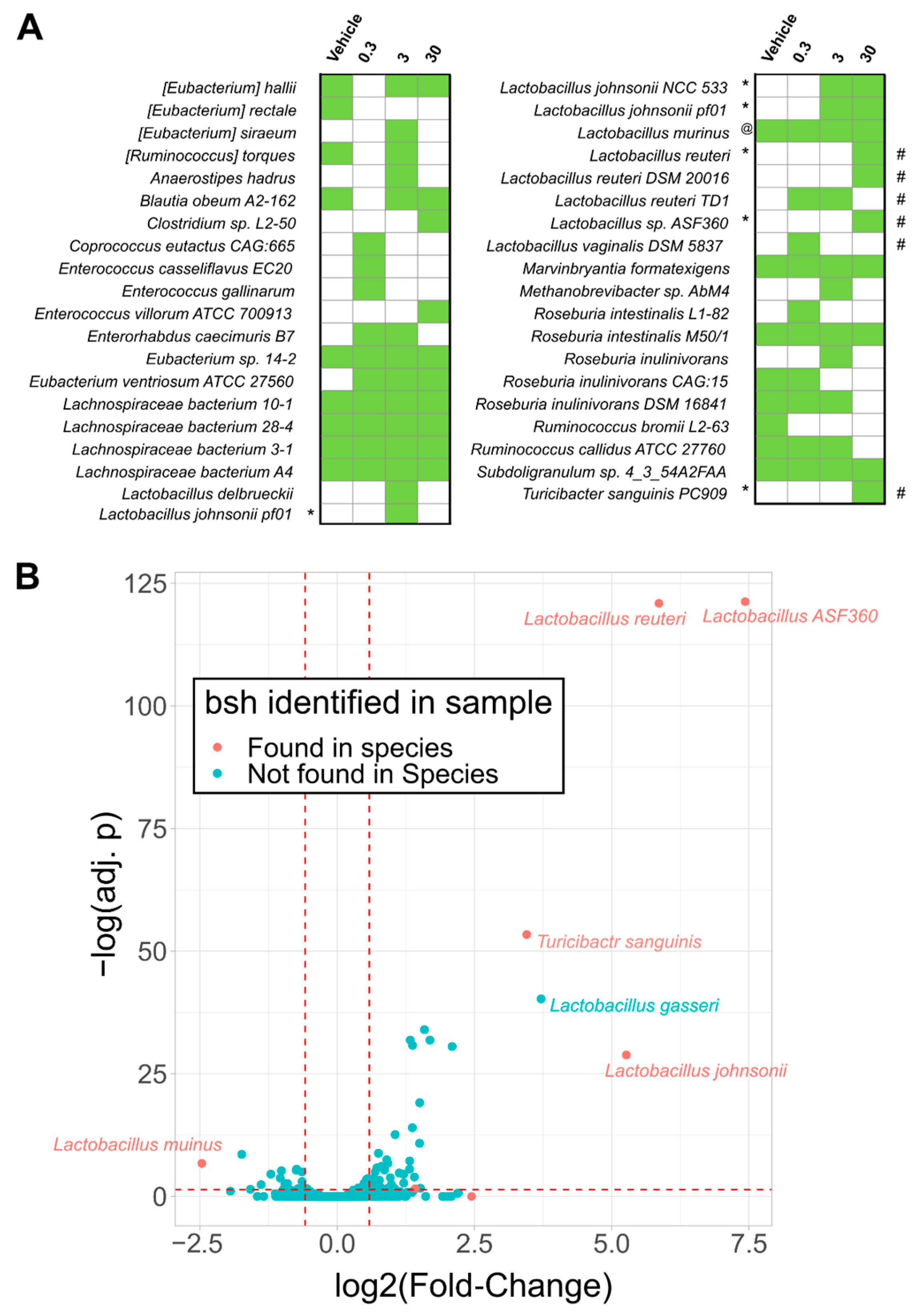
2.1. TCDD-Elicited Toxicity Enriched for Lactobacillus Species
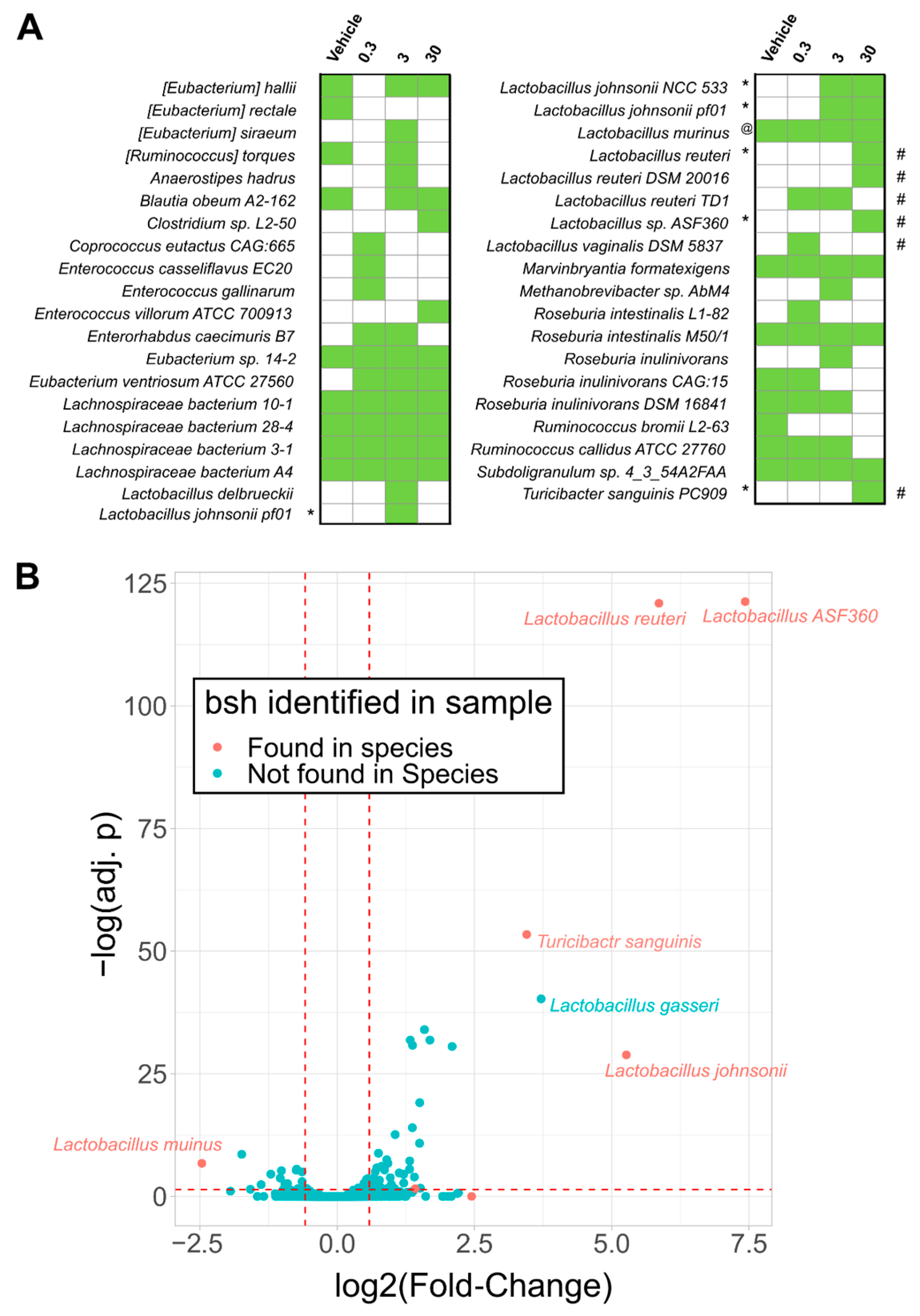
2.2. Bile Salt Hydrolase (Bsh) Levels Correlated with Significantly Enriched Species
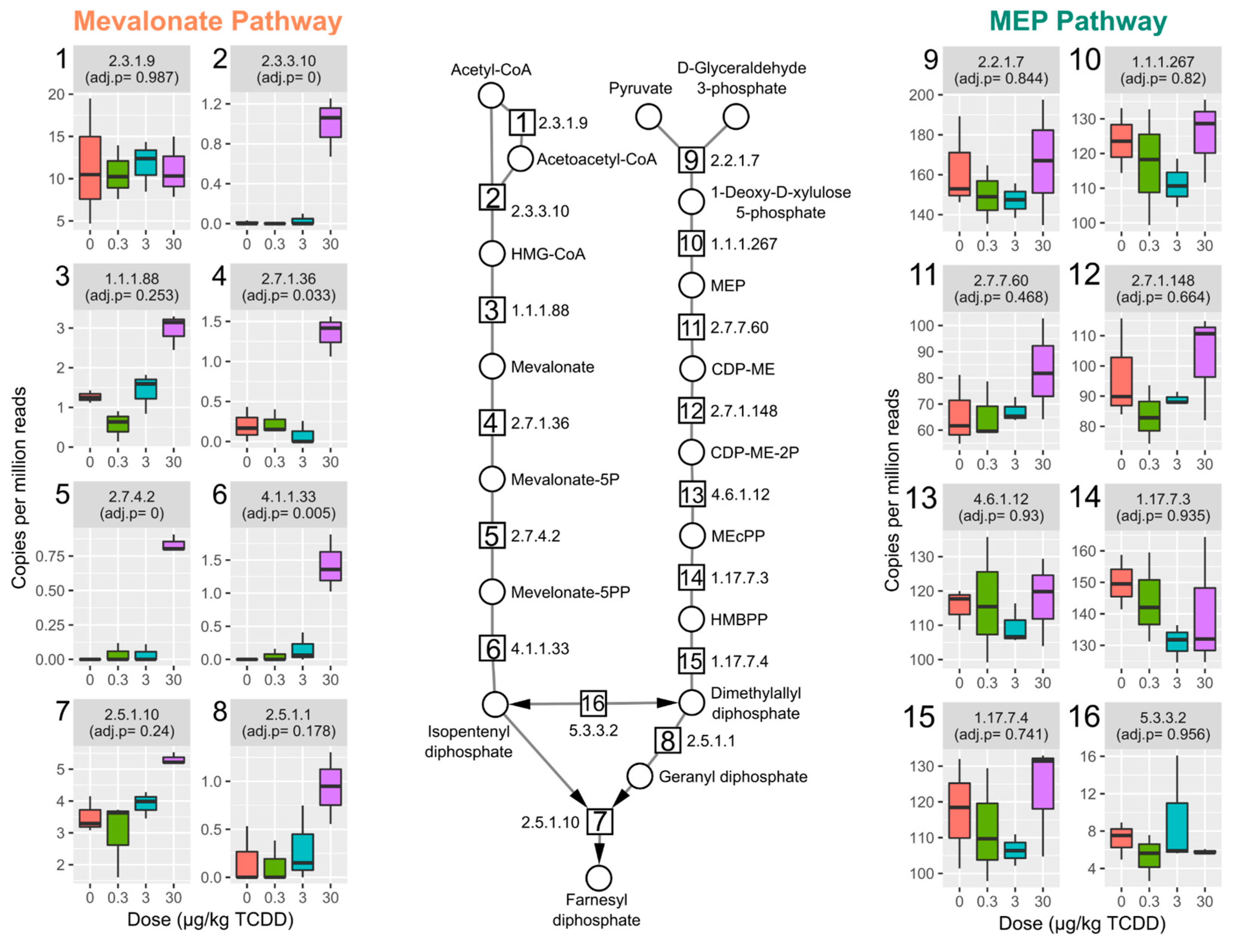
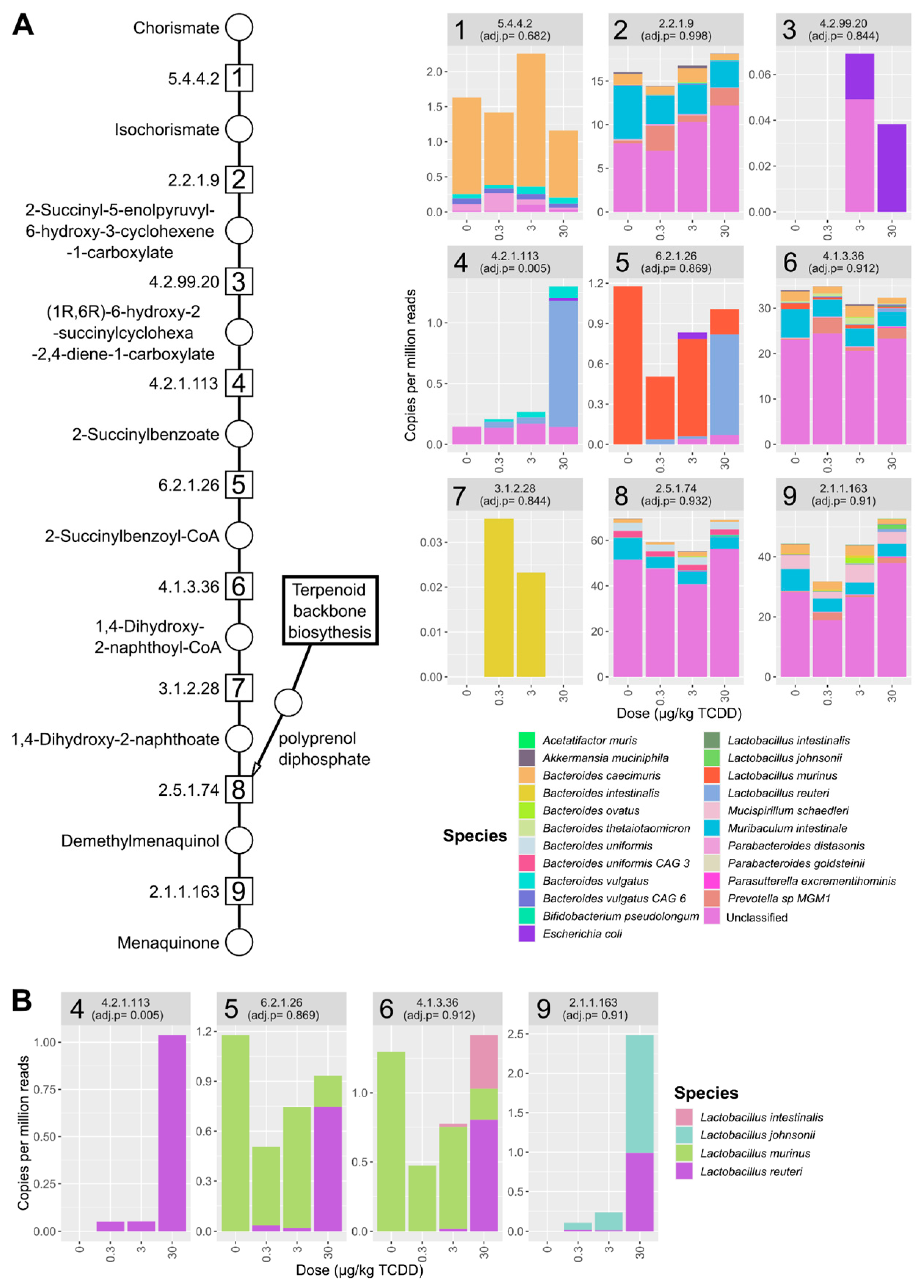
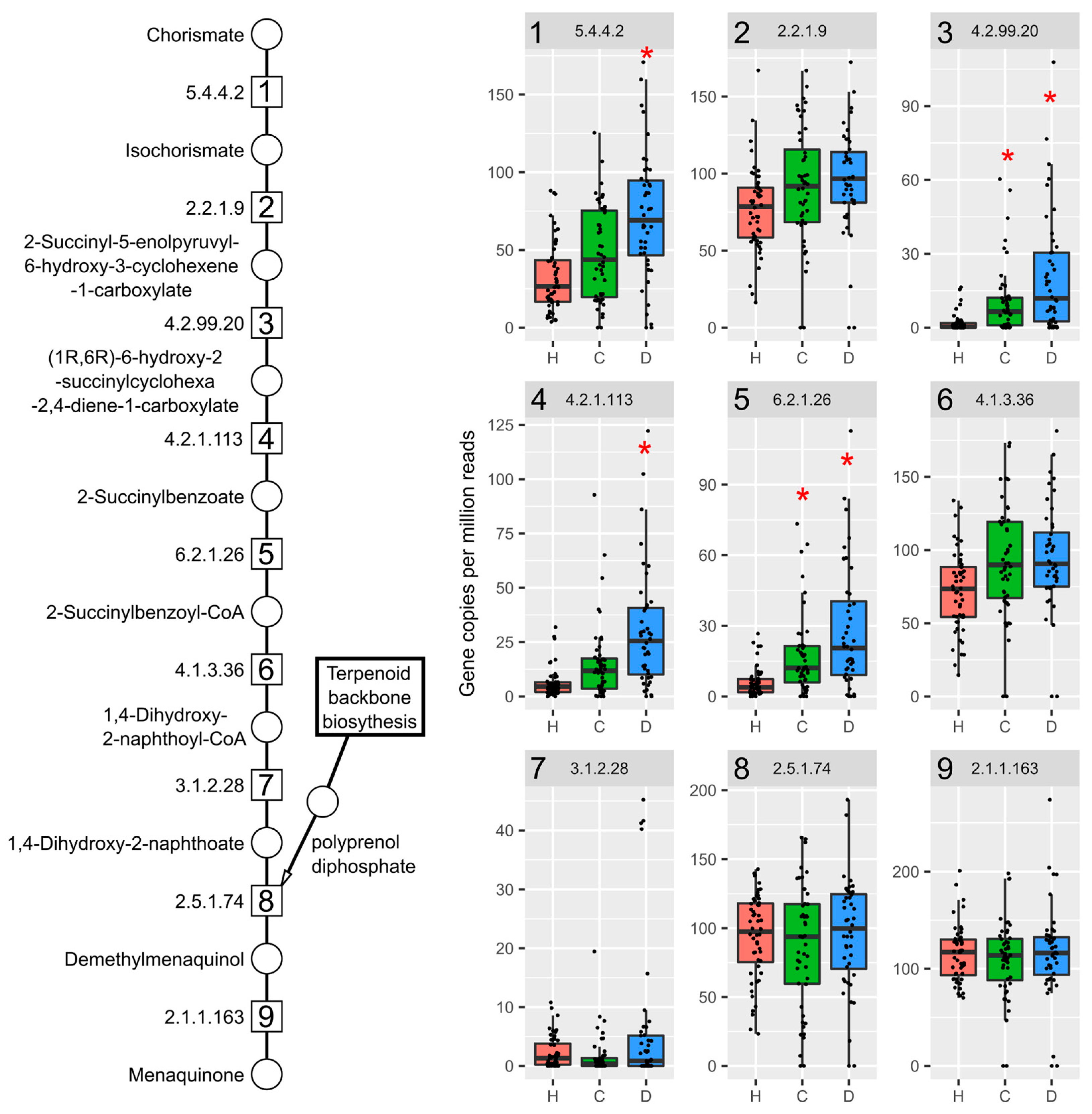
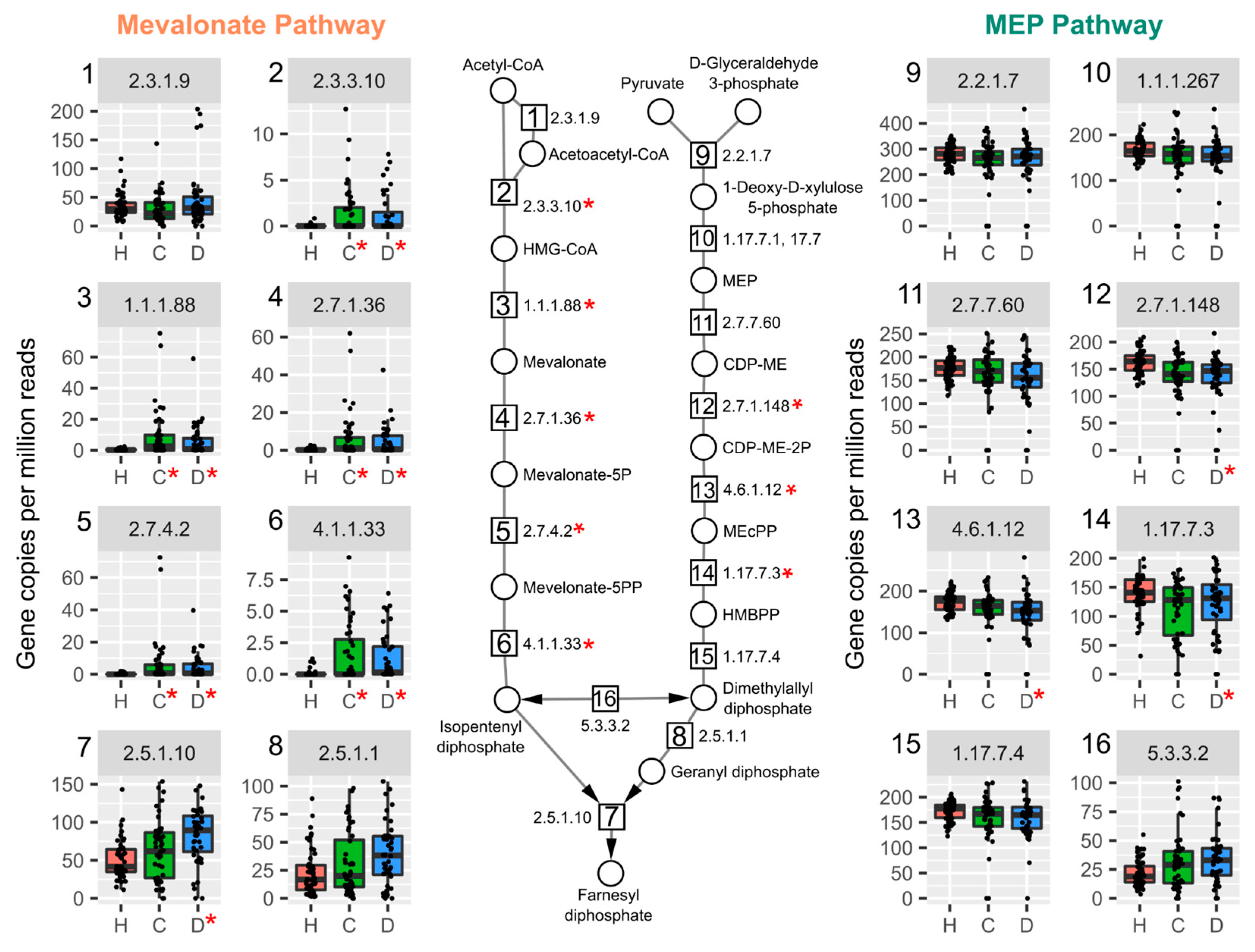
2.3. TCDD Enriched for Mevalonate-Dependent Isoprenoid Biosynthesis
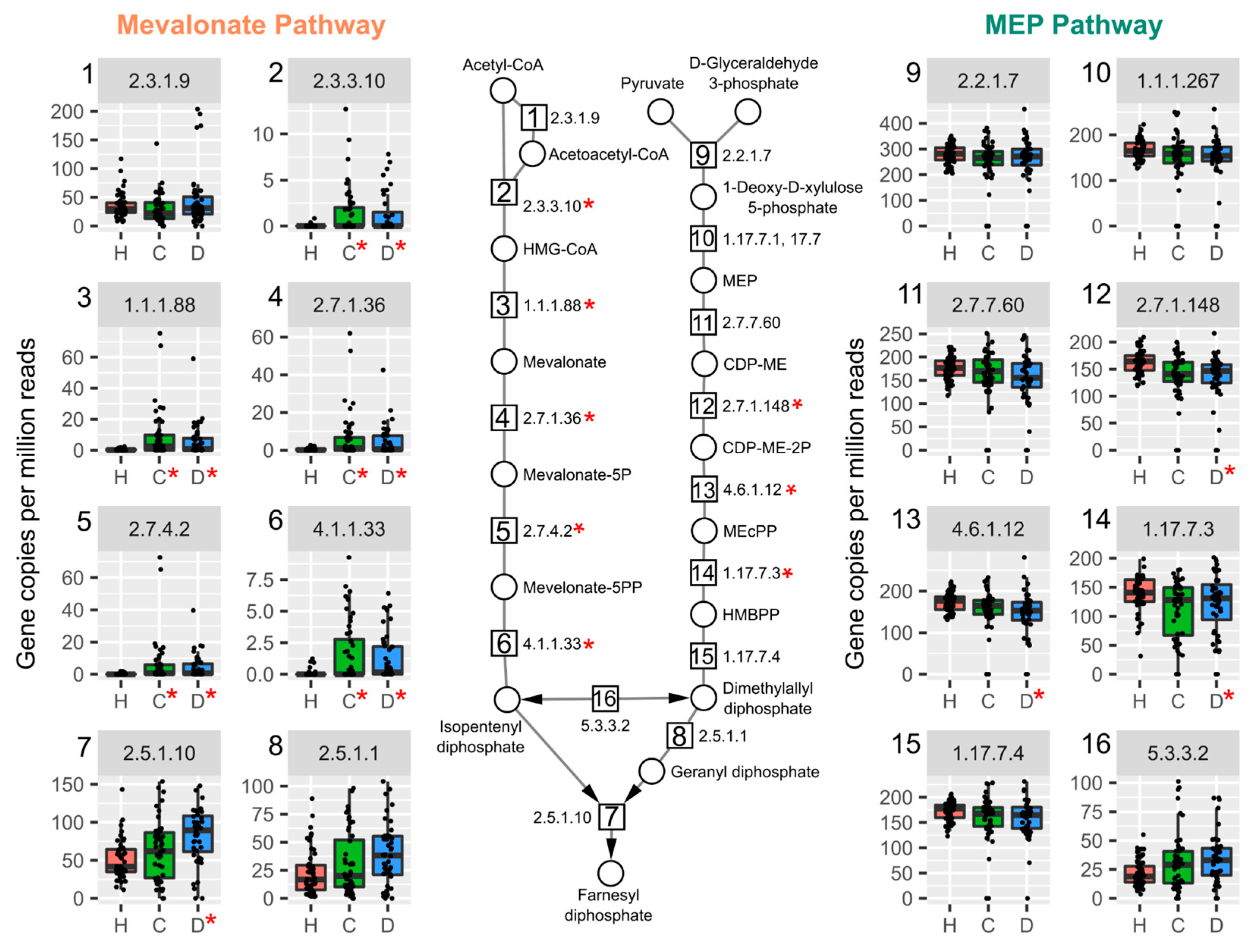
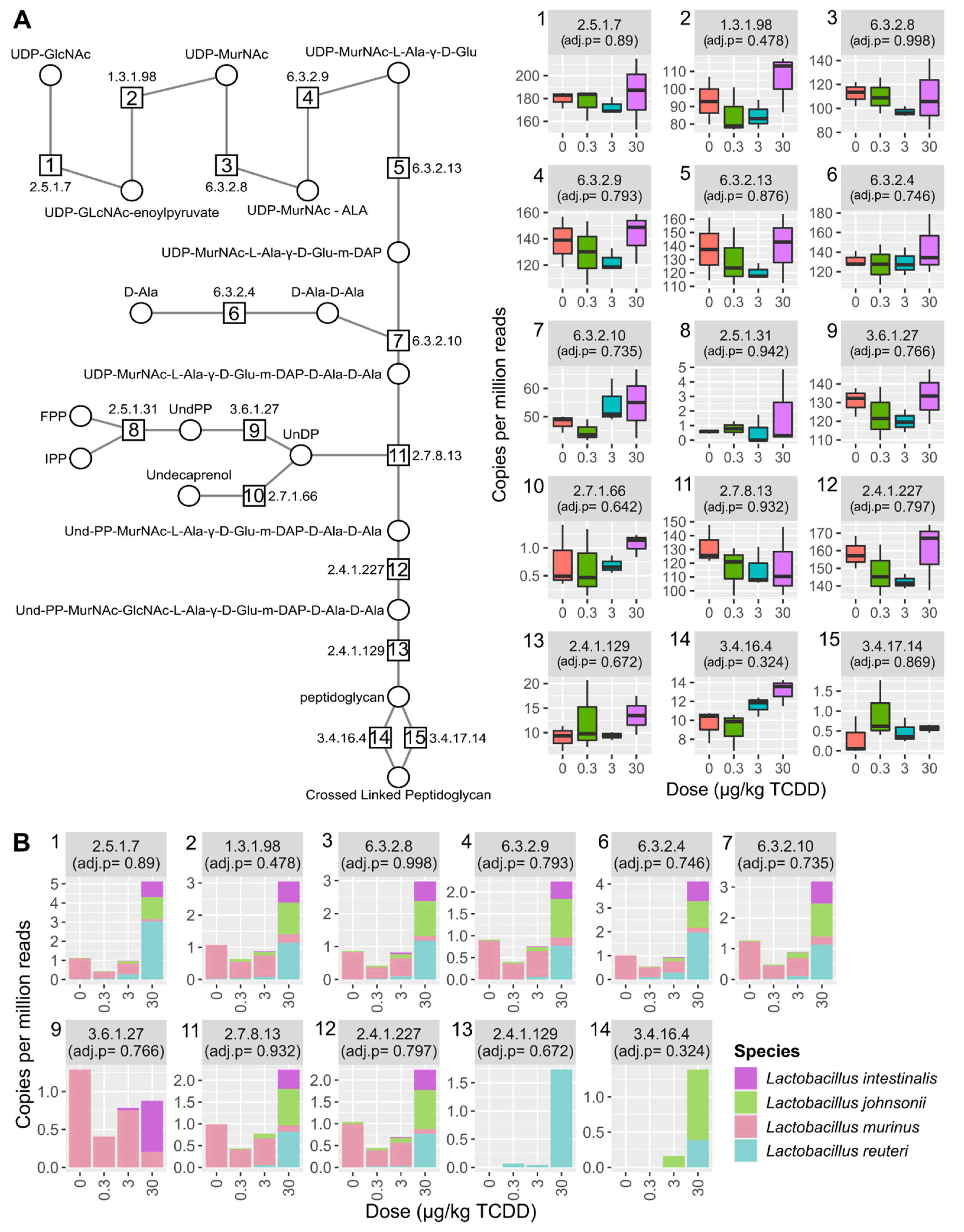
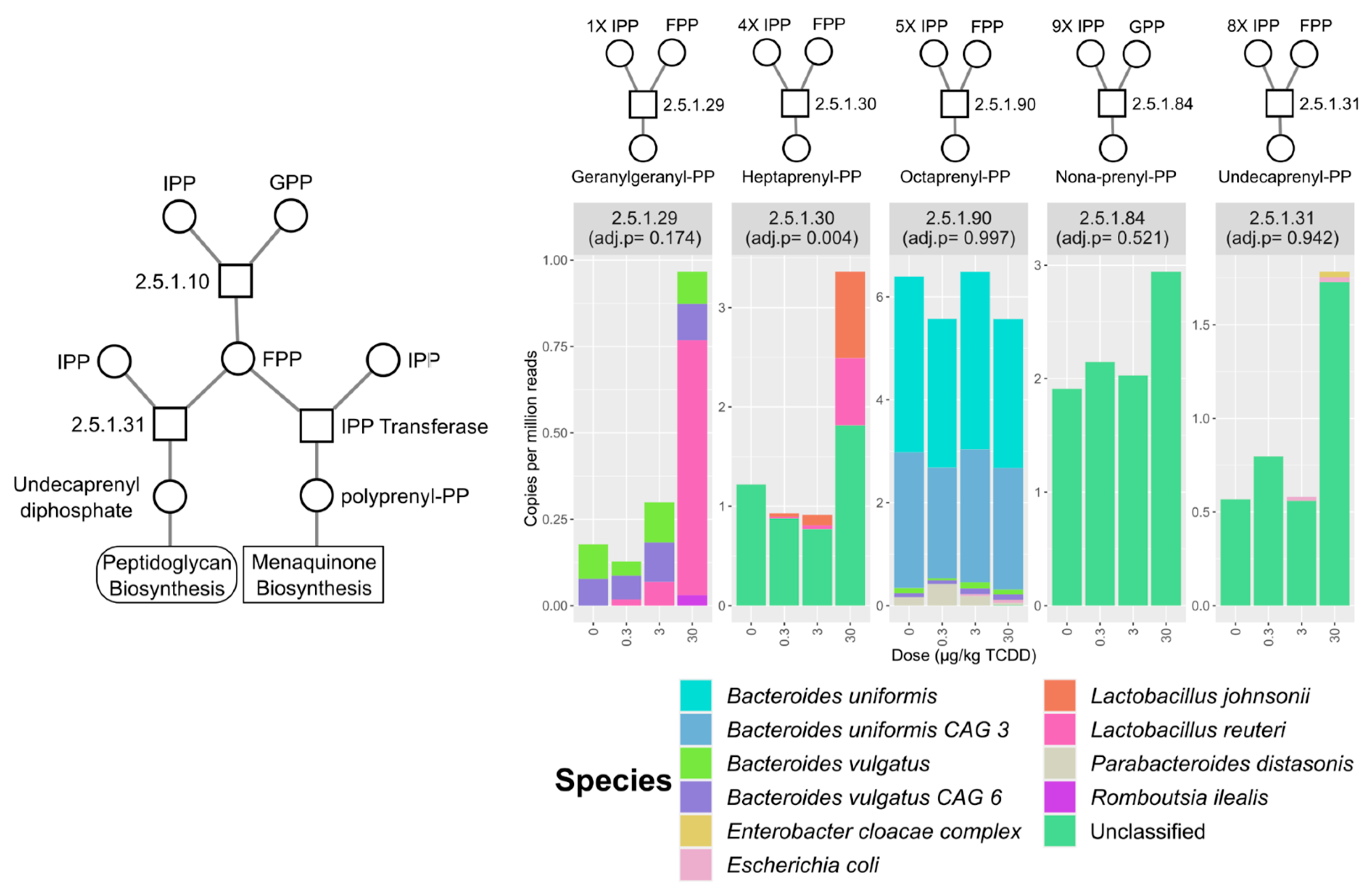
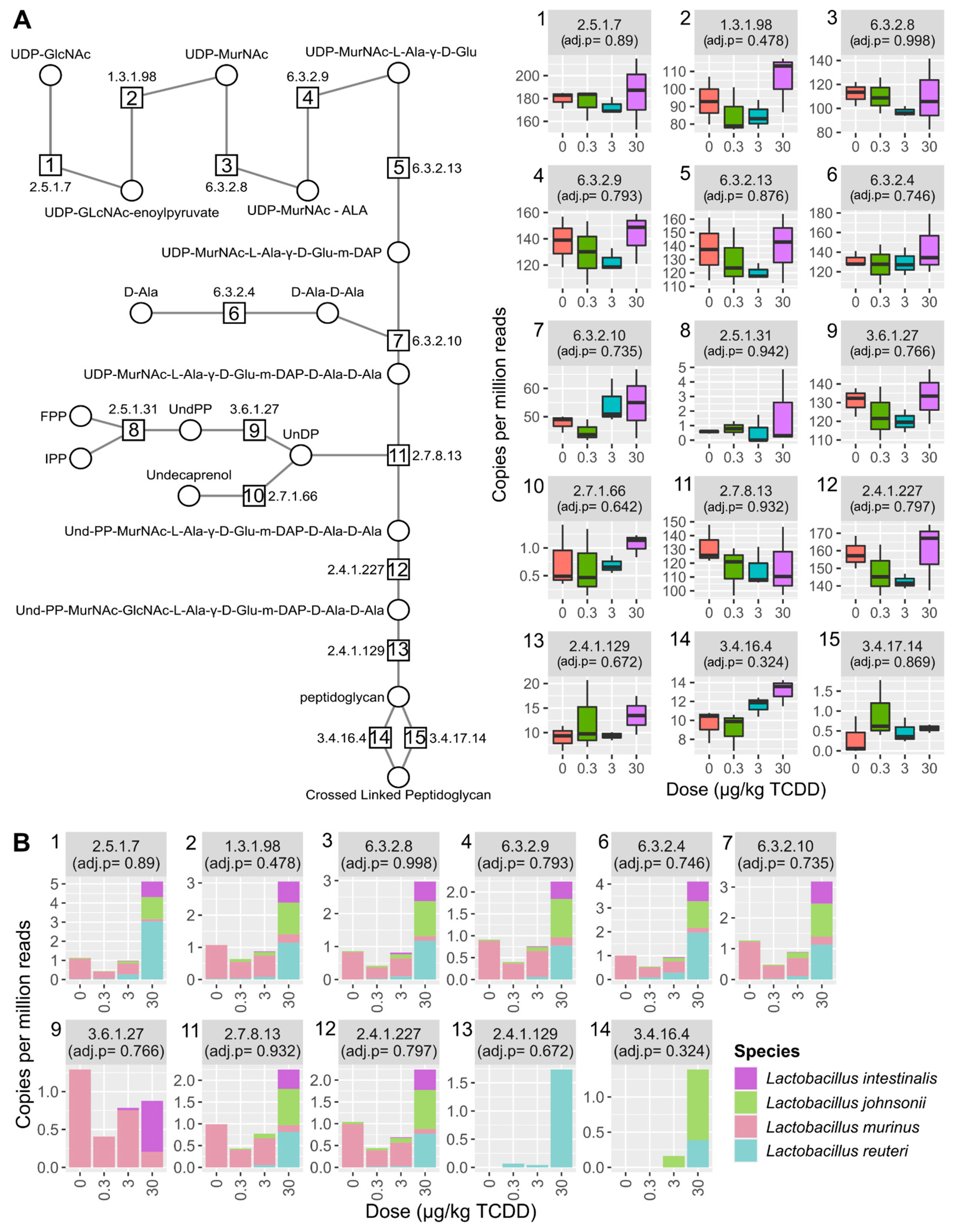
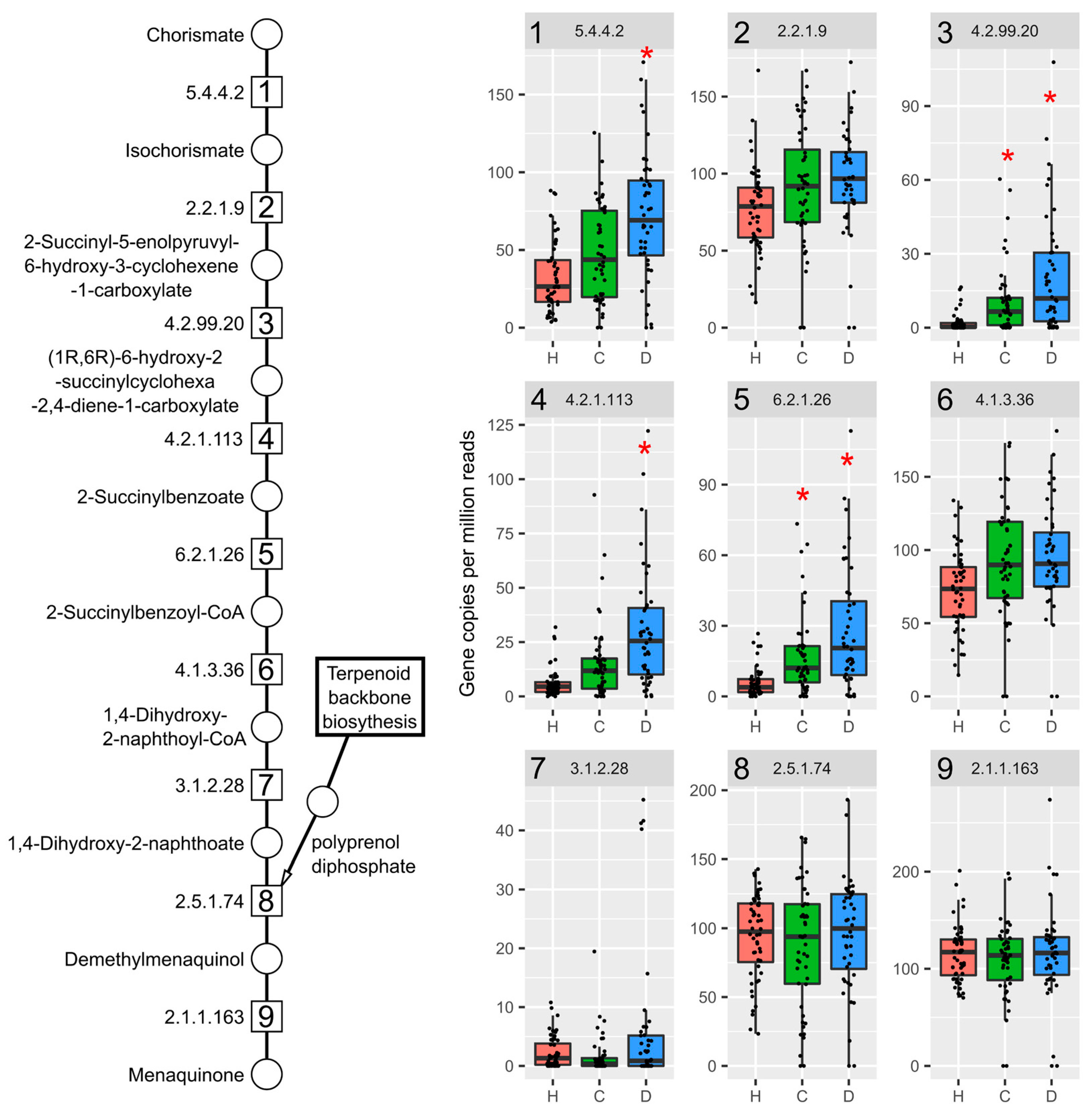
2.4. Vitamin K2 (Menaquinone) and Peptidoglycan Biosynthesis Pathways in Mouse NAFLD-Phenotypes and Gut Microbiomes of Cirrhosis Patients
3. Discussion
4. Materials and Methods
4.1. Animal Treatment
4.2. Metagenomic Sequencing
4.3. Metagenomic Taxonomic Analysis
4.4. Metagenomic Functional Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to Diabetes Mellitus, Cardiovascular Disease or Cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of Inflammation in Nonalcoholic Fatty Liver Disease: The Multiple Parallel Hits Hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Cave, M.; Appana, S.; Patel, M.; Falkner, K.C.; McClain, C.J.; Brock, G. Polychlorinated Biphenyls, Lead, and Mercury Are Associated with Liver Disease in American Adults: NHANES 2003–2004. Environ. Health Perspect. 2010, 118, 1735–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, S.; Yang, Y.; Wen, C.; Liu, W.; Cao, L.; Feng, X.; Chen, J.; Wang, H.; Tang, Y.; Tian, L.; et al. Effects of Environmental Contaminants in Water Resources on Nonalcoholic Fatty Liver Disease. Environ. Int. 2021, 154, 106555. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Lind, L.; Salihovic, S.; van Bavel, B.; Ingelsson, E.; Lind, P.M. Persistent Organic Pollutants and Liver Dysfunction Biomarkers in a Population-Based Human Sample of Men and Women. Environ. Res. 2014, 134, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Rantakokko, P.; Männistö, V.; Airaksinen, R.; Koponen, J.; Viluksela, M.; Kiviranta, H.; Pihlajamäki, J. Persistent Organic Pollutants and Non-Alcoholic Fatty Liver Disease in Morbidly Obese Patients: A Cohort Study. Environ. Health 2015, 14, 79. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Nichols, R.G.; Correll, J.; Murray, I.A.; Tanaka, N.; Smith, P.B.; Hubbard, T.D.; Sebastian, A.; Albert, I.; Hatzakis, E.; et al. Persistent Organic Pollutants Modify Gut Microbiota–Host Metabolic Homeostasis in Mice through Aryl Hydrocarbon Receptor Activation. Environ. Health Perspect. 2015, 123, 679–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fader, K.A.; Nault, R.; Zhang, C.; Kumagai, K.; Harkema, J.R.; Zacharewski, T.R. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD)-Elicited Effects on Bile Acid Homeostasis: Alterations in Biosynthesis, Enterohepatic Circulation, and Microbial Metabolism. Sci. Rep. 2017, 7, 5921. [Google Scholar] [CrossRef]
- Petriello, M.C.; Hoffman, J.B.; Vsevolozhskaya, O.; Morris, A.J.; Hennig, B. Dioxin-like PCB 126 Increases Intestinal Inflammation and Disrupts Gut Microbiota and Metabolic Homeostasis. Environ. Pollut. 2018, 242, 1022–1032. [Google Scholar] [CrossRef]
- Stedtfeld, R.D.; Chai, B.; Crawford, R.B.; Stedtfeld, T.M.; Williams, M.R.; Xiangwen, S.; Kuwahara, T.; Cole, J.R.; Kaminski, N.E.; Tiedje, J.M.; et al. Modulatory Influence of Segmented Filamentous Bacteria on Transcriptomic Response of Gnotobiotic Mice Exposed to TCDD. Front. Microbiol. 2017, 8, 1708. [Google Scholar] [CrossRef] [Green Version]
- Nault, R.; Fader, K.A.; Lydic, T.A.; Zacharewski, T.R. Lipidomic Evaluation of Aryl Hydrocarbon Receptor-Mediated Hepatic Steatosis in Male and Female Mice Elicited by 2,3,7,8-Tetrachlorodibenzo-p-dioxin. Chem. Res. Toxicol. 2017, 30, 1060–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nault, R.; Fader, K.A.; Ammendolia, D.A.; Dornbos, P.; Potter, D.; Sharratt, B.; Kumagai, K.; Harkema, J.R.; Lunt, S.Y.; Matthews, J.; et al. Dose-Dependent Metabolic Reprogramming and Differential Gene Expression in TCDD-Elicited Hepatic Fibrosis. Toxicol. Sci. 2016, 154, 253–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fader, K.A.; Nault, R.; Ammendolia, D.A.; Harkema, J.R.; Williams, K.J.; Crawford, R.B.; Kaminski, N.E.; Potter, D.; Sharratt, B.; Zacharewski, T.R. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Alters Lipid Metabolism and Depletes Immune Cell Populations in the Jejunum of C57BL/6 Mice. Toxicol. Sci. 2015, 148, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angrish, M.M.; Dominici, C.Y.; Zacharewski, T.R. TCDD-Elicited Effects on Liver, Serum, and Adipose Lipid Composition in C57BL/6 Mice. Toxicol. Sci. 2013, 131, 108–115. [Google Scholar] [CrossRef] [Green Version]
- Katsiki, N.; Mikhailidis, D.P.; Mantzoros, C.S. Non-Alcoholic Fatty Liver Disease and Dyslipidemia: An Update. Metabolism 2016, 65, 1109–1123. [Google Scholar] [CrossRef]
- Cholico, G.N.; Fling, R.R.; Zacharewski, N.A.; Fader, K.A.; Nault, R.; Zacharewski, T. Thioesterase Induction by 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Results in a Futile Cycle That Inhibits Hepatic β-Oxidation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Fernandez-Salguero, P.M.; Hllbert, D.M.; Rudikoff, S.; Ward, J.M.; Gonzalez, F.J. Aryl-Hydrocarbon Receptor-Deficient Mice Are Resistant to 2,3,7,8-Tetrachlorodibenzo-p-Dioxin-Induced Toxicity. Toxicol. Appl. Pharmacol. 1996, 140, 173–179. [Google Scholar] [CrossRef]
- Bock, K.W. Aryl Hydrocarbon Receptor (AHR)-Mediated Inflammation and Resolution: Non-Genomic and Genomic Signaling. Biochem. Pharmacol. 2020, 182, 114220. [Google Scholar] [CrossRef]
- Schiering, C.; Wincent, E.; Metidji, A.; Iseppon, A.; Li, Y.; Potocnik, A.J.; Omenetti, S.; Henderson, C.J.; Wolf, C.R.; Nebert, D.W.; et al. Feedback Control of AHR Signalling Regulates Intestinal Immunity. Nature 2017, 542, 242–245. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, F.; Grander, C.; Effenberger, M.; Adolph, T.E.; Tilg, H. Gut Dysfunction and Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2019, 10, 611. [Google Scholar] [CrossRef] [Green Version]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The Role of the Gut Microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef]
- Tian, J.; Feng, Y.; Fu, H.; Xie, H.Q.; Jiang, J.X.; Zhao, B. The Aryl Hydrocarbon Receptor: A Key Bridging Molecule of External and Internal Chemical Signals. Environ. Sci. Technol. 2015, 49, 9518–9531. [Google Scholar] [CrossRef] [Green Version]
- Rothhammer, V.; Quintana, F.J. The Aryl Hydrocarbon Receptor: An Environmental Sensor Integrating Immune Responses in Health and Disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Qiu, J.; Heller, J.J.; Guo, X.; Chen, Z.M.E.; Fish, K.; Fu, Y.X.; Zhou, L. The Aryl Hydrocarbon Receptor Regulates Gut Immunity through Modulation of Innate Lymphoid Cells. Immunity 2012, 36, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; DeLuca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan Catabolites from Microbiota Engage Aryl Hydrocarbon Receptor and Balance Mucosal Reactivity via Interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, I.A.; Nichols, R.G.; Zhang, L.; Patterson, A.D.; Perdew, G.H. Expression of the Aryl Hydrocarbon Receptor Contributes to the Establishment of Intestinal Microbial Community Structure in Mice. Sci. Rep. 2016, 6, 33969. [Google Scholar] [CrossRef] [Green Version]
- Lefever, D.E.; Xu, J.; Chen, Y.; Huang, G.; Tamas, N.; Guo, T.L. TCDD Modulation of Gut Microbiome Correlated with Liver and Immune Toxicity in Streptozotocin (STZ)-Induced Hyperglycemic Mice. Toxicol. Appl. Pharmacol. 2016, 304, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neamah, W.H.; Busbee, P.B.; Alghetaa, H.; Abdulla, O.A.; Nagarkatti, M.; Nagarkatti, P. AhR Activation Leads to Alterations in the Gut Microbiome with Consequent Effect on Induction of Myeloid Derived Suppressor Cells in a CXCR2-Dependent Manner. Int. J. Mol. Sci. 2020, 18, 9613. [Google Scholar] [CrossRef] [PubMed]
- Stedtfeld, R.D.; Brett Sallach, J.; Crawford, R.B.; Stedtfeld, T.M.; Williams, M.R.; Waseem, H.; Johnston, C.T.; Li, H.; Teppen, B.J.; Kaminski, N.E.; et al. TCDD Administered on Activated Carbon Eliminates Bioavailability and Subsequent Shifts to a Key Murine Gut Commensal. Appl. Microbiol. Biotechnol. 2017, 101, 7409–7415. [Google Scholar] [CrossRef]
- Shao, J.-W.; Ge, T.-T.; Chen, S.-Z.; Wang, G.; Yang, Q.; Huang, C.-H.; Xu, L.-C.; Chen, Z. Role of Bile Acids in Liver Diseases Mediated by the Gut Microbiome. World J. Gastroenterol. 2021, 27, 3010–3021. [Google Scholar] [CrossRef] [PubMed]
- Marion, S.; Desharnais, L.; Studer, N.; Dong, Y.; Notter, M.D.; Poudel, S.; Menin, L.; Janowczyk, A.; Hettich, R.L.; Hapfelmeier, S.; et al. Biogeography of Microbial Bile Acid Transformations along the Murine Gut. J. Lipid Res. 2020, 61, 1450–1463. [Google Scholar] [CrossRef] [PubMed]
- Foley, M.H.; O’Flaherty, S.; Allen, G.; Rivera, A.J.; Stewart, A.K.; Barrangou, R.; Theriot, C.M. Lactobacillus Bile Salt Hydrolase Substrate Specificity Governs Bacterial Fitness and Host Colonization. Proc. Natl. Acad. Sci. USA 2021, 118, e2017709118. [Google Scholar] [CrossRef]
- Matsubara, T.; Li, F.; Gonzalez, F.J. FXR Signaling in the Enterohepatic System. Mol. Cell. Endocrinol. 2013, 368, 17–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, H.; Kolodziejczyk, A.A.; Halstuch, D.; Elinav, E. Bile Acids in Glucose Metabolism in Health and Disease. J. Exp. Med. 2018, 215, 383–396. [Google Scholar] [CrossRef]
- Pols, T.W.H.; Noriega, L.G.; Nomura, M.; Auwerx, J.; Schoonjans, K. The Bile Acid Membrane Receptor TGR5: A Valuable Metabolic Target. Dig. Dis 2011, 29, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Fiorucci, S.; Mencarelli, A.; Palladino, G.; Cipriani, S. Bile-Acid-Activated Receptors: Targeting TGR5 and Farnesoid-X-Receptor in Lipid and Glucose Disorders. Trends Pharmacol. Sci. 2009, 30, 570–580. [Google Scholar] [CrossRef]
- O’Flaherty, S.; Briner Crawley, A.; Theriot, C.M.; Barrangou, R. The Lactobacillus Bile Salt Hydrolase Repertoire Reveals Niche-Specific Adaptation. mSphere 2018, 3, e00140-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, L.; Ling, Z.; Chen, D.; Liu, Y.; Yang, F.; Li, L. Disorganized Gut Microbiome Contributed to Liver Cirrhosis Progression: A Meta-Omics-Based Study. Front. Microbiol. 2018, 9, 3166. [Google Scholar] [CrossRef] [Green Version]
- Fazili, T.; Riddell, S.; Kiska, D.; Endy, T.; Giurgea, L.; Sharngoe, C.; Javaid, W. Streptococcus Anginosus Group Bacterial Infections. Am. J. Med. Sci. 2017, 354, 257–261. [Google Scholar] [CrossRef]
- Workman, S.D.; Strynadka, N.C.J. A Slippery Scaffold: Synthesis and Recycling of the Bacterial Cell Wall Carrier Lipid. J. Mol. Biol. 2020, 432, 4964–4982. [Google Scholar] [CrossRef]
- Johnston, J.M.; Bulloch, E.M. Advances in Menaquinone Biosynthesis: Sublocalisation and Allosteric Regulation. Curr. Opin. Struct. Biol. 2020, 65, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Hernández, S.B.; Cava, F.; Pucciarelli, M.G.; García-del Portillo, F.; de Pedro, M.A.; Casadesús, J. Bile-Induced Peptidoglycan Remodelling in Salmonella enterica: Bile-Induced Peptidoglycan Remodelling. Environ. Microbiol. 2015, 17, 1081–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravcheev, D.A.; Thiele, I. Genomic Analysis of the Human Gut Microbiome Suggests Novel Enzymes Involved in Quinone Biosynthesis. Front. Microbiol. 2016, 7, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut Microbiota and Human NAFLD: Disentangling Microbial Signatures from Metabolic Disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Sun, J.; Xia, S.; Li, L.; Li, Y.; Wang, P.; Shi, Y.; Le, G. Effects of Different Lactobacillus Reuteri on Inflammatory and Fat Storage in High-Fat Diet-Induced Obesity Mice Model. J. Funct. Foods 2015, 14, 424–434. [Google Scholar] [CrossRef]
- Ting, W.-J.; Kuo, W.-W.; Hsieh, D.; Yeh, Y.-L.; Day, C.-H.; Chen, Y.-H.; Chen, R.-J.; Padma, V.; Chen, Y.-H.; Huang, C.-Y. Heat Killed Lactobacillus Reuteri GMNL-263 Reduces Fibrosis Effects on the Liver and Heart in High Fat Diet-Hamsters via TGF-β Suppression. Int. J. Mol. Sci. 2015, 16, 25881–25896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreasen, A.S.; Larsen, N.; Pedersen-Skovsgaard, T.; Berg, R.M.G.; Møller, K.; Svendsen, K.D.; Jakobsen, M.; Pedersen, B.K. Effects of Lactobacillus acidophilus NCFM on Insulin Sensitivity and the Systemic Inflammatory Response in Human Subjects. Br. J. Nutr. 2010, 104, 1831–1838. [Google Scholar] [CrossRef] [Green Version]
- Khare, A.; Gaur, S. Cholesterol-Lowering Effects of Lactobacillus Species. Curr. Microbiol. 2020, 77, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Roh, Y.S.; Cho, A.; Cha, Y.-S.; Oh, S.-H.; Lim, C.W.; Kim, B. Lactobacillus Aggravate Bile Duct Ligation-Induced Liver Inflammation and Fibrosis in Mice. Toxicol. Res. 2018, 34, 241–247. [Google Scholar] [CrossRef]
- Martoni, C.J.; Labbé, A.; Ganopolsky, J.G.; Prakash, S.; Jones, M.L. Changes in Bile Acids, FGF-19 and Sterol Absorption in Response to Bile Salt Hydrolase Active L. reuteri NCIMB 30242. Gut Microbes 2015, 6, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Grover, S.; Batish, V.K. Hypocholesterolaemic Effect of Dietary Inclusion of Two Putative Probiotic Bile Salt Hydrolase-Producing Lactobacillus plantarum Strains in Sprague–Dawley Rats. Br. J. Nutr. 2011, 105, 561–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, N.Y.; Shin, M.J.; Youn, G.S.; Yoon, S.J.; Choi, Y.R.; Kim, H.S.; Gupta, H.; Han, S.H.; Kim, B.K.; Lee, D.Y.; et al. Lactobacillus Attenuates Progression of Nonalcoholic Fatty Liver Disease by Lowering Cholesterol and Steatosis. Clin. Mol. Hepatol. 2021, 27, 110–124. [Google Scholar] [CrossRef]
- Kemis, J.H.; Linke, V.; Barrett, K.L.; Boehm, F.J.; Traeger, L.L.; Keller, M.P.; Rabaglia, M.E.; Schueler, K.L.; Stapleton, D.S.; Gatti, D.M.; et al. Genetic Determinants of Gut Microbiota Composition and Bile Acid Profiles in Mice. PLoS Genet. 2019, 15, e1008073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, T.C.; Vuong, H.E.; Luna, C.D.G.; Pronovost, G.N.; Aleksandrova, A.A.; Riley, N.G.; Vavilina, A.; McGinn, J.; Rendon, T.; Forrest, L.R.; et al. Intestinal Serotonin and Fluoxetine Exposure Modulate Bacterial Colonization in the Gut. Nat. Microbiol. 2019, 4, 2064–2073. [Google Scholar] [CrossRef]
- Tannock, G.W.; Dashkevicz, M.P.; Feighner, S.D. Lactobacilli and Bile Salt Hydrolase in the Murine Intestinal Tract. Appl. Environ. Microbiol. 1989, 55, 1848–1851. [Google Scholar] [CrossRef] [Green Version]
- Winston, J.A.; Rivera, A.; Cai, J.; Patterson, A.D.; Theriot, C.M. Secondary Bile Acid Ursodeoxycholic Acid Alters Weight, the Gut Microbiota, and the Bile Acid Pool in Conventional Mice. PLoS ONE 2021, 16, e0246161. [Google Scholar] [CrossRef]
- Chae, J.P.; Valeriano, V.D.; Kim, G.-B.; Kang, D.-K. Molecular Cloning, Characterization and Comparison of Bile Salt Hydrolases from Lactobacillus johnsonii PF01. J. Appl. Microbiol. 2013, 114, 121–133. [Google Scholar] [CrossRef]
- De Smet, I.; Van Hoorde, L.; Vande Woestyne, M.; Christiaens, H.; Verstraete, W. Significance of Bile Salt Hydrolytic Activities of Lactobacilli. J. Appl. Bacteriol. 1995, 79, 292–301. [Google Scholar] [CrossRef]
- Bustos, A.Y.; Saavedra, L.; de Valdez, G.F.; Raya, R.R.; Taranto, M.P. Relationship between Bile Salt Hydrolase Activity, Changes in the Internal PH and Tolerance to Bile Acids in Lactic Acid Bacteria. Biotechnol. Lett. 2012, 34, 1511–1518. [Google Scholar] [CrossRef]
- Sato, H.; Macchiarulo, A.; Thomas, C.; Gioiello, A.; Une, M.; Hofmann, A.F.; Saladin, R.; Schoonjans, K.; Pellicciari, R.; Auwerx, J. Novel Potent and Selective Bile Acid Derivatives as TGR5 Agonists: Biological Screening, Structure−Activity Relationships, and Molecular Modeling Studies. J. Med. Chem. 2008, 51, 1831–1841. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Vranová, E.; Coman, D.; Gruissem, W. Network Analysis of the MVA and MEP Pathways for Isoprenoid Synthesis. Annu. Rev. Plant Biol. 2013, 64, 665–700. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, V.; Rezzi, S.; Eggersdorfer, M.; Galli, F. Metabolic and Functional Interplay between Gut Microbiota and Fat-Soluble Vitamins. Crit. Rev. Food Sci. Nutr. 2020, 61, 3211–3232. [Google Scholar] [CrossRef] [PubMed]
- Brooijmans, R.; Smit, B.; Santos, F.; van Riel, J.; de Vos, W.M.; Hugenholtz, J. Heme and Menaquinone Induced Electron Transport in Lactic Acid Bacteria. Microb Cell Fact. 2009, 8, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianniello, R.G.; Zheng, J.; Zotta, T.; Ricciardi, A.; Gänzle, M.G. Biochemical Analysis of Respiratory Metabolism in the Heterofermentative Lactobacillus spicheri and Lactobacillus reuteri. J. Appl. Microbiol. 2015, 119, 763–775. [Google Scholar] [CrossRef]
- Plichta, D.R.; Somani, J.; Pichaud, M.; Wallace, Z.S.; Fernandes, A.D.; Perugino, C.A.; Lähdesmäki, H.; Stone, J.H.; Vlamakis, H.; Chung, D.C.; et al. Congruent Microbiome Signatures in Fibrosis-Prone Autoimmune Diseases: IgG4-Related Disease and Systemic Sclerosis. Genome Med. 2021, 13, 35. [Google Scholar] [CrossRef]
- Quinn, R.A.; Whiteson, K.; Lim, Y.W.; Zhao, J.; Conrad, D.; LiPuma, J.J.; Rohwer, F.; Widder, S. Ecological Networking of Cystic Fibrosis Lung Infections. NPJ Biofilms Microbiomes 2016, 2, 4. [Google Scholar] [CrossRef]
- Chu, N.-N.; Chen, W.-L.; Xu, H.-R.; Li, X.-N. Pharmacokinetics and Safety of Ezetimibe/Simvastatin Combination Tablet: An Open-Label, Single-Dose Study in Healthy Chinese Subjects. Clin. Drug Investig. 2012, 32, 791–798. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Ahmadi, Z. Effects of Statins on Gut Microbiota (Microbiome). Rev. Clin. Med. 2019, 6, 5. [Google Scholar]
- Whitaker, E.; Alshammari, A. Bacteriostatic Effect of Simvastatin on Selected Oral Streptococci in Vitro. Contemp. Clin. Dent. 2017, 8, 59. [Google Scholar] [CrossRef]
- Wu, S.; Jiang, P.; Zhao, X.-M.; Chen, W.-H. Treatment Regimens May Compromise Gut-Microbiome-Derived Signatures for Liver Cirrhosis. Cell Metab. 2021, 33, 455–456. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-Invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef]
- McCoy, K.D.; Ohland, C.L. Innate Responses to Gut Microbiota; Critical Assessment of the Necessary Experimental Controls. Curr. Opin. Microbiol. 2021, 59, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Zeevi, D.; Levy, M.; Zilberman-Schapira, G.; Suez, J.; Tengeler, A.C.; Abramson, L.; Katz, M.N.; Korem, T.; Zmora, N.; et al. Transkingdom Control of Microbiota Diurnal Oscillations Promotes Metabolic Homeostasis. Cell 2014, 159, 514–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fader, K.A.; Nault, R.; Doskey, C.M.; Fling, R.R.; Zacharewski, T.R. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Abolishes Circadian Regulation of Hepatic Metabolic Activity in Mice. Sci. Rep. 2019, 9, 6514. [Google Scholar] [CrossRef] [PubMed]
- Nault, R.; Fader, K.A.; Harkema, J.R.; Zacharewski, T. Loss of Liver-Specific and Sexually Dimorphic Gene Expression by Aryl Hydrocarbon Receptor Activation in C57BL/6 Mice. PLoS ONE 2017, 12, e0184842. [Google Scholar] [CrossRef]
- Fling, R.R.; Doskey, C.M.; Fader, K.A.; Nault, R.; Zacharewski, T.R. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) Dysregulates Hepatic One Carbon Metabolism during the Progression of Steatosis to Steatohepatitis with Fibrosis in Mice. Sci. Rep. 2020, 10, 14831. [Google Scholar] [CrossRef]
- Lee, G.; You, H.J.; Bajaj, J.S.; Joo, S.K.; Yu, J.; Park, S.; Kang, H.; Park, J.H.; Kim, J.H.; Lee, D.H.; et al. Distinct Signatures of Gut Microbiome and Metabolites Associated with Significant Fibrosis in Non-Obese NAFLD. Nat. Commun. 2020, 11, 4982. [Google Scholar] [CrossRef]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J.; et al. Suppressed Hepatic Bile Acid Signalling despite Elevated Production of Primary and Secondary Bile Acids in NAFLD. Gut 2018, 67, 1881–1891. [Google Scholar] [CrossRef]
- Aranha, M.M.; Cortez-Pinto, H.; Costa, A.; da Silva, I.B.M.; Camilo, M.E.; de Moura, M.C.; Rodrigues, C.M.P. Bile Acid Levels Are Increased in the Liver of Patients with Steatohepatitis. Eur. J. Gastroenterol. Hepatol. 2008, 20, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Kakiyama, G.; Pandak, W.M.; Gillevet, P.M.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; Takei, H.; Muto, A.; Nittono, H.; Ridlon, J.M.; et al. Modulation of the Fecal Bile Acid Profile by Gut Microbiota in Cirrhosis. J. Hepatol. 2013, 58, 949–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, N.; Loomba, R.; Yang, Z.-H.; Wu, D.; Fang, S.; Bettencourt, R.; Lan, P.; Zhu, R.; Zhu, L. Alterations in Bile Acid Metabolizing Gut Microbiota and Specific Bile Acid Genes as a Precision Medicine to Subclassify NAFLD. Physiol. Genom. 2020, 53, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Plant, L.; Conway, P. Association of Lactobacillus spp. with Peyer’s Patches in Mice. Clin. Diagn Lab. Immunol. 2001, 8, 320–324. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Li, Y.; Xiao, H.; Shi, Y.; Le, G.; Sun, J. Isolation of Lactobacillus reuteri from Peyer’s Patches and Their Effects on SIgA Production and Gut Microbiota Diversity. Mol. Nutr. Food Res. 2016, 60, 2020–2030. [Google Scholar] [CrossRef] [PubMed]
- Fader, K.A.; Nault, R.; Raehtz, S.; McCabe, L.R.; Zacharewski, T.R. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Dose-Dependently Increases Bone Mass and Decreases Marrow Adiposity in Juvenile Mice. Toxicol. Appl. Pharmacol. 2018, 348, 85–98. [Google Scholar] [CrossRef]
- Collins, F.L.; Irwin, R.; Bierhalter, H.; Schepper, J.; Britton, R.A.; Parameswaran, N.; McCabe, L.R. Lactobacillus Reuteri 6475 Increases Bone Density in Intact Females Only under an Inflammatory Setting. PLoS ONE 2016, 11, e0153180. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable Association Discovery in Population-Scale Meta-Omics Studies. Biorxiv 2021. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzosa, E.A.; Morgan, X.C.; Segata, N.; Waldron, L.; Reyes, J.; Earl, A.M.; Giannoukos, G.; Boylan, M.R.; Ciulla, D.; Gevers, D.; et al. Relating the Metatranscriptome and Metagenome of the Human Gut. Proc. Natl. Acad. Sci. USA 2014, 111, E2329–E2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Fish, J.A.; Gilman, M.; Sun, Y.; Brown, C.T.; Tiedje, J.M.; Cole, J.R. Xander: Employing a Novel Method for Efficient Gene-Targeted Metagenomic Assembly. Microbiome 2015, 3, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, J.A.; Chai, B.; Wang, Q.; Sun, Y.; Brown, C.T.; Tiedje, J.M.; Cole, J.R. FunGene: The Functional Gene Pipeline and Repository. Front. Microbiol. 2013, 4, 291. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fling, R.R.; Zacharewski, T.R. Aryl Hydrocarbon Receptor (AhR) Activation by 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) Dose-Dependently Shifts the Gut Microbiome Consistent with the Progression of Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2021, 22, 12431. https://doi.org/10.3390/ijms222212431
Fling RR, Zacharewski TR. Aryl Hydrocarbon Receptor (AhR) Activation by 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) Dose-Dependently Shifts the Gut Microbiome Consistent with the Progression of Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2021; 22(22):12431. https://doi.org/10.3390/ijms222212431
Chicago/Turabian StyleFling, Russell R., and Timothy R. Zacharewski. 2021. "Aryl Hydrocarbon Receptor (AhR) Activation by 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) Dose-Dependently Shifts the Gut Microbiome Consistent with the Progression of Non-Alcoholic Fatty Liver Disease" International Journal of Molecular Sciences 22, no. 22: 12431. https://doi.org/10.3390/ijms222212431